
INTRODUCTION
Tumours not only interrupt local tissue function but have been shown to produce an additional systemic effect on the entire metabolism of the body. A current paradigm of cancer pathology has inflammatory processes as central to many of the paraneoplastic symptoms observed in cancer patients, such as cancer cachexia, which is observed in more than 50% of advanced cancer patients [1, 2]. It is characterised by progressive weight loss associated with skeletal muscle atrophy and depletion of adipose tissue, irrespective of nutritional intake [3]. Lower performance status and global quality of life with higher fatigue scores and pain have been observed in patients who lost weight with gastrointestinal cancer or non-small cell lung cancer [4, 5]. At least 20% of deaths are caused by cachexia in part due to impaired respiratory or cardiac function [6, 7]. Moreover, the loss of body weight has been observed in a number of patients with various cancers with median survival of less than 6 months, and hence is an indicator of terminal disease [8]. Weight loss has also been associated with poor prognosis, higher recurrence and worse response to chemotherapy [9–11]. Importantly, it has been shown that the attenuation of weight loss would significantly improve overall survival in patients with gastrointestinal cancer [12]. Taken together, cancer cachexia significantly impairs cancer patient quality of life and prognosis, and reversal of this condition would be beneficial to the management of malignancy in general.
An effective therapeutic option is currently not available to manage cancer cachexia. This is partly due to a limited understanding of the molecular mechanisms underlying this disease process, highlighting the need to better define mediators and factors implicated in the pathology. Research in the field has largely been driven by candidate gene approaches in which specific molecules are selected for study based on their cellular roles. However, it is becoming apparent that cancer cachexia is a complex multi-factorial disease which may involve multiple pathways, and candidate molecular approaches may therefore have limited value in providing insights for overall molecular interactions which are likely to be networked. Moreover, paucity of data on the effects of cancer cachexia on heart muscle highlights the need to investigate this tissue in parallel to skeletal muscle for the purposes of identifying tissue-specific or global muscle responses to cancer cachexia.
We have recently undertaken a genomics approach to studying the multiple changes occurring in muscle during cancer cachexia [13]. While we were able to identify a broad variety of changes at the genomic level, whether these translate into functional changes depends on the realisation of downstream effects. Protein expression levels provide further information on whether the effects observed at the gene level are in fact translated, together with experimental validation of the observed genomic changes. Differential proteomics represents an effective analytical tool to gather information on multiple proteins in a single experimental setup. So far few studies have utilised this approach to investigate aspects of cancer: one study focused on the identification of urine biomarkers associated with cancer cachexia in patients and the other study reported the proteomic profile in skeletal muscle of cachectic rats [14, 15]. These MALDI-TOF experiments were either indirect (urinary proteins) or else focussed on protein oxidation/carbonylation specifically and precluded analysis of differentially expressed proteins. Two very recent studies on proteomic changes in cachexic muscle used label-free approaches [16, 17]. To our knowledge the current work represents the first use of iTRAQ quantitative mass spectrometry to investigate the proteomic profiles of cachexia affected skeletal (fast [gastrocnemius] and slow [soleus] twitch) and heart muscles. Isobaric tags for relative and absolute quantification (iTRAQ) followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) were used [18]. The iTRAQ approach allows multiple protein changes to be revealed at once as well as providing a validation tool for observed changes at the morphological and gene levels. Use of isobaric tags as internal standards provides higher data accuracy and reliability, and has been successfully applied to studies of proteomic changes associated with liver regeneration and tumour phenotypes of different cancers [19–22].
RESULTS
Proteomics analysis of different muscle types
Proteomics analysis identified and quantified more than 300 proteins, with > 95% confidence in correct sequence identification (unused score of > 1.3 and FDR of 1%), in each of two iTRAQ experiments. Spectral, peptide and protein summary data from the two experiments are shown in Table 1, and the full lists of proteins relating to each of the muscle types, gastrocnemius, soleus and heart are shown in the Supplementary Materials (Supplementary Tables 1, 2 and 3). Approximately 5–10% of proteins from the full lists were significantly differentially expressed, at a p-value of 0.05 (27 proteins from gastrocnemius, 34 proteins from soleus and 24 proteins from heart), and are shown in Tables 2, 3 and 4. When the full protein lists across muscle types were compared, 199 proteins were common across all three groups, 75 were common to gastrocnemius and heart, 83 were common to soleus and gastrocnemius, and no proteins were unique to just a single muscle type (Figure 1A). However, when a similar comparison was made based on the much shorter significantly differentially expressed protein lists (i.e., those shown in Tables 2, 3 and 4), then there was only a single protein that was common across all three muscle types (myosin-4 or Myh4), with 3–6 proteins common across pairs of muscle types, and the majority of differentially expressed proteins unique to each muscle type (Figure 1B).
Table 1: Summary data from Protein Pilot v5.0 for the two ITRAQ proteomics experiments
iTRAQ Experiment 1 |
iTRAQ Experiment 2 |
|
|---|---|---|
Spectra |
50,805 |
30,437 |
Distinct peptides |
7,388 |
5,917 |
Proteins (before grouping) |
2,659 |
1,434 |
Proteins (after grouping) |
369 |
329 |
Protein ID yield with Local FDR (1%) |
357 |
331 |
Peptide ID yield with Local FDR (1%) |
4,333 |
3,196 |
Spectral ID yield with Local FDR (1%) |
36,171 |
19,312 |
Data was processed with Protein Pilot™ 5.0 software using the Paragon™ Algorithm for the identification of spectra, peptides and proteins. The data represents a protein detection threshold cutoff > 1.3 (unused score), representing a confidence of correct sequence identification of > 95%.
Table 2: Proteins dysregulated in mouse gastrocnemius muscle during cancer cachexia, quantified by iTRAQ based discovery proteomics
Unused |
%Cov(95) |
gi Accession |
Protein Name (protein term for STRING bioinformatics analysis) |
Biol Rep |
Peptides (95%) |
cachexia/control iTRAQ tag area ratio |
P Val |
|---|---|---|---|---|---|---|---|
6.27 |
9.565 |
160358829 |
hemopexin precursor (Hpx) |
BR3 |
4 |
1.935 |
0.00847 |
6.27 |
9.565 |
BR2 |
4 |
1.603 |
0.00119 |
||
25.65 |
36.520 |
BR5 |
16 |
1.565 |
0.00466 |
||
65.16 |
77.450 |
439253893 |
actin, alpha skeletal muscle (Acta1) |
BR2 |
254 |
1.724 |
0.00718 |
65.16 |
77.450 |
BR3 |
254 |
1.515 |
0.04439 |
||
16.02 |
23.400 |
568909414 |
myosin-binding protein H isoform X1 (MybpH) |
BR1 |
8 |
1.539 |
0.00461 |
16.02 |
23.400 |
BR3 |
8 |
1.233 |
0.03045 |
||
61.71 |
8.521 |
568913099 |
nebulin isoform X9 (Nebl) |
BR3 |
38 |
1.500 |
0.00023 |
61.71 |
8.521 |
BR2 |
38 |
1.367 |
2.39E-07 |
||
61.71 |
8.521 |
BR1 |
38 |
1.257 |
3.21E-05 |
||
11.62 |
20.640 |
755519288 |
troponin T, slow skeletal muscle isoform X2 (Tnnt1) |
BR3 |
8 |
1.282 |
0.02597 |
11.62 |
20.640 |
BR2 |
8 |
1.245 |
0.03467 |
||
27.66 |
72.180 |
11875203 |
tropomyosin beta chain isoform Tpm2.2st [Tpm2] |
BR5 |
49 |
1.268 |
7.61E-05 |
123.22 |
92.610 |
BR3 |
116 |
1.136 |
0.00018 |
||
13.92 |
52.380 |
68226433 |
histone H2B type 2-B (Hist2h2bb) |
BR3 |
9 |
1.250 |
0.00047 |
13.92 |
52.380 |
BR1 |
9 |
1.207 |
0.00161 |
||
163.01 |
77.090 |
205830428 |
myosin heavy chain Iia (Myh2) |
BR3 |
604 |
1.208 |
0.04663 |
163.01 |
77.090 |
BR1 |
604 |
1.172 |
0.02887 |
||
163.01 |
77.090 |
BR2 |
604 |
1.134 |
0.00084 |
||
349.71 |
67.970 |
67189167 |
myosin-4 [Myh4] |
BR5 |
254 |
1.189 |
2.01E-06 |
517.79 |
79.940 |
BR1 |
917 |
1.253 |
7.19E-05 |
||
517.79 |
79.940 |
BR2 |
917 |
1.073 |
0.01459 |
||
26.32 |
61.210 |
300069034 |
PDZ and LIM domain protein 5 isoform ENH4 (Pdlim5) |
BR3 |
17 |
1.199 |
0.00136 |
26.32 |
61.210 |
BR2 |
17 |
1.146 |
0.02435 |
||
32.06 |
0.891 |
755499204 |
titin isoform X3 (Ttn) |
BR1 |
23 |
1.177 |
0.03356 |
32.06 |
0.891 |
BR2 |
23 |
1.155 |
0.00572 |
||
204.45 |
67.600 |
18859641 |
myosin-7 (Myh7) |
BR2 |
348 |
1.176 |
7.31E-09 |
204.45 |
67.600 |
BR1 |
348 |
1.111 |
0.00018 |
||
136.63 |
65.380 |
568947734 |
myosin-binding protein C, fast-type isoform X1 [Mybpc2] |
BR1 |
79 |
1.159 |
5.04E-06 |
85.11 |
39.260 |
BR5 |
41 |
1.166 |
0.00515 |
||
75.05 |
61.190 |
157951643 |
alpha-actinin-2 [Actn2] |
BR2 |
66 |
1.074 |
0.00484 |
75.05 |
61.190 |
BR1 |
66 |
1.091 |
0.02410 |
||
97.56 |
80.260 |
163310765 |
serum albumin precursor [Alb] |
BR4 |
79 |
0.76 |
1.04E-09 |
95.64 |
79.610 |
BR1 |
65 |
0.94 |
0.05683 |
||
34.76 |
41.040 |
254553458 |
glucose-6-phosphate isomerase (Gpi1) |
BR2 |
31 |
0.909 |
0.00992 |
34.76 |
41.040 |
BR1 |
31 |
0.855 |
0.02262 |
||
36.56 |
43.710 |
33563250 |
desmin (Des) |
BR1 |
21 |
0.902 |
0.00300 |
36.56 |
43.710 |
BR2 |
21 |
0.894 |
0.00139 |
||
18.09 |
66.920 |
6753810 |
fatty acid-binding protein, heart (Fabp3) |
BR4 |
26 |
0.891 |
0.04207 |
15.38 |
63.910 |
BR2 |
16 |
0.874 |
0.01664 |
||
65.9 |
81.680 |
6679937 |
glyceraldehyde-3-phosphate dehydrogenase isoform 2 (Gapdh) |
BR2 |
96 |
0.888 |
0.03505 |
58.71 |
84.080 |
BR4 |
78 |
0.804 |
0.03664 |
||
52.99 |
48.750 |
227330633 |
phosphoglucomutase-2 (Pgm2) |
BR2 |
32 |
0.888 |
0.00047 |
52.99 |
48.750 |
BR1 |
32 |
0.881 |
0.00351 |
||
63.56 |
45.130 |
18079339 |
aconitate hydratase, mitochondrial precursor (Aco2) |
BR1 |
35 |
0.882 |
0.00050 |
63.56 |
45.130 |
BR2 |
35 |
0.882 |
0.00038 |
||
19.28 |
37.040 |
568912066 |
ATP synthase subunit gamma, mitochondrial isoform X1 (Atp5c1) |
BR2 |
14 |
0.878 |
0.00898 |
19.41 |
33.670 |
BR5 |
11 |
0.819 |
0.00172 |
||
37.66 |
67.750 |
31982186 |
malate dehydrogenase, mitochondrial precursor (Mdh2) |
BR1 |
26 |
0.875 |
0.00132 |
37.66 |
67.750 |
BR2 |
26 |
0.788 |
2.89E-06 |
||
138.32 |
68.170 |
6755256 |
glycogen phosphorylase, muscle form (Pygm) |
BR1 |
103 |
0.863 |
9.37E-05 |
138.32 |
68.170 |
BR2 |
103 |
0.840 |
1.72E-08 |
||
34.34 |
54.360 |
148747424 |
ADP/ATP translocase 1 (Slc25a4) |
BR1 |
23 |
0.826 |
0.00918 |
34.34 |
54.360 |
BR2 |
23 |
0.785 |
0.00854 |
||
12.45 |
32.390 |
568889869 |
ATP synthase subunit O, mitochondrial [Atp5o] |
BR2 |
7 |
0.734 |
0.00203 |
10.26 |
27.700 |
BR4 |
6 |
0.802 |
0.00818 |
||
9.98 |
55.920 |
6679078 |
nucleoside diphosphate kinase B [Nme2] |
BR1 |
8 |
0.752 |
0.00190 |
9.98 |
55.920 |
BR2 |
8 |
0.800 |
0.01394 |
Two technical replicates (n = 2) and five biological replicates (n = 5) therefore n = 10 samples were used and a total of 377 proteins identified and quantified by 2D LCMSMS (QStar Elite) and ProteinPilot v5.0 analysis. The proteins which were differentially expressed in a consistent direction in the majority of biological replicates, and that were also statistically significant in two or more biological replicates, are listed in this table (27 proteins in all). The full table of identified proteins is provided in Supplementary Table 1. This short-list of differentially expressed proteins was processed using the STRING v10 WEB based bioinformatics tool, and the identified clusters and protein enrichment shown in Figure 2A and Table 5 respectively.
