
INTRODUCTION
Breast cancer mainly affects post-menopausal women, however, it is estimated that 4.8-5.0% of cases occur in young adults, less than 40 years [1]. Even at this early age the disease can be highly fatal. In the USA, where cancer is the second leading cause of total deaths in young women aged less than 40 years, breast cancer is the leading cause of cancer deaths in this age group [2].
There is evidence that some cancers in very young adults have differential biology, and probably etiology/pathogenesis, compared to older persons [3]. Surprisingly, only a few studies have explored this question. In breast cancer, germline mutations in BRCA1 and BRCA2 genes may support the carcinogenic process in around 20% of the young patients [4–7], but only in 1-4% of post-menopausal women [8]. Mutations in other cancer predisposing genes such as TP53, PTEN, CHEK2, may explain an additional 4% of early onset cases [9]
Younger age has been associated with a less favorable prognosis in breast cancer partly because early onset cases comprise a lower proportion of the relatively good outcome luminal A subtype and higher proportion of the more aggressive triple negative subtype. Moreover, within each subtype, women diagnosed at an early age may have worse outcomes than those diagnosed at more advanced ages in any breast cancer subtype, i.e., luminal [10–12], triple negative and HER2 [13]. Although young age seems to be a poor prognostic factor, different age cut offs have been used, varying from 35 [10, 13] to 40 years [11, 12]. It is interesting to observe that women aged less than 35 years old seems to have similar disease-free survival among themselves, which is worse compared with women aged 35 to 50 years old [10, 13].
Accordingly, mRNA abundance analysis revealed a differential transcriptional profile in tumors arising in young women, with enrichment of biological processes related to immature mammary cell populations and growth factor signaling [11]. Molecular signatures of breast cancer subtypes, irrespective of age, have been examined and great differences have been shown between basal-like and luminal tumors, the former presenting a higher rate of genomic rearrangements than the latter [14]. In addition, numerous subtype-associated and novel gene mutations have been described [15–18]. These studies however, have not focused on somatic point mutations (single nucleotide variants, SNVs) that may distinguish early onset breast cancer. Hence, our aims were to characterize a group of Brazilian patients with early onset breast cancer for BRCA1 and BRCA2 germline mutations, as well as for somatic SNVs arising in luminal subtype tumors.
RESULTS
Family history suggestive of hereditary breast and ovarian cancer syndrome (HBOCS) and germline BRCA1 and BRCA2 mutations
Our first aim was to evaluate family history of cancer and to detect BRCA1 and BRCA2 mutations in very young Brazilian patients. For this purpose, 79 young women were interviewed, among whom 17 (21.5%) were not able to provide family history for one or both sides of the family. Thirty (48.3%) out of 62 patients with informative family history reported at least one close relative (until 3rd degree) with breast, ovarian, pancreatic or prostate cancer, among whom 10 (16.2%) reported at least one affected first degree family member (Supplementary Table 1).
Thirteen out of 79 patients presented pathogenic mutations (16.5%) in BRCA1 or BRCA2 genes. These, represent 12 distinct types of mutations: three frameshift and one missense in BRCA1 and four frameshift, three nonsense and one missense in BRCA2. Only one mutation (frameshift mutation in BRCA2 c.2808_2811delACAA (p.Ala938Profs) was detected in two women; one nonsense mutation c.483T>A (C161X) on exon 6 of BRCA2 was detected for the first time (Supplementary Tables 2 and 3).
Twenty-nine variants of uncertain significance (VUS) were also identified, including 13 distinct missense variants, each one detected only once: two in BRCA1 and 11 in BRCA2 gene. Two patients presented more than one (missense) VUS in BRCA2 gene, one of them, diagnosed with a triple negative tumor reported a positive family history (c.3349A>G; c.5414A>G; c.8092G>A); the other one, diagnosed with a luminal B tumor, presented a limited family history (c.2837A>G; c.7418G>A). In addition, one VUS characterized as an in-frame deletion in exon 23 of BRCA1 gene (c.5425_5430delGTTGTG) (Supplementary Table 4), was observed in a patient with positive family history, diagnosed with triple negative breast cancer. The other VUS were characterized as intronic or synonymous variants.
