
INTRODUCTION
Gain-of-function mutations of KRAS, NRAS, BRAF, PIK3CA genes, or loss of tumor suppressor function of PTEN, resulting in continuous activation of RAS-mitogen-activated protein kinase (MAPK) or phosphoinositide 3-kinase (PI3K) pathways, characterize most colorectal cancers (CRC) [1]. More, 12–15% CRC show high mutational load and microsatellite instability [2]. Specific mutations of the different genes involved in MAPK/PI3K pathway may confer different biological aggressiveness and effectiveness of treatment strategies.
Overall, KRAS exons 2–4 (KRAS2-4), NRAS exons 2–4 (NRAS2–4), BRAF exon 15 (BRAF15) mutant (mut) MCRC patients are prevalent [3–6]. KRAS2 mut characterize 45–55% MCRC, mostly consisting of codon 12 (80%) c.35 G>A (G12D) and c.35 G>T (G12V) transversions [7, 8], and codon 13, prevalently c.38 G>A (G13D) mut, and impair the intrinsic GTPase activity of RAS, leading to constitutive, growth-factor-receptor independent activation of downstream signaling [9–11]. In MCRC patients, the reported prevalence of KRAS2–4 mut is 52.8%, NRAS2–4 mut 5.3%, BRAF15 mut 4.7–8.7%, prevalently c.1799 T>A (V600E) [3, 12]. Massive parallel sequencing of multiple genes by different NGS platforms enables mutation detection highly accurate and it is able to detect mutations at 5% allelic fraction (AF) [13, 14]. PGM/Colon Lung Cancer Panel identified all point mutations, and failed in 4.4% CRC mutant samples. In early CRC, KRAS2–4 37.8%, and NRAS2–4 4.6% mut were reported [13, 14]. KRAS, NRAS mut were mutually exclusive, BRAF mut (mostly not V600E) occasionally, PIK3CA mut frequently coexisted with RAS mut. Detection of multiple gene mutations and dynamic molecular characterization in the individual patient, could help monitoring biological evolution of metastatic disease, with prognostic and predictive clinical implications.
Treatment strategy of MCRC patients differs according to patient’s fitness (age, comorbidity), metastatic extension (liver-limited (L-L) or other/multiple metastatic sites (O/MM)), and KRAS2–4/NRAS2–4/BRAF15 genotype [9, 6, 15]. KRAS2–4/NRAS2–4 wt or mut genotype addresses the addiction of anti-Vascular Endothelial Growth Factor (VEGF) or anti-Epidermal Growth Factor Receptor (EGFR) to first line triplet or doublet chemotherapy of MCRC [3, 16–18]: anti-EGFR drugs in RAS wt [16–18]; anti-VEGF drugs in RAS wt and mut patients [3]. KRAS wt genotype predicted favourable clinical outcomes of anti-EGFR or anti-VEGF molecules added to doublet chemotherapy [19].
Clinical outcome of MCRC patients treated with BEV-containing chemotherapy is not significantly affected by KRAS2 status, wt or mut; median OS ranges between 29.9–38 months, and 19.9–21 months, respectively [19, 20, 9]. In KRAS2 mut patients, retrospective analysis showed that BEV addition to irinotecan, 5-fluorouracil, leucovorin (IFL) was predictive of significantly prolonged PFS, compared to IFL [19, 20]; in KRAS2 wt and mut MCRC patients treated with BEV added to IFL, median OS was 27.7 and 19.9 months, respectively, not significantly different [19, 20]. The prognostic relevance of KRAS2 and BRAF15 genotype was not significantly different, even though the hazard ratio (HR) was 0.64 and 0.38, respectively. A significantly better prognosis was reported only when KRAS2/BRAF15 wt patients were compared with patients harboring mutations in KRAS2 or BRAF15 genes (HR 0.51) [19]. Intensive first line treatment adding BEV to triplet chemotherapy, according to FOLFOXIRI plus BEV and FIr-B/FOx, consisting of 5-fluorouracil associated to alternating irinotecan/BEV or oxaliplatin, according to previously reported weekly schedule [5], increased activity and efficacy of MCRC patients: objective response rate (ORR) 77–82%, PFS 13.1 and 12 months, OS 30.9 and 28 months, not significantly different in KRAS2 wt and mut patients [5, 3]. Median OS of patients treated with FIr-B/FOx in KRAS2 wt and mut patients was 38 months and 21 months, respectively, not significantly different [9]; the prevalent KRAS2 c.35 G>A (G12D) mutant genotype may significantly affect worse OS [10, 11]. More recently, KRAS2 genotype was reported as affecting significantly different PFS and OS in patients treated with XelOx/BEV [21]. Retrospective analysis of clinical outcome according to KRAS2–4/NRAS2–4/BRAF15 genotype in PRIME, FIRE-3, PEAK, TRIBE randomized trials showed that EGFR- and VEGF-inhibitors are more and equivalently effective in KRAS2–4/NRAS2–4 wt patients [16–18, 3]. In the TRIBE study, FOLFOXIRI-Bev may predict a favourable effect in BRAF15 mut patients, compared to FOLFIRI-BEV [3].
The present study evaluated the prevalence, individual distribution and prognostic relevance of KRAS2–4/NRAS2–4/BRAF15 mutations, detected by next generation sequencing, in MCRC patients treated with FIr-B/FOx intensive first line treatment.
RESULTS
KRAS2–4/NRAS2–4/BRAF15 genotype was evaluated in 67 tumoral samples of 87 MCRC patients treated with FIr-B/FOx first line treatment (77%) (Table 1): 58 (86.6%) primary tumours, 9 (13.4%) metastatic sites (3 liver, 2 peritoneal carcinomatosis, 2 ovary, 1 local recurrence, and 1 lymph node); 60 (89.6%) obtained before first line metastatic treatment, 7 (10.4%) after first line treatment. Demographic and baseline features of evaluated patients were representative of the overall treated population [9].
