
INTRODUCTION
Several aspects of ovarian cancer (OvCa) deem it a very challenging malignancy to diagnose and treat. Even though the five-year survival rate is high when women are diagnosed in the early stages of OvCa, most women are rarely diagnosed in early stages as the symptoms are merely vague abdominal discomfort or bloating [1]. The five-year survival rate of women diagnosed with OvCa drops precipitously when women are diagnosed with stage III or IV as aggressive metastasis to neighboring abdominal organs has already progressed [1, 2]. Patients’ survival is strongly correlated with the outcome of the surgical debulking [3]. The standard clinical management for advanced stage OvCa includes debulking surgery followed by adjuvant chemotherapy or neoadjuvant chemotherapy followed by surgery in patients who present with un-resectable disease [4]. However, after initial response, tumor recurrence from residual disease is encountered in about 70% of patients who will eventually die of a progressively chemo-resistant cancer [4]. Indeed, optimal surgical debulking of tumors (< 1 cm of residual tumor) significantly improves patients’ survival compared to sub-optimal debulking [4]. Unfortunately, suboptimal debulking (>1 cm of residual tumor) is frequently encountered due to widespread microscopic and inaccessible lesions throughout the abdomen preventing complete removal of the tumor [4]. There remains a need for improved OvCa treatment that addresses the current limitations of surgical debulking and reduces treatment resistance that often arises in response to chemotherapy [4].
OvCa is heterogeneous and affected by epigenetic and genetic factors [5]. A major reason for the lack of success in effectively eradicating OvCa can be due to the complex interconnected signaling networks coupled with the distinctive peritoneal tumor microenvironment (TME) [6]. Several immune cells, including tumor associated macrophages (TAMs), T cells, natural killer (NK) cells in addition to fibroblasts and a wide host of chemokines and cytokines all interact with each other to promote the growth and metastasis of OvCa cells [7]. Therefore, understanding the pathobiology of OvCa and its unique TME that hosts this malignancy, is crucial in our development of more sensitive diagnostic tools and enhanced treatment options.
Epidemiology
Epithelial OvCa (EOC) is the leading cause of gynecologic cancer-related death in the USA. More than 70% of patients are diagnosed with advanced disease (Stage III or IV) [4, 8]. In the year 2017 alone, a growing 22,240 new cases and 14,080 cancer-related deaths occured in USA [9]. The incidence is higher in Caucasians than in Hispanics, American Indian/Alaska Natives, Blacks, and Asian/Pacific Islanders [10]. The mean age of diagnosis of ovarian cancer is 63 years. Differences in age distribution are summarized in (http://seer.cancer.gov/statfacts/html/ovary.html).
Risk factors
Genetic risk factors
The lifetime risk of developing OvCa in the USA is currently 1.4%. The risk increases with mutations in several genes namely, BRCA1/2, mismatch repair and ARID1 genes [11]. Women with BRCA mutations have an increased risk of developing ovarian, fallopian tube and peritoneal cancer, specifically, 20–50% in BRCA1 and 10–20% in BRCA2 [12]. Women with mutations in mismatch repair genes associated with type 2 Lynch syndrome are at higher risk of developing colon, endometrial and ovarian cancers [13, 14]. Similarly, women with mutations in ARID1A are at risk of developing endometrial or clear cell ovarian carcinoma [11].
Non-genetic risk factors
Include incessant ovulation as well as repeated rupture and repair of ovarian follicles with continued exposure to gonadotropins [15]. Nulliparity and infertility, both interfere with protective hormone release, are considered risk factors. In support of this, multiparous women who are pregnant after the age of 35 years are at a reduced risk of developing EOC [16–18]. Gynecologic diseases such as endometriosis, polycystic ovary syndrome (PCOS), and postmenopausal hormonal therapy perturb the estrogen and progesterone cycle and increase the risk of OvCa [19, 20]. Intrauterine devices, obesity, cigarette smoking, exposure to talc and asbestos have also been shown to increase the risk of OvCa [21–24].
Protective factors
Include oral contraceptives, multiparity, salpingo-oophrectomy, tubal ligation, hysterectomy, breast feeding, nonsteroidal anti-inflammatory (NSAID) drugs and acetaminophen.
Pathological subtypes
Many pathological subtypes are described for EOC. These are further classified into serous, clear cells, mucinous, and endometrioid.
Serous cancer
High-grade serous cancer (HGSC) compromise 70 to 80% of all cases and typically arises from either the surface of the ovary or from the distal fallopian tube [25]. HGSC strongly and diffusely expresses p53 and p16. HGSC also expresses Wilm’s tumor-1 (WT-1), estrogen receptor (ER), and Paired Box-8 (PAX-8) in most cases as well as a high Ki67 proliferative index. Genetic alterations in BRCA1 or BRCA2 germline mutations are present in up to 10% of women with HGSC. Additional associated genetic alterations that are specific to HGSC are TP53 (as high as 96%), NF1, RB1, CDK12, PTEN and PIK3CA [5, 26, 27].
Low-grade serous cancer (LGSC) comprise less than 5% of OvCa. LGSC has a low Ki67 proliferative rate with normal p53 expression, and commonly expresses WT-1, ER, and progesterone receptor (PR). Mutations in BRAF and KRAS typically lead to LGSC [2, 28].
Endometrioid carcinoma
Comprises 10% of epithelial types. Cancer cells express vimentin, ER, PR, PAX-8, and CA125. Genetic mutations of CTNNB-1 (β-catenin), PTEN, PIK3CA and ARID1A with microsatellite instability are also present [29, 30]. Endometrioid carcinoma as with clear cell carcinoma, typically arise from endometriosis which is linked to the theory of retrograde menstruation [25].
Clear cell carcinoma
Comprises 10% of epithelial cases. Hypoxia-inducible factor 1 alpha (HIF-1 α) [31], glypican-3 [32], and hepatocyte nuclear factor 1-beta (HNF-1 beta) are highly expressed [33]. Genetic alterations include mutations in ARID1A KRAS, PTEN, and PIK3CA [34].
Mucinous carcinoma
Comprises 3% of epithelial cases. Gastrointestinal markers CK20, CDX2, CK7 and molecular mutations as KRAS as well as mucin genes MUC2, MUC3, and MUC17 are commonly expressed [35, 36].
Molecular pathogenesis
Tumors of the ovary have been divided into type I which includes LGSC, mucinous, low grade endometrioid and clear cell carcinoma and type II which includes HGSC, high grade endometrioid and undifferentiated/malignant mixed carcinomas. Type I tumors include mutations in KRAS, BRAF, PTEN and CTNNB-1, while type II tumors are typically associated with p53 mutations. However, evidence from mouse models of OvCa indicated that p53 mutations alone are not sufficient to drive invasive carcinoma. As HGSC composes 70% of all ovarian serous tumors, its pathogenesis is especially important [35].
The pathogenesis of OvCa can also be segregated by anatomical origins namely ovarian or tubal (fallopian tube) derived [37]. The ovarian subtypes arise from ovarian surface epithelia which are derived from mesothelial coelomic epithelia covering the ovaries. They are often a result of the repeated formation of cortical inclusion cysts and endometriosis. It is hypothesized that ovarian carcinoma of this subtype arises from ovarian inclusions cysts that underwent Mullerian metaplasia [38]. The Fallopian or tubal subtypes originate from the coelom, namely the Mullerian or paramesonephric ducts. The majority of serous tumors appear to arise from the secretory cells in the distal fallopian tube. Serous tubal intraepithelial cancer (STIC) is the precursor lesion with TP53 mutations similar to those found in HGSC [39–41]. More recent studies theorize that the exact origin of tubal subtypes is at the junctional zone between the fallopian tube epithelium and the mesothelium of the tubal serosa. This site in particular, confers extensive connections with the lymphatic system making it relatively easy to invade and metastasize to the abdominal cavity. Thus, prophylactic salpingectomy is encouraged for women undergoing hysterectomy for benign conditions [42].
Clinical features
OvCa can manifest as either acute or subacute in nature. Acute forms are usually due to advanced stages of cancer spread leading to bowel obstruction and pleural effusion. Subacute cases will manifest as a unilateral or bilateral adnexal mass, pelvic pain or abdominal pain, postmenopausal bleeding, rectal bleeding or atypical glandular cells on cervical cytology [1, 43].
Diagnosis of EOC
Diagnostic studies are categorized into two phases: an initial evaluation of the presumed adnexal or abdominal mass by imaging studies such as abdominal ultrasound and MRI followed by surgical evaluation, and pathological identification of the subtype, grading, and staging. Pathological grading and identification of the subtypes are achieved after percutaneous fine needle biopsy or after cytoreduction [44]. Several tumor markers are considered in the diagnosis such as CA125 (cancer antigen 125), human epididymis protein 4 (HE4) and carcinoembryonic antigen (CEA) that are increased in advanced EOC and are considered as prognostic markers, though they lack specificity and sensitivity [44].
Determinants of peritoneal metastasis
Cancer cells
EOC is unique among cancers in that cancer cells have diverse progenitors, ovarian surface epithelium (OSE) and fimbrial epithelia, that express common epithelial markers as keratins, EpCAM and E-cadherin as well as mesenchymal markers as vimentin and N-cadherin [45–47]. Malignant cells are shed from the primary tumor into the peritoneal cavity where they survive as free-floating single cells or aggregate as spheroids in the peritoneal fluid “malignant ascites”. Single cells and spheroids can not only survive anchorage-independent apoptosis “anoikis”, but also can proliferate in suspension and seed onto the mesothelial cells lining the peritoneal cavity resulting in extensive peritoneal dissemination [48, 49]. Phenotypic characterization of single and multicellular malignant cells isolated from ascitic fluid revealed that these cells exhibit dual “hybrid” as well as heterogeneous E-and N-Cadherin expression [49]. The latter study also reported cadherin-dependent diversity in cell-cell interactions, spheroid formation, and ultrastructure. This is further supported by an elegant report from the same group [50] implicating cadherin-plasticity in mesothelial adhesion, clearance and collagen invasion. Cadherin plasticity is also implicated in the dynamic switch between epithelial-mesenchymal transition (EMT) and mesenchymal-epithelial transition (MET). EMT-MET switch is regulated by complex sequential transcriptional machinery with early induction of the transcription factors SNAIL (SNAI1) and slug; whereas SNAI2, ZEB1/2 and TWIST were induced at later phases [51–55]. EMT- transcription factors are induced by a plethora of upstream factors that act individually or synergistically to induce an invasive phenotype of EOC cells. The expression of EMT-inducing transcription factors (Snail, Slug, Twist and Zeb1/2) is associated with metastatic, recurrent and chemo-resistant tumors and poor prognosis [51, 53, 55–58]. Correlation between EMT and aggressiveness of OvCa is supported by the downregulation of E-cadherin [59] and overexpression of mesenchymal signatures specifically transforming growth factor beta and its receptors (TGFβ/TGFβRs), CD44 [60], bone morphogenetic proteins and their receptors (BMPs/BMPRs), receptor tyrosine kinases and their ligands [54], Wnt [61, 62] and Notch [53] signaling pathways. In addition to intrinsic EMT inducers activated in cancer cells, cues from the peritoneal TME strongly induce EMT. For example, mesothelial cells, adipocytes and ascitic fluid rich in growth factors, bioactive lipids, matrix metalloproteases (MMPs), as well as inflammatory and immune cells; all induce hypoxia, inflammation and oxidative stress and corroborate to induce EMT [63–66].