Table 3: Proteins dysregulated in mouse soleus muscle during cancer cachexia, quantified by iTRAQ based discovery proteomics
Unused |
%Cov(95) |
gi Accession |
Name (protein term for STRING bioinformatics analysis) |
Peptides (95%) |
cachexia/control iTRAQ tag area ratio |
P Val |
|---|---|---|---|---|---|---|
2.33 |
1.648 |
167555029 |
fibrinogen alpha chain isoform 1 precursor (Fga) |
1 |
7.622 |
0.03395 |
163.01 |
77.090 |
205830428 |
myosin heavy chain Iia (Myh2) |
604 |
2.317 |
7.33E-09 |
36.51 |
66.550 |
116517336 |
four and a half LIM domains protein 1 isoform 2 (Fhl1) |
26 |
2.190 |
2.49E-06 |
204.45 |
67.600 |
18859641 |
myosin-7 (Myh7) |
348 |
2.179 |
7.61E-14 |
10.47 |
16.640 |
6754254 |
heat shock protein HSP 90-alpha (Hsp90aa1) |
15 |
2.108 |
0.00198 |
5.09 |
21.650 |
7304987 |
cysteine and glycine-rich protein 3 (Csrp3) |
3 |
1.949 |
0.04617 |
35.4 |
30.930 |
84875544 |
LIM domain-binding protein 3 isoform c (Ldb3) |
29 |
1.947 |
0.00235 |
52.65 |
26.890 |
568966329 |
myosin-binding protein C, slow-type isoform X14 (Mybpc2) |
32 |
1.878 |
6.31E-07 |
6.76 |
10.760 |
755517050 |
heterogeneous nuclear ribonucleoproteins A2/B1 isoform X1 (Hnrnpa2b1) |
5 |
1.766 |
0.00248 |
40.83 |
9.432 |
568939784 |
filamin-C isoform X1 (Flnc) |
23 |
1.755 |
0.00031 |
36.56 |
43.710 |
33563250 |
desmin (Des) |
21 |
1.648 |
4.46E-05 |
7.46 |
22.600 |
6680836 |
calreticulin precursor (Calr) |
5 |
1.493 |
0.01178 |
13.92 |
52.380 |
68226433 |
histone H2B type 2-B (Hist2h2bb) |
9 |
1.487 |
0.00297 |
55.41 |
77.090 |
82524274 |
myosin-1 (Myh1) |
648 |
1.483 |
0.00235 |
32.06 |
0.891 |
755499204 |
titin isoform X3 (Ttn) |
23 |
1.333 |
0.00746 |
41.06 |
37.460 |
31981690 |
heat shock cognate 71 kDa protein (Hspa8) |
24 |
1.330 |
0.02401 |
13.5 |
2.518 |
254675244 |
plectin isoform 1 (Plec) |
9 |
1.324 |
0.02871 |
73.3 |
90.850 |
256000780 |
tropomyosin alpha-1 chain isoform Tpm1.1st (Tpm1) |
134 |
1.316 |
0.00037 |
517.79 |
79.940 |
67189167 |
myosin-4 (Myh4) |
917 |
1.278 |
1.73E-05 |
27.09 |
65.000 |
6678371 |
troponin C, skeletal muscle (Tnnc2) |
39 |
1.254 |
0.02125 |
127.94 |
42.870 |
568953176 |
myomesin-2 isoform X2 (Myom2) |
78 |
1.208 |
0.00377 |
136.63 |
65.380 |
568947734 |
myosin-binding protein C, fast-type isoform X1 (Mybpc2) |
79 |
1.207 |
0.00239 |
131.92 |
55.140 |
568949470 |
sarcoplasmic/endoplasmic reticulum calcium ATPase 1 isoform X1 (Atp2a1) |
106 |
1.160 |
0.03768 |
13.31 |
20.390 |
568966988 |
phosphate carrier protein, mitochondrial isoform X1 (Slc25a3) |
9 |
0.751 |
0.01161 |
9.46 |
13.540 |
18700024 |
isocitrate dehydrogenase 3, beta subunit (Idh3b) |
5 |
0.741 |
0.02699 |
12.9 |
20.000 |
6679261 |
pyruvate dehydrogenase E1 component subunit alpha, somatic form, mitochondrial precursor (Pdha1) |
9 |
0.740 |
0.02822 |
31.64 |
24.640 |
33859811 |
trifunctional enzyme subunit alpha, mitochondrial precursor (Hadha) |
15 |
0.728 |
0.00343 |
53.27 |
95.270 |
7949078 |
myosin regulatory light chain 2, skeletal muscle isoform (Mylpf) |
114 |
0.717 |
0.00636 |
20.82 |
78.910 |
498752597 |
hemoglobin subunit beta-1 (Hbb-b1) |
18 |
0.646 |
0.00448 |
18.25 |
44.170 |
6755963 |
voltage-dependent anion-selective channel protein 1 (Vdac1) |
9 |
0.625 |
0.03542 |
3.5 |
20.930 |
13385090 |
cytochrome c oxidase subunit 6B1 (Cox6b1) |
3 |
0.589 |
0.00206 |
40.28 |
88.960 |
255708425 |
myoglobin (Mb) |
45 |
0.387 |
0.00094 |
13.55 |
53.400 |
568893484 |
histone H4 (Hist2h4) |
12 |
0.305 |
0.00360 |
26.34 |
77.720 |
568936906 |
myosin regulatory light chain 2, ventricular/cardiac muscle isoform isoform X1 (Myl2) |
21 |
0.282 |
1.15E-06 |
Two technical replicates (n = 2) and a single biological replicate (n = 1), therefore n = 2 samples were used. A total of 285 proteins were identified and quantified by LCMSMS (QStar Elite) and ProteinPilot v5.0 analysis. Significantly dysregulated proteins are listed in this table (34 proteins). The full table of identified/quantified proteins is provided in Supplementary Table 2. This list of proteins was also processed using the STRING v10 WEB based bioinformatics tool, and the identified clusters and protein enrichment shown in Figure 2B and Table 5 respectively.
Table 4: Proteins dysregulated in mouse heart muscle during cancer cachexia, quantified by iTRAQ based discovery proteomics
Unused |
%Cov(95) |
gi Accession |
Protein Name (protein term for STRING bioinformatics analysis) |
Biol Rep |
Peptides (95%) |
cachexia/control iTRAQ tag area ratio |
P Val |
|---|---|---|---|---|---|---|---|
2.71 |
4.358 |
19527078 |
fibrinogen gamma chain precursor (Fgg) |
BR2 |
2 |
5.434 |
0.02153 |
3.87 |
10.43 |
7304875 |
alpha-2-HS-glycoprotein isoform 1 precursor (Ahsg) |
BR2 |
3 |
1.698 |
0.02759 |
42.25 |
37.59 |
20330802 |
serotransferrin precursor (Tf) |
BR2 |
24 |
1.558 |
1.66E-06 |
55.67 |
96.36 |
568991776 |
parvalbumin alpha isoform X1 (Pvalb) |
BR1 |
60 |
1.556 |
1.46E-05 |
5.38 |
36.59 |
568973615 |
cytochrome b-c1 complex subunit 8 isoform X1 (Uqcrq) |
BR1 |
5 |
1.539 |
0.02216 |
18.58 |
2.393 |
568913135 |
nebulin isoform X26 (Nebl) |
BR1 |
11 |
1.445 |
2.77E-05 |
7.68 |
22.5 |
568384815 |
four and a half LIM domains protein 1 isoform 3 (Fhl1) |
BR1 |
5 |
1.393 |
0.04835 |
22.73 |
8.355 |
568924485 |
glycogen debranching enzyme isoform X1 (Agl) |
BR1 |
12 |
1.334 |
0.03097 |
25.23 |
62.09 |
6678391 |
troponin I, fast skeletal muscle (Tnni2) |
BR1 |
21 |
1.310 |
0.02100 |
7.9 |
4.348 |
110347469 |
alpha-2-macroglobulin precursor (A2m) |
BR2 |
6 |
1.306 |
0.03769 |
349.71 |
67.97 |
67189167 |
myosin-4 (Myh4) |
BR1 |
254 |
1.290 |
1.01E-08 |
22.03 |
65 |
6678371 |
troponin C, skeletal muscle (Tnnc2) |
BR1 |
31 |
1.263 |
0.00660 |
22.03 |
65 |
6678371 |
BR2 |
31 |
1.175 |
0.03371 |
|
44.94 |
95.27 |
7949078 |
myosin regulatory light chain 2, skeletal muscle isoform (Mylpf) |
BR1 |
63 |
1.223 |
0.00132 |
90.83 |
50.89 |
7304855 |
alpha-actinin-3 (Actn3) |
BR1 |
46 |
1.216 |
0.00119 |
34.69 |
64.88 |
226958349 |
triosephosphate isomerase (Tpi1) |
BR2 |
26 |
1.180 |
0.02520 |
119.92 |
71.97 |
6755256 |
glycogen phosphorylase, muscle form (Pygm) |
BR1 |
106 |
1.165 |
0.00628 |
77.95 |
33.24 |
568953176 |
myomesin-2 isoform X2 (Myom2) |
BR1 |
40 |
1.147 |
0.02854 |
77.95 |
33.24 |
568953176 |
BR2 |
40 |
1.104 |
0.04321 |
|
71.57 |
55.88 |
6680748 |
ATP synthase subunit alpha, mitochondrial precursor (Atp5a1) |
BR1 |
45 |
0.813 |
0.01146 |
71.57 |
55.88 |
6680748 |
BR2 |
45 |
0.896 |
0.02872 |
|
17.54 |
28.21 |
6679261 |
pyruvate dehydrogenase E1 component subunit alpha, somatic form, mitochondrial precursor (Pdha1) |
BR2 |
10 |
0.861 |
0.00378 |
31.14 |
33.73 |
226823367 |
fumarate hydratase, mitochondrial precursor (Fh) |
BR2 |
19 |
0.815 |
0.00233 |
10.46 |
46.09 |
9845265 |
ubiquitin-60S ribosomal protein L40 (Uba52) |
BR1 |
6 |
0.722 |
0.00579 |
10.46 |
46.09 |
9845265 |
BR2 |
6 |
0.813 |
0.00761 |
|
13.34 |
55.92 |
6679078 |
nucleoside diphosphate kinase B (Nme2) |
BR1 |
9 |
0.752 |
0.00838 |
13.34 |
55.92 |
6679078 |
BR2 |
9 |
0.719 |
0.02891 |
|
4.08 |
27.18 |
31980744 |
ATP synthase subunit g, mitochondrial (Atp5l) |
BR2 |
2 |
0.676 |
0.04705 |
27.32 |
73.27 |
568936906 |
myosin regulatory light chain 2, ventricular/cardiac muscle isoform isoform X1 (Myl2) |
BR1 |
30 |
0.671 |
0.00436 |
Two technical replicates (n = 2) and two biological replicates (n = 2), therefore n = 4 samples were used and a total of 278 proteins identified and quantified by 2D LCMSMS (QStar Elite) and ProteinPilot v5.0 analysis. The proteins which were significantly differentially expressed and in a consistent direction, in both biological replicates, are listed in this table (24 proteins). The full table of identified proteins is provided in Supplementary Table 3. This list of proteins was also processed using the STRING v10 WEB based bioinformatics tool, and the identified clusters and protein enrichment shown in Figure 2C and Table 5 respectively.