Table 1: Distribution of tumoral samples of MCRC patients according to site (primary or metastatic) and timing of sampling (pre- or post-treatment)
Tumoral Samples |
||||||
|---|---|---|---|---|---|---|
Timing of sampling referred to FIr-B/FOx treatment |
Total |
Primary tumor |
Metastatic site |
|||
No. |
% |
No. |
% |
No. |
% |
|
Total No. |
67 |
100 |
58 |
86.6 |
9 |
13.4 |
Pre-treatment |
60 |
89.6 |
53 |
79.1 |
7 |
10.4 |
Post-treatment |
7 |
10.4 |
5 |
7.5 |
2 |
3.0 |
Abbreviations: No., number.
Table 2 shows the prevalence of KRAS2–4/NRAS2–4/BRAF15 wild-type (wt) and mutant (mut) samples: in 63 patients evaluable for KRAS2–4/NRAS2–4/BRAF15 (94.0%), triple wt were 14 (22.2%), and mut 49 (77.8%); KRAS2–4 wt patients were 21 (33.3%), mut 42 (66.7%), 4 samples not evaluable for KRAS2–4 genotype, because the coverage of specific amplicons was not sufficient for accurate mutation detection; KRAS2 wt patients were 23 (36.5%), mut 40 (63.5%); NRAS2–4 wt patients were 56 (83.6%), mut 11 (16.4%); BRAF15 wt patients were 62 (92.5%), mut 5 (7.5%). KRAS exon 2 mutational status was discordant between previous monogenic evaluation and NGS data in 10 patients (15.9%): 5 (7.9%) evaluated as mut by monogenic assay were wt by NGS; 4 (6.3%) wt by monogenic assay showed a KRAS exon 2 mutation by NGS; a different KRAS exon 2 mutation was reported in 1 patient (1.6%).
Table 2: Prevalence of KRAS/NRAS/BRAF wild-type and mutant samples
Samples |
Patients/Samples |
||
|---|---|---|---|
All |
wild-type |
mutant |
|
No. (%) |
No. (%) |
No. (%) |
|
KRAS2-4/NRAS2-4/BRAF15 |
63 (94) |
14 (22.2) |
49 (77.8) |
KRAS2-4 |
63 (94) |
21 (33.3) |
42 (66.7) |
KRAS 2 |
63 (94) |
23 (36.5) |
40 (63.5) |
NRAS2-4 |
67 (100) |
56 (83.6) |
11 (16.4) |
BRAF15 |
67 (100) |
62 (92.5) |
5 (7.5) |
Table 3 shows the prevalence of mutant samples, with single and multiple mutations, according to the mutation detection criteria. Overall, 60 mutations were detected in 49 mut samples, range 1–3 mut (median 1.22 mut/mut sample) at >500×cov and >1% AF detection criteria: 43 (71.7%) at >1000×cov/>5% AF; 9 (15%) at >500×cov/>5% AF; 8 (13.3%) at >1000×cov/<5% AF. The distribution of mut samples according to the mutation detection criteria, >1000×cov and >5% AF, >500×cov and >5%, and >1000×cov and <5% AF was, respectively: in 49 KRAS2–4/NRAS2–4/BRAF15 mut (77.8%), 40 (63.5%), 8 (12.7%), 1 (1.6%); in 42 (66.7%) KRAS2–4 mut, 32 (50.8%), 9 (14.3%), 1 (1.6%); in 40 (63.5%) KRAS2 mut, 30 (47.6%), 9 (14.3%), 1 (1.6%); in 11 (16.4%) NRAS2–4 mut, 7 (10.4%), 1 (1.5%), 3 (4.5%); in 5 (7.5%) BRAF15 mut, 1 (1.5%) at >500×cov and >5% AF, 4 (6%) at >1000×cov and <5% AF.
Table 3: Prevalence of single and multiple KRAS/NRAS/BRAF mutations according to mutation detection criteria
Mutations |
Mutation detection criteria |
|||||
|---|---|---|---|---|---|---|
Single |
≥2 |
Coverage >1000 Allelic fraction >5% |
Coverage 500-1000 Allelic fraction >5% |
Coverage >1000 Allelic fraction >1<5% |
||
No. (%) |
No. (%) |
No. (%) |
No. (%) |
No. (%) |
No. (%) |
|
Mutations |
60 |
43 (71.7) |
9 (15.0) |
8 (13.3) |
||
KRAS2-4/NRAS2-4/BRAF15 |
49 (77.8) |
41 (65.1) |
8 (12.7) |
40 (63.5) |
8 (12.7) |
1 (1.6) |
KRAS2 |
40 (63.5) |
33 (52.4) |
7 (11.1) |
30 (47.6) |
9 (14.3) |
1 (1.6) |
KRAS2-4 |
42 (66.7) |
35 (55.6) |
7 (11.1) |
32 (50.8) |
9 (14.3) |
1 (1.6) |
NRAS2-4 |
11 (16.4) |
6 (8.9) |
5 (7.5) |
7 (10.4) |
1 (1.5) |
3 (4.5) |
BRAF15 |
5 (7.5) |
0 (-) |
5 (7.5) |
0 (-) |
1 (1.5) |
4 (6.0) |
The distribution of KRAS2–4/NRAS2–4/BRAF15 mutations, single and multiple, in individual mutant sample was, respectively: in 49 KRAS2–4/NRAS2–4/BRAF15 mut samples, 41 single (65.1%) and 8 (12.7%) ≥2 mut; in 42 KRAS2–4 mut, 35 (55.6%) single and 7 (11.1%) ≥2 mut; in 40 KRAS2 mut, 33 (52.4%) single and 7 (11.1%) ≥2 mut; in 11 NRAS2–4 mut, 6 (8.9%) single and 5 (7.5%) ≥2 mut; in 5 BRAF15 mut, all ≥2 mut (7.5%).