Mesothelial cells
Mesothelial cells are the first barrier that faces metastatic OvCa cells. They are organized as a single layer of simple epithelium covering the sub extracellular matrix (ECM). rich in collagen I that covers abdominal, pelvic as well as visceral organs including the omentum [67–69]. Apically, mesothelial cells secrete glycosaminoglycans, surfactant and proteoglycans to establish an anti-adhesive surface. The bidirectional cross-talk between cancer and mesothelial Cells promotes cancer cell chemotaxis to the mesothelial cells, followed by integrin-mediated adhesion and invasion with subsequent increase in MMPs, and urokinase type plasminogen activator (uPA) and its receptor (uPAR) [70–73]; eventually leading to mesothelial clearance and invasion of the sub-mesothelial layers [74–76]. In addition, clinical reports show that cancer cells preferentially bind to regions of disrupted mesothelium at sites of entry of lymphatic and blood vessels [67–69, 77].
The propensity of OvCa to metastasize to the mesothelial cells is initially instigated by cancer cell secretome that preconditions the mesothelial cell niche to induce the expression of multiple pro-inflammatory mediators as bioactive lipids (e.g. LPA)/inflammatory cytokines/chemokines [78–80], ECM/integrins [67, 68, 81–86], cell adhesion molecules as VCAM1, ICAM1, CD44/HA [87–89], and uPA/uPAR [71, 90]. OvCa cell adhesion to mesothelial cells is mediated by bidirectional binding of ECM, integrins and cell adhesion molecules. This binding activates multiple downstream signaling pathways that in corroboration with activation of oncogenic signaling pathways, promote cancer cell colonization, invasiveness, and metastasis.
The omentum and omental adipocytes
The omentum, which is subdivided into lesser and greater, is a double layered peritoneal fold that covers the intestines and abdominal organs. Physiologically, it functions as a fat and energy depot due to the abundance of white adipocytes [91, 92]. It also has a role in immune surveillance via aggregates of macrophages known as milky spots that play an important role in containing intraperitoneal infections [93, 94]. Milky spots also play an important role in the tropism of OvCa cells to the omentum [95–97]. Importantly, omental adipocytes release cytokines/chemokines “adipokines”, which contribute to OvCa cell homing, invasion and metastasis [91, 92, 95, 98]. The bidirectional interaction between omental adipocytes and cancer cells causes dedifferentiation and reprogramming of adipocytes into cancer-associated adipocytes (CAA) [98]. In this process, cancer cells secrete cytokines and chemokines that induce lipolysis in adipocytes, breaking their lipids (triglycerides), and releasing fatty acids and glycerol. Consequently, adipocytes undergo de-differentiation into a pre-adipocyte stage (fibroblastoid) and secrete adipokines [91, 98] (Figure 1). In turn, the uptake of fatty acids by cancer cells increases where they are used for generation of energy by beta oxidation [91] to meet the increasing demands of the rapidly proliferating OvCa cells.
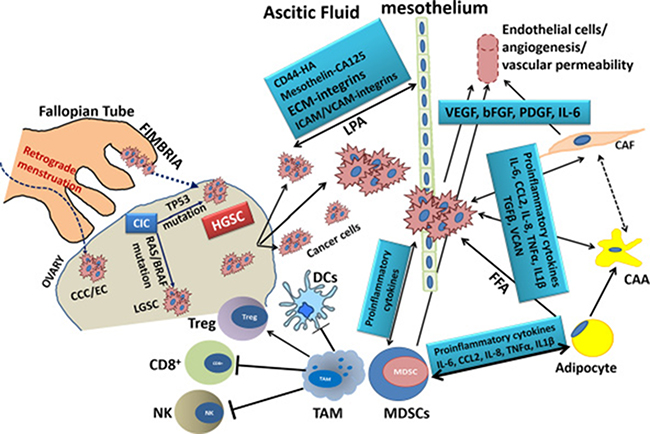
Figure 1: Schematic Representation of the key cell types in Ovarian Cancer Microenvironment and the molecules involved in their interactions. HGSC: high grade serous cancer; LGSC: low grade serous cancer; CCC: clear cell carcinoma; EC: endometrial carcinoma; CIC: carcinoma in situ; CAA: cancer-associated adipocyte; CAF: cancer-associated fibroblast; FFA: free fatty acids; VEGF: vascular endothelial growth factor; bFGF: basic fibroblast growth factor; PDGF: platelet-derived growth factor; VCAN: versican; CD8+, cytotoxic T cell; Treg: regulatory T cell; DCs: dendritic cells; MDSCs: myeloid-derived suppressor cell; ECM, extracellular matrix; IL-x, interleukin-x; ICAM/VCAM: intercellular/vascular adhesion molecule; HA: hyaluronic acid; CA125: cancer antigen 125; LPA: lysophosphatidic acid; NK: natural killer cell; TAM: tumor-associated macrophage; TGFβ: growth transforming growth factor β; TNFα: tumor necrosis factor-α.
Fibroblasts
Cancer associated fibroblasts (CAFs) play an important role in the EOC progression. In the peritoneal milieu, the origin of CAFs is unclear. The activation of resident fibroblasts and mesenchymal stem cells has long been considered the main origin of CAFs in the tumor microenvironment [99]. However, mesothelial cells have been shown as an important source of activated fibroblasts in inflammatory and fibrotic peritoneal pathologies as peritoneal dialysis, in which mesothelial cells are converted into myofibroblasts through mesothelial to mesenchymal transition (MMT) [100]. This hypothesis was supported in clinical specimens from patients with peritoneal metastases from ovarian and colon cancers, in which submesothelial fibroblasts expressing both mesothelial (calretinin, cytokeratins, mesothelin) and myofibroblasts (α-SMA) markers were detected by immunostaining [101]. More recently, mechanistic studies by Rynne-Vidal and colleagues [102] demonstrated that mesothelial cells isolated from ascitic fluid of OvCa patients with peritoneal metastases underwent MMT and promoted in vivo growth of xenografts through TGF-β-Smad-dependent MMT program, highlighting the crucial impact of the TGF-β-mediated bidirectional communication between OvCa cells and mesothelial cell-derived CAFs to form a suitable metastatic niche [102].
Another source of CAFs in the unique peritoneal TME is the omental adipocytes that have undergone delipidation and de-differentiation into pre-adipocyte fibroblastoid or stem-like stage [98, 103, 104]. Although this hypothesis has not yet been reported in OvCa, our unpublished data as well as reports in breast and pancreatic cancer [98, 103, 104] support its implication in OvCa. This is further supported by earlier reports of adipocyte de-differentiation into fibroblasts in inflammatory fibrotic changes encountered in dysfunctional adipose tissues in obesity and type 2 diabetes [105]. Moreover, endothelial to mesenchymal transition that has been reported in vasculopathies and atherosclerotic plaques [106] was suggested as a source of CAFs in OvCa [107].
The CAF phenotype is induced by environmental cues (Figure 2) characterized by inflammation, and hypoxia activating fibroblasts to exhibit characteristics of both myofibroblasts and secretory phenotype further contributing to inflammatory TME, cancer invasiveness and metastasis [101, 102, 108–111]. Increased number of CAFs corresponds with a more advanced OvCa stage, higher frequency of lymph node metastases, and amplified lymphatic and micro-vessel density [111]. CAFs can be activated by multiple mechanisms triggered by secreted factors from OvCa cells as TGF-β1, inflammatory cytokines and chemokines, reactive oxygen species (ROS) as well as MMPs [108]. The activation of CAF by TNFα has been shown to upregulate transforming growth factor α (TGF-α) through an inflammatory process activating NFkB. In turn CAF-derived TGFα induces epidermal growth factor (EGFR) signaling in cancer cells which stimulates cancer cell growth [112]. CAFs can also be activated through increased expression of progranulin (PGRN) peptide that stimulates EMT in cancer cells and upregulates the expression of smooth muscle actin α (α-SMA) in fibroblasts. High levels of both PGRN and α-SMA and low E-cadherin levels were associated with poor prognosis [108, 113]. Importantly, the molecular cross-talk between cancer cells and CAFs in the ovarian TME has been shown to be regulated by TGFβ/TGFβRs/SMAD pathway in CAFs with subsequent overexpression and secretion of target genes as versican [110]. The latter mediates tumor migration and invasion through binding to CD44 with subsequent activation of the NFkB and JNK signaling pathways in OvCa cells further supporting a pro-inflammatory TME and tumor progression [110].
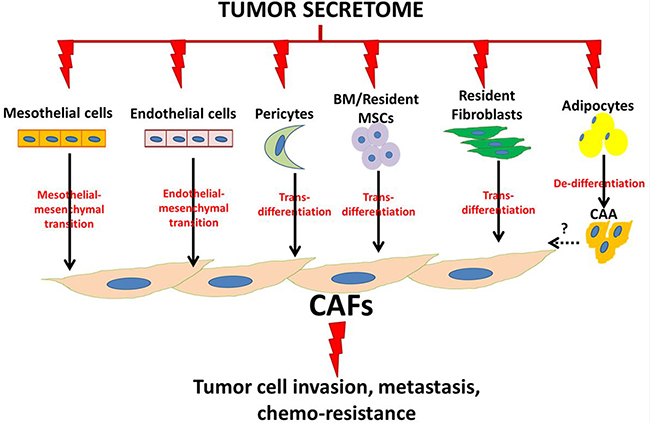
Figure 2: Schematic illustration of ovarian cancer cell-induced phenotypic commitment of stromal cells into cancer-associated phenotype. CAFs: cancer-associated fibroblasts, CAA: cancer-associated adipocytes, BM: bone marrow, MSCs: mesenchymal stem cells.
Tumor associated macrophages (TAMs)
The pro-inflammatory peritoneal TME characterized by increased LPA is continuously produced by cancer and mesothelial cells with subsequent increase in several pro-inflammatory cytokines and chemokines including macrophage chemoattractant protein-1 (MCP-1/CCL2) which recruits macrophages to the peritoneal TME promoting tumor growth, invasiveness, vascular permeability and angiogenesis. The cross-talk between cancer cells and TAMs upregulates the secretion of MMPs, uPA/uPAR, and prostaglandin E2 (PGE2) [79, 80, 114–117] which influence tumor migration and invasion through activation of NFkB, the key regulator of pro-inflammatory molecules in both TAMs and cancer cells, making them even more important in tumor progression [79, 80]. In addition, the increased infiltration of TAMs in the peritoneal TME not only promotes cancer cell invasiveness but also contributes to an immunosuppressive environment that suppresses the function of T cells, dendritic cells (DCs) and natural killer (NK) cells [7]. TAMs also contribute to the phenotypic switch of fibroblasts into cancer associated fibroblasts (CAFs), and in turn activate multiple pathways specifically TGFβ pathway that leads to inflammation and immune suppression, and the associated chemo-resistance, recurrence and poor prognosis [118, 119]. In addition, increased TAMs in the OvCa milieu was found to decrease the sensitivity to VEGF targeting therapy [120]. The augmented inflammatory TME promoted many clinical trials targeting inflammatory cytokines/chemokines and their receptors, TGFβ/TGFβRs as well as COX-2 inhibitors (Table 1).