Neither of these patients presented large deletions/amplifications in BRCA1 and BRCA2, nor CHEK2 mutations (c.1100delC).
Somatic SNVs detected by whole exome sequencing
Eight patients, who were BRCA1 and BRCA2 wild type carriers with luminal HER2 negative tumors, had their tumor and normal exomes sequenced. These patients mainly reported Brazilian ancestry in both sides of the family, which means that their parents and grandparents were born in Brazil, but they were not aware from where did more ancient ancestries had come from. One patient reported one maternal grandmother with Amerindian ancestry and a second patient reported grandparents from the paternal side with European ancestry.
Whole exome sequencing of these eight tumors and matched blood samples was performed to a mean sequencing depth of 35.8x for tumors and 36.3x for corresponding blood samples (Supplementary Table 5). The mean total mutation rate across all samples was 1.9/Mbp. The mean non-silent mutation rate was 1.8/Mbp (Supplementary Table 6) and the most frequent events were C to T transitions, mainly seen in trinucleotides ACG>ATG and CCT>CTG (Figure 1).
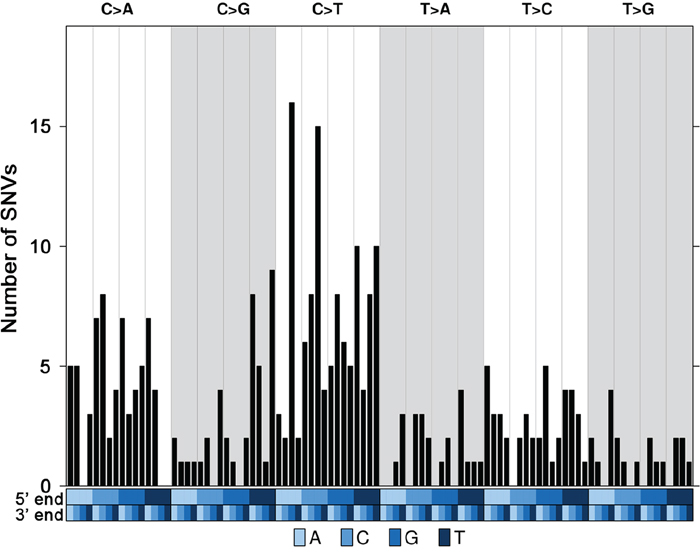
Figure 1: Trinucleotide mutational profile of current luminal samples. Trinucleotide barplot showing the number of Single Nucleotide Variants (SNVs) in the context of each of the 96 trinucleotide mutation types. The blue covariates at the bottom of the plot represent the 5' and 3' ends. All the 310 SNVs were considered.
We identified 310 somatic single nucleotide variants (SNVs), comprising of 303 unique variants (five SNVs were detected in two patients each; one SNV was detected in three patients), and mainly comprising intergenic regions, 3 prime UTR, missense, intron and synonymous variants (Supplementary Table 7). The median mutation load was 37.5 and varied from 19-74 SNVs per tumor.
SeqSig analysis revealed 55 likely driver non-synonymous mutations in 53 genes (false discovery rate, (FDR) < 10%); (Figure 2) and PIK3CA was the only recurrent finding, which was detected in three different tumors. Somatic SNVs were then verified by performing an independent capillary sequencing (except for GLI3, LONRF3 and EPPK1 that were not tested) and 81% (42/52) were confirmed (Supplementary Tables 8, 8a). Confirmed SNVs included nonsense mutations in four genes, GRHL2, GRIN1, NOL9 and TTC21B, as well as 38 missense mutations in 36 different genes, including known tumor suppressor genes, such as TP53 and POLD1, and protein kinases like PRKD1, PRKAR1A and AK8.