Table 4 shows the panel of detected mutations, and of the >2 mut samples according to mutation detection criteria. Distribution of KRAS2 mut was: codon 12, 35 (55.6%); codon 13, 5 (7.9%). KRAS2 c.35 G>A (G12D) was detected in 16 (25.4%) samples, 13 (20.6%) single and 3 (4.8%) ≥2 mut: 1 associated with NRAS2 c.34 G>A (G12S) and BRAF15 c.1756 G>A (E586K) mutations; 1 associated with NRAS2 c.38 G>T (G13V) and BRAF15 c.1405 G>A (G469R) mutations; 1 associated with NRAS2 c.182 A>G (Q61R) mutation. KRAS2 c.35 G>T (G12V) was detected in 15 (23.8%) samples, 13 (20.6%) single and 2 (3.2%) ≥2 mut: 1 associated with a KRAS4 c.436 G>A (A146T) mutation; 1 associated with a BRAF15 c.1756 G>A (E586K) mutation. Other KRAS2 mutations were: c.35 G>C (G12A) 1 (1.6%); c.34 G>C (G12R) 1 (1.6%); c.34 G>A (G12S) 2 (3.2%), including 1 associated with KRAS c.37 G>A (G13S) and NRAS c.35 G>A (G12D); c.38 G>A (G13D), 5 (7.9%), 4 (6.4%) single and 1 (1.6%) ≥2 mut, associated with a BRAF15 c.1406 G>C (G469A) mutation. KRAS3 codon 61 mut were detected in 2 patients (3.2%), specifically c.182 A>T (Q61L) and c.182 A>G (Q61R), 1 patient each (1.6%). Distribution of NRAS mutations was: NRAS codon 12, 2 (3.0%), specifically c.34 G>A (G12S) associated to KRAS c.35 G>A and BRAF c.1756 G>A mutations, and c.35 G>A (G12D) associated to double KRAS mutations; NRAS codon 13, 4 (6%), c.37 G>A (G13S) 1 patient (1.5%), c.38 G>A (G13D) 2 patients (3%), and c.38 G>T (G13V) associated to KRAS c.35 G>A and BRAF c.1405 G>A mutations, 1 patient (1.5%); NRAS codon 61, 5 patients (7.5%), specifically c.182 A>T (Q61L) 1 (1.5%), and c.182 A>G (Q61R) 4 (6%) patients, 2 single and 2 associated to KRAS c.35 G>A, and to BRAF c.1796 C>T (T599I) mutations, respectively. All 5 (7.5%) BRAF15 mut were atypical and associated to KRAS and/or NRAS mutations: c.1405 G>A (G469R) associated to KRAS c.35 G>A and NRAS c.38 G>T mutations; c.1406 G>C (G469A) associated to KRAS c.38 G>A mutation; c.1756 G>A (E586K) 2 samples, associated to KRAS c.35 G>A/NRAS c.34 G>A, and to KRAS c.35 G>T mutations, respectively; c.1796 C>T (T599I) associated to NRAS c.182 A>G mutation.
Table 4: Panel of single and multiple KRAS/NRAS/BRAF mutations in individual mutant samples, according to mutation detection criteria
Mutations |
≥ 2 mutant samples |
Mutation detection criteria |
|||||||
|---|---|---|---|---|---|---|---|---|---|
1 |
≥2 |
KRAS |
NRAS |
BRAF |
Cov >1000 AF >5% |
Cov 500-1000 AF >5% |
Cov >1000 AF >1<5% |
||
No. (%) |
No. (%) |
No. (%) |
No. |
No. |
No. |
No. (%) |
No. (%) |
No. (%) |
|
KRAS |
|||||||||
35 G>A |
16 (25.4) |
13 (20.6) |
3 (4.8) |
35 G>A |
34 G>A |
1756 G>A |
2 |
0 |
1 (BRAF) |
35 G>T |
15 (23.8) |
13 (20.6) |
2 (3.2) |
35 G>T + |
1756 G>A |
1 |
0 |
1 (KRAS) |
|
35 G>C |
1 |
1 |
0 |
||||||
34 G>C |
1 |
1 |
0 |
||||||
34 G>A |
2 |
1 |
1 (1.6) |
34 G>A + |
35 G>A |
3 |
0 |
0 |
|
38 G>A |
5 |
4 |
1 (1.6) |
38 G>A |
1406 G>C |
1 |
1 |
0 |
|
182 A>T |
1 |
1 |
0 |
||||||
182 A>G |
1 (1.6) |
1 |
0 |
||||||
NRAS |
|||||||||
37 G>A |
1 (1.5) |
1 |
0 |
||||||
38 G>A |
2 (3.0) |
2 |
0 |
||||||
182 A>T |
1 (1.5) |
1 |
0 |
||||||
182 A>G |
4 (6.0) |
2 |
2 |
182 A>G |
1796 C>T |
0 |
0 |
2 (NRAS,BRAF) |
|
Abbreviations: Cov, coverage; AF, allelic fraction.
All the 8 mutations detected at >1000×cov and <5% AF (13.3%) and consisting of 1 KRAS, 3 NRAS, 4 BRAF mutations, were detected in 6 mutant samples harboring ≥2 mutations: 5 associated to KRAS mutations detected at >1000×cov and >5% AF; 1 (1.6%) mut sample with a NRAS c.182 A>G (Q61R) associated to BRAF c.1796 C>T (T599I) mutation represented the only sample with mutations detected at >1000×cov and <5% AF.
