
INTRODUCTION
Breast cancer is the leading cause of cancer-related deaths among women worldwide [1]. Over 1.7 million women are diagnosed with breast-cancer annually [1]. Despite efforts at early detection, 30-40% of women are diagnosed with metastatic cancer and die from therapy-resistant disease [2]. Over 70% of breast cancers are estrogen receptor positive (ER+), with well-known estrogen-driven malignant transformation and therapy resistance [3–5]. Anterior Gradient 2 (AGR2) is a protein disulfide isomerase in the endoplasmic reticulum first discovered in ER+ breast cancer cells [6–8]. AGR2 inhibits the tumor suppressor p53, promotes cell survival and proliferation, and mediates metastatic spread in breast cancer cells [9–12]. AGR2 expression is associated with decreased survival among women with ER+ breast cancer as well as tamoxifen and fulvestrant resistance [12–14].
A substantial body of recent experimental research has shown that estrogen-mediated activation of estrogen receptor directly targets AGR2 for active gene transcription. To begin with, AGR2 and ESR1 protein expression has been shown by immunohistochemistry to be positively correlated in both ER+ breast cancer cell lines and ER+ breast tumors [8, 11, 15, 16]. AGR2 knockdown by siRNA, shRNA, and miRNA in ER+ breast cancer lines reduces growth, survival, and migration, as well as fulvestrant and tamoxifen resistance [12, 17–19]. Estradiol treatment of ER+ breast cancer cell lines stimulates both AGR2 expression and a twofold increase in ESR1 binding to the AGR2 promoter region as detected by chromatin immunoprecipitation (ChIP) [12, 20]. ESR1 was shown to increase AGR2 expression from a transiently transfected AGR2 promoter reporter plasmid [12]. ChIP-Seq and chromatin interaction analysis by paired-end tag sequencing (ChIA-Pet) studies defining global ER-binding sites further support the targeted binding and transcriptional activation of AGR2 by ER in cell lines and primary tumor tissue, and an increased number of occupied ER-binding sites may correlate with poor prognosis [14, 21–23]. Altogether, these studies provide strong experimental evidence that estrogen-mediated activation of ESR1 directly upregulates AGR2 gene transcription.
Two studies have reported an association of increased protein-level AGR2 expression and decreased breast cancer survival using tumor immunohistochemistry. One analysis is adjusted for some clinical variables, and the other is not confounder-adjusted [12, 24]. A third retrospective study of AGR2 as a predictor of disease-free survival reports a significant association of decreased survival and increased tumor AGR2 mRNA expression using qRT-PCR among 78 women with tamoxifen-treated ER+ breast cancer but without adjustment for potential confounders [16].
Now, large breast cancer cohorts with clinical follow-up and multiplatform –omics primary tumor data have been reported. These studies expand our understanding of molecular subtypes of breast cancer as well as genetic prognostic factors, such as specific tumor mutations, in breast cancer [25–27]. These large -omics datasets utilize the gold standard design, the prospective observational cohort study, for the discovery and validation of disease prognostic factors. Therefore, they allow us to confirm and further explore previously experimentally demonstrated gene associations with actual patient data, not only with breast tumor gene expression but also with investigation of other clinical variables such as stage, tumor size, and lymph node metastasis. Here, primary tumor mRNA data from women in the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) breast cancer cohort were analyzed for AGR2 expression and disease-specific survival. Differentially expressed genes and their cellular pathways between women with tumors having either high and low AGR2 expression were also explored.
RESULTS
Validation of the AGR2 survival association
Clinical and primary tumor mRNA expression data from 2,000 fresh-frozen breast cancer specimens from the METABRIC study were analyzed for prognostic gene associations. Cohort selection for analysis excluded tumor specimens with benign or rare histological types or missing clinical data; Table 1 shows baseline characteristics of the cohort. Single-gene and multigene survival analyses identified AGR2 and Estrogen Receptor 1 (ESR1) as significantly associated with DSS in highly significant Cox proportional-hazards regression models (Table 2). Each one standard deviation increase in relative expression of AGR2 was associated with 14% increased hazard of death from disease (HR 1.14 (1.01-1.29), P = 0.03). Conversely, each one standard deviation increase in relative expression of ESR1 was associated with 18% decreased hazard of death from disease (HR 0.82 (0.69-0.99), P = 0.04). Increased AGR2 mRNA expression was correlated with increased ESR1 mRNA expression (Spearman’s rho = 0.547, P < 0.001).
Table 1: Patient characteristics of METABRIC sample for Disease-specific survival multivariate model
Patient characteristic |
Patient sub-characteristic |
n = 1341 |
|---|---|---|
Disease-specific survival (months) |
117.60 [62.33, 188.73] |
|
Age at diagnosis (years) |
61.12 [50.92, 69.83] |
|
Tumor stage (%) |
1 |
455 (33.9) |
2 |
769 (57.3) |
|
3 |
110 (8.2) |
|
4 |
7 (0.5) |
|
Lymph Node Positivity Status (%) |
Negative |
714 (53.2) |
Positive |
627 (46.8) |
|
Tumor size (cm) |
2.20 [1.70, 3.00] |
|
ER Positivity Status (%) |
Negative |
298 (22.2) |
Positive |
1043 (77.8) |
|
HER2 Positivity Status (%) |
Negative |
1176 (87.7) |
Positive |
165 (12.3) |
|
Breast surgery (%) |
Breast-conserving |
574 (42.8) |
Mastectomy |
767 (57.2) |
Survival model n = 1341. Results for Disease-specific survival, Age at diagnosis, and Tumor size reported as “median [interquartile range].” All others reported as “absolute value (percentage).”