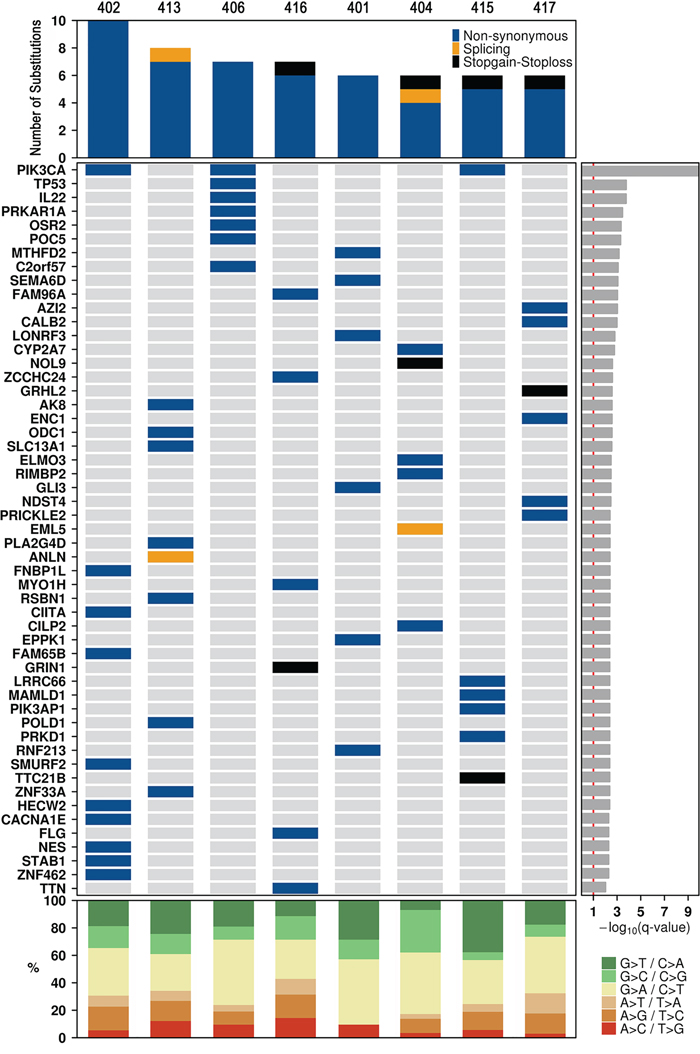
Figure 2: Landscape of coding somatic SNVs. Each of the 54 genes in which at least one significant SNV was identified is listed down the left hand side. The genes are listed by their significant SeqSig q-value (FDR adjusted p-value). Type and number of mutations (top panel), significantly mutated genes (middle panel) and percentage of Single Nucleotide Variants (SNVs) (bottom panel) per tumor sample.
We compared our results with the gene list from the “Cancer Gene Census” database (http://cancer.sanger.ac.uk/census) [19] and detected five genes, PIK3CA, TP53, PRKAR1A, POLD1 and CIITA, which were already causally implicated in cancer.
We have then examined more closely this list of candidates to identify potentially cancer driver genes, using the score system described in methods, mainly based on detection in databases of mouse insertional mutagenesis experiments and causal relationship mutation function assessment algorithms, Kaplan-Meier (KM) plotter [20] (to assess the effect of the genes on breast cancer prognosis) (Supplementary Figure 1) and literature, among others.
Excluding the five genes included in the “Cancer Census Gene” database, another 18 genes were already reported as candidate cancer genes through transposon-based forward genetic screens in mice [21] (Table 1). Seven genes were relatively frequently mutated (≥1%) in cancer in general or in breast cancer specifically: CACNA1E, HECW2, STAB1, ZNF462, FLG, TTN and NDST4. Finally, using the above ranking system, 20 genes, scoring at least 2, were considered possible cancer drivers (Supplementary Tables 9, 9a), such as PIK3AP1, GRHL2, CACNA1E, SMURF2, SEMA6D, RSBN1, MTHFD2, among others.