Efficacy according to KRAS/NRAS/BRAF genotype and specific mutations
At median follow-up 21 months, clinical outcome of 67 patients evaluated for KRAS2–4/NRAS2–4/BRAF15 genotype was consistent with previously reported in overall population treated with FIr-B/FOx intensive first line (Table 5) [9, 10, 22]: PFS 13 months (3–113+ months), OS 27 months (4–119+ months); in KRAS2 wt and mut, median PFS was 14 (3–93+ months) and 12 months (3–113+ months), median OS 28 (6+−97 months) and 21 months (4–119+ months), respectively, not significantly different [23] (Figure 1A, 1B); in c.35 G>A KRAS mut, median PFS was 8 months (3–113+ months), median OS 14 months (4–119+ months), not significantly different compared with wt (Figure 1C, 1D). Among KRAS2–4 wt and mut, median PFS was 13 (3–93+ months) and 12 months (3–113+ months), median OS 27 months equivalently (6+−97 and 4–119+ months), respectively, not significantly different (Figure 2A). Among NRAS2–4 wt and mut, median PFS was 16 (3–113+ months) and 12 months (6–93+ months), median OS 28 (4–119+ months) and 22 months (8–93+ months), respectively, not significantly different (Figure 2B). Among BRAF15 wt and mut, median PFS was 14 (3–113+ months) and 8 months (6–17 months), median OS was 28 (4–119+ months) and 11 months (8–94+ months), respectively (Figure 3A), not significantly different. Among KRAS2–4/NRAS2–4/BRAF15 wt and mut, median PFS was 18 (3–33 months) and 12 months (3–113+ months), median OS was 28 (6+−97 months) and 22 months (4–119+ months), respectively (Figure 3B). PFS and OS of wt compared to mut patients was not significantly different (P = 0.866 and 0.956, respectively). Among mutant patients with ≥2 compared to 1 mut gene, PFS was 11 and 14 months, OS was 22 and 37 months, not significantly different. PFS was trendy worse in patients harboring ≥2 mut KRAS2–4/NRAS2–4/BRAF15 genes compared with triple wt (P 0.059).
Table 5: Efficacy according to KRAS/NRAS/BRAF genotype
Patients |
Progression-free survival (months) Range |
Overall survival (months) Range |
||
|---|---|---|---|---|
No. |
% |
|||
Overall evaluated population |
67 |
100 |
13 |
27 |
KRAS exon 2 wild-type |
23 |
37.5 |
14 |
28 |
KRAS exon 2 mutant |
40 |
63.5 |
12 |
21 |
c.35 G > A KRAS mutant |
16 |
25.3 |
8 |
14 |
KRAS exon 2-4 wild-type |
21 |
33.3 |
13 |
27 |
KRAS exon 2-4 mutant |
42 |
66.7 |
12 |
27 |
NRAS exon 2-4 wild-type |
56 |
83.6 |
16 |
28 |
NRAS exon 2-4 mutant |
11 |
16.4 |
12 |
22 |
BRAF wild-type |
62 |
92.5 |
14 |
28 |
BRAF mutant |
5 |
7.5 |
8 |
11 |
KRAS/NRAS/BRAF wild-type |
14 |
22.2 |
18 |
28 |
KRAS/NRAS/BRAF mutant |
49 |
77.8 |
12 |
22 |
Single mutant gene |
40 |
63.5 |
14 |
37 |
≥2 mutant genes |
9 |
14.3 |
11 |
22 |
Abbreviations: No, number.
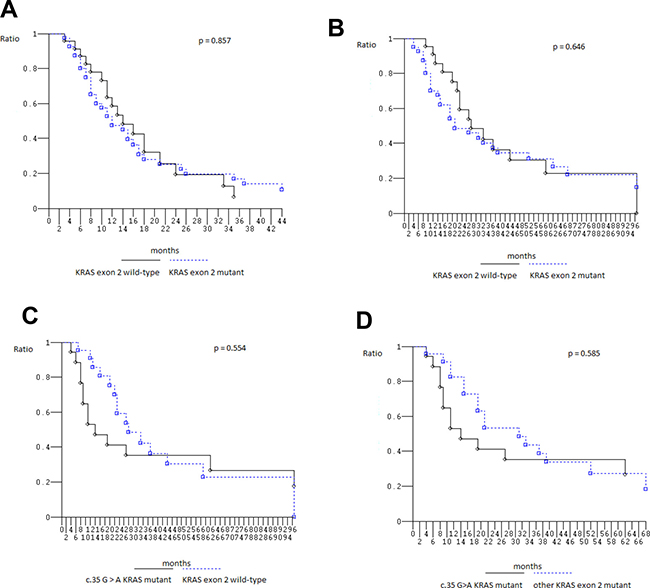
Figure 1: Kaplan–Meier survival estimate. (A) Progression-free Survival KRAS exon 2 wild-type versus mutant; (B) Overall Survival KRAS exon 2 wild-type versus mutant; (C) Overall Survival c.35 G > A KRAS mutant patients versus KRAS exon 2 wild-type patients; (D) Overall Survival c.35 G > A KRAS mutant patients versus other KRAS exon 2 mutant patients.
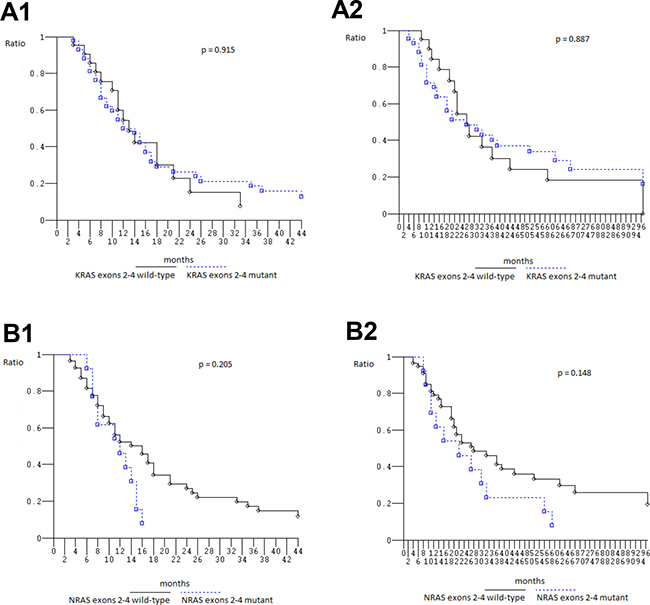
Figure 2: Overall Survival, Kaplan–Meier survival estimate. (A) KRAS2-4 wild type versus mutant; (B) NRAS2-4 wild-type versus mutant; 1, Progression-free survival; 2, Overall survival.