Table 2: Hazards of death from single and multivariate Cox regression of disease-specific survival
Affymetrix U133A microarray mRNA expression dataset |
HR (95% CI), P |
|---|---|
DSS (n = 1341) |
|
Multivariate model |
|
AGR2 |
1.14 (1.01-1.29), 0.03 |
ESR1 |
0.82 (0.69-0.99), 0.04 |
Ageper year |
1.01 (1.00-1.02), 0.004 |
Mastectomy, compared to breast-conserving surgery |
1.31 (1.07-1.61), 0.01 |
Lymph node metastasis |
1.73 (1.34-2.24), <0.001 |
Tumor Sizeper cm |
1.12 (1.06-1.19), <0.001 |
GATA3 mutation |
0.67 (0.47-0.96), <0.03 |
TP53 mutation |
1.55 (1.23-1.95), <0.001 |
Stratification variables |
ER IHC status, HER2 expression status, stage |
Model P |
< 1.3 x 10-14 |
HR (95% CI), P: Hazard ratio (95% confidence interval) and p-value, scaled to one standard deviation of gene expression for AGR2 and ESR1; DSS: Disease-specific survival.
Clinical and molecular covariates were included in the single and multigene Cox proportional-hazards models to adjust for potential confounders of DSS. Clinical covariates significantly associated with DSS were age, breast surgery (breast-conserving vs. mastectomy), presence of positive lymph nodes, and tumor size. Molecular covariates significantly associated with DSS were AGR2 and ESR1 mRNA expression levels, and GATA3 and TP53 mutation statuses, both tumor mutations with known survival associations (Table 2). Table 3 shows predicted five-year survival differences comparing women with high (z > 1.5) versus low (z < -1.5) AGR2 tumor mRNA expression while modeling various stages and lymph node status combinations from the Cox regression. Figure 1 shows characteristic, predicted disease-specific survival curves.
Table 3: Disease-specific survival (DSS) rates of ER+/HER2- mastectomy patients with high or low primary breast tumor AGR2 mRNA microarray expression (METABRIC)
Tumor Stage |
Tumor Size (cm) |
Lymph Node Positivity Status |
AGR2 mRNA expression |
DSS at 60 mo (%) |
Difference in DSS at 60 mo between high and low AGR2 (%) |
DSS at 120 mo (%) |
Difference in DSS at 60 mo between high and low AGR2 (%) |
|---|---|---|---|---|---|---|---|
I |
1.7 |
Negative |
High |
91.63% |
-3.99% |
77.66% |
-10.19% |
Low |
95.62% |
87.85% |
|||||
1.7 |
Positive |
High |
85.94% |
-6.59% |
64.50% |
-15.37% |
|
Low |
92.53% |
79.87% |
|||||
II |
2.2 |
Negative |
High |
89.15% |
-5.13% |
75.86% |
-10.94% |
Low |
94.28% |
86.80% |
|||||
2.5 |
Negative |
High |
88.76% |
-5.31% |
75.08% |
-11.26% |
|
Low |
94.07% |
86.34% |
|||||
2.5 |
Positive |
High |
81.32% |
-8.62% |
60.83% |
-16.68% |
|
Low |
89.95% |
77.51% |
|||||
III |
4.5 |
Positive |
High |
72.75% |
-12.20% |
52.94% |
-19.24% |
Low |
84.96% |
72.18% |
|||||
IV |
3.1 |
Negative |
High |
24.80% |
-24.14% |
24.80% |
-24.14% |
Low |
48.94% |
48.94% |
|||||
3.1 |
Positive |
High |
8.91% |
-20.04% |
8.91% |
-20.04% |
|
Low |
28.95% |
28.95% |
Disease-specific survival rates of ER+/HER2- mastectomy patients were calculated with ESR1 mRNA microarray expression set to median 10.7, and age at diagnosis set to median 61 years. AGR2 mRNA expression levels “High” are those for which z-score > 1.5, and “Low” are those for which z-score < -1.5.
Tumor size for each respective stage represents the median tumor size for patients with that stage. The DSS rates calculated for Stage II and tumor size = 2.2 cm represent that with the most common stage and the median tumor size across all stages.
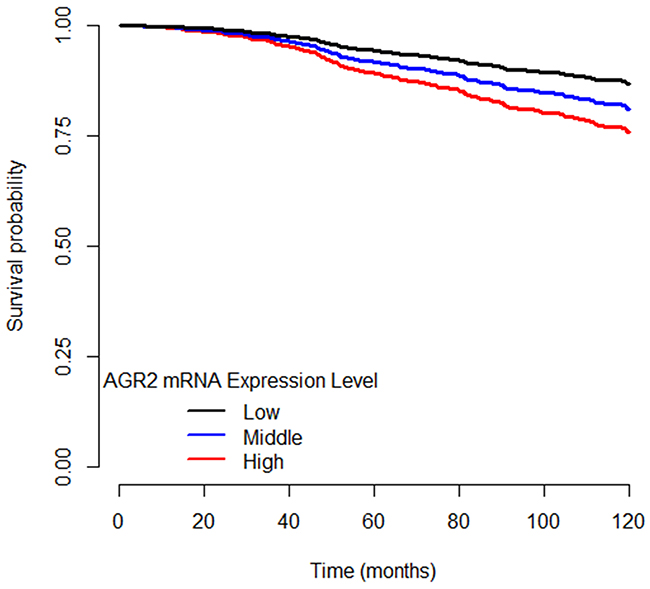
Figure 1: Disease-specific survival curves of ER+ breast cancer patients separated by AGR2 mRNA expression level. High AGR2 mRNA expression level is designated as z ≥ 1.5. Low AGR2 mRNA expression level is designated as z ≤ -1.5. Middle AGR2 mRNA expression level is designated as -1.5 < z < 1.5.