Table 1: Clinical trials targeting the ovarian cancer cells and their interactions with tumor microenvironment
Drug |
Target |
Clinical Trial |
NCT Trial |
|---|---|---|---|
Aflibercept (VEGF trap) |
vascular endothelial growth factor (VEGF) |
Phase 2 |
NCT00327171 |
Bevacizumab + paclitaxel and carboplatin |
VEGF-A |
Phase 3 |
NCT01239732 |
Bevacizumab and Erlotinib |
VEGF-A + EGFR |
Phase 2 |
NCT00130520 |
Bevacizumab + Carboplatin |
VEGF-A |
Phase 2 |
NCT00937560 |
Chiauranib |
serine-threonine kinases: aurora kinase B (aurora B), VEGF receptors (VEGFRs), stem cell factor receptor (c-KIT), and platelet-derived growth factor receptors (PDGFRs) |
Phase 1 |
NCT03166891 |
Nintedanib + Bevacizumab |
VEGFR1/2/3, FGFR1/2/3 and PDGFRα/β (angiogenesis and fibrosis) |
Phase 1 |
NCT02835833 |
INCB062079 |
fibroblast growth factor receptor 4 (FGFR4) |
Phase 1 |
NCT03144661 |
Sorafinib + paclitaxel and carboplatin |
Multi-targeted Receptor Tyrosine Kinase Inhibitor (RTKi) |
Phase 2 |
NCT00390611 |
Sunitinib (SU11248) |
Multi-targeted RTKi |
Phase 2 |
NCT00543049 |
Tocilizumab and Interferon alpha 2-b (IFN-α2b)+ Carboplatin and Caelyx or doxorubicin |
Interleukin-6 receptor (IL-6R) |
Phase 1 |
NCT01637532 |
Siltuximab (CNTO 328) |
IL-6R |
Phase 2 |
NCT00841191 |
Plerixafor |
CXCR4 |
Phase 1 |
NCT02179970 |
PD 0360324+ cyclophosphamide |
Macrophage colony stimulating factor (M-CSF) |
Phase 2 |
NCT02948101 |
Celecoxib + cyclophosphamide |
cyclooxygenases (COX-1 and COX-2) |
Phase 2 |
NCT00538031 |
Ketorolac |
COX-1 and COX-2/GTPase inhibition |
Phase 0 |
NCT02470299 |
Metformin + paclitaxel and carboplatin |
Antidiabetic medication/metabolism |
Phase 1 |
NCT02312661 |
Metformin |
Antidiabetic medication/metabolism |
Phase 2 |
NCT01579812 |
Metformin+ atorvastatin + doxycycline+ mebendazole |
Antidiabetic medication/metabolism (glucose and lipid levels) |
Phase 3 |
NCT02201381 |
INCAGN01876 + Nivolumab + Ipilimumab |
Tumor necrosis factor α (TNFα), Programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocytes’ CTLA-4. |
Phase 1 |
NCT03126110 |
MK-3475 (pembrolizumab) + Gemcitabine and cisplatin |
PD-1 |
Phase 2 |
NCT02608684 |
Oregovomab and Nivolumab |
Cancer Antigen 125 (CA-125) and PD-1 |
Phase 1 |
NCT03100006 |
Durvalumab (MEDI4736 + motolimod) + pegylated liposomal doxorubicin |
Programmed cell death ligand 1(PD-L1) and Toll like receptor 8 (TLL8) |
Phase1 |
NCT02431559 |
Autologous Monocytes + Sylatron (PegIFNα)+ Actimmune (IFNγ-1b) |
Immunotherapy |
Phase 1 |
NCT02948426 |
Vigil bi-shRNA furin and GMCSF (FANG) Augmented Autologous Tumor Cell Immunotherapy |
TGFβ1 and TGFβ2 (tumor)+ Immune stimulation |
Phase 2 |
NCT02346747 |
Vigil (Adjuvant FANG) |
TGFβ1 and TGFβ2 (tumor) + Immune stimulation |
Phase 2 |
NCT01309230 |
Atezolizumab and Vigil |
PDL1 and TGFβ1 and TGFβ2 (tumor) |
Phase 2 |
NCT03073525 |
NK immunotherapy |
Combination of Cryosurgery and NK Immunotherapy |
Phase 2 |
NCT02849353 |
Therapeutic autologous Antigen-Specific CD4+ lymphocytes |
Immunotherapy |
Phase 1 |
NCT00101257 |
Myeloid-derived suppressor cells (MDSCs)
These represent a heterogeneous population of cells of myeloid origin that, in the steady immature state, are present in the bone marrow, but not in secondary lymphoid organs and lack suppressive activity. The phenotype of MDSCs is Lin-HLA−DR-CD33+ or CD11b+CD14−CD33+ and have also been identified within a CD15+ population in human peripheral blood [121]. When activated by ROS, reactive nitrogen species (RNS), or arginase, arginase they become potent suppressors of various T-cell functions. MDSCs accumulate in lymphoid organs and in tumors in response to growth factors and inflammatory cytokines/chemokines [122] as well as PGE2 enriched in the OvCa TMDC. Obermajer and colleagues [123] showed that PGE-2 attracts MDSC into ascites of OvCa patients by inducing expression of functional CXCR4 in cancer-associated MDSCs, and plays a role in the production of its ligand CXCL12 [123]. These studies provided a rationale for targeting COX-2, and CXCR4 in cancer therapy [123] (Table 1). In tumor tissues, MDSCs can be distinguished from TAMs by their granulocytic morphology, high expression of arginase, inducible nitric oxide synthase (iNOS), and Gr1 (which are not expressed by TAMs) as well as low expression of F4/80 (expressed by TAMs) [121]. In addition to suppressing T-cell functions, high numbers of immunosuppressive MDSCs in the OvCa TME were identified and were shown to promote and maintain the OvCa stem cell pool by stimulating miR-101 expression and targeting co-repressor CtBP2 in OvCa cells [124].
Dendritic cells (DCs)
Dendritic cells (DCs) are specialized antigen-presenting mononuclear cells, that in their immature state exhibit phagocytic ability, and when become functionally mature, they produce cytokines and exhibit immunostimulatory capacity [125]. DCs are sensitized after exposure to tumor antigen, migrate to regional lymph nodes, and stimulate the proliferation of naive T cells to initiate the immune response [126]. Based on the expression of cluster of differentiation (CD) markers, DCs exhibit antigen cross-presentation via MHC class I or class II molecules to activate CD8+ or CD4+ T cells [125]. Increased number of tumor-infiltrating DCs in tumor tissue was correlated with favorable prognosis [126]. In addition, substantial numbers of functional plasmacytoid (tolerogenic) dendritic cells (PDCs) were detected in malignant ascites of patients with OvCa and mechanistically, they significantly induced tumor angiogenesis [125]. The ability of DCs to process and present antigens and stimulate anti-tumor immune response promoted the development of clinical trials using DCs vaccines in which autologous DCs pulsed with either tumor cell lysates in combination with bevacizumab and oral metronomic cyclophosphamide for patients with recurrent stage III/IV OvCa in whom anti-tumor responses and clinical benefit for patients were observed [127, 128].
Lymphocytes
Tumor associated lymphocytes (TILs) comprise T-cells, and regulatory T cells (T-regs) that have left the intravascular compartment and localized in tumor stroma (stromal TILs) or inside the tumor islets (intraepithelial TILs). In particular, intraepithelial TILs play an extensive role in the control of tumor growth in almost all solid tumors including OvCa (summarized in [129]). CD8+ or CD4+T-lymphocytes can recognize cancer antigens or over-expressed self-antigens and inhibit cancer development [129]. TILs play a key role in tumor immune surveillance through T cell receptor (TCR)-mediated recognition of tumor antigens that have been processed by antigen presenting DCs [130]. Upon recognition of tumor antigens by TCR/MHC engagement, activated CD8+ cytotoxic T cells are able to directly kill malignant cells by mechanisms including perforin/granzyme secretion and FasL/Fas binding [131]. Studies indicated that the expression of the death Fas ligand (FasL/CD95L) was exclusively expressed in tumor vasculature and created a barrier that suppressed normal T cell function, allowing tumor cells to grow unrecognized by the immune system. Blocking this selective FasL expression may lead to effective immunotherapy targeting tumor progression [132–134]. Along with CD4+ helper T cells, cytotoxic CD8+ T-cells can secrete various cytokines/chemokines to direct the activities of other immune cells [135]. Several clinical studies in OvCa, reported a positive correlation between patient survival and the presence of intra-epithelial TILs [135–137]. CD3+ TILs were reported in treatment-naïve OvCa specimens, but survival advantage was associated only with intraepithelial but not stromal TILs [135, 138, 139]. Meta-analysis of the majority of reports that investigated the prognostic value of TILs in OvCa [140] using the CD8+ marker to specifically evaluate cytotoxic T cells, found that intraepithelial CD8+ TILs exhibited a consistent and stronger association with patients’ survival than CD3+ TILs [140].
In the inflammatory OvCa TME, TILs’ function is suppressed by regulatory T cells (Tregs), MDSCs, and TAMs, with their secreted plethora of soluble inhibitory factors as IL-6, IL-10, arginase (Arg)1, and TGFβ, various metabolites like adenosine, and depleted tryptophan due to increased indoleamine 2,3-dioxygenase 1 (IDO-1) activity [141, 142]. Suppression of T cell functions occurs through downregulation of MHC molecules and co-stimulatory ligands, with upregulation of inhibitory receptors like programmed cell death protein ligand 1 (PD-L1) on tumor cells and cytotoxic T-lymphocyte antigen-4 (CTLA-4, CD152) [143]. Hamanishi and colleagues [144] reported that high expression of programmed death 1 (PD-1/CD279) on OvCa cells was associated with poorer patients’ survival and with reduced CD8+ TILs suggesting that PD-L1 expression promotes an immunosuppressive TME by inhibiting T-cell infiltration [144]. These observations promoted clinical trials targeting PD1 or PDL-1 as well as CTLA-4 in OvCa (Table 1). The efficacy of single or dual blockade of PD-1 and/or CTLA-4 in combination with standard of care therapy was demonstrated in multiple models of OvCa with synergistic effects [143, 145, 146]. However the efficacy in treatment of OvCa patients is still being evaluated in clinical trials.
Regulatory T-cells (Treg) cells. CD4 Tregs are T-cell subpopulation that suppresses the function of activated T-cells. They can be divided into naturally occurring thymus-generated T-regs with a phenotype of CD4+CD25+FOXP3+ and the adaptive Tr1 Treg and Th3 Tregs which have variable CD25 expression. Moreover, patients with OvCa expressed Treg subsets with upregulated cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and downregulated expression of CD28 [147, 148]. Additionally, in vitro induced CD8 Tregs block CD4 T-cells proliferation via TGF-β1 and IFN-ɣ. Tumors are not only known to increase the number of Tregs in peripheral blood of OvCa patients, but also recruit and stimulate Treg tumor infiltration and localization [147, 148].