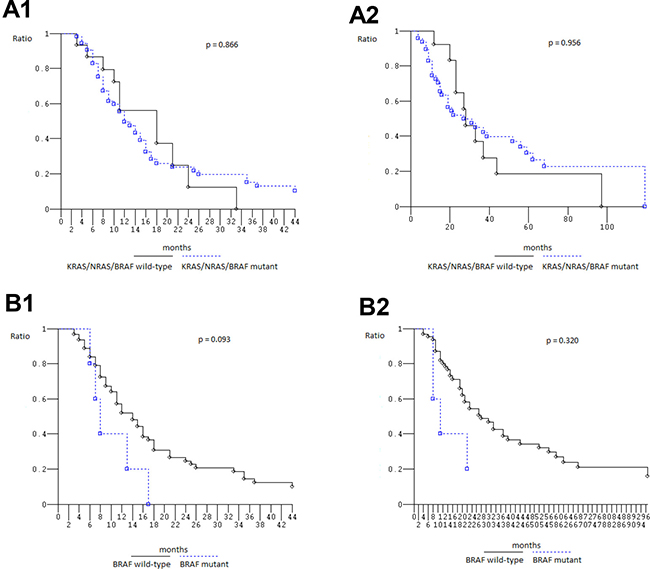
Figure 3: Overall Survival, Kaplan–Meier survival estimate. (A) KRAS/NRAS/BRAF wild-type versus mutant patients; (B) BRAF wild-type versus BRAF mutant patients; 1, Progression-free survival; 2, Overall survival.
Among 22 L-L (32.8%) patients, median PFS was 16 months (3–113+), median OS 33 months (6+−113+): in KRAS2–4/NRAS2–4/BRAF15 wt and mut, median PFS was 18 (3–33 months) and 16 months (5–113+ months), median OS was 33 (6+-97 months) and 39 months (8–113+ months), respectively, not significantly different (P 0.685 and 0.480 respectively). Among 45 O/MM patients (67.2%), median PFS was 12 months (3–59+), median OS 23 months (4–119+): in KRAS2–4/NRAS2–4/BRAF15 wt and mut, median PFS was 18 (5–24 months) and 12 months (3–59+ months), median OS was 28 (10+-70+ months) and 21 months (4–119+ months), respectively, not significantly different (P 0.624 and 0.538 respectively). Among 13 right side patients (19.4%), median PFS was 14 months (5–113+), median OS 20 months (6+−113+): only 1 patient was KRAS2–4/NRAS2–4/BRAF15 wt with PFS 11 months and OS 44 months; among KRAS2–4/NRAS2–4/BRAF15 mut median PFS 16 months (5–113+) and median OS 19 months (6–113+). Among 54 left side patients (81.6%), median PFS was 13 months (3–88+), median OS 27 months (4–119+); among KRAS2–4/NRAS2–4/BRAF15 wt median PFS 18 months (3–33) and OS 28 months (6+−97); among KRAS2–4/NRAS2–4/BRAF15 mut median PFS 12 months (3–88+) and median OS 27 months (4–119+). In the overall analysed population, PFS and OS of right compared with left tumors were not significantly different (P 0.675 and 0.751, respectively).
DISCUSSION
The present study evaluated the prevalence and individual distribution of KRAS2–4/NRAS2–4/BRAF15 mutations by NGS in MCRC patients treated with intensive first line treatment adding BEV to triplet chemotherapy according to previously reported FIr-B/FOx schedule [5]. Next generation sequencing by Ion Torrent platform, mostly performed before first line metastatic treatment (89.6% of samples) and in primary tumor samples (79.1%), detected overall 77.8% KRAS2–4/NRAS2–4/BRAF15 mut MCRC patients at molecular diagnostic criteria of target sequence coverage >500×cov and >1% mutant AF; specifically, KRAS2–4 66.7%, NRAS2–4 16.4%, BRAF15 7.5% mut MCRC patients. KRAS2 and KRAS2–4/NRAS2–4/BRAF15 mut at >1000×cov and >5% AF were 47.6% and 63.5%, respectively, consistent with that previously reported in the same MCRC cohort by our group using direct sequencing [9, 10], and in the range of reported 45–55% KRAS2 and 65.6% KRAS2–4/NRAS2–4/BRAF15 mut [3]. In the retrospective evaluation of TRIBE trial [3], KRAS2–4, NRAS2–4, BRAF15 mutations were detected in 52.8%, 5.3% and 7.5% of patients, respectively; all wild-type were 34.4%. The application of >500×cov and >1% AF mutation criteria may increase detectable mutations due to the increased NGS diagnostic accuracy, that may detect tumoral clonal heterogeneity: KRAS2–4 mut were prevalently detected at >1000×cov/>5% AF (50.8%), while NRAS2–4 mut (16.4%) were frequently detected at 500–1000xcov and <5%AF (6%), and BRAF15 mut (7.5%) were all detected at 500–1000×cov and <5%AF. Thus, KRAS2–4/NRAS2–4/BRAF15 mutations were frequently detected (28.3%) at 500–1000×cov and <5%AF.
Present findings show that RAS mutations were enriched in MCRC patients, compared to early CRC where KRAS2–4 37.8%, and NRAS2–4 4.6% mut were reported by the same NGS platform [13, 14].