Identification of DEGs and major molecular pathways by AGR2 prognostic groups
Relative hazard of disease-specific death by AGR2 expression suggested z-score values of ± 1.5 as cut-point values to define AGR2 prognostic groups as high and low AGR2 expression breast tumor groups. The Benjamini-Hochberg (BH) q-value is a widely accepted, stringent adjustment of the standard p-value to decrease the false discovery rate (FDR) in the circumstance of multiple comparisons [28, 29], and we have used multiple BH q-value cutoffs to most effectively minimize the false discovery rate. Differentially expressed genes (DEGs) between these high (mRNA expression z-score levels of AGR2 > 1.5) and low (z-score < -1.5) AGR2 expression tumor groups from the METABRIC study were identified using the cutoff of BH q-value < 0.05. To verify our results from the METABRIC study, we also identified DEGs from RNA sequencing data from tumor mRNA expression from the TCGA study of human breast tumors with BH q-value < 0.05. Gene enrichment studies provided lists of more than 5,000 DEGs from each cohort, METABRIC and TCGA. We performed modular enrichment analysis of the 3,000 most statistically significant DEGs in each cohort; significantly enriched KEGG pathways are shown in Table 4. We used a second cutoff, the log2 of gene expression fold-change of 1.5 to generate a subset of the DEGs that are most differentially expressed from each cohort (resulting in 790 genes from METABRIC, and 1143 genes from TCGA) for further pathway analyses by Ingenuity Pathway Analysis (IPA). IPA of the DEGs from each cohort yielded significantly overrepresented pathways and functional networks between high and low AGR2 groups (Table 5), with cutoffs for pathway significance set to BH q-values < 0.050 and <0.200 (Supplementary Table 1). Although, IPA does not recommend a strict cutoff, q-value < 0.250 is an accepted standard for gene set analysis tools like DAVID and gene set enrichment analysis [30, 31].
Table 4: KEGG pathways over-represented in the high AGR2 group (z > 1.5) vs. the low AGR2 group (z < -15)
Breast Cancer Study |
Enriched KEGG pathways |
Benjamini-Hochberg q-value, regulation changea |
|---|---|---|
METABRIC |
Pathogenic Escherichia coli infection |
0.01, down |
Cell cycle |
0.01, down |
|
HTLV-I infection |
0.01, down |
|
TNF signaling pathway |
0.03, down |
|
NF-kappa B signaling pathway |
0.03, down |
|
TCGA |
Cell cycle |
0.05, down |
Regulation change: Listed as up or down if ≥ 2/3rd genes in cluster share the same up or down directional expression change.
Table 5: Top IPA pathways over-represented in the high AGR2 group (z > 1.5) vs. low AGR2 group (z < -15)
Breast Cancer Study |
Enriched IPA canonical pathways |
Benjamini Hochberg q-valuea, directional regulation changeb |
|---|---|---|
METABRIC |
Estrogen-mediated S-phase Entry |
0.005, down |
Cyclins and Cell Cycle Regulation |
0.005, mixed |
|
Glioblastoma Multiforme Signaling |
0.010, down |
|
Cell Cycle: G1/S Checkpoint Regulation |
0.010, down |
|
Glioma Signaling |
0.012, down |
|
TCGA |
Wnt/Beta-catenin Signaling |
0.079, mixed |
Estrogen-mediated S-phase Entry |
0.199, down |
|
Antiproliferative Role of TOB in T Cell Signaling |
0.199, down |
|
Cyclins and Cell Cycle Regulation |
0.199, mixed |
|
Anandamide Degradation |
0.199, up |
aCutoffs for pathway significance were set to BH q-values < 0.050 and < 0.200. The full list of significant pathways for METABRIC and TCGA can be found in Supplementary Table 1.
bRegulation change: Listed as up or down if ≥ 2/3rd genes in cluster share the same up or down directional expression change, otherwise listed as mixed if general directional change is unclear.
We also performed permutation analysis to verify that these IPA pathways were dependent on the specific list of genes and not enriched by chance. Supplementary Table 2 shows the number of pathways enriched to q-value < 0.050 and < 0.200, and Supplementary Table 3 shows the number of times that the original significantly enriched METABRIC IPA pathways (Table 5) were significantly enriched in IPA with random permutation of gene names to fixed columns of gene expression fold change. Importantly, the majority of the top IPA pathways originally significantly enriched in METABRIC and TCGA were not found to be significantly enriched in permutation analyses (Supplementary Table 3, Supplementary Table 4). Specifically, IPA pathways with direct roles in breast cancer pathophysiology such as Estrogen-mediated S-phase Entry, Cyclins and Cell Cycle Regulation, and Cell Cycle: G1/S Checkpoint Regulation were not significantly enriched in permutation analyses. Our results strongly support that the METABRIC and TCGA significantly enriched IPA pathways were not enriched by chance and were dependent on the list of differentially expressed genes between high and low AGR2 expression groups.
DISCUSSION
We validated that AGR2 and ESR1 mRNA expression levels are significantly associated with DSS (AGR2: HR 1.14 (1.01-1.29), P = 0.03; ESR1: HR 0.82 (0.69-0.99), P = 0.04), and that AGR2 and ESR1 mRNA expression were correlated. We then determined differentially expressed genes between tumors grouped by high or low AGR2 mRNA expression and retrieved significantly enriched KEGG and IPA pathways, which represented those involved in cell cycle progression, inflammation, and immune response.
AGR2 is an endoplasmic reticulum-resident protein disulfide isomerase with increased expression levels that are implicated in several cancers, including breast, ovarian, prostate, bladder, and pancreatic cancer [8, 11, 14–16, 32–37]. It is hypothesized that increasing AGR2 within the endoplasmic reticulum allows cancer cells to adapt to higher secretory protein synthesis demands during tumorigenesis and metastasis [6]. For example, AGR2 is overexpressed and secreted by both bladder and breast cancer cells [36, 38]. Intracellular versus secreted AGR2 had different roles in enhancing breast cancer fulvestrant resistance [18]. These studies provided mechanistic biological plausibility to a meaningful role of AGR2 in metastasis and hormone therapy resistance.
We validated the negative correlation between AGR2 primary tumor mRNA expression levels and disease-specific survival using data from a larger cohort of women with available information for confounder-adjustment by a variety of potential clinical and molecular confounders. AGR2 expression was inversely associated with DSS, while ESR1 expression was directly associated with DSS. Since ESR1 signaling increases AGR2 expression but estrogen receptor expression itself is associated with increased survival, it may be difficult to observe the independent negative prognostic association of AGR2 with decreased survival. Furthermore, because the expression of these two genes is correlated, and collinearity tends to decrease observed statistical significance in regression models, this may explain why the observed p-values are not lower even in this relatively large cohort. Predictably, increased ESR1 expression was associated with increased DSS, consistent with ER+ breast cancers being more differentiated, less aggressive, and often responsive to hormone therapy [39, 40]. The positive Spearman’s correlation between AGR2 and ESR1 agreed with prior literature showing that AGR2 expression is directly stimulated by estrogen signaling [14]. The opposing associations of AGR2 and ESR1 expression with DSS could reflect the role of AGR2 in hormone therapy- resistant ER+ breast cancer: ER+ breast cancers may develop resistance by overexpressing AGR2 [12–14]. Indeed, the downregulation of the “Estrogen-mediated S phase entry” pathway in breast tumors with high AGR2 mRNA expression—suggested by IPA analyses in both the METABRIC and TCGA datasets—may represent breast tumors that overexpress AGR2 to become resistant to estrogen therapy (Table 5). Furthermore, while ESR1 can initially upregulate AGR2 expression, breast cancer cells that achieve tamoxifen resistance no longer require ESR1 for AGR2 expression, likely due to an alteration in the activity of the transcription factor FOXA1 [19]. A combined strategy to block AGR2 expression or function in combination with anti-estrogenic hormonal therapy may be a novel strategy for the treatment of ER+ breast cancers.