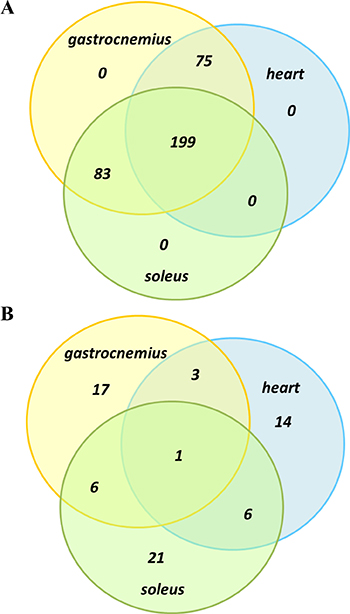
Figure 1: Venn diagram comparison of protein overlap between muscle groups. Full protein lists, as shown in Supplementary Tables 1, 2 and 3, were compared (A) as well as significantly differentially expressed proteins lists, as shown in Tables 2, 3 and 4 (B).
Bioinformatics analysis
The significantly differentially expressed protein lists (i.e., Tables 2, 3 and 4) were used to explore enrichment of gene ontology (GO) biological processes and molecular functions (Table 5), and protein clusters (Figure 2). Three main processes or functions emerged across muscle types; muscle contraction, metabolites/energy and cytoskeletal protein binding (Table 5). A few processes/functions were uniquely identified in just a single muscle type, including; unfolded protein binding (soleus) and complement and coagulation cascades (heart). Protein group clustering is shown in Figure 4 for each muscle type, with the specific protein involved in enriched biological processes and molecular functions highlighted. In general, muscle contraction and cytoskeletal binding proteins were upregulated in the C26 cachectic mice, whereas the energy/metabolism protein group was generally downregulated in the C26 mice (Tables 2, 3, 4). Ingenuity Pathway Analysis (IPA) was used to explore relationships of the differentially expressed proteins to each other and to diseases and functions (Figure 3). The top ID associated network identified by IPA in each of the muscle types was “skeletal and muscular system development and function”. Other networks were also identified for each of the specific muscle types, including; organismal injury and abnormalities in gastrocnemius (IPA statistically significant diseases and functions sub-categories frequently show cancers of various kinds). For soleus, additional significant ID associated networks included cardiovascular system development and function and embryonic development. For heart, additional significant ID associated network functions included cardiovascular system development and function and cellular assembly and organization.
Table 5: Enrichment of biological processes, molecular functions and KEGG pathways of protein lists that are differentially expressed in mouse gastrocnemius, soleus and heart muscle during cancer cachexia
GO id |
Term |
Number of Genes |
p-value |
p-value FDR |
p-value Bonferroni |
||
Gastrocnemius |
Biological Process Enrichment |
GO:0006936 |
muscle contraction |
8 |
2.59E-12 |
3.48E-08 |
3.48E-08 |
GO:0003012 |
muscle system process |
8 |
2.20E-11 |
1.48E-07 |
2.96E-07 |
||
GO:0006091 |
generation of precursor metabolites and energy |
6 |
6.77E-08 |
3.04E-04 |
9.11E-04 |
||
GO:0044724 |
single-organism carbohydrate catabolic process |
4 |
8.49E-07 |
2.85E-03 |
1.14E-02 |
||
GO:0016052 |
carbohydrate catabolic process |
4 |
1.31E-06 |
3.52E-03 |
1.76E-02 |
||
Molecular Function Enrichment |
GO:0008092 |
cytoskeletal protein binding |
11 |
1.46E-10 |
6.89E-07 |
6.89E-07 |
|
GO:0003779 |
actin binding |
7 |
8.28E-08 |
1.95E-04 |
3.89E-04 |
||
GO:0005200 |
structural constituent of cytoskeleton |
4 |
4.85E-07 |
7.60E-04 |
2.28E-03 |
||
GO:0046933 |
proton-transporting ATP synthase activity, rotational mechanism |
2 |
3.81E-05 |
4.48E-02 |
1.79E-01 |
||
KEGG Pathways Enrichment |
4530 |
Tight junction |
4 |
1.84E-05 |
5.24E-03 |
5.24E-03 |
|
5410 |
Hypertrophic cardiomyopathy (HCM) |
3 |
1.30E-04 |
1.46E-02 |
3.69E-02 |
||
5414 |
Dilated cardiomyopathy |
3 |
1.55E-04 |
1.46E-02 |
4.39E-02 |
||
Soleus |
Biological Process Enrichment |
GO:0003012 |
muscle system process |
13 |
1.91E-19 |
2.57E-15 |
2.57E-15 |
GO:0006936 |
muscle contraction |
12 |
5.93E-19 |
3.98E-15 |
7.97E-15 |
||
GO:0006941 |
striated muscle contraction |
6 |
8.75E-10 |
3.92E-06 |
1.18E-05 |
||
GO:0030029 |
actin filament-based process |
9 |
2.10E-09 |
7.05E-06 |
2.82E-05 |
||
GO:0007517 |
muscle organ development |
7 |
6.10E-08 |
1.64E-04 |
8.20E-04 |
||
GO:0061061 |
muscle structure development |
8 |
1.22E-07 |
2.73E-04 |
1.64E-03 |
||
GO:0060048 |
cardiac muscle contraction |
4 |
5.49E-07 |
9.98E-04 |
7.38E-03 |
||
GO:0030239 |
myofibril assembly |
4 |
6.02E-07 |
9.98E-04 |
8.09E-03 |
||
GO:0055002 |
striated muscle cell development |
5 |
6.68E-07 |
9.98E-04 |
8.98E-03 |
||
GO:0055001 |
muscle cell development |
5 |
1.25E-06 |
1.52E-03 |
1.68E-02 |
||
GO:0055003 |
cardiac myofibril assembly |
3 |
1.39E-06 |
1.52E-03 |
1.86E-02 |
||
GO:0003008 |
system process |
14 |
1.43E-06 |
1.52E-03 |
1.92E-02 |
||
GO:0060047 |
heart contraction |
4 |
1.47E-06 |
1.52E-03 |
1.97E-02 |
||
GO:0055008 |
cardiac muscle tissue morphogenesis |
4 |
1.94E-06 |
1.86E-03 |
2.61E-02 |
||
GO:0003015 |
heart process |
4 |
2.07E-06 |
1.86E-03 |
2.79E-02 |
||
GO:0070252 |
actin-mediated cell contraction |
3 |
2.94E-06 |
2.47E-03 |
3.95E-02 |
||
GO:0014706 |
striated muscle tissue development |
6 |
3.18E-06 |
2.52E-03 |
4.28E-02 |
||
GO:0060415 |
muscle tissue morphogenesis |
4 |
3.61E-06 |
2.66E-03 |
4.85E-02 |
||
Molecular Function Enrichment |
GO:0005200 |
structural constituent of cytoskeleton |
7 |
1.49E-12 |
7.00E-09 |
7.00E-09 |
|
GO:0003779 |
actin binding |
10 |
3.19E-11 |
7.49E-08 |
1.50E-07 |
||
GO:0008092 |
cytoskeletal protein binding |
12 |
1.55E-10 |
2.42E-07 |
7.27E-07 |
||
GO:0008307 |
structural constituent of muscle |
4 |
9.90E-09 |
1.13E-05 |
4.66E-05 |
||
GO:0005198 |
structural molecule activity |
9 |
1.21E-08 |
1.13E-05 |
5.67E-05 |
||
GO:0032403 |
protein complex binding |
9 |
7.30E-06 |
5.73E-03 |
3.44E-02 |
||
GO:0042805 |
actinin binding |
3 |
1.10E-05 |
7.37E-03 |
5.16E-02 |
||
GO:0031433 |
telethonin binding |
2 |
1.31E-05 |
7.69E-03 |
6.15E-02 |
||
GO:0051371 |
muscle alpha-actinin binding |
2 |
6.08E-05 |
3.18E-02 |
2.86E-01 |
||
GO:0005516 |
calmodulin binding |
4 |
1.01E-04 |
4.75E-02 |
4.75E-01 |
||
GO:0051082 |
unfolded protein binding |
3 |
1.15E-04 |
4.92E-02 |
5.42E-01 |
||
KEGG Pathways Enrichment |
4530 |
Tight junction |
6 |
4.48E-08 |
1.27E-05 |
1.27E-05 |
|
5410 |
Hypertrophic cardiomyopathy (HCM) |
5 |
1.49E-07 |
1.89E-05 |
4.23E-05 |
||
5414 |
Dilated cardiomyopathy |
5 |
2.00E-07 |
1.89E-05 |
5.68E-05 |
||
4260 |
Cardiac muscle contraction |
4 |
4.75E-06 |
3.38E-04 |
1.35E-03 |
||
4612 |
Antigen processing and presentation |
3 |
1.71E-04 |
9.73E-03 |
4.87E-02 |
||
Heart |
Biological Process Enrichment |
GO:0006936 |
muscle contraction |
6 |
4.92E-09 |
6.61E-05 |
6.61E-05 |
GO:0003012 |
muscle system process |
6 |
2.41E-08 |
1.62E-04 |
3.24E-04 |
||
GO:0044724 |
single-organism carbohydrate catabolic process |
4 |
5.17E-07 |
2.32E-03 |
6.96E-03 |
||
GO:0016052 |
carbohydrate catabolic process |
4 |
7.98E-07 |
2.68E-03 |
1.07E-02 |
||
Molecular Function Enrichment |
GO:0003779 |
actin binding |
5 |
2.03E-05 |
9.53E-02 |
9.53E-02 |
|
KEGG Pathways Enrichment |
4530 |
Tight junction |
4 |
1.13E-05 |
3.21E-03 |
3.21E-03 |
|
4610 |
Complement and coagulation cascades |
3 |
6.97E-05 |
9.89E-03 |
1.98E-02 |
Proteins were identified and quantified by iTRAQ based discovery proteomics (see Tables 2, 3 and 4) and processed using the STRING v10 WEB based bioinformatics tool to identify protein enrichment categories. Cellular compartment enrichment is shown in the supplementary materials (Supplementary Table 4).