Natural killer cells (NK) are lymphocytes of the innate immune system and are defined by the expression of cell adhesion markers CD56 and CD16 and the lack of T-cell receptor CD3 [149]. They target cells with low MHC Class-I expression including tumor cells by using perforins to puncture the membranes of target cells leading to activation of the apoptotic cascade by granzymes. In addition to the aforementioned mechanism, members of the tumor necrosis factor receptor family such as Fas/CD59 also contribute to NK cytotoxicity when activated [150]. In the face of such immune-surveillance, tumor cells have adeptly managed to go undetected via several mechanisms. For instance, studies showed that MUC16, a high molecular weight mucin overexpressed by OvCa has the ability to inhibit NK cell and downregulate CD16. NK cells lose their CD16 by a metalloprotease called ADAM17. Inhibition of this metalloprotease enhances CD16-mediated NK cell killing ability through antibody-mediated cellular cytotoxicity by maintaining the CD16 on the cell surface [151]. This phenomenon is not exclusive to OvCa cells; some leukemia cell lines inhibit NK cells by up-regulating MHC Class-I expression which sends an inhibitory signal to NK cells [150].
Endothelial cells: In the ovarian TME, two factors are critical to modulate the blood vessel structure: cellular permeability and angiogenesis [152]. In normal tissue, the endothelial cells are composed of a single layer of continuous uniform cells with few cytoplasmic projections, while tumor endothelial cells are of abnormal shape and size [152, 153]. OvCa cells secrete a plethora of factors to induce phenotypic changes in the omental microvasculature. Initially identified as vascular permeability factor [154], vascular endothelial growth factor (VEGF) has been long considered as the key regulator of angiogenesis that drives endothelial cell survival, proliferation, and migration while increasing vascular permeability [155]. VEGF is not only produced by cancer cells, but it is also produced by TAMs, CAAs, as well as CAFs [155, 156]. VEGF contributes to the development of peritoneal carcinomatosis associated with malignant ascites formation, the hallmarks of advanced OvCa [156, 157]. Preclinical and clinical studies showed that VEGF levels inversely correlates with disease prognosis and patients’ survival [155–157]. VEGF inhibition has been shown to inhibit tumor growth, invasion, metastasis, and ascites production. These findings promoted the clinical evaluation of agents targeting VEGF/VEGFRs with approval of anti-VEGF humanized antibody (Avastin, bevacizumab) in patients with OvCa [156] as single agents or in combination with standard of care therapy (https://www.cancer.gov/about-cancer/treatment/drugs/ovarian). In addition to VEGF, deregulation of normal endothelium in the peritoneal TME is also induced by proangiogenic factors such as IL-6, IL-8, PDGF, FGF, CCL2, CXCR4, uPA/uPAR, angiopiotein-1, bioactive lipids and neuroendocrine hormones produced by OvCa cells and the other cellular components in the peritoneal TME [120, 156]. This upregulation of the proangiogenic factors and their interconnected signaling pathways not only contributes to increased vascular permeability, tumor growth and angiogenesis, but also contributes to the suboptimal response to VEGF/VEGFR targeting therapy [120, 156]. Therefore, clinical trials targeting these proangiogenic factors, their receptors including receptor tyrosine kinases in OvCa patients, are currently underway (Table 1).
Ascitic fluid. In normal physiologic conditions, the movement of proteins from the intravascular space to the peritoneal fluid is tightly regulated by 5 layer-barrier namely, capillary endothelium, capillary basement membrane, interstitial stroma, mesothelial basement membrane and mesothelial cells of the peritoneal lining [158]. This barrier is maintained by intact tight junctions (at the mesothelial and endothelial interface) and intravascular anionic macromolecules which create a difference in oncotic pressure across the peritoneal membrane [158]. In patients with OvCa, this barrier is breached with increased vascular and mesothelial permeability and transudation of high protein fluid from intravascular compartment to peritoneal cavity. Along with the high protein concentration, increased inflammatory cytokines and chemokines and reduced lymphatic flow also contribute to the buildup of ascitic fluid [6, 25, 156, 158, 159]. In addition, ascites is rich in bioactive lipids as lysophosphatidic acid (LPA), that has been long identified as OvCa promoting factor [160]. LPA is produced by OvCa cells and the cellular components in the peritoneal TME. High levels of LPA in ascitic fluid lead to aberrant receptor signaling and activation of pro-inflammatory and pro-survival pathways as well as transactivation of receptor tyrosine kinases [161–164] that further OvCa progression and are associated with poor prognosis [7, 160, 165]. At the cellular level, ascitic fluid contains floating cancer cells, macrophages and immune cells; all contribute to the malignant aggressive phenotype of OvCa [120, 155–157].
Treatment
Standard treatment of OvCa is composed of initial surgical management with exploratory laparotomy followed by total hysterectomy, bilateral salpingo-oophrectomy, omentectomy, and retroperitoneal lymphadenectomy depending on the extent and spread of the primary tumor. Initial treatment options are primary surgery followed by chemotherapy and neoadjuvant chemotherapy followed by surgery. Standard chemotherapy involves carboplatin and paclitaxel. Various targeted therapies are being studied in combination with carboplatin/paclitaxel in hopes of improving survival (Table 1). In addition to the recently FDA approved targeted therapies as poly (ADP-ribose) polymerase PARP inhibitors and VEGF inhibitors, other targeted therapies currently in clinical trials include other angiogenesis inhibitors (VEGF/VEGFRs, FGFRs, PDGFRα/β), multi-target receptor tyrosine kinase inhibitors (RTKi), cyclooxygenase-2 (Cox-2) inhibitors, and inhibitors of cytokines and their receptors (IL-6/IL-6R, SDF1/CXCR4, M-CSF, TGFβ1/2/3).
In recent years, immunotherapy for advanced stage OvCa was introduced in clinical trials using immune check points inhibitors targeting programmed cell death protein (PD1) and its ligand (PDL1) as well as cytotoxic T-lymphocyte-associated protein 4 (CTL4) with neutralizing antibodies that restore the functions of cytotoxic T-lymphocytes to recognize and eradicate tumor cells. More recently, personalized therapy with autologous tumor and immune cells reprogrammed ex-vivo to stimulate the immune system and overcome immune evasion of OvCa cells are currently in clinical trials. However, emerging data suggests limited survival advantages. In addition, targeting tumor metabolism has recently gained more appreciation as evidenced by clinical trials of metformin in advanced HGSC either alone or in combination of standard of care therapy (Table 1).
Despite the changing nature of chemotherapeutic regimens, OvCa resistance and recurrence still remain a common problem mainly due to suboptimal resection of microscopic and/or lesions that cannot be removed due to their site in the peritoneal cavity, tumor heterogeneity, evolution of chemo-resistant tumor cells and the unique site of OvCa spread in the peritoneal cavity.
CONCLUSIONS
Advanced epithelial OvCa carries the largest burden of disease mortality among all gynecologic malignancies. Although the majority of patients show substantial initial response to first-line therapy (e.g., surgery and combined platinum plus paclitaxel based therapies), almost 70% of patients experience recurrence of their cancer within 18 months. Our ability to effectively treat recurrent OvCa is the single greatest impediment to improve disease outcome. Current challenges in curing patients with OvCa are: 1) late-stage detection for the majority of OvCa 2) suboptimal debulking surgery mostly due to infiltrative nature of the disease, and 3) recurrence of chemo-resistant cancer. Indeed successful treatment of OvCa can be achieved by improving surgical approaches to precisely excise the tumor with minimal residual disease, and enhancing our understanding of the complex interplay of cancer cells within the unique peritoneal TME. The ultimate goals are: 1) identifying and validating diagnostic biomarkers for early stage disease, 2) identifying and validating prognostic biomarkers in the primary tumor that predict response to therapy and recurrence, and 3) targeting the determinants of cancer cell-TME interactions in neoadjuvant or adjuvant settings.
ACKNOWLEDGMENTS
This work was supported by Marsha Rivkin Pilot award and R01 CA193437 to N.S.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Goff BA, Mandel LS, Drescher CW, Urban N, Gough S, Schurman KM, Patras J, Mahony BS, Andersen MR. Development of an ovarian cancer symptom index: possibilities for earlier detection. Cancer. 2007; 109:221–27. https://doi.org/10.1002/cncr.22371.
2. Gershenson DM, Sun CC, Lu KH, Coleman RL, Sood AK, Malpica A, Deavers MT, Silva EG, Bodurka DC. Clinical behavior of stage II-IV low-grade serous carcinoma of the ovary. Obstet Gynecol. 2006; 108:361–68. https://doi.org/10.1097/01.AOG.0000227787.24587.d1.
3. Borley J, Wilhelm-Benartzi C, Yazbek J, Williamson R, Bharwani N, Stewart V, Carson I, Hird E, McIndoe A, Farthing A, Blagden S, Ghaem-Maghami S. Radiological predictors of cytoreductive outcomes in patients with advanced ovarian cancer. BJOG. 2015; 122:843–49. https://doi.org/10.1111/1471-0528.12992.
4. Borley J, Wilhelm-Benartzi C, Brown R, Ghaem-Maghami S. Does tumour biology determine surgical success in the treatment of epithelial ovarian cancer? A systematic literature review. Br J Cancer. 2012; 107:1069–74. https://doi.org/10.1038/bjc.2012.376.
5. Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, Godwin AK, Gross J, Hartmann L, et al, and Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–15. https://doi.org/10.1038/nature10166.
6. Ahmed N, Stenvers KL. Getting to know ovarian cancer ascites: opportunities for targeted therapy-based translational research. Front Oncol. 2013; 3:256. https://doi.org/10.3389/fonc.2013.00256.
7. Worzfeld T, Pogge von Strandmann E, Huber M, Adhikary T, Wagner U, Reinartz S, Müller R. The Unique Molecular and Cellular Microenvironment of Ovarian Cancer. Front Oncol. 2017; 7:24. https://doi.org/10.3389/fonc.2017.00024.
8. Rizzuto I, Stavraka C, Chatterjee J, Borley J, Hopkins TG, Gabra H, Ghaem-Maghami S, Huson L, Blagden SP. Risk of Ovarian Cancer Relapse score: a prognostic algorithm to predict relapse following treatment for advanced ovarian cancer. Int J Gynecol Cancer. 2015; 25:416–22. https://doi.org/10.1097/IGC.0000000000000361.
9. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67:7–30. https://doi.org/10.3322/caac.21387.
10. Howell EA, Egorova N, Hayes MP, Wisnivesky J, Franco R, Bickell N. Racial disparities in the treatment of advanced epithelial ovarian cancer. Obstet Gynecol. 2013; 122:1025–32. https://doi.org/10.1097/AOG.0b013e3182a92011.
11. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, Yang W, Heravi-Moussavi A, Giuliany R, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010; 363:1532–43. https://doi.org/10.1056/NEJMoa1008433.