mRNA expression fold changes in DEGs of breast tumors with high AGR2 compared to those with low AGR2 were consistent with prior findings highlighting the roles of intracellular and extracellular AGR2 in tumorigenesis pathways mediated by estrogen signaling and Insulin Growth Factor 1 (IGF-1) [38]. Indeed, IGF1 receptor (IGF1R) was significantly upregulated in tumors with high AGR2 mRNA expression compared to those with low AGR2 mRNA expression (METABRIC microarray IGF1R mRNA fold change = 3.03, TCGA RNA Seq IGF1R mRNA fold change = 3.13). Increased AGR2 mRNA expression was correlated with increased IGF1R mRNA expression (Spearman’s rho = 0.451, p < 0.001). Recent studies have shown that particular IGF1R polymorphisms significantly increase the risk of early tumor progression in tamoxifen-treated ER+ breast cancer, and that inhibition of IGF1R activity enhances response to trastuzumab therapy [41, 42]. Therefore, IGF1R-mediated signaling likely plays a role in AGR2-mediated trastuzumab and tamoxifen resistance in ER+ breast cancer tumors and is worthy of further investigation.
The association of increased AGR2 mRNA expression and decreased disease-specific survival may also be because higher expression of the endoplasmic reticulum-resident protein disulfide isomerase AGR2 allows for homeostatic adaptation to an increased demand on protein synthesis and secretion in oncogenesis. Tumor cells are challenged by hypoxia, nutrient deficiency, and increased proteomic demand. If the tumor cell cannot counteract these pathological conditions successfully, proper protein folding and endoplasmic reticulum homeostasis will be disturbed, ultimately causing endoplasmic reticulum stress. Endoplasmic reticulum stress activates the unfolded protein response (UPR), which is tailored to re-establish endoplasmic reticulum homeostasis by shutting down certain types of protein translation, upregulating endoplasmic reticulum folding machinery components, and boosting endoplasmic reticulum quality control mechanisms such as endoplasmic reticulum-associated degradation [43]. AGR2 expression is modulated by endoplasmic reticulum stress-induced UPR and in turn maintains endoplasmic reticulum homeostasis. Basal levels of intracellular AGR2 are controlled by IRE1-alpha and ATF6-alpha, two of the three key arms of the UPR [44]. In the gastrointestinal tract, AGR2 is critically required for Mucin 2 production, and in its absence, the intestinal epithelium exhibits increased endoplasmic reticulum stress markers such as Grp78 and Xbp1 splicing, as well as elevated pro-inflammatory cytokines [45–47]. Consequently, AGR2 knockout mice spontaneously develop severe ileocolitis that histopathologically resembles human Crohn’s disease [47]. Furthermore, endoplasmic reticulum stress induces AGR2 expression in inflammatory pre-neoplastic pancreatic tissue. By enhancing endoplasmic reticulum folding capacity, AGR2 allows pre-cancerous cells to accommodate increased protein demand both before and after oncogenic mutations such as that of Kras, ultimately leading to pancreatic cancer progression [48]. In normal breast development, AGR2 is maximally expressed during late pregnancy and lactation to accommodate the increased demand on protein synthesis and secretion during these specific physiological conditions [20]. It is therefore plausible that AGR2 is involved in the UPR to resolve endoplasmic reticulum stress, and that breast cancer cells can utilize the homeostatic effects of AGR2 to overcome their own pathologically elevated need for protein synthesis and secretion. When endoplasmic reticulum stress is too severe, the UPR turns from a pro-survival to a pro-death response [49]. Pro-death UPR signals, including those mediated by CHOP, GADD34, ERO1-alpha, and Bcl-2, tip the balance towards apoptosis as well as activate inflammatory signaling cascades [50–53]. It is possible that breast cancer cells that fail to upregulate AGR2 likewise fail to mount a successful pro-survival UPR that restores endoplasmic reticulum homeostasis, thus succumbing to pro-death UPR signals that promote apoptosis and inflammation. UPR-induced apoptosis includes apoptosis mediated by the transcription factor CHOP, which can promote the transcription of pro-apoptotic BH3-only proteins such as Noxa and Puma through p53-dependent mechanisms [54, 55]. Since AGR2 has been shown to inhibit p53 activation through a DUSP10-mediated pathway, it is likely that high AGR2 levels relieve endoplasmic reticulum stress and thus stave off pro-death UPR-mediated apoptosis [10].
Additionally, pro-death UPR signals activate classical inflammatory signaling cascades, including the production of pro-inflammatory molecules like IL-6, IL-8, and TNF-alpha, as well as acute-phase response neutrophilia and associated production of cytokines like IL-1beta and IL-2R [53, 56–58]. NF-kB carries out pro-death UPR-mediated inflammation, and it is a known activator of p53 [56, 59, 60]. The KEGG NF-kB signaling pathway is downregulated in breast tumors with high AGR2 expression compared to those with low AGR2 expression within the METABRIC microarray dataset (Table 4). Furthermore, this NF-kB pathway downregulation, and the fact that AGR2 can inhibit p53 activity, suggest that AGR2 prevents both pro-death UPR-mediated apoptosis and inflammation, ultimately promoting tumor cell survival and proliferation.