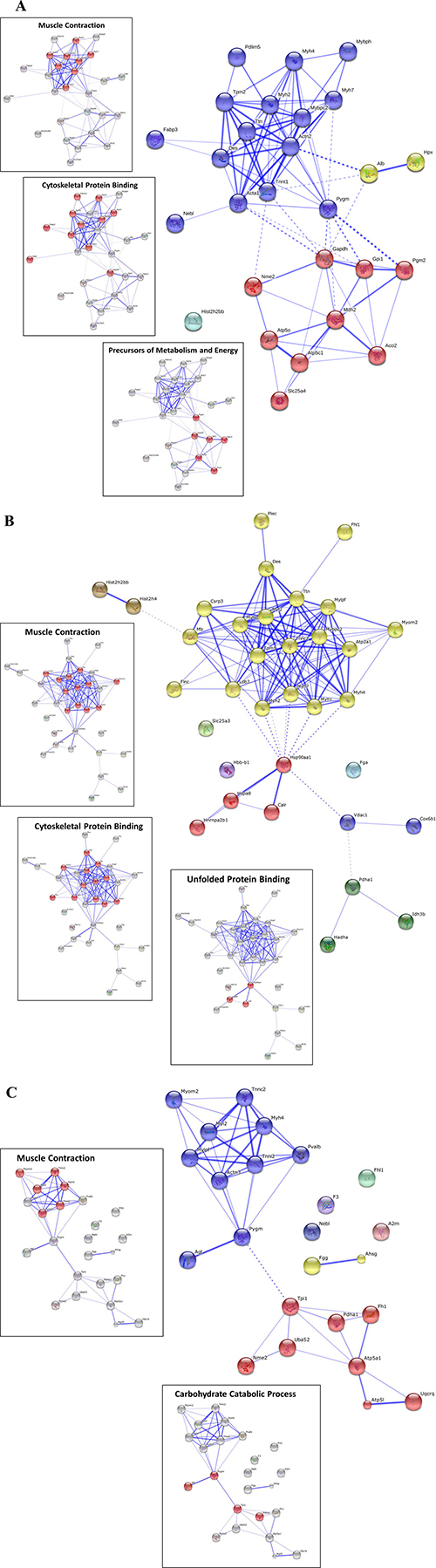
Figure 2: STRING v10 bioinformatics analysis of proteins dysregulated during cancer cachexia in gastrocnemius (27 proteins) (A), soleus (34 proteins) (B) and heart (24 proteins) (C) muscle. MCL 2 point clustering was used for the STRING analysis, and the differentially expressed proteins used for clustering are shown in Tables 2, 3 and 4. The insets show some of the significantly enriched biological processes and molecular functions highlighted in red against grey clusters (the full list of significant enrichment is shown in Table 5).
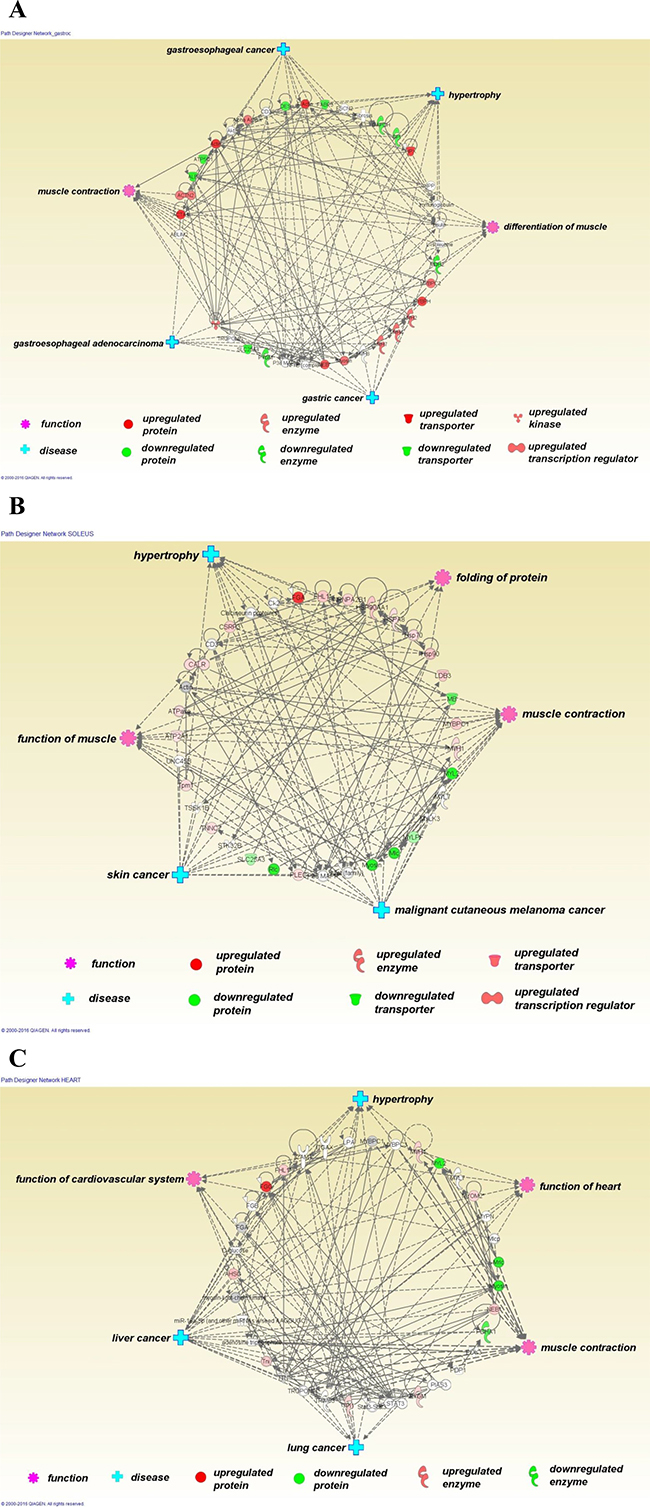
Figure 3: Ingenuity Pathway Analysis (Qiagen 2015) was used to explore relationships of the differentially expressed proteins to each other and to diseases and functions. Differentially expressed proteins (derived from the lists shown in Tables 2, 3 and 4) are shown in a circular layout (upregulated proteins in red, downregulated proteins in green and non-coloured molecules predicted by the software to have interactions). Six statistically significantly related functions (pink cog) and diseases (blue cross) are displayed at points around the central circle. The top ID associated network identified by IPA for each of the muscle types is shown in this figure; gastrocnemius (A), soleus (B) and heart (C). For gastrocnemius, the most significant ID associated network functions (IPA top network score = 53) are skeletal and muscular system development and function, cardiovascular disease, organismal injury and abnormalities (IPA statistically significant diseases and functions sub-categories frequently show cancers of various kinds). For soleus, the most significant ID associated network functions (IPA top network score = 44) are skeletal and muscular system development and function, cardiovascular system development and function, embryonic development. For heart, the most significant ID associated network functions (IPA top network score = 21) were skeletal and muscular system development and function, cardiovascular system development and function, cellular assembly and organization.
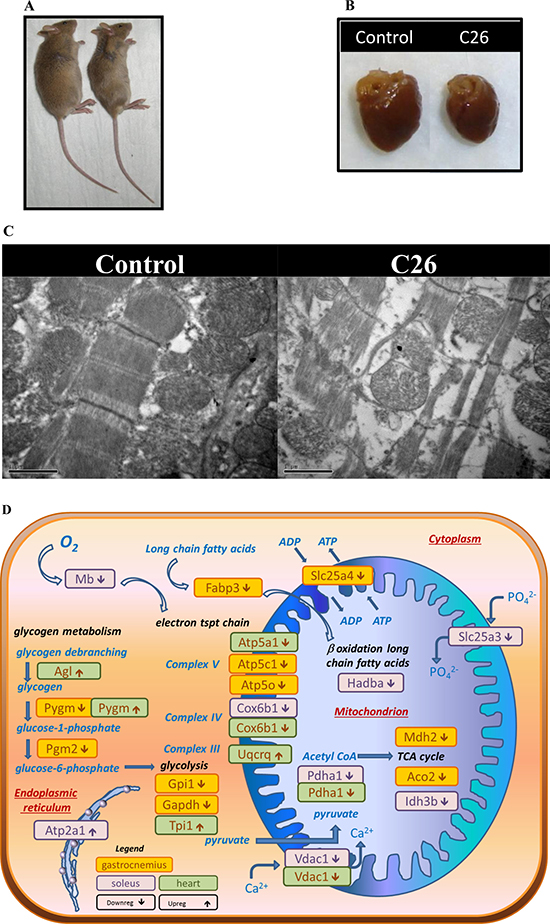
Figure 4: (A) Tumour-mediated effect on whole body size. (B) Tumour-mediated effect on heart. (C) Electron micrographs showing structural and mitochondrial alterations in cardiac muscle of NTB control and C26-bearing mice. Increased intercellular spaces were seen and appeared to be associated with a loss of myofibrils and reduced density of mitochondrial matrix (Magnification = 25,000×). (D) Schematic of the role of muscle proteins differentially expressed during cancer-induced cachexia in the cellular energy/metabolism pathways. Differentially expressed proteins are indicated as small rectangles, with colour representing the muscle type in which differential expression was observed (yellow = gastrocnemius, purple = soleus, green = heart) and the arrows indicating up ( ) or down (
) or down ( ) regulation. The displayed proteins were those identified in the proteomics experiments and listed in Tables 2, 3 and 4.
) regulation. The displayed proteins were those identified in the proteomics experiments and listed in Tables 2, 3 and 4.
Electron microscopy of cardiac muscle
Sarcomere structure
Disrupted ultrastructural features were evident in cardiac muscle from C26-bearing mice. Hearts from C26-bearing mice were significantly smaller and the absolute weight was less than hearts from NTB controls (Table 6, Figures 4A and 4B). Analysis of muscle from control mice using electron microscopy showed the parallel register of myofibrils consisting of sarcomeres in series (Figure 4C). The sarcomere is the functional and structural unit for contraction and is demarcated by two Z-discs. Sections from cachectic muscle harvested at the endpoint (20% weight loss, day 14) exhibited altered ultrastructure to varying degrees in different myocytes. Vesicle-like structures were apparent at the Z-discs, and invaded into surrounding myofibrils [23]. Sarcomere disintegration was also evident, and extreme changes seen where the sarcomeres appeared ‘torn’ at the M-line (Figure 4C).