12. King MC, Marks JH, Mandell JB, Grp NY, and New York Breast Cancer Study Group. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003; 302:643–46. https://doi.org/10.1126/science.1088759.
13. Crijnen TE, Janssen-Heijnen ML, Gelderblom H, Morreau J, Nooij MA, Kenter GG, Vasen HF. Survival of patients with ovarian cancer due to a mismatch repair defect. Fam Cancer. 2005; 4:301–05. https://doi.org/10.1007/s10689-005-6573-2.
14. Watson P, Bützow R, Lynch HT, Mecklin JP, Järvinen HJ, Vasen HF, Madlensky L, Fidalgo P, Bernstein I, and International Collaborative Group on HNPCC. The clinical features of ovarian cancer in hereditary nonpolyposis colorectal cancer. Gynecol Oncol. 2001; 82:223–28. https://doi.org/10.1006/gyno.2001.6279.
15. Salehi F, Dunfield L, Phillips KP, Krewski D, Vanderhyden BC. Risk factors for ovarian cancer: an overview with emphasis on hormonal factors. J Toxicol Environ Health B Crit Rev. 2008; 11:301–21. https://doi.org/10.1080/10937400701876095.
16. Whiteman DC, Siskind V, Purdie DM, Green AC. Timing of pregnancy and the risk of epithelial ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2003; 12:42–46.
17. Stewart LM, Holman CD, Aboagye-Sarfo P, Finn JC, Preen DB, Hart R. In vitro fertilization, endometriosis, nulliparity and ovarian cancer risk. Gynecol Oncol. 2013; 128:260–64. https://doi.org/10.1016/j.ygyno.2012.10.023.
18. Ness RB, Cramer DW, Goodman MT, Kjaer SK, Mallin K, Mosgaard BJ, Purdie DM, Risch HA, Vergona R, Wu AH. Infertility, fertility drugs, and ovarian cancer: a pooled analysis of case-control studies. Am J Epidemiol. 2002; 155:217–24. https://doi.org/10.1093/aje/155.3.217.
19. Chittenden BG, Fullerton G, Maheshwari A, Bhattacharya S. Polycystic ovary syndrome and the risk of gynaecological cancer: a systematic review. Reprod Biomed Online. 2009; 19:398–405. https://doi.org/10.1016/S1472-6483(10)60175-7.
20. Anderson GL, Judd HL, Kaunitz AM, Barad DH, Beresford SA, Pettinger M, Liu J, McNeeley SG, Lopez AM, and Women’s Health Initiative Investigators. Effects of estrogen plus progestin on gynecologic cancers and associated diagnostic procedures: the Women’s Health Initiative randomized trial. JAMA. 2003; 290:1739–48. https://doi.org/10.1001/jama.290.13.1739.
21. Tworoger SS, Gertig DM, Gates MA, Hecht JL, Hankinson SE. Caffeine, alcohol, smoking, and the risk of incident epithelial ovarian cancer. Cancer. 2008; 112:1169–77. https://doi.org/10.1002/cncr.23275.
22. Huncharek M, Geschwind JF, Kupelnick B. Perineal application of cosmetic talc and risk of invasive epithelial ovarian cancer: a meta-analysis of 11,933 subjects from sixteen observational studies. Anticancer Res. 2003; 23:1955–60.
23. Makowski L, Zhou C, Zhong Y, Kuan PF, Fan C, Sampey BP, Difurio M, Bae-Jump VL. Obesity increases tumor aggressiveness in a genetically engineered mouse model of serous ovarian cancer. Gynecol Oncol. 2014; 133:90–97. https://doi.org/10.1016/j.ygyno.2013.12.026.
24. Zhang Y, Nowicka A, Solley TN, Wei C, Parikh A, Court L, Burks JK, Andreeff M, Woodward WA, Dadbin A, Kolonin MG, Lu KH, Klopp AH. Stromal Cells Derived from Visceral and Obese Adipose Tissue Promote Growth of Ovarian Cancers. PLoS One. 2015; 10:e0136361. https://doi.org/10.1371/journal.pone.0136361.
25. Vaughan S, Coward JI, Bast RC Jr, Berchuck A, Berek JS, Brenton JD, Coukos G, Crum CC, Drapkin R, Etemadmoghadam D, Friedlander M, Gabra H, Kaye SB, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011; 11:719–25. https://doi.org/10.1038/nrc3144.
26. Kurman RJ. Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Ann Oncol. 2013 (Suppl 10); 24:x16–21. https://doi.org/10.1093/annonc/mdt463.
27. Vang R, Shih IM, Kurman RJ. Ovarian low-grade and high-grade serous carcinoma: pathogenesis, clinicopathologic and molecular biologic features, and diagnostic problems. Adv Anat Pathol. 2009; 16:267–82. https://doi.org/10.1097/PAP.0b013e3181b4fffa.
28. chmeler KM, Sun CC, Malpica A, Deavers MT, Bodurka DC, Gershenson DM. Low-grade serous primary peritoneal carcinoma. Gynecol Oncol. 2011; 121:482-6.
29. Catasús L, Bussaglia E, Rodrguez I, Gallardo A, Pons C, Irving JA, Prat J. Molecular genetic alterations in endometrioid carcinomas of the ovary: similar frequency of beta-catenin abnormalities but lower rate of microsatellite instability and PTEN alterations than in uterine endometrioid carcinomas. Hum Pathol. 2004; 35:1360–68. https://doi.org/10.1016/j.humpath.2004.07.019.
30. Moreno-Bueno G, Gamallo C, Pérez-Gallego L, de Mora JC, Suárez A, Palacios J. beta-Catenin expression pattern, beta-catenin gene mutations, and microsatellite instability in endometrioid ovarian carcinomas and synchronous endometrial carcinomas. Diagn Mol Pathol. 2001; 10:116–22. https://doi.org/10.1097/00019606-200106000-00008.
31. Lee S, Garner EI, Welch WR, Berkowitz RS, Mok SC. Over-expression of hypoxia-inducible factor 1 alpha in ovarian clear cell carcinoma. Gynecol Oncol. 2007; 106:311–17. https://doi.org/10.1016/j.ygyno.2007.03.041.
32. Stadlmann S, Gueth U, Baumhoer D, Moch H, Terracciano L, Singer G. Glypican-3 expression in primary and recurrent ovarian carcinomas. Int J Gynecol Pathol. 2007; 26:341–44. https://doi.org/10.1097/pgp.0b013e31802d692c.
33. Kato N, Sasou S, Motoyama T. Expression of hepatocyte nuclear factor-1beta (HNF-1beta) in clear cell tumors and endometriosis of the ovary. Mod Pathol. 2006; 19:83–89. https://doi.org/10.1038/modpathol.3800492.
34. Tan DS, Kaye S. Ovarian clear cell adenocarcinoma: a continuing enigma. J Clin Pathol. 2007; 60:355–60. https://doi.org/10.1136/jcp.2006.040030.
35. Kurman RJ, Shih IM. Pathogenesis of ovarian cancer: lessons from morphology and molecular biology and their clinical implications. Int J Gynecol Pathol. 2008; 27:151–60. https://doi.org/10.1097/PGP.0b013e318161e4f5.
36. Vang R, Gown AM, Wu LS, Barry TS, Wheeler DT, Yemelyanova A, Seidman JD, Ronnett BM. Immunohistochemical expression of CDX2 in primary ovarian mucinous tumors and metastatic mucinous carcinomas involving the ovary: comparison with CK20 and correlation with coordinate expression of CK7. Mod Pathol. 2006; 19:1421–28. https://doi.org/10.1038/modpathol.3800698.
37. Karnezis AN, Cho KR, Gilks CB, Pearce CL, Huntsman DG. The disparate origins of ovarian cancers: pathogenesis and prevention strategies. Nat Rev Cancer. 2017; 17:65–74. https://doi.org/10.1038/nrc.2016.113.
38. Koshiyama M, Matsumura N, Konishi I. Subtypes of Ovarian Cancer and Ovarian Cancer Screening. Diagnostics (Basel). 2017; 7:E12. https://doi.org/10.3390/diagnostics7010012.
39. Casagrande JT, Louie EW, Pike MC, Roy S, Ross RK, Henderson BE. “Incessant ovulation” and ovarian cancer. Lancet. 1979; 2:170–73. https://doi.org/10.1016/S0140-6736(79)91435-1.
40. Kindelberger DW, Lee Y, Miron A, Hirsch MS, Feltmate C, Medeiros F, Callahan MJ, Garner EO, Gordon RW, Birch C, Berkowitz RS, Muto MG, Crum CP. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: evidence for a causal relationship. Am J Surg Pathol. 2007; 31:161–69. https://doi.org/10.1097/01.pas.0000213335.40358.47.
41. Ng A, Barker N. Ovary and fimbrial stem cells: biology, niche and cancer origins. Nat Rev Mol Cell Biol. 2015; 16:625–38. https://doi.org/10.1038/nrm4056.
42. Seidman JD, Yemelyanova A, Zaino RJ, Kurman RJ. The fallopian tube-peritoneal junction: a potential site of carcinogenesis. Int J Gynecol Pathol. 2011; 30:4–11. https://doi.org/10.1097/PGP.0b013e3181f29d2a.
43. Bankhead CR, Kehoe ST, Austoker J. Symptoms associated with diagnosis of ovarian cancer: a systematic review. BJOG. 2005; 112:857–65. https://doi.org/10.1111/j.1471-0528.2005.00572.x.
44. Prat J, Oncology FC, and FIGO Committee on Gynecologic Oncology. Staging classification for cancer of the ovary, fallopian tube, and peritoneum. Int J Gynaecol Obstet. 2014; 124:1–5. https://doi.org/10.1016/j.ijgo.2013.10.001.
45. Auersperg N. The stem-cell profile of ovarian surface epithelium is reproduced in the oviductal fimbriae, with increased stem-cell marker density in distal parts of the fimbriae. Int J Gynecol Pathol. 2013; 32:444–53. https://doi.org/10.1097/PGP.0b013e3182800ad5.
46. Auersperg N. Ovarian surface epithelium as a source of ovarian cancers: unwarranted speculation or evidence-based hypothesis? Gynecol Oncol. 2013; 130:246–51. https://doi.org/10.1016/j.ygyno.2013.03.021.
47. Auersperg N. The origin of ovarian cancers—hypotheses and controversies. Front Biosci (Schol Ed). 2013; 5:709–19. https://doi.org/10.2741/S401.
48. Latifi A, Luwor RB, Bilandzic M, Nazaretian S, Stenvers K, Pyman J, Zhu H, Thompson EW, Quinn MA, Findlay JK, Ahmed N. Isolation and characterization of tumor cells from the ascites of ovarian cancer patients: molecular phenotype of chemoresistant ovarian tumors. PLoS One. 2012; 7:e46858. https://doi.org/10.1371/journal.pone.0046858.
49. Klymenko Y, Kim O, Stack MS. Complex Determinants of Epithelial: Mesenchymal Phenotypic Plasticity in Ovarian Cancer. Cancers (Basel). 2017; 9:104. https://doi.org/10.3390/cancers9080104.