Table 1: Cancer-related analysis of confirmed gene variants detected in breast cancer samples in the current analysis
ID |
Gene |
Alteration |
CGC |
CCGD (mice) |
Mutation domain |
Same variant in BC/Other Cancers |
SNVs Frequency in all cancers |
SNVs Frequency in BC |
SNVs in BC young patients/all ages |
FATHMM |
PolyPhen |
SIFT |
GV/GD |
CRAVAT - CHASM |
KM - OS |
Literature |
Score |
Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
401 |
MTHFD2 |
p.P17L c.50C>T |
No |
low_complexitySource: segmasker |
No/No |
38/37401 (0.10%) |
1/2137 (0.05%) |
0/1 |
Pathogenic (0.87) |
Benign |
Tolerated |
0.00/97.78 (C65) |
0.2851 |
p = 0.0027 OE |
2.5 |
pd |
||
401 |
SEMA6D |
p.E553A c.1658A>C |
No |
Plexin Repeat |
No/No |
317/37626 (0.84%) |
17/2159 (0.78%) 1 FS |
2/17 |
Pathogenic (0.74) |
ND |
Tolerated |
0.00/106.71 (C65) |
0.3967 |
p = 0.0091 n≤200 |
[8] |
3 |
pd |
|
402 |
CACNA1E |
p.R590W c.1768C>T |
No |
No results |
Ion transport Domain |
No/Yes |
902/37516 (2.40%) |
54/2116 (2.55%) 2 NS |
3/45 |
Pathogenic (0.89) |
Deleterious |
Not Tolerated |
0.00/101.29 (C65) |
0.0523 |
p = 0.11 n≤200 |
3 |
pd |
|
402 |
CIITA |
p.P443T c.1327C>A |
Yes |
No results |
NACHT domain |
No/No |
2/37750 (0.005%) |
0 |
- |
Pathogenic (0.58) |
Deleterious |
Tolerated |
0.00/37.56 (C35) |
0.3563 |
p = 0.085 |
4.5 |
CGC |
|
402 |
FAM65B/RIPOR2 |
p.E718D c.2154G>C |
No |
Blood - C [14] |
No Pfam annotations found |
No/No |
130/37401 (0.35%) |
7/2114 (0.33%) 1 FS |
1/7 |
Neutral (0.26) |
ND |
Tolerated |
0.00/44.60 (C35) |
0.8042 |
p = 0.14 |
NO |
1 |
Neutral |
402 |
HECW2 |
p.D265G c.794A>G |
No |
Liver - D [15] |
C2 Domain |
No/No |
422/38016 (1.11%) |
20/2312 (0.86%) |
1/20 |
Pathogenic (0.98) |
Deleterious |
Not Tolerated |
0.00/93.77 (C65) |
0.058 |
p = 0.42 n≤200 |
[16] |
3 |
pd |
402 |
NES |
p.E340V c.1019A>T |
No |
No results |
No fuctional domain |
No/No |
346/37419 (0.92%) |
17/2137 (0.79%) 1 NS |
0/17 |
Pathogenic (0.59) |
Deleterious |
Not Tolerated |
0.00/121.33 (C65) |
0.1896 |
p = 0.028 UE |
3 |
pd |
|
402 |
PIK3CA |
p.E545K c.1633G>A |
Yes |
Blood - D [1] Gastric - D [22] Liver - C [23, 24] Nervous system - D [25] Skin - B [26] |
PIK domain |
Yes/Yes |
10271/107457 (9.56%) |
4098/15384 (26.64%) |
- |
Pathogenic (0.97) |
Deleterious |
Tolerated |
0.00/56.87 (C55) |
0.0002 |
p = 0.057 |
Oncogene |
7.5 |
CGC |
402 |
SMURF2 |
p.S193C c.578C>G |
No |
Blood - A [1] Colorectal - C [2, 27] Gastric - C [22] Liver - A [15, 23] Pancreatic – D [28] |
disorderSource: IUPred |
No/No |
117/38086 (0.30%) |