Table 6: Weight of carcass (whole body minus tumor) and tumor; absolute and normalized weights of heart at the endpoint
Carcass Weight (g) |
TumourWeight (g) |
Heart Weight (mg) |
|
|---|---|---|---|
Control |
27 ± 0.3 |
--- |
130.6 ± 1.8 |
C26 |
20 ± 0.2 |
0.7 ± 0.1 |
107.4 ± 2.6 |
Significant weight loss was observed in C26-bearing mice on day 14 after tumor inoculation (endpoint). Absolute heart weight was reduced in C26-bearing mice. Statistical significance of weight was determined by two-tailed unpaired t-test, *p < 0.05, **p < 0.01, ***p < 0.001 compared to the controls (n = 4).
Mitochondrial structure: The normal appearance of mitochondria with densely packed cristae and homogenous matrix was diminished in muscles of C26-bearing mice (Figure 4C). Mitochondrial changes such as the presence of electron-lucent areas and swelling are evident. Some mitochondria are associated with vesicle-like structures which may represent autophagic bodies in the myocyte, with a reduced proportion of normal mitochondria observed. By contrast, the proportion of mitochondria with locally electron-lucent matrix increased in the C26 cachectic group and swelling and/or fragmentation features of cristae were more apparent in mitochondria of C26-bearing mice compared to the controls (Figure 4C).
These changes in mitochondria were observed to be similar to the features seen in the electron micrographs of cachectic skeletal muscle described in our previous work [24], and were consistent with the proteomic changes observed in both the myofibril/cytoskeletal and the energy/metabolism protein groups. When the energy/metabolism group of differentially expressed proteins were superimposed on the various cellular metabolic pathways (Figure 4D), it was evident that multiple metabolic pathways were affected by downregulated protein expression, including glycogen metabolism, glycolysis, electron transport chain, TCA cycle and fatty acid metabolism/β oxidation. Furthermore, several proteins involved in transport of ions and ADP/ATP across the mitochondrial lumen were also downregulated (Vdac1, Slc25a3 and Slc25a4). Proteins from the gastrocnemius have the largest number of differentially expressed proteins related to energy/metabolism, and are represented across all of the metabolic pathways (Figure 4D).
DISCUSSION
This work represents the first study into muscle proteomic changes in the C26 mouse model of cancer cachexia. It follows our published genomics study on the same model [13] and has the further advantage that we were able to identify downstream effects in the form of protein expression changes relating to muscle function (i.e., functional genomics). Furthermore, effects across slow twitch, fast twitch and heart muscle types have been compared.
Proteomic similarities and differences across muscle types
In all muscle samples, the majority of differentially expressed proteins fell into three main groups; muscle function, energy/metabolism and cytoskeleton. A small number of proteins had muscle specific enrichment, such as proton transporting ATP synthase activity in gastrocnemius, unfolded protein binding in soleus and complement and coagulation cascades in cardiac muscle (Table 5).
Proteins were identified and quantified by iTRAQ based discovery proteomics (see Tables 2, 3 and 4) and processed using the STRING v10 WEB based bioinformatics tool to identify protein enrichment categories. Cellular compartment enrichment is shown in the supplementary materials (Supplementary Table 4).
There was considerable overlap of the full list of proteins identified across the three muscle types (199 proteins identified across all three muscle types). However, there was very little overlap of specific proteins that were differentially expressed (see Venn diagrams in Figure 1A and 1B). Even though muscle function and energy/metabolism processes were represented across all three muscle types, only one differentially expressed protein occurred across all three muscle types (Myh4, upregulated in all 3 muscle types) and just a small amount of overlap was found between each of the muscle types (Figure 1B). Three proteins overlapped between gastrocnemius and heart and these were: Nebl (upregulated in both), Nme2 (downregulated in both) and Pygm (upregulated in heart and downregulated in gastrocnemius). Between soleus and heart, six proteins were differentially expressed in both: Fhl1, Myom2 and Tnnc2 being upregulated in both muscle types, Pdha1 and Myl2 being downregulated in both and Mylpf upregulated in heart and downregulated in soleus. In soleus and gastrocnemius six proteins overlap, with upregulation of Hist2h2bb, Mybpc2, Myh2, Myh7 and Ttn in both muscle types, with Des upregulated in soleus but downregulated in gastrocnemius. Nonetheless, most of the differentially expressed proteins across muscle types were involved in the same pathways/processes. This is well illustrated in Figure 4D which shows common metabolic pathways affected across muscle types, even though specific proteins within these pathways may differ.
The sarcomere and disruption of muscle architecture in cancer cachexia
Loss of sarcomeres, disintegration of Z-discs and swollen mitochondria have been reported in skeletal muscle of sepsis- or denervation-induced myopathies [25, 26]. The molecular structure and components of the Z-disc have previously been described [27], and consist largely of titin, α-actinin and nebulin. Since the Z-disc is an anchor point for myofibrillar proteins such as myosin (via large protein titin) and actin, disrupted Z-discs could result in the release of these myofibrillar proteins. This was evident in a sepsis model with higher myofibrillar protein release rate [25]. In this study, all of the main Z-disc proteins were identified in the proteomics lists (Tables 2–4), and were consistently upregulated across all three muscle types, likely reflecting the disintegration of the Z-disc and release of its constituent proteins. There is a possibility that this upregulation of sarcomeric proteins in the soluble extracts is due to upregulated gene expression, however unlikely for the following reasons: (1) our data represent comparisons of C26 with control muscle samples that underwent identical extraction processes. By comparison with the soluble protein fraction, and even membrane proteins, the normal/intact sarcomere is a robust structure, containing some particularly high molecular weight proteins and protein complexes which are difficult to extract [28, 29]. So weakening of the sarcomere in diseases/disorders such as muscular dystrophy, cachexia and sarcopaenia are likely to facilitate extraction of sarcomere associated proteins. In fact muscle wasting in cachexia has previously been causally linked to proteasome-mediated protein degradation [30], (2) cancer cachexia is a catabolic rather than an anabolic process with weakened sarcomeres and muscle ultrastructural abnormalities, as supported by the electron microscopy data (Figure 4C), our previous work [24] and also data from other groups [30–33], (3) our previous genomics study has identified upregulation of protein catabolic processes, particularly those related to ubiquitin [13], and increased protein ubiquitination has also been reported by others [30] so degradation of muscle proteins is likely at play – which would also be consistent with facilitating release/upregulation of sarcomeric proteins during the protein extraction protocol, by comparison with normal controls, (4) gene expression of some muscle structural proteins, such as troponin are reduced in cachexia [33, 34], likely further weakening the sarcomere. We therefore argue that the balance of evidence supports the likelihood that upregulation of sarcomere associated peptides in the C26 samples indicates sarcomere breakdown rather than a homeostatic response to sarcomere weakening. This is the simplest explanation given that the key feature of cachexia is muscle breakdown. It aligns with our observation which is methodologically consistent with extraction being easier and more peptides now appearing in solution upon extraction. Moreover, the M-line has also been suggested to be an important site for the regular register of thick filaments and contributes to the stability of sarcomeres during continuous contraction [35], which consists largely of the protein myomesin [36]. Myomesin overexpression was observed in both soleus and heart (Tables 3 and 4), consistent with disintegration of the M-line and dissolution of its constituents. Therefore, the disrupted sarcomeric structure observed in the skeletal muscle of C26-bearing mice could accelerate muscle wasting and indeed reflect a common response to catabolic conditions including cancer cachexia.
Consistent with these morphological changes to myocyte architecture, our proteomics experiments revealed upregulated soluble levels of a variety of muscle fibre proteins, consistent with disintegration of the sarcomere in the C26 mice. These included a variety of myosins (Myh4, Myh7, Mylpf, Myl12, MybpH, Myh2, MybpC2, Myl2), troponins (Tnnt1, Tnni2, Tnnc2), tropomyosins (Tpm2), actins (Acta1, Actn2, Actn3), titin (Ttn), filamin (flnc) and myomesin (MyoM2). The emerging picture is of disintegration of the sarcomere along with loss of its normal connections to the cytoskeleton and organelles such as the mitochondrion. In particular, dissolution of Z-disc and M-line proteins, titin, α-actinin, nebulin and myomesin is likely to be particularly disruptive to sarcomeric integrity.
Downregulation of energy, metabolism and mitochondrial function in cachexia
Morphological alterations were evident in the mitochondria of cachectic heart muscle (Figure 4C), that could be related to disruption of energy homeostasis in skeletal muscle, and is consistent with our previous observations on cachectic skeletal muscle [24]. These changes included electron–lucent areas which corresponded to a loss of cristae in these mitochondria; suggesting defective oxidative phosphorylation for generating ATP. In addition, swollen mitochondria, which are reported to be associated with cellular ATP depletion, were present [37]. Vesicle-like structures, likely to be autophagic bodies were also observed in some mitochondria which suggested eventual mitochondrial loss via autophagy in cachectic muscle [23]. Therefore, it appears that during cancer cachexia, not only is the sarcomeric structure disrupted, but muscle energy homeostasis may also be disturbed. Such an ultrastructural phenotype may partly explain the reduced grip strength reported in the C26 model [38].
The energy metabolism protein group is almost consistently downregulated across all three muscle types, explaining the lack of energy/strength and fatigue which are hallmarks of cachexia (Tables 2, 3, 4 and Figure 4D). Differentially expressed proteins are representative of multiple pathways, including glycogen metabolism, glycolysis, fatty acid metabolism, TCA cycle, and the electron transport chain. Furthermore myoglobin, a major oxygen reserve in muscle is significantly downregulated in soleus muscle, indicating a further level of compromise of the electron transport chain. The largest number of proteomic changes are focused on the various mitochondrial pathways, with all three muscle types represented (Figure 4D). However, the fast twitch gastrocnemius muscle also has differentially expressed proteins in the glycogen metabolism, glycolysis and fatty acid transport pathways, and therefore appears to be affected across the largest number of metabolic pathways, including glycolysis, glycogen metabolism, TCA cycle, electron transport chain and fatty acid transport/metabolism (Figure 4D). Overall, downregulation of the majority of energy/metabolism proteins not-withstanding several proteins in this group, are upregulated in heart muscle, including Ag1, Pygm, Tpi1 and Uqcrq (Table 4 and Figure 4D), suggesting an attempt to activate homeostatic mechanisms to protect this vital organ.
A multifunctional protein which may be related to energy insufficiency in cancer cachexia is Nme2, which is involved in the synthesis of nucleoside triphosphates other than ATP, and is downregulated in both gastrocnemius and cardiac muscles (Tables 2 and 4). Nucleoside triphosphate kinases are believed to provide GTP to dynamins, which in turn regulate membrane processes such as vesical formation, fusion to other organelles and mitochondrial inner membrane fusion, and knockdown of nucleoside triphosphate kinases resulting in mitochondrial fragmentation [39]. Furthermore, Nme2 also negatively regulates Rho activity, which has a role in cytoskeletal dynamics [40].
Regulatory proteins and connectivity of the sarcomere to subcellular structures
Given that this is a mouse model of cancer cachexia, it is interesting that the muscle function group of proteins is upregulated in all three muscle types. This effect is a likely result of muscle catabolic processes weakening the muscle fibre structure and solubilising muscle fibre proteins, which would normally be fixed within the sarcomere. Consistent with this possibility, the intermediate filament protein desmin, which regulates sarcomere architecture, and links it to mitochondria and the cell nucleus, is differentially expressed in both gastrocnemius and soleus, being downregulated and upregulated respectively. Desmin-null mice have perturbed muscle architecture, involving both myofibrils and mitochondria, and develop hypertrophic cardiomyopathy [41, 42]. Desmin was also downregulated in cardiac muscle in both biological replicates (Supplementary Materials; Table 3), however did not quite achieve the p = 0.05 statistical significance level. Disintegration of the sarcomere during cachexia may be further compounded by desmin dysregulation, and thereby loss of connectivity to mitochondria. Plectin is another protein which associates with the periphery of Z-discs and promotes mechanical integrity of myocytes by connectivity of the myofibril to the cytoskeleton and mitochondria [43–45]. Plectin was upregulated in soleus (Table 3) consistent with loss of sarcomeric integrity. Other proteins within our differentially expressed proteins lists, which may be involved with cytoskeletal assembly/integrity include Pdlim5 [46] (upregulated in gastrocnemius) and Ldb3 (also called ZASP, upregulated in soleus), which interacts with α-actinin and mutations of which are associated with myopathy [47].