UPR-induced inflammation can be critical for anti-tumor immunity, since activated leukocytes can present tumor antigens to CD4+ T cells [61, 62]. Consequently, tumor-infiltrating lymphocytes have been shown to be favorable for breast cancer survival, and UPR activation is associated with tumor-infiltrating lymphocytes in breast cancer [63, 64]. In both the METABRIC and TCGA datasets, downregulation of genes such as CD4 and CD40 may indicate decreased CD4+ T cell response or decreased CD40-mediated apoptosis, both of which are correlated with tumor regression, better response to therapy, and higher overall breast cancer survival [65, 66]. Furthermore, breast tumors that overexpress AGR2 exhibit downregulation of KEGG pathways epitomized by the inflammatory response, including Pathogenic Escherichia Coli infection, HTLV-1 infection, and TNF pathway (Table 4). UPR activation in immune cells and various stromal cells can induce TNF-alpha secretion, and conversely TNF-alpha can trigger UPR activation in the liver to amplify the inflammatory response [53, 58, 67, 68]. The close interplay between the UPR and the TNF pathway, plus recent findings that show that TNF-alpha can prevent in vivo breast cancer development, suggest that downregulation of the TNF pathway in AGR2-overexpressing breast cancers may contribute to their hormone therapy resistance [69].
The downregulation of mRNA expression of cell cycle pathway genes in tumors with higher AGR2 expression may be due to preservation technique of the NCDB database tumor samples, a technique which may under-represent extracellular AGR2, whose function has been proven to be distinct from that of intracellular AGR2 protein. While extracellular AGR2 promotes cell cycle progression, intracellular AGR2 functionally interacts with ER [18]. Furthermore, AGR2 has been shown to promote cell cycle progression via induction of cell cycle proteins such as cyclin D1, and extracellular AGR2 knockdown using anti-AGR2 antibodies in three ER+ breast cancer cell lines significantly reduces cyclin D1 protein levels and cell growth [17]. Thus, downregulation of cell cycle pathway genes in high AGR2 mRNA expression tumors may reflect the differing roles of extracellular and intracellular AGR2 and the under-measurement of extracellular AGR2 in our analyses.
Limitations of our study include the inherent possibility of causal inference and the risks of selection biases and unmeasured confounding that are relevant to observational cohort studies, with consequent concerns for generalizability, although we used the largest breast cancer study with available –omics data, adequate clinical follow-up, and a disease-specific survival outcome. The TCGA breast cancer cohort was not useful for a validation of the AGR2 survival association due to inadequately short follow-up time of the cohort. With a large sample size of n = 1,341, multivariable adjustment for several known clinical and molecular confounders, and consistency of our results with prior literature, we believe that the survival association is unlikely to represent a false-positive signal. Given consistency with prior studies using a better powered and more rigorous analysis, we see these results as an independent cohort validation of the previously reported AGR2 survival association. Another limitation is that the overall dataset used has missing data for some cases. Finally, the discussion above highlights the significant body of laboratory research that has established the biological plausibility for AGR2 expression as a prognostic biomarker and potential therapeutic target in breast cancer, lending credence to our findings.
In conclusion, we validated the AGR2 survival association in breast cancer and explored various pathways through which AGR2 mediates poor response to treatment and thus decreased disease-specific survival. These pathways involve ESR1, IGF1R, and pro-death UPR-mediated apoptosis and inflammation with subsequent recruitment of tumor-infiltrating lymphocytes. As therapy inefficacy and subsequent metastases drive breast cancer mortality, further elucidation of both intracellular and extracellular AGR2 biology and its ability to confer hormone therapy resistance may lead to improved understanding of breast cancer mortality. Simultaneous suppression of AGR2 and ESR1 activity represents a potentially promising concept to be further investigated for breast cancer.
MATERIALS AND METHODS
Data source
We performed an observational retrospective cohort analysis of women with breast cancer with publicly available clinical and primary tumor mRNA expression and mutation data from the METABRIC study. Data was retrieved using the cBioPortal implementation in R, package CGDS-R [25, 26, 70]. The METABRIC study included over 2,000 fresh-frozen breast cancer specimens and a subset of normal breast tissue from tumor banks in the UK and Canada. The METABRIC study reported 2,136 primary tumors with expression array (Affymetrix U133A microarray) data and 2,433 primary tumors with somatic mutation testing by sequencing for 173 genes. We also analyzed RNA sequencing data for mRNA expression from 1,100 primary breast cancer tumors from The Cancer Genome Atlas (TCGA) to validate our results for differentially expressed genes from the METABRIC study [27].
Cohort selection
Women with publicly available clinical, mutation, and mRNA expression data were selected (n = 1,889) from the Curtis et. al. METABRIC cohort [26]. Women with intraductal or intralobular histological types were included; those with benign or rare histological types were excluded (new n = 1,854). Women who did not undergo surgery (breast-conserving or mastectomy) were excluded (new n = 1,835). Women without reported disease stage were also excluded (new n = 1,370). Survival analyses were limited to women with no missing data for any potential confounder that was included in Cox model (final n = 1,341).
Covariate selection and definitions
Initial covariates for survival analysis included age at diagnosis, breast surgery, cellularity, treatment with adjuvant chemotherapy, claudin subtype, ER IHC status, grade, HER2 expression status, HER2 copy number alterations, hormone therapy, menopause status, integrative cluster (METABRIC study molecular subtype), laterality, number of positive lymph nodes, Nottingham prognostic index, PR expression status, treatment with adjuvant radiotherapy, tumor size, and tumor stage. Additional specific molecular covariates included the somatic tumor mutation status of genes with known breast cancer survival associations among women in the METABRIC cohort: TP53, GATA3, MAP3K1, NF1, PIK3CA, SMAD4, and USP9X. Also included were the mRNA expression levels of genes that regulate AGR2 or are potentially similar to AGR2 including ESR1, ESR2, and AGR3 [6, 7, 10, 12, 14, 15, 24, 25, 71–75]. Molecular covariates utilized for final analyses on different subgroups within breast cancer tumors included ESR1, TP53 mutation, and GATA3 mutation.