KRAS2–4/NRAS2–4/BRAF15 mut were often detected at <5% AF (13.3%), all involving MCRC patients harbouring >2 KRAS and/or NRAS mut and prevalently involving atypical BRAF mutations, different from that typically reported (codon 600) in CRC [14]. All BRAF15 mut harboured >2 mut, associated with other KRAS2–4/NRAS2–4 mut, all atypical. More, KRAS, NRAS, BRAF mutations were not mutually exclusive: tumoral samples prevalently harboured single gene (KRAS2–4, NRAS2–4) mutations (65.1%); individual MCRC patients often harboured double or triple KRAS2–4/NRAS2–4/BRAF15 mutations (12.7%), frequently involving NRAS2–4 and all BRAF15 mut, as recently reported [13, 14]. One MCRC patient was only detected at 500–1000×cov and <5%AF (1.6%), and harbored NRAS c.182 A>G (Q61R) associated to BRAF c.1796 C>T (T599I) mutations. Thus, massive parallel sequencing by Ion torrent platform can increase mutation detection by increasing diagnostic accuracy, if >1% mutant AF with >1000×cov is included to specifically detect clonal heterogeneity involving KRAS, NRAS and atypical BRAF mutations, thus increasing the detection of multiple genes mutations in individual MCRC patients. To this aim, NGS is able to detect mutant alleles at the 5% level [13, 14].
Specific mutations of different genes involved in the same signalling pathway (BRAF and RAS mutations) can confer different biological aggressiveness and effectiveness of treatment strategies. Preliminary analysis of differential clinical outcome in overall MCRC patients treated with FIr-B/FOx intensive first line treatment according to KRAS2 genotype confirmed previously reported median PFS 13 months and OS 27 months, a trendy worse OS 21 months in KRAS2 mut, and PFS 8 months and OS 14 months in the prevalent KRAS2 c.35G > A mut MCRC patients [11]. Clinical outcome was not significantly different in KRAS2–4, NRAS2–4, BRAF15 mut and wt MCRC patients. The 5 BRAF15 mut, all atypical and associated to other KRAS2–4 and/or NRAS2–4 mut, compared to wt MCRC patients showed trendy worse, not significantly different, PFS 8 months and OS 11 months, even if treated with BEV added to triplet chemotherapy. Worse prognosis was previously shown by the prevalent BRAF15 c.1799 T > A (V600E) mutation, characterizing 4.7–8.7% CRC, in MCRC patients treated with doublet chemotherapy alone or added to cetuximab, BEV, and cetuximab plus BEV, with median PFS 5.6–8 months and median OS 10.3–15.9 months [24–26]. The favourable predictive effect of cetuximab or BEV addiction to chemotherapy in KRAS exon 2 wild-type patients was not significantly confirmed in BRAF mutant MCRC patients [20, 24, 25]. Mutations in BRAF gene occur in two regions of the BRAF kinase domain, exon 15, the activation segment (which protects the substrate binding site), and, less commonly, exon 11, the G loop (which mediates ATP-binding). Less common BRAF mutations at codons 594 and 596 correlated with longer OS when compared with BRAF V600E mutations (62 vs 12.6 months, P = 0.002) [27], and trendy longer compared to BRAF wild-type (35.9 months) in MCRC patients treated with FOLFOXIRI/BEV [3].
Differential clinical outcome according to KRAS2–4/NRAS2–4/BRAF15 genotype mut and wt was not significantly different (PFS 12 vs 18 months; OS 22 vs 28 months). The retrospective evaluation of TRIBE trial also reported no significant interaction between RAS or BRAF status and treatment effect in PFS or OS [3]. RAS mutant patients treated with first line FOLFOXIRI plus BEV achieved median OS 25.6 months, BRAF mutant 13.4 months, and triple wild-type 37.1 months.
In the 22.2% KRAS2–4/NRAS2–4/BRAF15 triple wt MCRC patients, the efficacy was trendy higher with a median PFS 18 months and median OS 28 months; furthermore, the efficacy of first line FIr-B/FOx, adding BEV to triplet chemotherapy is close to be significantly different in KRAS2–4/NRAS2–4/BRAF15 MCRC triple wt compared with MCRC patients harbouring ≥2 mutant genes (PFS 11 months) and it requires further prospective validation.
Prospective studies should be developed to better evaluate differential clinical outcome in MCRC patients harbouring KRAS c.35 G > A (G12D), BRAF c.1799 T > A (V600E) and atypical, less common mutations, as well as in KRAS2–4/NRAS2–4/BRAF15 wt and in patients harbouring ≥2 mutant genes.
More, high sensitive KRAS2–4/NRAS2–4/BRAF15 multigenic analysis performed in metastatic tissues and/or liquid biopsies could dynamically be helpful to monitor the evolution of mutant genes spectrum, more closely evaluate prognostic implications and individually predict targeted treatment.
MATERIALS AND METHODS
Patients and samples
Eighty-seven consecutive, unselected, MCRC patients were enrolled in previously reported phase II study and the expanded clinical program proposing FIr-B/FOx as first line treatment [5, 9]. KRAS2–4/NRAS2–4/BRAF15 genotype was evaluated in tumoral samples of 67 (77%) patients (Table 1), specifically primary tumours or metastatic sites, pre- or post-treatment.
Study was approved by Local Ethical Committee (Comitato Etico, Azienda Sanitaria Locale n.4 L’Aquila, Regione Abruzzo, Italia) and conducted in accordance with Declaration of Helsinki. All patients provided written, informed consent.
FIr-B/FOx association consisted of 5-fluorouracil associated to alternating irinotecan/BEV or oxaliplatin, according to previously reported weekly schedule [5].
Mutational analysis
KRAS2–4/NRAS2–4/BRAF15 analyses were performed on paraffin-embedded tissue blocks from primary tumor and/or metastatic site. Genotype status was previously analyzed for KRAS2 (codon 12–13) mutations and BRAF c.1799 T>A (V600E) mutations, according to genotype analyses recommendations for clinical implications [28], by SNaPshot® multiplex, and/or direct sequencing, as previously reported [9].