Proteomics and pathology triggers in cancer cachexia
One of the limitations of this case-control study is that it is not clear what the upstream triggers or processes that initiate cancer related muscle wasting are. Additional work, such as a longitudinal study of cancer cachexia progression will be required to determine what these may be. However, there is evidence in our data of several mechanisms which could impact both sarcomere structure and the cellular energy metabolism pathways; (1) protein folding machinery may not be up to the demand placed on it. In the soleus, two heat shock proteins (Hsp90aa1 and Hspa8) and the sarcoplasmic/endoplasmic reticulum calcium ATPase (Atp2a1) are all upregulated, suggesting the possibility that the ER may be under stress. Furthermore, several proteins which transport ATP/ADP, Ca2+ and inorganic phosphate (Slc25a4, Vdac1 and Slc25a3, respectively) are downregulated, possibly further exacerbating any ER stress, with potentially wide-ranging effects on cellular function, (2) several inflammation related proteins (acute phase) are differentially expressed across muscle types, but especially in cardiac muscle. These include; Ahsg (fetuin-A), which is increased in heart and is a protein which is known to increase during inflammation [48], acute phase proteins are upregulated, including fibrinogen chains in soleus (Fga) and heart (Fgg) and α-2-macroglobulin in the heart. Inflammation can be a driver of muscle catabolic processes [49], (3) a variety of ion or small molecule binding and transport proteins are differentially expressed. Some of these may be upregulated as an attempt to maintain homeostasis, such as several iron carrier proteins; hemopexin (upregulated in gastrocnemius), serotransferrin (upregulated in heart) and the calcium binding protein parvalbumin (upregulated in heart). However, downregulation of others may drive pathological processes, including the ion channel Vdac1 which is downregulated in both soleus and heart and the inorganic phosphate transporter Slc25a3 which is downregulated in soleus. Four and a half LIM domains protein 1 (FHL1) is upregulated in heart (Table 4). This protein is abundant in skeletal muscle and to a lesser extent heart, and is a multifunctional protein likely to be involved with ion channel binding. Defects in the FHL1 gene are associated with myopathy [50], (4) several proteins in our differentially expressed protein lists may be involved with DNA binding and transcription regulation, including the histone proteins Hist2h2bb and Hist2h4 (differentially expressed in both gastrocnemius and soleus muscle), and Nme2 (earlier discussed in the context of GTP synthesis and downregulated in both gastrocnemius and cardiac muscles) is a transcriptional activator of the MYC gene and can also bind DNA non-specifically [51, 52].
Limitations
A limitation of the current study is the absence of functional analysis which could provide a mechanistic link between cachexia related protein dysregulation and hypothesised metabolic changes associated with cancer progression. However, it does provide a basis/rationale for future work into sarcomere and mitochondrial dysfunction in cachexia. A number of reviews speculating on the molecular mechanisms of muscle wasting in cancer cachexia appear in the literature [34, 49, 53–61] indicating the importance of improving knowledge in this area. Furthermore, studies in the field addressing functional issues in the context of cancer cachexia are increasing [31, 62, 63]. We suggest that discovery based studies such as genomics and proteomics (i.e., functional genomics) may not only support/refute current hypotheses, but may also be drivers of new hypotheses. The advantages of “omics” and systems biology approaches have recently been reviewed by Gallagher et al. [60]. Such studies are not only complementary to, but also important precursors to functional studies and are beginning to provide important insights into the molecular drivers of cancer cachexia [16, 17]. We expect that our current study will provide the groundwork for future mechanistic investigations.
MATERIALS AND METHODS
Establishment of the colon 26 (C26) carcinoma mouse model of cancer cachexia
The C26 mouse model was established and managed as previously described [13, 24]. Briefly, male Balb/c-DBAj hybrid mice (10–11 weeks old) were obtained from the Animal Resources Centre (Perth, WA, Australia) and housed at the Molecular Physiology Unit (ANZAC Research Institute, Concord, NSW, Australia). Mice were maintained at a constant temperature (22°C) on a 12 hr light: 12 hr dark cycle. Standard rodent chow and water ad libitum were freely accessible. All animal work prior to culling was conducted under ethics approval (Sydney South West Area Health Service Animal Welfare Committee; protocol number 2007/006) and has therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
For tumour inoculation, 1 × 106 C26 cells in 100 μl RPMI/PS were subcutaneously inoculated in the upper right flank of the Balb/c-DBAj mice (day 0). Non-tumour-bearing (NTB) control mice were injected with 100 μl RPMI medium/PS. Mice were physically restrained by hand holding for the inoculation process, and total body weight of animals was monitored daily. Mice were sacrificed by cervical dislocation at 2 pm to minimise diurnal variation. For the identification of cachexia-specific effects (single endpoint), the experimental endpoint for cachexia was selected so that percentage weight loss in C26-bearing mice ranged from 13–18%, which was within the ethical limit of 20%. Mice were culled on days 14 and 16 post-tumour inoculation to account for the inter-individual variability of weight loss. Lower hindlimbs were selected for study due to enrichment in both slow and fast muscles which could be used for comparison of changes across muscle types in cancer cachexia. Individual muscles soleus and gastrocnemius skeletal muscles and heart were harvested, snap-frozen in liquid nitrogen and stored at −80°C.
Electron microscopy (EM)
The electron microscopy procedures have previously been described [24]. In brief, muscle tissue was fixed in 2% PFA/2.5% glutaraldehyde in 0.1M sodium cacodylate buffer straight after excision (n = 3 for both NTB and C26 groups). Prior to post-fixation in osmium tetroxide/0.2M sodium cacodylate buffer, 2 washes of 1 hr each were done in 0.1M sodium cacodylate buffer. Samples were then progressively dehydrated in 70%, 90% and 100% ethanol, before an overnight incubation in LR white resin (London Resin Company, London, UK). LR white resin was replaced the next day for another hr of infiltration. Samples were then embedded in closed gelatine capsules with fresh resin and polymerised at 60°C for 24 hrs. Ultrathin sections (70 nm) were cut from fixed muscle and stained with 2% uranyl acetate (aqueous) and 2% lead citrate and examined under a Jeol1400 transmission electron microscope (Electron Microscopic Unit, UNSW, NSW, Australia) at a voltage of 100 kV.
Protein extraction
Muscle extracts were prepared by homogenising muscles in ice-cold buffer [0.15% SDS, 1 mM EDTA, 1 mM EGTA, 1 mM sodium orthovanadate (Na3VO4), 10 mM sodium fluoride (NaF), 15 mM sodium chloride (NaCl), 20 mM HEPES, 10 mM PMSF, pH 7.5] supplemented with 10 mM PMSF and 1 complete™, EDTA-free protease inhibitor cocktail tablet (Roche Diagnostics, Sydney, Australia) using a Pro200 homogeniser (PRO Scientific Inc, Sydney, Australia). Samples were incubated on ice for 1 hr and supernatant lysates were collected following centrifugation at 14,000g for 10 min at 4°C. Protein samples were divided into aliquots and kept at –80°C before subsequent experimental procedures.
iTRAQ labeling and 2-D LC-MS/MS
For quantitative MS, iTRAQ isobaric labelling was used (Applied Biosystems, Foster City, CA, USA). iTRAQ labelling, 2-D LC-MS/MS and data analysis was performed as described previously [64, 65]. In brief, protein concentration was determined, and protein (100 μg) from skeletal muscle (gastrocnemius and soleus) or cardiac muscle (whole heart) extracts was precipitated with acetone containing 0.1% v/v HCl at –20°C, centrifuged at 15 000 g, air dried and resuspended in a final concentration of 50 mM NaHCO3/0.1% SDS (pH 8.8). For subsequent steps, including trypsinisation, the iTRAQ reagents protocol (Applied Biosystems) was followed, except that 200 mM iodoacetamide (Pierce, Sydney, Australia) was used to alkylate cysteines, and immediately prior to adding iTRAQ reagents, 2.5 μL of 500 mM Na2CO3 was added to ensure a basic pH 8–9.
Labelled peptides were passed through a strong cation exchange column (SCX), dried under vacuum and passed through a C18 column to desalt the sample (Peptide MacroTrap, Michrom Bioresources, Auburn, CA, USA), eluting with 500 μL CH3CN : water : formic acid (50:50:0.1, v:v:v), followed by 200 μL CH3CN. To ensure that any peptides present in the SCX flow through would also be captured, an Oasis cartridge (Waters, Sydney, Australia) was used to desalt the SCX flow through component, and the eluent from this step was pooled with the sample from the final C18 step. For LCMSMS analysis the extracted iTRAQ labelled peptides (5 μg) were captured onto a strong cation exchange micro column (0.75 x ~20 mm, Poros S10, Applied Biosystems) and each of the ammonium acetate eluent steps (12 in total) was captured and desalted on a C18 precolumn cartridge (Michrom Bioresources). After a 10 min wash the pre-column was switched (Switchos, Amsterdam, The Netherlands) into line with an analytical column containing C18 reverse phase packing material (Magic, 5 μm, 200Å, Michrom Bioresources) [66]. Peptides were eluted using a 45 min linear gradient of buffer A (H2O:CH3CN of 98:2 containing 0.1% formic acid-buffer) to buffer B (H2O:CH3CN of 20:80 containing 0.1% formic acid-buffer) at 300 nL/min. High voltage (2300 V) was applied through a low volume tee (Upchurch Scientific, Oak Harbor, WA, USA) at the column inlet and the outlet positioned 1 cm from the orifice of an API QStar Pulsar i hybrid tandem mass spectrometer (Applied Biosystems). Positive ions were generated by electrospray and the QStar operated in information- dependent acquisition mode. A Tof MS survey scan was acquired (m/z 350–1700, 0.75 s) and the three largest multiply charged ions (counts 420, charge state z2 to z4) sequentially selected by Q1 for MS/MS analysis. Nitrogen was used as collision gas and an optimum collision energy automatically chosen (based on charge state and mass). Tandem mass spectra were accumulated for up to 2.5 s (m/z 65–2000) with two repeats.