Single and multiple-gene AGR2 prognostic biomarker evaluation
AGR2 was evaluated as a potential prognostic biomarker using single and multiple-gene multivariable Cox proportional-hazards models of disease-specific survival (DSS) as functions of continuous mRNA expression (n = 1,341). Clinical and molecular covariates were included to adjust for potential confounders of DSS. The proportional hazards assumption was checked and the regression model was stratified by estrogen receptor (ER) immunohistochemistry (IHC) status, HER2 expression status, and stage to maintain proportional hazards. Hazard ratios (HR) are reported per each one standard deviation increase in gene expression to aid between gene comparisons of relative effect estimates in terms of change in HR values per each standard deviation increase in mRNA expression. R and the “survival” and “rms” packages were used for statistical computing [76–78].
Identification of differential gene expression patterns and major molecular pathways by high and low AGR2 tumor expression groups
First, we used the final Cox model for DSS and modeled the restricted cubic splines of tumor AGR2 mRNA expression as a continuous variable to verify that the relationship of AGR2 mRNA expression with DSS is linear. Next, differentially expressed genes (DEGs) between high and low AGR2, with high (mRNA expression z-score levels of AGR2 > 1.5) vs. low (z-score < -1.5), were determined using Linear Models for Microarray with the limma package in R [79, 80]. We further verified our results for differentially expressed genes using RNA sequencing data for tumor mRNA expression from the TCGA study of human breast tumors. Significant differences were deemed those with a Benjamini-Hochberg procedure q-value < 0.05, to account for multiple comparisons. Gene enrichment studies provided lists of more than 5000 DEGs from each cohort, METABRIC and TCGA. Modular enrichment analyses of the most statistically significant (by q-value) 3,000 DEGs were performed using DAVID [30, 81]. Significantly enriched functional annotation clustering and KEGG pathways were determined and ranked to show overrepresented pathways coincident with higher AGR2 prognostic expression levels [82]. Ingenuity Pathway Analysis (IPA) (Version 01-07) was performed on 790 and 1143 genes, respectively, from the DEG lists obtained from the METABRIC and TCGA datasets, with an BH q-value < 0.001 and a log2 fold change cutoff of 1.5. Cutoff for IPA pathway significance was set to BH q-values < 0.050 and <0.200. Permutation analysis of IPA was performed to validate that IPA pathways were not enriched by chance. IPA was run with random permutation of gene names to fixed columns of gene expression fold change. Ensembl and GeneCards were used to gather background information about relevant gene functions and expression [83, 84].
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
FUNDING
This work was supported in part by the Phebe Novakovic Fund.
REFERENCES
1. Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends--an update. Cancer Epidemiol Biomarkers Prev. 2016; 25:16–27.
2. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005; 55:74–108.
3. Arpino G, Weiss H, Lee AV, Schiff R, De Placido S, Osborne CK, Elledge RM. Estrogen receptor-positive, progesterone receptor-negative breast cancer: association with growth factor receptor expression and tamoxifen resistance. J Natl Cancer Inst. 2005; 97:1254–61.
4. Jorns JM, Healy P, Zhao L. Review of estrogen receptor, progesterone receptor, and HER-2/neu immunohistochemistry impacts on treatment for a small subset of breast cancer patients transferring care to another institution. Arch Pathol Lab Med. 2013; 137:1660–1663.
5. Xu CY, Jiang ZN, Zhou Y, Li JJ, Huang LM. Estrogen receptor alpha roles in breast cancer chemoresistance. Asian Pac J Cancer Prev. 2013; 14:4049–4052.
6. Brychtova V, Mohtar A, Vojtesek B, Hupp TR. Mechanisms of anterior gradient-2 regulation and function in cancer. Semin Cancer Biol. 2015; 33:16–24.
7. Thompson DA, Weigel RJ. hAG-2, the human homologue of the Xenopus laevis cement gland gene XAG-2, is coexpressed with estrogen receptor in breast cancer cell lines. Biochem Biophys Res Commun. 1998; 251:111–116.
8. Kuang WW, Thompson DA, Hoch RV, Weigel RJ. Differential screening and suppression subtractive hybridization identified genes differentially expressed in an estrogen receptor-positive breast carcinoma cell line. Nucleic Acids Res. 1998; 26:1116–1123.
9. Vitello EA, Quek SI, Kincaid H, Fuchs T, Crichton DJ, Troisch P, Liu AY. Cancer-secreted AGR2 induces programmed cell death in normal cells. Oncotarget. 2016; 7:49425–34. https://doi.org/10.18632/oncotarget.9921.
10. Hrstka R, Bouchalova P, Michalova E, Matoulkova E, Muller P, Coates PJ, Vojtesek B. AGR2 oncoprotein inhibits p38 MAPK and p53 activation through a DUSP10-mediated regulatory pathway. Mol Oncol. 2016; 10:652–662.
11. Liu D, Rudland PS, Sibson DR, Platt-Higgins A, Barraclough R. Human homologue of cement gland protein, a novel metastasis inducer associated with breast carcinomas. Cancer Res. 2005; 65:3796–3805.
12. Hrstka R, Nenutil R, Fourtouna A, Maslon MM, Naughton C, Langdon S, Murray E, Larionov A, Petrakova K, Muller P, Dixon MJ, Hupp TR, Vojtesek B. The pro-metastatic protein anterior gradient-2 predicts poor prognosis in tamoxifen-treated breast cancers. Oncogene. 2010; 29:4838–4847.
13. Wang Z, Hao Y, Lowe AW. The adenocarcinoma-associated antigen, AGR2, promotes tumor growth, cell migration, and cellular transformation. Cancer Res. 2008; 68:492–497.
14. Salmans ML, Zhao F, Andersen B. The estrogen-regulated anterior gradient 2 (AGR2) protein in breast cancer: a potential drug target and biomarker. Breast Cancer Res. 2013; 15:204.
15. Fletcher GC, Patel S, Tyson K, Adam PJ, Schenker M, Loader JA, Daviet L, Legrain P, Parekh R, Harris AL, Terrett JA. hAG-2 and hAG-3, human homologues of genes involved in differentiation, are associated with oestrogen receptor-positive breast tumours and interact with metastasis gene C4.4a and dystroglycan. Br J Cancer. 2003; 88:579–585.