9/2288 (0.39) 3 FS |
1/9 |
Pathogenic (0.91) |
Benign |
Not Tolerated |
0.00/111.67 (C65) |
0.4405 |
p = 0.074 n≤200 |
4 |
PD |
|
402 |
STAB1 |
p.G1381R c.4141G>A |
No |
No results |
No fuctional domain |
No/No |
502/37566 (1.34%) |
23/2158 (1.06%) |
0/22 |
Pathogenic (0.72) |
Deleterious |
Tolerated |
0.00/125.13 (C65) |
0.6908 |
p = 0.071 |
2.5 |
pd |
|
402 |
ZNF462 |
p.G2426C c.7276G>T |
No |
Breast - C [34] |
No fuctional domain |
No/No |
555/37476 (1.48%) |
36/2137 (1.68%) |
3/36 |
Pathogenic (0.89) |
Deleterious |
Tolerated |
0.00/158.23 (C65) |
0.0004 |
p = 0.028 n≤200 |
NO |
2.5 |
pd |
404 |
CILP2 |
p.R472G c.1414C>G |
No |
Blood - D [1] |
No fuctional domain |
No/No |
309/37401 (0.83%) |
5/2137 (0.23%) |
0/5 |
Neutral (0.1) |
Deleterious |
Not Tolerated |
0.00/125.13 (C65) |
0.661 |
p = 0.17 n≤200 |
NO |
1.5 |
Neutral |
404 |
ELMO3 |
p.L251F c.753G>C |
No |
No results |
No Pfam annotations found |
No/No |
117/37401 (0.31%) |
2/2137 (0.09%) |
1/2 |
Pathogenic (0.66) |
Benign |
Tolerated |
0.00/21.82 (C15) |
0.0675 |
p = 0.000052 OE |
2 |
pd |
|
404 |
NOL9 |
p.S283* c.848C>G |
No |
No results |
low_complexitySource: segmasker |
No/No |
111/37401 (0.30%) |
5/2137 (0.23%) |
1/5 |
Neutral (0.13) |
ND |
ND |
- |
- |
p = 0.15 |
NO |
1.5 |
Neutral |
406 |
C2orf57/TEX44 |
p.T265M c.794C>T |
No |
Blood - D [1] |
Domain of unknown function |
No/Yes |
89/37401 (0.24%) |
1/2137 (0.05%) |
0/1 |
Neutral (0.00) |
Benign |
Tolerated |
0.00/81.04 (C65) |
0.5572 |
p = 0.14 n≤200 |
NO |
0.5 |
Neutral |
406 |
IL22 |
p.R73C c.217C>T |
No |
No results |
Interleukin 22 domain |
No/No |
60/37402 (0.16%) |
3/2137 (0.14%) |
0/3 |
Pathogenic (0.57) |
Deleterious |
Not Tolerated |
0.00/179.53 (C65) |
0.6159 |
p = 0.4 n≤200 |
2.5 |
pd |
|
406 |
OSR2 |
p.G262E c.785G>A |
No |
No results |
Zinc Finger domain |
No/No |
98/37312 (0.26%) |
5/2126 (0.24%) |
0/5 |
Pathogenic (0.94) |
Deleterious |
Not Tolerated |
0.00/97.85 (C65) |
0.0922 |
p = 0.2 |
[41] |
2.5 |
pd |
406 |
PIK3CA |
p.H1047R c.3140A>G |
Yes |
Blood - D [1] Gastric - D [22] Liver - C [23, 24] Nervous system - D [25] Skin - B [26] |
PI3K/PI4K domain |
Yes/Yes |
10271/107457 (9.56%) |
4098/15384 (26.64%) |
- |
Pathogenic (0.96) |
Deleterious |
Tolerated |
0.00/28.82 (C25) |
0 |
p = 0.057 |
Oncogene |
7.5 |
CGC |
406 |
POC5 |
p.R541Q c.1622G>A |
No |
No results |
No Pfam annotations found |
No/No |
87/37355 (0.23%) |
3/2137 (0.14%) |
2/3 |
Pathogenic (0.94) |
Deleterious |
Not Tolerated |
0.00/48.81 (C35) |
0.4111 |
p = 0.06 n≤200 |