Quantitative proteomics data analysis
All of the data was collected using two 8-plex iTRAQ experiments and each run was performed in duplicate (two technical replicates). Automated inline 2-D LC-MS/MS was carried out using 5 biological replicates (i.e., 5 control mice and 5 cachexia mice, so 10 mice in total) for gastrocnemius muscle, 2 biological replicates (i.e., 2 control mice and 2 cachexia mice so 4 mice in total) for cardiac muscle and a single biological sample for soleus muscle (i.e., 1 control mouse and 1 cachexia mouse, so 2 mice in total, as a skeletal muscle comparison tissue). Combined technical replicates were processed using Protein Pilot™ 5.0 software (Applied Biosystems/MDS Sciex), using the Paragon™ Algorithm and selecting thorough ID search effort and biological modifications selected in ID focus. The database used was NCBInr (mouse data current at 20–10–15). ProteinPilot identification and quantitation parameters were set as follows: A detect protein threshold cutoff > 1.3 (unused score) was used, representing a confidence of > 95% in correct protein identification and autobias correction applied to the reporter ion ratios. The iTRAQ reporter labels were applied to the samples as follows: Experiment 1 gastrocnemius C26-bearing mice/NTB control reporter ion ratios were 118/115, 119/116 and 121/117 (gastrocnemius biological replicates 1, 2 and 3); soleus C26-bearing mice/NTB control reporter ion ratio was 114/113; Experiment 2 cardiac C26-bearing mice/NTB control reporter ion ratios were 115/113, 116/114 (cardiac biological replicates 1 and 2) and gastrocnemius C26-bearing mice/NTB control reporter ion ratios were 119/117 and 121/118 (gastrocnemius biological replicates 4 and 5). The background correction option was not used, however autobias correction values were as follows for each of the reporter ion ratios: Experiment 1 gastrocnemius 118/115 (0.8851), 119:116 (0.7827), 121:117 (0.8455), soleus 114/113 (0.5979); Experiment 2 cardiac muscle 115:113 (0.4845), 116:114 (0.7605), gastrocnemius 119/117 (1.0335), 121:118 (0.9808).
Bioinformatics analysis; STRING v10 and ingenuity pathway
STRING v10 [67] was used to perform network analysis and enrichment of protein groups on the proteins which were found to be differentially expressed in each of the muscle types. For this analysis the default settings were used, the MCL clustering tool was set to a value of 2 and the confidence view selected for output presentation. The protein terms used for each analysis are shown in Tables 2, 3 and 4. STRING v10 was also used to explore enrichment of biological processes, molecular function, cellular components and KEGG pathways against the mouse genome background (only FDR and/or Bonferroni corrected significant values are shown). Ingenuity Pathway Analysis (IPA) software (Qiagen 2015) was used to compare the level of overlap of protein lists between the three muscle types and to explore biochemical networks, diseases and functions which were significantly related to the differentially expressed proteins.
Proteomics terminology
Use of the terms “up-regulated”, “down-regulated”, and “differential expression level”, are widespread in the proteomics literature and are a convention adopted from the genomics field. Consequently they are sometimes taken to imply genetic regulation. However, in the proteomics context they can only ever represent protein level/s relative to a reference or control sample. Such levels may change due to transcriptional, translational, post-translational or disease mechanisms, and proteomics methods alone cannot establish the mechanism/s by which protein levels change. To help clarify our current usage of this terminology and to avoid false inferences, our use of these terms in the current context only indicates protein levels in the cachexia samples relative to the control samples. Any mechanistic inferences that we make are based on a combination of some or all of our proteomic data, muscle ultrastructural observations and our previously published genomics data.
CONCLUSIONS
Complementary findings from our morphological and proteomic analyses provide a global picture of the molecular events that underlie skeletal muscle wasting during cancer cachexia; disintegrated sarcomeric structure, increased soluble protein expression of Z-disc components, and disruption of the M-line with increased expression of soluble myomesin. This model for muscle breakdown, triggered by disruption of the Z-line which releases myofibrillar proteins, presents the Z-disc as the most likely initial and rate-limiting step of muscle breakdown in cancer-induced muscle wasting. These processes are further exacerbated by disrupted energy homeostasis, which is another recurrent feature of the pathophysiology of cancer-induced muscle cachexia. Altered mitochondrial morphology, in combination with the reduced expression of proteins regulating substrate and energy metabolism, suggest that muscle cells are likely to be undergoing a state of energy crisis which ultimately results in cancer-induced cachexia. A wide range of biological processes could potentially be affected by the lack of cellular fuel, including muscle contraction, protein turnover and substrate metabolism. However, based on the results presented, it is still unclear if the observed disrupted energy metabolism during cancer cachexia is a consequence or a cause of muscle wasting. The data generated by this iTRAQ proteomics study shows that the genomic changes which we have previously identified are indeed translated into multiple functional downstream changes, which in turn account for muscle wasting in cancer-induced cachexia. Future work should aim to elucidate what the primary triggers are for these changes, and to this purpose, longitudinal studies may be the way forward.
Author contributions
PP, TCT and AMYS conceived and planned the project. These authors also performed editorial work on the text. AMYS, AP and NLB performed experimental work (tissue collection and processing, iTRAQ proteomics analysis, mass spectrometry, 1D SDS/PAGE and Western blotting) and wrote the main manuscript text. All authors contributed to data interpretation. All authors reviewed the manuscript.
ACKNOWLEDGMENTS
We thank A/Prof. Graham Robertson and Prof. Stephen Clarke for providing support for the animal experiments. We thank Ms Sigrid Fraser and the Electron Microscope Unit UNSW for her invaluable assistance with the ultrastructural analysis. We thank A/Prof Mark J. Raftery for providing subsidized access to the Bioanalytical Mass Spectrometry Facility, UNSW Australia, with technical and analytical support to perform iTRAQ mass spectrometry at this facility.
CONFLICTS OF INTEREST
No conflicts of interest, financial or otherwise, are declared by any of the authors.
FUNDING
This project was supported by a UNSW Australia Faculty of Medicine Research Grant (P. Polly), and A. M. Y. Shum was the recipient of an Australian Postgraduate Award and the UNSW Research Excellence Scholarship. We thank these organisations for their financial support.
REFERENCES
1. Bossola M, Pacelli F, Tortorelli A, Doglietto GB. Cancer cachexia: it’s time for more clinical trials. Ann Surg Oncol. 2007; 14:276–85. https://doi.org/10.1245/s10434-006-9179-5.
2. Shum AM, Polly P. Cancer cachexia: molecular targets and pathways for diagnosis and drug intervention. Endocr Metab Immune Disord Drug Targets. 2012; 12:247–59.
3. Muscaritoli M, Bossola M, Aversa Z, Bellantone R, Rossi Fanelli F. Prevention and treatment of cancer cachexia: new insights into an old problem. Eur J Cancer. 2006; 42:31–41. https://doi.org/10.1016/j.ejca.2005.07.026.
4. Persson C, Glimelius B. The relevance of weight loss for survival and quality of life in patients with advanced gastrointestinal cancer treated with palliative chemotherapy. Anticancer Res. 2002; 22:3661–8.
5. Scott HR, McMillan DC, Brown DJ, Forrest LM, McArdle CS, Milroy R. A prospective study of the impact of weight loss and the systemic inflammatory response on quality of life in patients with inoperable non-small cell lung cancer. Lung Cancer. 2003; 40:295–9.
6. Tisdale MJ. Molecular pathways leading to cancer cachexia. Physiology (Bethesda). 2005; 20:340–8. https://doi.org/10.1152/physiol.00019.2005.
7. Fearon KC. Cancer cachexia: developing multimodal therapy for a multidimensional problem. Eur J Cancer. 2008; 44:1124–32. https://doi.org/10.1016/j.ejca.2008.02.033.
8. Salpeter SR, Malter DS, Luo EJ, Lin AY, Stuart B. Systematic review of cancer presentations with a median survival of six months or less. J Palliat Med. 2012; 15:175–85. https://doi.org/10.1089/jpm.2011.0192.
9. Dewys WD, Begg C, Lavin PT, Band PR, Bennett JM, Bertino JR, Cohen MH, Douglass HO Jr, Engstrom PF, Ezdinli EZ, Horton J, Johnson GJ, Moertel CG, et al, and Eastern Cooperative Oncology Group. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am J Med. 1980; 69:491–7.
10. Ross PJ, Ashley S, Norton A, Priest K, Waters JS, Eisen T, Smith IE, O’Brien ME. Do patients with weight loss have a worse outcome when undergoing chemotherapy for lung cancers? Br J Cancer. 2004; 90:1905–11. https://doi.org/10.1038/sj.bjc.6601781.
11. Thivat E, Therondel S, Lapirot O, Abrial C, Gimbergues P, Gadea E, Planchat E, Kwiatkowski F, Mouret-Reynier MA, Chollet P, Durando X. Weight change during chemotherapy changes the prognosis in non metastatic breast cancer for the worse. BMC Cancer. 2010; 10:648. https://doi.org/10.1186/1471-2407-10-648.
12. Andreyev HJ, Norman AR, Oates J, Cunningham D. Why do patients with weight loss have a worse outcome when undergoing chemotherapy for gastrointestinal malignancies? Eur J Cancer. 1998; 34:503–9.
13. Shum AM, Fung DC, Corley SM, McGill MC, Bentley NL, Tan TC, Wilkins MR, Polly P. Cardiac and skeletal muscles show molecularly distinct responses to cancer cachexia. Physiol Genomics. 2015; 47:588–99. https://doi.org/10.1152/physiolgenomics.00128.2014.
14. Skipworth RJ, Stewart GD, Bhana M, Christie J, Sturgeon CM, Guttridge DC, Cronshaw AD, Fearon KC, Ross JA. Mass spectrometric detection of candidate protein biomarkers of cancer cachexia in human urine. Int J Oncol. 2010; 36:973–82.
15. Marin-Corral J, Fontes CC, Pascual-Guardia S, Sanchez F, Olivan M, Argiles JM, Busquets S, Lopez-Soriano FJ, Barreiro E. Redox balance and carbonylated proteins in limb and heart muscles of cachectic rats. Antioxid Redox Signal. 2010; 12:365–80. https://doi.org/10.1089/ars.2009.2818.
16. Barreto R, Mandili G, Witzmann FA, Novelli F, Zimmers TA, Bonetto A. Cancer and Chemotherapy Contribute to Muscle Loss by Activating Common Signaling Pathways. Front Physiol. 2016; 7:472. https://doi.org/10.3389/fphys.2016.00472.
17. Ebhardt HA, Degen S, Tadini V, Schilb A, Johns N, Greig CA, Fearon KCH, Aebersold R, Jacobi C. Comprehensive proteome analysis of human skeletal muscle in cachexia and sarcopenia: a pilot study. J Cachexia Sarcopenia Muscle. 2017; 8:567–82. https://doi.org/10.1002/jcsm.12188.
18. Zieske LR. A perspective on the use of iTRAQ reagent technology for protein complex and profiling studies. J Exp Bot. 2006; 57:1501–8. https://doi.org/10.1093/jxb/erj168.
19. Hsieh HC, Chen YT, Li JM, Chou TY, Chang MF, Huang SC, Tseng TL, Liu CC, Chen SF. Protein profilings in mouse liver regeneration after partial hepatectomy using iTRAQ technology. J Proteome Res. 2009; 8:1004–13. https://doi.org/10.1021/pr800696m.
20. Keshamouni VG, Michailidis G, Grasso CS, Anthwal S, Strahler JR, Walker A, Arenberg DA, Reddy RC, Akulapalli S, Thannickal VJ, Standiford TJ, Andrews PC, Omenn GS. Differential protein expression profiling by iTRAQ-2DLC-MS/MS of lung cancer cells undergoing epithelial-mesenchymal transition reveals a migratory/invasive phenotype. J Proteome Res. 2006; 5:1143–54. https://doi.org/10.1021/pr050455t.
21. Glen A, Evans CA, Gan CS, Cross SS, Hamdy FC, Gibbins J, Lippitt J, Eaton CL, Noirel J, Wright PC, Rehman I. Eight-plex iTRAQ analysis of variant metastatic human prostate cancer cells identifies candidate biomarkers of progression: An exploratory study. Prostate. 2010; 70:1313–32. https://doi.org/10.1002/pros.21167.