50. Klymenko Y, Kim O, Loughran E, Yang J, Lombard R, Alber M, Stack MS. Cadherin composition and multicellular aggregate invasion in organotypic models of epithelial ovarian cancer intraperitoneal metastasis. Oncogene. 2017; 36:5840–51. https://doi.org/10.1038/onc.2017.171.
51. Davidson B, Tropé CG, Reich R. Epithelial-mesenchymal transition in ovarian carcinoma. Front Oncol. 2012; 2:33. https://doi.org/10.3389/fonc.2012.00033.
52. Rafehi S, Ramos Valdes Y, Bertrand M, McGee J, Préfontaine M, Sugimoto A, DiMattia GE, Shepherd TG. TGFβ signaling regulates epithelial-mesenchymal plasticity in ovarian cancer ascites-derived spheroids. Endocr Relat Cancer. 2016; 23:147–59. https://doi.org/10.1530/ERC-15-0383.
53. Zhou J, Jain S, Azad AK, Xu X, Yu HC, Xu Z, Godbout R, Fu Y. Notch and TGFβ form a positive regulatory loop and regulate EMT in epithelial ovarian cancer cells. Cell Signal. 2016; 28:838–49. https://doi.org/10.1016/j.cellsig.2016.03.016.
54. Huang RY, Wong MK, Tan TZ, Kuay KT, Ng AH, Chung VY, Chu YS, Matsumura N, Lai HC, Lee YF, Sim WJ, Chai C, Pietschmann E, et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013; 4:e915. https://doi.org/10.1038/cddis.2013.442.
55. Xu Z, Jiang Y, Steed H, Davidge S, Fu Y. TGFβ and EGF synergistically induce a more invasive phenotype of epithelial ovarian cancer cells. Biochem Biophys Res Commun. 2010; 401:376–81. https://doi.org/10.1016/j.bbrc.2010.09.059.
56. Davidowitz RA, Selfors LM, Iwanicki MP, Elias KM, Karst A, Piao H, Ince TA, Drage MG, Dering J, Konecny GE, Matulonis U, Mills GB, Slamon DJ, et al. Mesenchymal gene program-expressing ovarian cancer spheroids exhibit enhanced mesothelial clearance. J Clin Invest. 2014; 124:2611–25. https://doi.org/10.1172/JCI69815.
57. Strobel T, Cannistra SA. Beta1-integrins partly mediate binding of ovarian cancer cells to peritoneal mesothelium in vitro. Gynecol Oncol. 1999; 73:362–67. https://doi.org/10.1006/gyno.1999.5388.
58. Jin H, Yu Y, Zhang T, Zhou X, Zhou J, Jia L, Wu Y, Zhou BP, Feng Y. Snail is critical for tumor growth and metastasis of ovarian carcinoma. Int J Cancer. 2010; 126:2102–11. https://doi.org/10.1002/ijc.24901.
59. Sawada K, Mitra AK, Radjabi AR, Bhaskar V, Kistner EO, Tretiakova M, Jagadeeswaran S, Montag A, Becker A, Kenny HA, Peter ME, Ramakrishnan V, Yamada SD, Lengyel E. Loss of E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. 2008; 68:2329–39. https://doi.org/10.1158/0008-5472.CAN-07-5167.
60. Lessan K, Aguiar DJ, Oegema T, Siebenson L, Skubitz AP. CD44 and beta1 integrin mediate ovarian carcinoma cell adhesion to peritoneal mesothelial cells. Am J Pathol. 1999; 154:1525–37. https://doi.org/10.1016/S0002-9440(10)65406-5.
61. Ford CE, Punnia-Moorthy G, Henry CE, Llamosas E, Nixdorf S, Olivier J, Caduff R, Ward RL, Heinzelmann-Schwarz V. The non-canonical Wnt ligand, Wnt5a, is upregulated and associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Gynecol Oncol. 2014; 134:338–45. https://doi.org/10.1016/j.ygyno.2014.06.004.
62. Mao Y, Xu J, Li Z, Zhang N, Yin H, Liu Z. The role of nuclear β-catenin accumulation in the Twist2-induced ovarian cancer EMT. PLoS One. 2013; 8:e78200. https://doi.org/10.1371/journal.pone.0078200.
63. Du J, Sun B, Zhao X, Gu Q, Dong X, Mo J, Sun T, Wang J, Sun R, Liu Y. Hypoxia promotes vasculogenic mimicry formation by inducing epithelial-mesenchymal transition in ovarian carcinoma. Gynecol Oncol. 2014; 133:575–83. https://doi.org/10.1016/j.ygyno.2014.02.034.
64. Ha JH, Ward JD, Radhakrishnan R, Jayaraman M, Song YS, Dhanasekaran DN. Lysophosphatidic acid stimulates epithelial to mesenchymal transition marker Slug/Snail2 in ovarian cancer cells via Gαi2, Src, and HIF1α signaling nexus. Oncotarget. 2016; 7:37664–79. https://doi.org/10.18632/oncotarget.9224.
65. Lin G, Gai R, Chen Z, Wang Y, Liao S, Dong R, Zhu H, Gu Y, He Q, Yang B. The dual PI3K/mTOR inhibitor NVP-BEZ235 prevents epithelial-mesenchymal transition induced by hypoxia and TGF-β1. Eur J Pharmacol. 2014; 729:45–53. https://doi.org/10.1016/j.ejphar.2014.02.011.
66. Wang Y, Ma J, Shen H, Wang C, Sun Y, Howell SB, Lin X. Reactive oxygen species promote ovarian cancer progression via the HIF-1α/LOX/E-cadherin pathway. Oncol Rep. 2014; 32:2150–58. https://doi.org/10.3892/or.2014.3448.
67. Burleson KM, Casey RC, Skubitz KM, Pambuccian SE, Oegema TR Jr, Skubitz AP. Ovarian carcinoma ascites spheroids adhere to extracellular matrix components and mesothelial cell monolayers. Gynecol Oncol. 2004; 93:170–81. https://doi.org/10.1016/j.ygyno.2003.12.034.
68. Burleson KM, Hansen LK, Skubitz AP. Ovarian carcinoma spheroids disaggregate on type I collagen and invade live human mesothelial cell monolayers. Clin Exp Metastasis. 2004; 21:685–97. https://doi.org/10.1007/s10585-004-5768-5.
69. Casey RC, Koch KA, Oegema TR Jr, Skubitz KM, Pambuccian SE, Grindle SM, Skubitz AP. Establishment of an in vitro assay to measure the invasion of ovarian carcinoma cells through mesothelial cell monolayers. Clin Exp Metastasis. 2003; 20:343–56. https://doi.org/10.1023/A:1024009131191.
70. Hirashima Y, Kobayashi H, Suzuki M, Tanaka Y, Kanayama N, Terao T. Transforming growth factor-beta1 produced by ovarian cancer cell line HRA stimulates attachment and invasion through an up-regulation of plasminogen activator inhibitor type-1 in human peritoneal mesothelial cells. J Biol Chem. 2003; 278:26793–802. https://doi.org/10.1074/jbc.M212187200.
71. Lizalek J, McKenna T, Huegel K, Marsh S, Carolan A, Kobliska A, Heying E, Gardner N, Miller G, Kotecki A, Henningsen M, Lundt A, Farley J, Ellerbroek SM. Lysophosphatidic Acid Stimulates Urokinase Receptor (uPAR/CD87) in Ovarian Epithelial Cancer Cells. Anticancer Res. 2015; 35:5263–70.
72. Mikuła-Pietrasik J, Uruski P, Matuszkiewicz K, Szubert S, Moszyński R, Szpurek D, Sajdak S, Tykarski A, Książek K. Ovarian cancer-derived ascitic fluids induce a senescence-dependent pro-cancerogenic phenotype in normal peritoneal mesothelial cells. Cell Oncol (Dordr). 2016; 39:473–81. https://doi.org/10.1007/s13402-016-0289-1.
73. Ray U, Roy SS, Chowdhury SR. Lysophosphatidic Acid Promotes Epithelial to Mesenchymal Transition in Ovarian Cancer Cells by Repressing SIRT1. Cell Physiol Biochem. 2017; 41:795–805. https://doi.org/10.1159/000458744.
74. Lengyel E. Ovarian cancer development and metastasis. Am J Pathol. 2010; 177:1053–64. https://doi.org/10.2353/ajpath.2010.100105.
75. Iwanicki MP, Davidowitz RA, Ng MR, Besser A, Muranen T, Merritt M, Danuser G, Ince TA, Brugge JS. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011; 1:144–57. https://doi.org/10.1158/2159-8274.CD-11-0010.
76. Kenny HA, Kaur S, Coussens LM, Lengyel E. The initial steps of ovarian cancer cell metastasis are mediated by MMP-2 cleavage of vitronectin and fibronectin. J Clin Invest. 2008; 118:1367–79. https://doi.org/10.1172/JCI33775.
77. Tsujimoto H, Takhashi T, Hagiwara A, Shimotsuma M, Sakakura C, Osaki K, Sasaki S, Shirasu M, Sakakibara T, Ohyama T, Sakuyama A, Ohgaki M, Imanishi T, et al. Site-specific implantation in the milky spots of malignant cells in peritoneal dissemination: immunohistochemical observation in mice inoculated intraperitoneally with bromodeoxyuridine-labelled cells. Br J Cancer. 1995; 71:468–72. https://doi.org/10.1038/bjc.1995.95.
78. Ren J, Xiao YJ, Singh LS, Zhao X, Zhao Z, Feng L, Rose TM, Prestwich GD, Xu Y. Lysophosphatidic acid is constitutively produced by human peritoneal mesothelial cells and enhances adhesion, migration, and invasion of ovarian cancer cells. Cancer Res. 2006; 66:3006–14. https://doi.org/10.1158/0008-5472.CAN-05-1292.
79. Said N, Najwer I, Motamed K. Secreted protein acidic and rich in cysteine (SPARC) inhibits integrin-mediated adhesion and growth factor-dependent survival signaling in ovarian cancer. Am J Pathol. 2007; 170:1054–63. https://doi.org/10.2353/ajpath.2007.060903.
80. Said NA, Elmarakby AA, Imig JD, Fulton DJ, Motamed K. SPARC ameliorates ovarian cancer-associated inflammation. Neoplasia. 2008; 10:1092–104. https://doi.org/10.1593/neo.08672.
81. Cannistra SA, Ottensmeier C, Niloff J, Orta B, DiCarlo J. Expression and function of beta 1 and alpha v beta 3 integrins in ovarian cancer. Gynecol Oncol. 1995; 58:216–25. https://doi.org/10.1006/gyno.1995.1214.
82. Carreiras F, Thiébot B, Leroy-Dudal J, Maubant S, Breton MF, Darbeida H. Involvement of alphavbeta 3 integrin and disruption of endothelial fibronectin network during the adhesion of the human ovarian adenocarcinoma cell line IGROV1 on the human umbilical vein cell extracellular matrix. Int J Cancer. 2002; 99:800–08. https://doi.org/10.1002/ijc.10433.
83. Leroy-Dudal J, Demeilliers C, Gallet O, Pauthe E, Dutoit S, Agniel R, Gauduchon P, Carreiras F. Transmigration of human ovarian adenocarcinoma cells through endothelial extracellular matrix involves alphav integrins and the participation of MMP2. Int J Cancer. 2005; 114:531–43. https://doi.org/10.1002/ijc.20778.