NO |
1 |
Neutral |
406 |
PRKAR1A |
p.L20F c.58C>T |
Yes |
Dimerization and phosphorylation region |
No/No |
112/40450 (0.28%) |
8/2379 (0.34%) |
0/8 |
Pathogenic (0.94) |
Benign |
Tolerated |
0.00/21.82 (C15) |
0.3232 |
p = 0.14 |
6 |
CGC |
||
406 |
TP53 |
p.T220C c.659A>G |
Yes |
Colorectal - C [2, 27, 45] Nervous system - NR [46] Skin- A [47] Liver - NR [24] |
P53 DNA-binding domain |
Yes/Yes |
31140/127779 (24.37%) |
3189/13359 (23.87%) |
- |
Pathogenic (0.99) |
Deleterious |
Not Tolerated |
- |
0.0012 |
p = 0.041 UE |
TSG |
9 |
CGC |
413 |
AK8 |
p.T101P c.301A>C |
No |
Blood - B [1] |
Adenylate kinase |
No/No |
106/37402 (0.28%) |
3/2114 (0.14%) |
1/3 |
Neutral (0.02) |
Benign |
Tolerated |
0.00/37.56 (C35) |
0.7606 |
p = 0.12 n≤200 |
NO |
2 |
pd |
413 |
PLA2G4D |
p.S173G c.517A>G |
No |
Blood - D [1] |
No fuctional domain |
No/No |
197/37446 (0.53%) |
10/2135 (0.47%) |
0/10 |
Neutral (0.04) |
Benign |
Tolerated |
0.00/55.27 (C55) |
0.3458 |
- |
NO |
0.5 |
Neutral |
413 |
POLD1 |
p.P146R c.437C>G |
Yes |
No results |
Exonucelase domain |
No/No |
263/37786 (0.70%) |
9/2137 (0.42%) |
0/9 |
Pathogenic (0.95) |
Deleterious |
Not Tolerated |
0.00/102.71 (C65) |
0.1793 |
p = 0.000042 OE |
6.5 |
CGC |
|
413 |
RSBN1 |
p.P148S c.442C>T |
No |
Blood - B [1] Liver - D [15] Colorectal - C [2] |
Pro-Rich domain |
No/No |
149/37401 (0.40%) |
12/2137 (0.52%) |
1/12 |
Neutral (0.27) |
Deleterious |
Not Tolerated |
0.00/73.35 (C65) |
0.1256 |
p = 0.011 UE |
NO |
4 |
PD |
413 |
SLC13A1 |
p.R277P c.830G>C |
No |
No results |
No Pfam annotations found |
No/No |
241/37402 (0.64%) |
15/2137 (0.70%) |
0/15 |
Neutral (0.02) |
Benign |
Tolerated |
0.0/102.71 (C65) |
0.6497 |
p = 0.064 |
NO |
0 |
Neutral |
413 |
ZNF33A |
p.G183V c.548G>T |
No |
No results |
No fuctional domain |
No/No |
166/37402 (0.44%) |
4/2137 (0.19%) |
0/4 |
Neutral (0.12) |
ND |
ND |
0.3098 |
p = 0.0081 n≤200 |
NO |
0 |
Neutral |
|
415 |
LRRC66 |
p.H434N c.1300C>A |
No |
No results |
disorderSource: IUPred |
No/No |
276/37403 (0.74%) |
7/2137 (0.33%) |
2/7 |
Neutral (0.00) |
Benign |
Tolerated |
0.00/68.35 (C65) |
0.577 |
p = 0.015 n≤200 |
NO |
0 |
Neutral |
415 |
MAMLD1 |
p.A775V c.2324C>T |
No |
No Pfam annotations found |
No/No |
172/37402 (0.46%) |
17/2137 (0.79%) |
0/17 |
Neutral (0.00) |
ND |
Tolerated |
0.00/64.43 (C65) |
0.7952 |
p = 0.23 |
NO |
1 |
Neutral |
|
415 |
PIK3AP1 |
p.Q285K c.853C>A |
No |
Dof, BCAP, and BANK (DBB) motif |
No/No |
194/37522 (0.51%) |
7/2137 (0.33%) |
0/7 |
Pathogenic (0.98) |
Deleterious |