22. Wang LN, Tong SW, Hu HD, Ye F, Li SL, Ren H, Zhang DZ, Xiang R, Yang YX. Quantitative proteome analysis of ovarian cancer tissues using a iTRAQ approach. J Cell Biochem. 2012; 113:3762–72. https://doi.org/10.1002/jcb.24250.
23. Arbustini E, Brega A, Narula J. Ultrastructural definition of apoptosis in heart failure. Heart Fail Rev. 2008; 13:121–35. https://doi.org/10.1007/s10741-007-9072-8.
24. Shum AM, Mahendradatta T, Taylor RJ, Painter AB, Moore MM, Tsoli M, Tan TC, Clarke SJ, Robertson GR, Polly P. Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer-induced skeletal muscle wasting. Aging (Albany NY). 2012; 4:133–43. https://doi.org/10.18632/aging.100436.
25. Williams AB, Decourten-Myers GM, Fischer JE, Luo G, Sun X, Hasselgren PO. Sepsis stimulates release of myofilaments in skeletal muscle by a calcium-dependent mechanism. FASEB J. 1999; 13:1435–43.
26. Tomanek RJ, Lund DD. Degeneration of different types of skeletal muscle fibres. I. Denervation. J Anat. 1973; 116:395–407.
27. Young P, Ferguson C, Banuelos S, Gautel M. Molecular structure of the sarcomeric Z-disk: two types of titin interactions lead to an asymmetrical sorting of alpha-actinin. EMBO J. 1998; 17:1614–24. https://doi.org/10.1093/emboj/17.6.1614.
28. Wang K, Ramirez-Mitchell R, Palter D. Titin is an extraordinarily long, flexible, and slender myofibrillar protein. Proc Natl Acad Sci U S A. 1984; 81:3685–9.
29. Ohlendieck K. Skeletal muscle proteomics: current approaches, technical challenges and emerging techniques. Skelet Muscle. 2011; 1:6. https://doi.org/10.1186/2044-5040-1-6.
30. Aulino P, Berardi E, Cardillo VM, Rizzuto E, Perniconi B, Ramina C, Padula F, Spugnini EP, Baldi A, Faiola F, Adamo S, Coletti D. Molecular, cellular and physiological characterization of the cancer cachexia-inducing C26 colon carcinoma in mouse. BMC Cancer. 2010; 10:363. https://doi.org/10.1186/1471-2407-10-363.
31. Puig-Vilanova E, Rodriguez DA, Lloreta J, Ausin P, Pascual-Guardia S, Broquetas J, Roca J, Gea J, Barreiro E. Oxidative stress, redox signaling pathways, and autophagy in cachectic muscles of male patients with advanced COPD and lung cancer. Free Radic Biol Med. 2015; 79:91–108. https://doi.org/10.1016/j.freeradbiomed.2014.11.006.
32. Tzika AA, Fontes-Oliveira CC, Shestov AA, Constantinou C, Psychogios N, Righi V, Mintzopoulos D, Busquets S, Lopez-Soriano FJ, Milot S, Lepine F, Mindrinos MN, Rahme LG, et al. Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. Int J Oncol. 2013; 43:886–94. https://doi.org/10.3892/ijo.2013.1998.
33. Tian M, Nishijima Y, Asp ML, Stout MB, Reiser PJ, Belury MA. Cardiac alterations in cancer-induced cachexia in mice. Int J Oncol. 2010; 37:347–53.
34. Pajak B, Orzechowska S, Pijet B, Pijet M, Pogorzelska A, Gajkowska B, Orzechowski A. Crossroads of cytokine signaling--the chase to stop muscle cachexia. J Physiol Pharmacol. 2008; 59:251–64.
35. Agarkova I, Ehler E, Lange S, Schoenauer R, Perriard JC. M-band: a safeguard for sarcomere stability? J Muscle Res Cell Motil. 2003; 24:191–203.
36. Tskhovrebova L, Trinick J. Making muscle elastic: the structural basis of myomesin stretching. PLoS Biol. 2012; 10:e1001264. https://doi.org/10.1371/journal.pbio.1001264.
37. Gupta A, Gupta S, Young D, Das B, McMahon J, Sen S. Impairment of ultrastructure and cytoskeleton during progression of cardiac hypertrophy to heart failure. Lab Invest. 2010; 90:520–30. https://doi.org/10.1038/labinvest.2010.43.
38. Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, Lacey DL, Goldberg AL, Han HQ. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010; 142:531–43. https://doi.org/10.1016/j.cell.2010.07.011.
39. Boissan M, Montagnac G, Shen Q, Griparic L, Guitton J, Romao M, Sauvonnet N, Lagache T, Lascu I, Raposo G, Desbourdes C, Schlattner U, Lacombe ML, et al. Membrane trafficking. Nucleoside diphosphate kinases fuel dynamin superfamily proteins with GTP for membrane remodeling. Science. 2014; 344:1510–5. https://doi.org/10.1126/science.1253768.
40. Truebestein L, Elsner DJ, Fuchs E, Leonard TA. A molecular ruler regulates cytoskeletal remodelling by the Rho kinases. Nat Commun. 2015; 6:10029. https://doi.org/10.1038/ncomms10029.
41. Milner DJ, Weitzer G, Tran D, Bradley A, Capetanaki Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol. 1996; 134:1255–70.
42. Milner DJ, Taffet GE, Wang X, Pham T, Tamura T, Hartley C, Gerdes AM, Capetanaki Y. The absence of desmin leads to cardiomyocyte hypertrophy and cardiac dilation with compromised systolic function. J Mol Cell Cardiol. 1999; 31:2063–76.
43. Hijikata T, Murakami T, Imamura M, Fujimaki N, Ishikawa H. Plectin is a linker of intermediate filaments to Z-discs in skeletal muscle fibers. J Cell Sci. 1999; 112:867–76.
44. Steinbock FA, Wiche G. Plectin: a cytolinker by design. Biol Chem. 1999; 380:151–8. https://doi.org/10.1515/BC.1999.023.
45. Reipert S, Steinbock F, Fischer I, Bittner RE, Zeold A, Wiche G. Association of mitochondria with plectin and desmin intermediate filaments in striated muscle. Exp Cell Res. 1999; 252:479–91. https://doi.org/10.1006/excr.1999.4626.
46. Krcmery J, Camarata T, Kulisz A, Simon HG. Nucleocytoplasmic functions of the PDZ-LIM protein family: new insights into organ development. Bioessays. 2010; 32:100–8. https://doi.org/10.1002/bies.200900148.
47. Lin X, Ruiz J, Bajraktari I, Ohman R, Banerjee S, Gribble K, Kaufman JD, Wingfield PT, Griggs RC, Fischbeck KH, Mankodi A. Z-disc-associated, alternatively spliced, PDZ motif-containing protein (ZASP) mutations in the actin-binding domain cause disruption of skeletal muscle actin filaments in myofibrillar myopathy. J Biol Chem. 2014; 289:13615–26. https://doi.org/10.1074/jbc.M114.550418.
48. Stefan N, Haring HU. The role of hepatokines in metabolism. Nat Rev Endocrinol. 2013; 9:144–52. https://doi.org/10.1038/nrendo.2012.258.
49. Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov. 2015; 14:58–74. https://doi.org/10.1038/nrd4467.
50. Windpassinger C, Schoser B, Straub V, Hochmeister S, Noor A, Lohberger B, Farra N, Petek E, Schwarzbraun T, Ofner L, Loscher WN, Wagner K, Lochmuller H, et al. An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. Am J Hum Genet. 2008; 82:88–99. https://doi.org/10.1016/j.ajhg.2007.09.004.
51. Postel EH, Berberich SJ, Flint SJ, Ferrone CA. Human c-myc transcription factor PuF identified as nm23-H2 nucleoside diphosphate kinase, a candidate suppressor of tumor metastasis. Science. 1993; 261:478–80.
52. Ji L, Arcinas M, Boxer LM. The transcription factor, Nm23H2, binds to and activates the translocated c-myc allele in Burkitt’s lymphoma. J Biol Chem. 1995; 270:13392–8.
53. Adams V, Anker SD, Schuler G. Muscle metabolism and exercise capacity in cachexia. Curr Pharm Des. 2011; 17:3838–45.
54. Hasselgren PO, Fischer JE. Muscle cachexia: current concepts of intracellular mechanisms and molecular regulation. Ann Surg. 2001; 233:9–17.
55. Wray CJ, Mammen JM, Hasselgren PO. Catabolic response to stress and potential benefits of nutrition support. Nutrition. 2002; 18:971–7.
56. Aversa Z, Costelli P, Muscaritoli M. Cancer-induced muscle wasting: latest findings in prevention and treatment. Ther Adv Med Oncol. 2017; 9:369–82. https://doi.org/10.1177/1758834017698643.
57. Loumaye A, Thissen JP. Biomarkers of cancer cachexia. Clin Biochem. 2017; 50:1281–88. https://doi.org/10.1016/j.clinbiochem.2017.07.011.
58. von Haehling S, Ebner N, Dos Santos MR, Springer J, Anker SD. Muscle wasting and cachexia in heart failure: mechanisms and therapies. Nat Rev Cardiol. 2017; 14:323–41. https://doi.org/10.1038/nrcardio.2017.51.
59. Petruzzelli M, Wagner EF. Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes Dev. 2016; 30:489–501. https://doi.org/10.1101/gad.276733.115.
60. Gallagher IJ, Jacobi C, Tardif N, Rooyackers O, Fearon K. Omics/systems biology and cancer cachexia. Semin Cell Dev Biol. 2016; 54:92–103. https://doi.org/10.1016/j.semcdb.2015.12.022.
61. Argiles JM, Busquets S, Stemmler B, Lopez-Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer. 2014; 14:754–62. https://doi.org/10.1038/nrc3829.
62. Tian M, Asp ML, Nishijima Y, Belury MA. Evidence for cardiac atrophic remodeling in cancer-induced cachexia in mice. Int J Oncol. 2011; 39:1321–6. https://doi.org/10.3892/ijo.2011.1150.
63. Bohnert KR, Gallot YS, Sato S, Xiong G, Hindi SM, Kumar A. Inhibition of ER stress and unfolding protein response pathways causes skeletal muscle wasting during cancer cachexia. FASEB J. 2016; 30:3053–68. https://doi.org/10.1096/fj.201600250RR.
64. Evans FF, Raftery MJ, Egan S, Kjelleberg S. Profiling the secretome of the marine bacterium Pseudoalteromonas tunicata using amine-specific isobaric tagging (iTRAQ). J Proteome Res. 2007; 6:967–75. https://doi.org/10.1021/pr060416x.
65. Lim YA, Rhein V, Baysang G, Meier F, Poljak A, Raftery MJ, Guilhaus M, Ittner LM, Eckert A, Gotz J. Abeta and human amylin share a common toxicity pathway via mitochondrial dysfunction. Proteomics. 2010; 10:1621–33. https://doi.org/10.1002/pmic.200900651.
66. Gatlin CL, Kleemann GR, Hays LG, Link AJ, Yates JR 3rd. Protein identification at the low femtomole level from silver-stained gels using a new fritless electrospray interface for liquid chromatography-microspray and nanospray mass spectrometry. Anal Biochem. 1998; 263:93–101. https://doi.org/10.1006/abio.1998.2809.
67. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015; 43:D447–52. https://doi.org/10.1093/nar/gku1003.