84. Niedbala MJ, Crickard K, Bernacki RJ. Interactions of human ovarian tumor cells with human mesothelial cells grown on extracellular matrix. An in vitro model system for studying tumor cell adhesion and invasion. Exp Cell Res. 1985; 160:499–513. https://doi.org/10.1016/0014-4827(85)90197-1.
85. Shen Y, Shen R, Ge L, Zhu Q, Li F. Fibrillar type I collagen matrices enhance metastasis/invasion of ovarian epithelial cancer via β1 integrin and PTEN signals. Int J Gynecol Cancer. 2012; 22:1316–24. https://doi.org/10.1097/IGC.0b013e318263ef34.
86. Kenny HA, Chiang CY, White EA, Schryver EM, Habis M, Romero IL, Ladanyi A, Penicka CV, George J, Matlin K, Montag A, Wroblewski K, Yamada SD, et al. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J Clin Invest. 2014; 124:4614–28. https://doi.org/10.1172/JCI74778.
87. Alkhamesi NA, Ziprin P, Pfistermuller K, Peck DH, Darzi AW. ICAM-1 mediated peritoneal carcinomatosis, a target for therapeutic intervention. Clin Exp Metastasis. 2005; 22:449–59. https://doi.org/10.1007/s10585-005-2893-8.
88. Gardner MJ, Jones LM, Catterall JB, Turner GA. Expression of cell adhesion molecules on ovarian tumour cell lines and mesothelial cells, in relation to ovarian cancer metastasis. Cancer Lett. 1995; 91:229–34. https://doi.org/10.1016/0304-3835(95)03743-G.
89. Slack-Davis JK, Atkins KA, Harrer C, Hershey ED, Conaway M. Vascular cell adhesion molecule-1 is a regulator of ovarian cancer peritoneal metastasis. Cancer Res. 2009; 69:1469–76. https://doi.org/10.1158/0008-5472.CAN-08-2678.
90. Al-Hassan NN, Behzadian A, Caldwell R, Ivanova VS, Syed V, Motamed K, Said NA. Differential roles of uPAR in peritoneal ovarian carcinomatosis. Neoplasia. 2012; 14:259–70. https://doi.org/10.1593/neo.12442.
91. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, Yamada SD, Peter ME, Gwin K, Lengyel E. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011; 17:1498–503. https://doi.org/10.1038/nm.2492.
92. Nieman KM, Romero IL, Van Houten B, Lengyel E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta. 2013; 1831:1533–41. https://doi.org/10.1016/j.bbalip.2013.02.010.
93. Buscher K, Wang H, Zhang X, Striewski P, Wirth B, Saggu G, Lütke-Enking S, Mayadas TN, Ley K, Sorokin L, Song J. Protection from septic peritonitis by rapid neutrophil recruitment through omental high endothelial venules. Nat Commun. 2016; 7:10828. https://doi.org/10.1038/ncomms10828.
94. Pond CM. Adipose tissue and the immune system. Prostaglandins Leukot Essent Fatty Acids. 2005; 73:17–30. https://doi.org/10.1016/j.plefa.2005.04.005.
95. Clark R, Krishnan V, Schoof M, Rodriguez I, Theriault B, Chekmareva M, Rinker-Schaeffer C. Milky spots promote ovarian cancer metastatic colonization of peritoneal adipose in experimental models. Am J Pathol. 2013; 183:576–91. https://doi.org/10.1016/j.ajpath.2013.04.023.
96. Gerber SA, Rybalko VY, Bigelow CE, Lugade AA, Foster TH, Frelinger JG, Lord EM. Preferential attachment of peritoneal tumor metastases to omental immune aggregates and possible role of a unique vascular microenvironment in metastatic survival and growth. Am J Pathol. 2006; 169:1739–52. https://doi.org/10.2353/ajpath.2006.051222.
97. Sodek KL, Murphy KJ, Brown TJ, Ringuette MJ. Cell-cell and cell-matrix dynamics in intraperitoneal cancer metastasis. Cancer Metastasis Rev. 2012; 31:397–414. https://doi.org/10.1007/s10555-012-9351-2.
98. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, Wang YY, Meulle A, Salles B, Le Gonidec S, Garrido I, Escourrou G, Valet P, Muller C. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011; 71:2455–65. https://doi.org/10.1158/0008-5472.CAN-10-3323.
99. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006; 6:392–401. https://doi.org/10.1038/nrc1877.
100. Yáñez-Mó M, Lara-Pezzi E, Selgas R, Ramírez-Huesca M, Domínguez-Jiménez C, Jiménez-Heffernan JA, Aguilera A, Sánchez-Tomero JA, Bajo MA, Álvarez V, Castro MA, del Peso G, Cirujeda A, et al. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N Engl J Med. 2003; 348:403–13. https://doi.org/10.1056/NEJMoa020809.
101. Sandoval P, Jiménez-Heffernan JA, Rynne-Vidal Á, Pérez-Lozano ML, Gilsanz Á, Ruiz-Carpio V, Reyes R, García-Bordas J, Stamatakis K, Dotor J, Majano PL, Fresno M, Cabañas C, López-Cabrera M. Carcinoma-associated fibroblasts derive from mesothelial cells via mesothelial-to-mesenchymal transition in peritoneal metastasis. J Pathol. 2013; 231:517–31. https://doi.org/10.1002/path.4281.
102. Rynne-Vidal A, Au-Yeung CL, Jiménez-Heffernan JA, Pérez-Lozano ML, Cremades-Jimeno L, Bárcena C, Cristóbal-García I, Fernández-Chacón C, Yeung TL, Mok SC, Sandoval P, López-Cabrera M. Mesothelial-to-mesenchymal transition as a possible therapeutic target in peritoneal metastasis of ovarian cancer. J Pathol. 2017; 242:140–51. https://doi.org/10.1002/path.4889.
103. Bochet L, Lehuédé C, Dauvillier S, Wang YY, Dirat B, Laurent V, Dray C, Guiet R, Maridonneau-Parini I, Le Gonidec S, Couderc B, Escourrou G, Valet P, Muller C. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013; 73:5657–68. https://doi.org/10.1158/0008-5472.CAN-13-0530.
104. Zoico E, Darra E, Rizzatti V, Budui S, Franceschetti G, Mazzali G, Rossi AP, Fantin F, Menegazzi M, Cinti S, Zamboni M. Adipocytes WNT5a mediated dedifferentiation: a possible target in pancreatic cancer microenvironment. Oncotarget. 2016; 7:20223–35. https://doi.org/10.18632/oncotarget.7936.
105. Lawler HM, Underkofler CM, Kern PA, Erickson C, Bredbeck B, Rasouli N. Adipose Tissue Hypoxia, Inflammation, and Fibrosis in Obese Insulin-Sensitive and Obese Insulin-Resistant Subjects. J Clin Endocrinol Metab. 2016; 101:1422–28. https://doi.org/10.1210/jc.2015-4125.
106. Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR, d’Escamard V, Li JR, Hadri L, Fujitani K, Moreno PR, Benard L, Rimmele P, et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. 2016; 7:11853. https://doi.org/10.1038/ncomms11853.
107. Madar S, Goldstein I, Rotter V. 'Cancer associated fibroblasts'--more than meets the eye. Trends Mol Med. 2013; 19:447-53. https://doi.org/10.1016/j.molmed.2013.05.004.
108. Cai J, Tang H, Xu L, Wang X, Yang C, Ruan S, Guo J, Hu S, Wang Z. Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness. Carcinogenesis. 2012; 33:20–29. https://doi.org/10.1093/carcin/bgr230.
109. Rynne-Vidal A, Jiménez-Heffernan JA, Fernández-Chacón C, López-Cabrera M, Sandoval P. The Mesothelial Origin of Carcinoma Associated-Fibroblasts in Peritoneal Metastasis. Cancers (Basel). 2015; 7:1994–2011. https://doi.org/10.3390/cancers7040872.
110. Yeung TL, Leung CS, Wong KK, Samimi G, Thompson MS, Liu J, Zaid TM, Ghosh S, Birrer MJ, Mok SC. TGF-β modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013; 73:5016–28. https://doi.org/10.1158/0008-5472.CAN-13-0023.
111. Zhang Y, Tang H, Cai J, Zhang T, Guo J, Feng D, Wang Z. Ovarian cancer-associated fibroblasts contribute to epithelial ovarian carcinoma metastasis by promoting angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer Lett. 2011; 303:47–55. https://doi.org/10.1016/j.canlet.2011.01.011.
112. Lau TS, Chan LK, Wong EC, Hui CW, Sneddon K, Cheung TH, Yim SF, Lee JH, Yeung CS, Chung TK, Kwong J. A loop of cancer-stroma-cancer interaction promotes peritoneal metastasis of ovarian cancer via TNFα-TGFα-EGFR. Oncogene. 2017; 36:3576–87. https://doi.org/10.1038/onc.2016.509.
113. Dong T, Yang D, Li R, Zhang L, Zhao H, Shen Y, Zhang X, Kong B, Wang L. PGRN promotes migration and invasion of epithelial ovarian cancer cells through an epithelial mesenchymal transition program and the activation of cancer associated fibroblasts. Exp Mol Pathol. 2016; 100:17–25. https://doi.org/10.1016/j.yexmp.2015.11.021.
114. Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008; 205:1261–68. https://doi.org/10.1084/jem.20080108.
115. Hagemann T, Robinson SC, Thompson RG, Charles K, Kulbe H, Balkwill FR. Ovarian cancer cell-derived migration inhibitory factor enhances tumor growth, progression, and angiogenesis. Mol Cancer Ther. 2007; 6:1993–2002. https://doi.org/10.1158/1535-7163.MCT-07-0118.
116. Hagemann T, Wilson J, Burke F, Kulbe H, Li NF, Plüddemann A, Charles K, Gordon S, Balkwill FR. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J Immunol. 2006; 176:5023–32. https://doi.org/10.4049/jimmunol.176.8.5023.
117. Kulbe H, Thompson R, Wilson JL, Robinson S, Hagemann T, Fatah R, Gould D, Ayhan A, Balkwill F. The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 2007; 67:585–92. https://doi.org/10.1158/0008-5472.CAN-06-2941.
118. Jammal MP, Martins-Filho A, Silveira TP, Murta EF, Nomelini RS. Cytokines and Prognostic Factors in Epithelial Ovarian Cancer. Clin Med Insights Oncol. 2016; 10:71–76. https://doi.org/10.4137/CMO.S38333.
119. Lau A, Kollara A, St John E, Tone AA, Virtanen C, Greenblatt EM, King WA, Brown TJ. Altered expression of inflammation-associated genes in oviductal cells following follicular fluid exposure: implications for ovarian carcinogenesis. Exp Biol Med (Maywood). 2014; 239:24–32. https://doi.org/10.1177/1535370213508216.