Not Tolerated |
0.00/53.23 (C45) |
0.1641 |
p = 0.17 n≤200 |
4.5 |
PD |
||
415 |
PIK3CA |
p.H1047L c.3140A>T |
Yes |
Blood - D [1] Gastric – D [22] Liver - C [23, 24] Nervous system - D [25] Skin - B [26] |
PI3K/PI4K domain |
Yes/Yes |
10271/107457 (9.56%) |
4098/15384 (26.64%) |
- |
Pathogenic (0.96) |
Benign |
Tolerated |
0.00/98.69 (C65) |
0 |
p = 0.057 |
Oncogene |
7.5 |
CGC |
415 |
PRKD1 |
p.Y800C c.2399A>G |
No |
No results |
Protein Kinase Domain |
No/No |
328/38363 (0.85%) |
11/2364 (0.46%) |
0/11 |
Pathogenic (0.98) |
Deleterious |
Not Tolerated |
0.00/193.72 (C65) |
0.0002 |
p = 0.075 |
2.5 |
pd |
|
415 |
TTC21B |
p.R898* c.2692C>T |
No |
No results |
Tetratricopeptide repeat |
No/No |
230/37405 (0.61%) |
13/2137 (0.61%) |
0/13 |
Pathogenic (0.90) |
ND |
ND |
- |
p = 0.15 |
NO |
1.5 |
Neutral |
|
416 |
FAM96A |
p.E75K c.223G>A |
No |
Colorectal - NR [2] |
Iron-sulfur cluster assembly protein |
No/No |
36/37402 (0.10%) |
2/2137 (0.09%) |
0/2 |
Pathogenic (0.94) |
Benign |
Tolerated |
0.00/56.87 (C55) |
0.6908 |
p = 0.039 N≤200 |
1.5 |
Neutral |
|
416 |
FLG |
p.R1166C c.3496C>T |
No |
No results |
disorderSource: IUPred |
No/Yes |
1467/37980 (3.87%) |
78/2139 (3.64%) |
- |
Neutral (0.01) |
ND |
Not Tolerated |
0.00/179.53 (C65) |
0.6299 |
p = 0.18 |
[63] |
1.5 |
Neutral |
416 |
GRIN1 |
p.Q910* c.2728C>T |
No |
Colorectal - NR [2] |
disorderSource: IUPred |
No/No |
162/37493 (0.43%) |
7/2137 (0.33%) |
0/7 |
Pathogenic (0.77) |
ND |
ND |
- |
- |
p = 0.24 |
[64] |
2.5 |
pd |
416 |
MYO1H |
p.E501G c.1502A>G |
No |
No results |
Myosin motor domain |
No/No |
237/37317 (0.63%) |
17/2126 (0.76%) |
1/17 |
Pathogenic (0.98) |
ND |
Tolerated |
0.00/97.85 (C65) |
0.0825 |
p = 0.14 n≤200 |
NO |
0.5 |
Neutral |
416 |
TTN |
p.L6228S c.18683T>C |
No |
Colorectal - B [65] |
IG-Like 43 domain |
No/No |
4470/37491 (11.92%) |
288/2105 (13.68%) |
- |
Pathogenic (0.81) |
Deleterious |
ND |
- |
0.1523 |
p = 0.019 OE |
4.5 |
PD |
|
417 |
AZI2 |
p.I66V c.196A>G |
No |
No results |
coiled_coilSource: ncoils |
No/No |
63/37401 (0.17%) |
5/2137 (0.23%) 1 FS |
0/5 |
Pathogenic (0.65) |
Benign |
Tolerated |
0.00/29.61 (C25) |
0.2633 |
p = 0.000061 n≤200 |
NO |
0 |
Neutral |
417 |
GRHL2 |
p.E32* c.94G>T |
No |
disorderSource: IUPred |
No/No |
171/37403 (0.46%) |
17/2138 (0.79%) |
0/17 |
Pathogenic (0.99) |
ND |
ND |
- |
- |
p = 0.39 |
3 |
pd |
||
417 |
NDST4 |
p.V313F c.937G>T |
No |
No results |
heparan sulfate-N-deacetylase domain |
No/No |
403/37402 (1.08%) |
6/2137 (0.28%) |
0/6 |
Pathogenic (0.98) |