16. Barraclough DL, Platt-Higgins A, de Silva Rudland S, Barraclough R, Winstanley J, West CR, Rudland PS. The metastasis-associated anterior gradient 2 protein is correlated with poor survival of breast cancer patients. Am J Pathol. 2009; 175:1848–1857.
17. Vanderlaag KE, Hudak S, Bald L, Fayadat-Dilman L, Sathe M, Grein J, Janatpour MJ. Anterior gradient-2 plays a critical role in breast cancer cell growth and survival by modulating cyclin D1, estrogen receptor-alpha, and survivin. Breast Cancer Res. 2010; 12:R32.
18. Li Z, Zhu Q, Chen H, Hu L, Negi H, Zheng Y, Ahmed Y, Wu Z, Li D. Binding of anterior gradient 2 and estrogen receptor-alpha: dual critical roles in enhancing fulvestrant resistance and IGF-1-induced tumorigenesis of breast cancer. Cancer Lett. 2016; 377:32–43.
19. Wright TM, Wardell SE, Jasper JS, Stice JP, Safi R, Nelson ER, McDonnell DP. Delineation of a FOXA1/ERalpha/AGR2 regulatory loop that is dysregulated in endocrine therapy-resistant breast cancer. Mol Cancer Res. 2014; 12:1829–1839.
20. Verma S, Salmans ML, Geyfman M, Wang H, Yu Z, Lu Z, Zhao F, Lipkin SM, Andersen B. The estrogen-responsive AGR2 gene regulates mammary epithelial proliferation and facilitates lobuloalveolar development. Dev Biol. 2012; 369:249–260.
21. Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlove YL, Velkov S, Ho A, Mei PH, Chew EG, Huang PY, Welboren WJ, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009; 462:58–64.
22. Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, Ali S, Chin SF, Palmieri C, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012; 481:389–393.
23. Welboren WJ, van Driel MA, Janssen-Megens EM, van Heeringen SJ, Sweep FC, Span PN, Stunnenberg HG. ChIP-Seq of ERalpha and RNA polymerase II defines genes differentially responding to ligands. EMBO J. 2009; 28:1418–1428.
24. Innes HE, Liu D, Barraclough R, Davies MP, O’Neill PA, Platt-Higgins A, de Silva Rudland S, Sibson DR, Rudland PS. Significance of the metastasis-inducing protein AGR2 for outcome in hormonally treated breast cancer patients. Br J Cancer. 2006; 94:1057–1065.
25. Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russel R, Sammut SJ, Tsui DW, Liu B, Dawson SJ, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016; 7:11479.
26. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Graf S, Ha G, Haffari G, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012; 486:346–352.
27. The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70.
28. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995; 57:289–300.
29. Benjamini Y, Drai D, Elmer G, Kafkaki N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001; 125:279–284.
30. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44–57.
31. Irizarry RA, Wang C, Zhou Y, Speed TP. Gene set enrichment analysis made simple. Stat Methods Med Res. 2009; 18:565–575.
32. Lacambra MD, Tsang JY, Ni YB, Chan SK, Tan PH, Tse GM. Anterior gradient 2 is a poor outcome indicator in luminal breast cancer. Ann Surg Oncol. 2015; 22:3489–3496.
33. Edgell TA, Barraclough DL, Rajic A, Dhulia J, Lewis KJ, Armes JE, Barraclough R, Rudland PS, Rice GE, Autelitano DJ. Increased plasma concentrations of anterior gradient 2 protein are positively associated with ovarian cancer. Clin Sci (Lond). 2010; 118:717–725.
34. Darb-Esfahani S, Fritzsche F, Kristiansen G, Weichert W, Sehouli J, Braicu I, Dietel M, Denkert C. Anterior gradient protein 2 (AGR2) is an independent prognostic factor in ovarian high-grade serous carcinoma. Virchows Arch. 2012; 461:109–116.
35. Zhang JS, Gong A, Cheville JC, Smith DI, Young CY. AGR2, an androgen-inducible secretory protein overexpressed in prostate cancer. Genes Chromosomes Cancer. 2005; 43:249–259.
36. Ho ME, Quek SI, True LD, Seiler R, Fleischmann A, Bagryanova L, Kim SR, Chia D, Goodglick L, Shimizu Y, Rosser CJ, Gao Y, Liu AY. Bladder cancer cells secrete while normal bladder cells express but do not secrete AGR2. Oncotarget. 2016; 7:15747–56. https://doi.org/10.18632/oncotarget.7400.
37. Ramachandran V, Arumugam T, Wang H, Logsdon CD. Anterior gradient 2 is expressed and secreted during the development of pancreatic cancer and promotes cancer cell survival. Cancer Res. 2008; 68:7811–7818.
38. Li Z, Wu Z, Chen H, Zhu Q, Gao G, Hu L, Negi H, Kamle S, Li D. Induction of anterior gradient 2 (AGR2) plays a key role in insulin-like growth factor-1 (IGF-1)-induced breast cancer cell proliferation and migration. Med Oncol. 2015; 32:577.
39. Parsa Y, Mirmalek SA, Kani FE, Aidun A, Salimi-Tabatabaee SA, Yadollah-Damvandi S, Jangholi E, Parsa T, Shahverdi E. A review of the clinical implications of breast cancer biology. Electron Physician. 2016; 8:2416–2424.
40. Shao W, Brown M. Advances in estrogen receptor biology: prospects for improvements in targeted breast cancer therapy. Breast Cancer Res. 2004; 6:39–52.
41. Winder T, Giamas G, Wilson PM, Zhang W, Yang D, Bohanes P, Ning Y, Gerger A, Stebbing J, Lenz HJ. Insulin-like growth factor receptor polymorphism defines clinical outcome in estrogen receptor-positive breast cancer patients treated with tamoxifen. Pharmacogenomics J. 2014; 14:28–34.
42. Browne BC, Crown J, Venkatesan N, Duffy MJ, Clynes M, Slamon D, O’Donovan N. Inhibition of IGF1R activity enhances response to trastuzumab in HER-2-positive breast cancer cells. Ann Oncol. 2011; 22:68–73.
43. Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005; 74:739–789.