120. Dalton HJ, Pradeep S, McGuire M, Hailemichael Y, Ma S, Lyons Y, Armaiz-Pena GN, Previs RA, Hansen JM, Rupaimoole R, Gonzalez-Villasana V, Cho MS, Wu SY, et al. Macrophages facilitate resistance to anti-VEGF therapy by altered VEGFR expression. Clin Cancer Res. 2017; 23:7034–46. https://doi.org/10.1158/1078-0432.CCR-17-0647.
121. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009; 9:162–74. https://doi.org/10.1038/nri2506.
122. Wu L, Deng Z, Peng Y, Han L, Liu J, Wang L, Li B, Zhao J, Jiao S, Wei H. Ascites-derived IL-6 and IL-10 synergistically expand CD14+HLA-DR-/low myeloid-derived suppressor cells in ovarian cancer patients. Oncotarget. 2017; 8:76843–56. https://doi.org/10.18632/oncotarget.20164.
123. Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 2011; 71:7463–70. https://doi.org/10.1158/0008-5472.CAN-11-2449.
124. Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, Vatan L, Szeliga W, Mao Y, Thomas DG, Kotarski J, Tarkowski R, Wicha M, et al. Myeloid derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity. 2013; 39:611-21. https://doi.org/10.1016/j.immuni.2013.08.025.
125. Curiel TJ, Cheng P, Mottram P, Alvarez X, Moons L, Evdemon-Hogan M, Wei S, Zou L, Kryczek I, Hoyle G, Lackner A, Carmeliet P, Zou W. Dendritic cell subsets differentially regulate angiogenesis in human ovarian cancer. Cancer Res. 2004; 64:5535–38. https://doi.org/10.1158/0008-5472.CAN-04-1272.
126. Zhang Z, Huang J, Zhang C, Yang H, Qiu H, Li J, Liu Y, Qin L, Wang L, Hao S, Zhang F, Wang X, Shan B. Infiltration of dendritic cells and T lymphocytes predicts favorable outcome in epithelial ovarian cancer. Cancer Gene Ther. 2015; 22:198–206. https://doi.org/10.1038/cgt.2015.7.
127. Kandalaft LE, Chiang CL, Tanyi J, Motz G, Balint K, Mick R, Coukos G. A Phase I vaccine trial using dendritic cells pulsed with autologous oxidized lysate for recurrent ovarian cancer. J Transl Med. 2013; 11:149. https://doi.org/10.1186/1479-5876-11-149.
128. Chiang CL, Kandalaft LE, Tanyi J, Hagemann AR, Motz GT, Svoronos N, Montone K, Mantia-Smaldone GM, Smith L, Nisenbaum HL, Levine BL, Kalos M, Czerniecki BJ, et al. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: from bench to bedside. Clin Cancer Res. 2013; 19:4801–15. https://doi.org/10.1158/1078-0432.CCR-13-1185.
129. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr Opin Immunol. 2014; 27:16–25. https://doi.org/10.1016/j.coi.2014.01.004.
130. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014; 14:135–46. https://doi.org/10.1038/nrc3670.
131. Peter ME, Hadji A, Murmann AE, Brockway S, Putzbach W, Pattanayak A, Ceppi P. The role of CD95 and CD95 ligand in cancer. Cell Death Differ. 2015; 22:549–59. https://doi.org/10.1038/cdd.2015.3.
132. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol. 2015; 33:55–63. https://doi.org/10.1016/j.coi.2015.01.011.
133. Melero I, Rouzaut A, Motz GT, Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014; 4:522–26. https://doi.org/10.1158/2159-8290.CD-13-0985.
134. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014; 20:607–15. https://doi.org/10.1038/nm.3541.
135. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, Rubin SC, Coukos G, Intratumoral T. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003; 348:203–13. https://doi.org/10.1056/NEJMoa020177.
136. Dadmarz RD, Ordoubadi A, Mixon A, Thompson CO, Barracchini KC, Hijazi YM, Steller MA, Rosenberg SA, Schwartzentruber DJ. Tumor-infiltrating lymphocytes from human ovarian cancer patients recognize autologous tumor in an MHC class II-restricted fashion. Cancer J Sci Am. 1996; 2:263–72.
137. Ma D, Gu MJ. Immune effect of tumor-infiltrating lymphocytes and its relation to the survival rate of patients with ovarian malignancies. J Tongji Med Univ. 1991; 11:235–39. https://doi.org/10.1007/BF02888158.
138. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, Kepner J, Odunsi T, Ritter G, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA. 2005; 102:18538–43. https://doi.org/10.1073/pnas.0509182102.
139. Santoiemma PP, Powell DJ Jr. Tumor infiltrating lymphocytes in ovarian cancer. Cancer Biol Ther. 2015; 16:807–20. https://doi.org/10.1080/15384047.2015.1040960.
140. Hwang WT, Adams SF, Tahirovic E, Hagemann IS, Coukos G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol. 2012; 124:192–98. https://doi.org/10.1016/j.ygyno.2011.09.039.
141. Chen Daniel S, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity. 39:1–10. https://doi.org/10.1016/j.immuni.2013.07.012.
142. Tanizaki Y, Kobayashi A, Toujima S, Shiro M, Mizoguchi M, Mabuchi Y, Yagi S, Minami S, Takikawa O, Ino K. Indoleamine 2,3-dioxygenase promotes peritoneal metastasis of ovarian cancer by inducing an immunosuppressive environment. Cancer Sci. 2014; 105:966–73. https://doi.org/10.1111/cas.12445.
143. Higuchi T, Flies DB, Marjon NA, Mantia-Smaldone G, Ronner L, Gimotty PA, Adams SF. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol Res. 2015; 3:1257–68. https://doi.org/10.1158/2326-6066.CIR-15-0044.
144. Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, Higuchi T, Yagi H, Takakura K, Minato N, Honjo T, Fujii S. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci USA. 2007; 104:3360–65. https://doi.org/10.1073/pnas.0611533104.
145. Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013; 73:3591–603. https://doi.org/10.1158/0008-5472.CAN-12-4100.
146. Wang L, Amoozgar Z, Huang J, Saleh MH, Xing D, Orsulic S, Goldberg MS. Decitabine Enhances Lymphocyte Migration and Function and Synergizes with CTLA-4 Blockade in a Murine Ovarian Cancer Model. Cancer Immunol Res. 2015; 3:1030–41. https://doi.org/10.1158/2326-6066.CIR-15-0073.
147. Preston CC, Goode EL, Hartmann LC, Kalli KR, Knutson KL. Immunity and immune suppression in human ovarian cancer. Immunotherapy. 2011; 3:539–56. https://doi.org/10.2217/imt.11.20.
148. Zhang S, Ke X, Zeng S, Wu M, Lou J, Wu L, Huang P, Huang L, Wang F, Pan S. Analysis of CD8+ Treg cells in patients with ovarian cancer: a possible mechanism for immune impairment. Cell Mol Immunol. 2015; 12:580–91. https://doi.org/10.1038/cmi.2015.57.
149. Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, Luo X, Cooley S, Verneris M, Walcheck B, Miller J. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood. 2013; 121:3599–608. https://doi.org/10.1182/blood-2012-04-425397.
150. Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene. 2008; 27:5932–43. https://doi.org/10.1038/onc.2008.267.
151. Belisle JA, Gubbels JA, Raphael CA, Migneault M, Rancourt C, Connor JP, Patankar MS. Peritoneal natural killer cells from epithelial ovarian cancer patients show an altered phenotype and bind to the tumour marker MUC16 (CA125). Immunology. 2007; 122:418–29. https://doi.org/10.1111/j.1365-2567.2007.02660.x.
152. Dudley AC. Tumor endothelial cells. Cold Spring Harb Perspect Med. 2012; 2:a006536. https://doi.org/ARTNa00653610.1101/cshperspect.a006536 https://doi.org/10.1101/cshperspect.a006536.
153. Brown MR, Blanchette JO, Kohn EC. Angiogenesis in ovarian cancer. Best Pract Res Clin Obstet Gynaecol. 2000; 14:901–18. https://doi.org/10.1053/beog.2000.0134.
154. Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002; 20:4368–80. https://doi.org/10.1200/JCO.2002.10.088.
155. Masoumi Moghaddam S, Amini A, Morris DL, Pourgholami MH. Significance of vascular endothelial growth factor in growth and peritoneal dissemination of ovarian cancer. Cancer Metastasis Rev. 2012; 31:143–62. https://doi.org/10.1007/s10555-011-9337-5.
156. Choi HJ, Armaiz Pena GN, Pradeep S, Cho MS, Coleman RL, Sood AK. Anti-vascular therapies in ovarian cancer: moving beyond anti-VEGF approaches. Cancer Metastasis Rev. 2015; 34:19–40. https://doi.org/10.1007/s10555-014-9538-9.
157. Winiarski BK, Wolanska KI, Rai S, Ahmed T, Acheson N, Gutowski NJ, Whatmore JL. Epithelial ovarian cancer-induced angiogenic phenotype of human omental microvascular endothelial cells may occur independently of VEGF signaling. Transl Oncol. 2013; 6:703–14. https://doi.org/10.1593/tlo.13529.
158. Sangisetty SL, Miner TJ. Malignant ascites: A review of prognostic factors, pathophysiology and therapeutic measures. World J Gastrointest Surg. 2012; 4:87–95. https://doi.org/10.4240/wjgs.v4.i4.87.
159. Kipps E, Tan DS, Kaye SB. Meeting the challenge of ascites in ovarian cancer: new avenues for therapy and research. Nat Rev Cancer. 2013; 13:273–82. https://doi.org/10.1038/nrc3432.
160. Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003; 3:582–91. https://doi.org/10.1038/nrc1143.
161. Gajewiak J, Tsukahara R, Tsukahara T, Fujiwara Y, Yu S, Lu Y, Murph M, Mills GB, Tigyi G, Prestwich GD. Alkoxymethylenephosphonate analogues of (Lyso) phosphatidic acid stimulate signaling networks coupled to the LPA2 receptor. ChemMedChem. 2007; 2:1789–98. https://doi.org/10.1002/cmdc.200700111.
162. Yu X, Zhang Y, Chen H. Lysophosphatidic acid (LPA) stimulates invasion and metastatic colonization of ovarian cancer cells through Rac activation. [Article in Chinese] Zhonghua Zhong Liu Za Zhi. 2015; 37:95-100.
163. Yu X, Zhang Y, Chen H. LPA receptor 1 mediates LPA-induced ovarian cancer metastasis: an in vitro and in vivo study. BMC Cancer. 2016; 16:846. https://doi.org/10.1186/s12885-016-2865-1.
164. Wang Z. Transactivation of Epidermal Growth Factor Receptor by G Protein-Coupled Receptors: Recent Progress, Challenges and Future Research. Int J Mol Sci. 2016; 17:95. https://doi.org/10.3390/ijms17010095.
165. Murph MM, Liu W, Yu S, Lu Y, Hall H, Hennessy BT, Lahad J, Schaner M, Helland A, Kristensen G, Børresen-Dale AL, Mills GB. Lysophosphatidic acid-induced transcriptional profile represents serous epithelial ovarian carcinoma and worsened prognosis. PLoS One. 2009; 4:e5583. https://doi.org/10.1371/journal.pone.0005583.