Mutation detection by massive parallel sequencing
Neoplastic cells were selected from highest density H&E slide; DNA was extracted by QIAamp Mini Kit from 2–5 10 μm serial sections, assessed by Qubit photometer, dsDNA High Sensitivity Assay Kit. Workflow consisted of generation of library of DNA amplified fragments flanked by Ion Torrent adapters, clonally amplified into Ion Sphere particles (ISP) by emulsion PCR, applied to Ion chip on Ion PGM sequencer and analyzed (Life Technologies): 10 ng DNA for library preparation with Ion AmpliSeq Library 96LV Kit 2.0, Colon/Lung Cancer Panel, including 207 amplicons covering 2800 hotspot regions in 50 genes with >500×sequence coverage; library barcoded with Ion Xpress Barcode Adapters 1–16 Kit; template prepared by emulsion PCR on Ion OneTouch 2; library quality control performed by Ion Sphere Quality Control Kit; sequencing primer, and polymerase added and loaded onto 316 chips; sequencing performed on PGM, data analysis with Torrent Suite Software V.3.2. Variant Caller plug-in applied by Colon/Lung hotspot file (http://www.thermofisher.com); Ion Reporter suite used to filter polymorphic variants, reviewed with Integrative Genomics Viewer (IGV V.2.1 Broad Institute Cambridge Massachusetts USA) [13, 14]. Molecular diagnostic criteria for mutation detection were: >500×sequence coverage; >1% mutant allelic fraction.
Study design
A retrospective analysis has been planned to evaluate prognostic relevance of KRAS2–4/NRAS2–4/BRAF15 genotype on clinical outcome of MCRC patients treated with first line FIr-B/FOx regimen. Clinical criteria of efficacy were PFS, OS, evaluated using Kaplan and Meier method [22]. Patients were evaluated according to involved metastatic sites, classified as L-L and O/MM. Log-rank test was used to compare PFS and OS in different subgroups [23]. PFS was defined as length of time between beginning of treatment and disease progression or death (resulting from any cause) or to last contact; OS as length of time between beginning of treatment and death or to last contact.
CONCLUSIONS
Next generation sequencing of multiple genes shows that most MCRC harbour KRAS2–4/NRAS2–4/BRAF15 mutations, prevalently as single gene mut, and frequently multiple gene mutations, at increased sensitivity due to clonal heterogeneity.
Clinical outcome of MCRC patients treated with intensive first line FIr-B/FOx regimen was not significantly affected by KRAS2–4/NRAS2–4/BRAF15 genotype status.
Differential prognosis and predictive effect of VEGF-inhibitors added to chemotherapy in MCRC patients harbouring KRAS2 c.35 G > A and BRAF15 mut, or KRAS2–4/NRAS2–4/BRAF15 wt should be prospectively evaluated by massive parallel sequencing, also according to other mutations differentially activating the downstream RAS-MAPK or PI3K pathways.
Abbreviations
AF: allelic fraction; anti-EGFR: anti-epidermal growth factor receptor; anti-VEGF: anti-vascular endothelial growth factor; BEV: bevacizumab; BRAF15: BRAF exon 15; cov: coverage; CRC: colorectal cancer; HR: hazard ratio; KRAS2–4: KRAS exons 2–4; IFL: irinotecan, 5-fluorouracil, and leucovorin; ISP: Ion Sphere particles; L-L: liver-limited; m: months; mitogen-activated protein kinase (MAPK); MCRC: metastatic colorectal cancer; mut: mutant; NGS: Next Generation Sequencing; NRAS2–4: NRAS exons 2–4; O/MM: other multiple metastatic sites; ORR: objective response rate; OS: overall survival; PFS: progression-free survival; phosphoinositide 3-kinase (PI3K); wt: wild-type.
Author contributions
GB contributed to the conception and design of the study, in the provision of study materials of patients, in the data analysis and interpretation, in the manuscript writing. ER contributed to the conception and design of the study, in the data analysis and interpretation, in the manuscript writing. GB, ER provided clinical management and data of patients. ADM, GC, DDG collected and assembled biological materials. FP, UM, PP, GT provided molecular genetic analysis, and contributed in the data analysis and interpretation. All authors participated in the collection and/or assembly of data. All authors read, revised and approved the final manuscript.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
REFERENCES
1. McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA, D’Assoro AB, Salisbury JL, Mazzarino MC, Stivala F, Libra M. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul. 2006; 46:249–279.
2. Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, Kovar CL, Lewis LR, Morgan MB, Newsham IF, Reid JG, Santibanez J, Shinbrot E, et al, and Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–337.
3. Cremolini C, Loupakis F, Antoniotti C, Lupi C, Sensi E, Lonardi S, Mezi S, Tomasello G, Ronzoni M, Zaniboni A, Tonini G, Carlomagno C, Allegrini G, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015; 16:1306–1315.
4. Ciardiello F, Normanno N, Maiello E, Martinelli E, Troiani T, Pisconti S, Giuliani F, Barone C, Cartenì G, Rachiglio AM, Montesarchio V, Tonini G, Rizzi D, et al. Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next-generation sequencing: findings from the CAPRI-GOIM trial. Ann Oncol. 2014; 25:1756–1761.
5. Bruera G, Santomaggio A, Cannita K, Baldi PL, Tudini M, De Galitiis F, Mancini M, Marchetti P, Antonucci A, Ficorella C, Ricevuto E. “Poker” association of weekly alternating 5-Fluorouracil, Irinotecan, Bevacizumab and Oxaliplatin (FIr-B/FOx) in first line treatment of metastatic colorectal cancer: a phase II study. BMC Cancer. 2010; 10:567.
6. Bruera G, Cannita K, Giordano AV, Vicentini R, Ficorella C, Ricevuto E. Prognostic relevance of KRAS genotype in metastatic colorectal cancer patients unfit for FIr-B/FOx intensive regimen. Int J Oncol. 2014; 44:1820–1830.
7. Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989; 49:4682–4689.
8. Andreyev HJ, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, Young J, Walsh T, Ward R, Hawkins N, Beranek M, Jandik P, Benamouzig R, et al. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Br J Cancer. 2001; 85:692–96.
9. Bruera G, Cannita K, Di Giacomo D, Lamy A, Troncone G, Dal Mas A, Coletti G, Frébourg T, Sabourin JC, Tosi M, Ficorella C, Ricevuto E. Prognostic value of KRAS genotype in metastatic colorectal cancer (MCRC) patients treated with intensive triplet chemotherapy plus bevacizumab (FIr-B/FOx) according to extension of metastatic disease. BMC Med. 2012; 10:135.