44. Higa A, Mulot A, Delom F, Bouchecareilh M, Nguyen DT, Boismenu D, Wise MJ, Chevet E. Role of pro-oncogenic protein disulfide isomerase (PDI) family member anterior gradient 2 (AGR2) in the control of endoplasmic reticulum homeostasis. J Biol Chem. 2011; 286:44855–44868.
45. Kaser A, Adolph TE, Blumberg RS. The unfolded protein response and gastrointestinal disease. Semin Immunopathol. 2013; 35:307–319.
46. Park SW, Zhen G, Verhaeghe C, Nakagami Y, Nguyenvu LT, Barczak AJ, Killeen N, Erle DJ. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc Natl Acad Sci U S A. 2009; 106:6950–6955.
47. Zhao F, Edwards R, Dizon D, Afrasiabi K, Mastroianni JR, Geyfman M, Ouellette AJ, Andersen B, Lipkin SM. Disruption of paneth and goblet cell homeostasis and increased endoplasmic reticulum stress in AGR2-/- mice. Dev Biol. 2010; 338:270–279.
48. Dumartin L, Alrawashdeh W, Trabulo SM, Radon TP, Steiger K, Feakins RM, di Magliano MP, Heeschen C, Esposito I, Lemoine NR, Crnogorac-Jurcevic T. ER stress protein AGR2 precedes and is involved in the regulation of pancreatic cancer initiation. Oncogene. 2017; 36:3094–103.
49. Schroder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci. 2008; 65:862–894.
50. Brush MH, Weiser DC, Shenolikar S. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol Cell Biol. 2003; 23:1292–1303.
51. McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. GADD153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001; 21:1249–1259.
52. Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007; 129:1337–1349.
53. Li Y, Schwabe RF, DeVries-Seimon T, Yao PM, Gerbod-Giannone MC, Tall AR, Davis RJ, Flavell R, Brenner DA, Tabas I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J Biol Chem. 2005; 280:21763–21772.
54. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004; 11:381–389.
55. Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem. 2006; 281:7260–7270.
56. Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008; 454:455–462.
57. Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000; 6:1355–1364.
58. Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006; 124:587–599.
59. Ryan KM, Ernst MK, Rice NR, Vousden KH. Role of NF-kappaB in p53-mediated programmed cell death. Nature. 2000; 404:892–897.
60. Wu H, Lozano G. NF-kappa B activation of p53. A potential mechanism for suppressing cell growth in response to stress. J Biol Chem. 1994; 269:20067–20074.
61. Garg AD, Nowis D, Golab J, Agostinis P. Photodynamic therapy: illuminating the road from cell death towards anti-tumour immunity. Apoptosis. 2010; 15:1050–1071.
62. Yang D, de la Rosa G, Tewary P, Oppenheim JJ. Alarmins link neutrophils and dendritic cells. Trends Immunol. 2009; 30:531–537.
63. Andre F, Dieci MV, Dubsky P, Sotiriou C, Curigliano G, Denkert C, Loi S. Molecular pathways: involvement of immune pathways in the therapeutic response and outcome in breast cancer. Clin Cancer Res. 2013; 19:28–33.
64. Kim JY, Heo SH, Song IH, Park IA, Kim YA, Gong G, Lee HJ. Activation of the PERK-eIF2alpha pathway is associated with tumor-infiltrating lymphocytes in HER2-positive breast cancer. Anticancer Res. 2016; 36:2705–2711.
65. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remedios C, Fend L, Hannani D, Aymeric L. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014; 20:1301–1309.
66. Slobodova Z, Ehrmann J, Krejci V, Zapletalova J, Melichar B. Analysis of CD40 expression in breast cancer and its relation to clinicopathological characteristics. Neoplasma. 2011; 58:189–197.
67. Meares GP, Liu Y, Rajbhandari R, Qin H, Nozell SE, Mobley JA, Corbett JA, Benveniste EN. PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol Cell Biol. 2014; 34:3911–3925.
68. Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol. 2016; 16:469–484.
69. Al-Zoubi M, Salem AF, Martinez-Outschoorn UE, Whitaker-Menezes D, Lamb R, Hulit J, Howel A, Gandara R, Sartini M, Arafat H, Bevilacqua G, Sotgia F, Lisanti MP. Creating a tumor-resistant microenvironment: cell-mediated delivery of TNFα completely prevents breast cancer tumor formation in vivo. Cell Cycle. 2013; 12:480–490.
70. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1.
71. Hrstka R, Brycthova V, Fabian P, Vojtesek B, Svoboda M. AGR2 predicts tamoxifen resistance in postmenopausal breast cancer patients. Dis Markers. 2013; 35:207–212.
72. Dong A, Wodziak D, Lowe AW. Epidermal growth factor receptor (EGFR) signaling requires a specific endoplasmic reticulum thioredoxin for the post-translational control of receptor presentation to the cell surface. J Biol Chem. 2015; 290:8016–8027.
73. Dong A, Gupta A, Pai RK, Tun M, Lowe AW. The human adenocarcinoma-associated gene, AGR2, induces expression of amphiregulin through hippo pathway co-activator YAP1 activation. J Biol Chem. 2011; 286:18301–18310.
74. Armes JE, Davies CM, Wallace S, Taheri T, Perrin LC, Autelitano DJ. AGR2 expression in ovarian tumours: a potential biomarker for endometrioid and mucinous differentiation. Pathology. 2013; 45:49–54.
75. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674.
76. R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing 2016. Available from: http://www.R-project.org.
77. Therneau R. A package for survival analysis in S. R package version 2.37-7. 2014; Available from: http://CRAN.R-project.org/package=survival.
78. Harrell FE Jr. rms: Regression modeling strategies. 2016.
79. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47.
80. Phipson B, Lee S, Majewski IJ, Alexander WS, Smyth GK. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann Appl Stat. 2016; 10:946–963.
81. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009; 37:1–13.
82. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27–30.
83. Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Gil L, Gordon L. Ensembl 2017. Nucleic Acids Res. 2017; 45:D635–42.
84. Fishilevich S, Zimmerman S, Kohn A, Iny Stein T, Olender T, Kolker E, Safran M, Lancet D. Genic insights from integrated human proteomics in genecards. Database (Oxford). 2016; 2016: baw030.