10. Bruera G, Cannita K, Di Giacomo D, Lamy A, Frébourg T, Sabourin JC, Tosi M, Ficorella C, Ricevuto E. Worse prognosis of KRAS c.35 G > A mutant metastatic colorectal cancer (MCRC) patients treated with intensive triplet chemotherapy plus bevacizumab (FIr-B/FOx). BMC Med. 2013; 11:59.
11. Bruera G, Cannita K, Tessitore A, Russo A, Alesse E, Ficorella C, Ricevuto E. The prevalent KRAS exon 2 c.35 G > A mutation in metastatic colorectal cancer patients: a biomarker of worse prognosis and potential benefit of bevacizumab-containing intensive regimens? Crit Rev Oncol Hematol. 2015; 93:190–202.
12. Artale S, Sartore-Bianchi A, Veronese SM, Gambi V, Sarnataro CS, Gambacorta M, Lauricella C, Siena S, Ward R, Hawkins N, Beranek M, Jandik P, Benamouzig R, et al. Mutations of KRAS and BRAF in primary and matched metastatic sites of colorectal cancer. J Clin Oncol. 2008; 26:4217–4219.
13. Malapelle U, Vigliar E, Sgariglia R, Bellevicine C, Colarossi L, Vitale D, Pallante P, Troncone G. Ion Torrent next-generation sequencing for routine identification of clinically relevant mutations in colorectal cancer patients. J Clin Pathol. 2015; 68:64–68.
14. Malapelle U, Pisapia P, Sgariglia R, Vigliar E, Biglietto M, Carlomagno C, Giuffrè G, Bellevicine C, Troncone G. Less frequently mutated genes in colorectal cancer: evidences from next-generation sequencing of 653 routine cases. J Clin Pathol. 2016; 69:767–771.
15. Bruera G, Cannita K, Giuliante F, Lanfiuti Baldi P, Vicentini R, Marchetti P, Nuzzo G, Antonucci A, Ficorella C, Ricevuto E. Effectiveness of liver metastasectomies in Metastatic Colorectal Cancer (MCRC) patients treated with triplet chemotherapy plus bevacizumab (FIr-B/FOx). Clin Colorectal Cancer. 2012; 11:119–126.
16. Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, et al. Panitumumab-FOLFOX4 treatment and RAS Mutations in Colorectal Cancer. N Engl J Med. 2013; 369:1023–1034.
17. Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon JL, Hecht JR, Yu H, Oliner KS, Go WY. PEAK: A randomized, multicenter phase II study of Panitumumab plus modified Fluorouracil, Leucovorin, and Oxaliplatin (mFOLFOX6) or Bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 Metastatic Colorectal Cancer. J Clin Oncol. 2014; 32:2240–2247.
18. Stintzing S, Modest DP, Rossius L, Lerch MM, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, Heintges T, Lerchenmüller C, Kahl C, Seipelt G, et al, and FIRE-3 investigators. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab for metastatic colorectal cancer (FIRE-3): a post-hoc analysis of tumour dynamics in the final RAS wild-type subgroup of this randomised open-label phase 3 trial. Lancet Oncol. 2016; 17:1426–1434.
19. Hurwitz HI, Yi J, Ince W, Novotny WF, Rosen O. The clinical benefit of bevacizumab in metastatic colorectal cancer is independent of K-ras mutation status: analysis of a phase III study of bevacizumab with chemotherapy in previously untreated metastatic colorectal cancer. Oncologist. 2009; 14:22–28.
20. Ince WL, Jubb AM, Holden SN, Holmgren EB, Tobin P, Sridhar M, Hurwitz HI, Kabbinavar F, Novotny WF, Hillan KJ, Koeppen H. Association of k-ras, b-raf, and p53 status with the treatment effect of Bevacizumab. J Natl Cancer Inst. 2005; 97:981–989.
21. Díaz-Rubio E, Gómez-España A, Massutí B, Sastre J, Reboredo M, Manzano JL, Rivera F, Safont MJ, Montagut C, González E, Benavides M, Marcuello E, Cervantes A, et al, and Spanish Cooperative Group for the Treatment of Digestive Tumors (TTD). Role of Kras status in patients with Metastatic Colorectal Cancer receiving first-line chemotherapy plus bevacizumab: a TTD group cooperative study. PLoS One. 2012; 7:e47345.
22. Kaplan EL, Meier P. Nonparametric estimation of incomplete observations. J Am Stat Assoc. 1958; 53:457–481.
23. Peto R, Peto J. Asymptomatically efficient rank invariant test procedures. J R Stat Soc Ser A. 1972; 135:185–206.
24. Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, de Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A. Wild-type BRAF is required for response to panitumumab and cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008; 26:5705–5712.
25. Van Cutsem E, Köhne CH, Làng I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, Schlichting M, Zubel A, Celik I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for Metastatic Colorectal Cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011; 29:2011–2019.
26. Bokemeyer C, Bondarenko I, Hartmann JT, de Braud F, Schuch G, Zubel A, Celik I, Schlichting M, Koralewski P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011; 22:1535–1546.
27. Cremolini C, Di Bartolomeo M, Amatu A, Antoniotti C, Moretto R, Berenato R, Perrone F, Tamborini E, Aprile G, Lonardi S, Sartore-Bianchi A, Fontanini G, Milione M, et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann Oncol. 2015; 26:2092–97.
28. Sepulveda AR, Hamilton SR, Allegra CJ, Grody W, Cushman-Vokoun AM, Funkhouser WK, Kopetz SE, Lieu C, Lindor NM, Minsky BD, Monzon FA, Sargent DJ, Singh VM, et al. Molecular biomarkers for the evaluation of colorectal cancer: guidelines from the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and the American Society of Clinical Oncology. J Clin Oncol. 2017; 35:1453–86.