Deleterious |
Not Tolerated |
0.00/49.94 (C45) |
0.0601 |
p = 0.1 |
2.5 |
pd |
|
417 |
PRICKLE2 |
p.P81L c.242C>T |
No |
Skin - D [26] |
PET Domain |
No/No |
213/37402 (0.57%) |
8/2138 (0.37%) |
0/8 |
Pathogenic (0.99) |
Deleterious |
Not Tolerated |
0.00/97.78 (C65) |
0.0751 |
p = 0.17 n≤200 |
3 |
pd |
SNVs Frequency in breast cancer*: frequency of SNVs (including synonymous) in breast cancer. SNVs (n) in breast cancer. Young pts/all ages: number of SNVs (excluding synonymous) in breast cancer (BC) patients ≤ 35 years/number of SNVs in BC patients with all ages (excluding patients who had unknown ages). CGC genes for which mutations have been causally implicated in cancer and which are catalogued at “Cancer Gene Census”. CCGD genes that are potential cancer drivers in genetic screens in mice and are catalogued at the “Candidate Cancer Gene Database”. NS: Nonsense; FS: Frameshift. KM-OS: (p) for overall survival evaluated through gene expression using KM plotter. Abbreviations: OE: Overexpression associated with longer survival; UE: Underexpression associated with longer survival. PD: Probably driver; pd: Possibly driver. The score system is described in Supplementary Table 9. Coments of literature is referenced in Supplementary Table 9a.
Each tumor sample was then individually explored to detect potential drivers. Three tumors presented SNVs in at least three potential cancer driver genes: 402, 406 and 415. In tumor 402, besides PIK3CA and CIITA, other candidate cancer genes harboring somatic SNVs were CACNA1E, NES, STAB1, HECW2, SMURF2 and ZNF462. In tumor 406, SNVs were detected in three known cancer genes reported in the “Cancer Gene Census” database [19]: PIK3CA, TP53 and PRKAR1A. However, the alteration detected in the latter was considered pathogenic in only one of the five mutation function assessment algorithms (Table 1). In addition, SNVs were observed in other two possibly driver genes, IL22 and OSR2. In tumor 415, besides PIK3CA, other potential cancer drivers affected by SNVs were PIK3AP1 and PRKD1.
In the other five tumors, SNVs were identified in one to three potential cancer driver genes: in tumor 413, RSBN1; in tumor 416: TTN and GRIN1; in tumor 401: SEMA6D [21, 22], as well as MTHFD2; in tumor 417: GRHL2, PRICKLE2 and NDST4. In tumor 404, a possible cancer driver is ELMO3, which KM plotter indicated that overexpression is associated with poor overall survival (Supplementary Figure 1).
To further explore somatic mutations in luminal tumors (HER2 negative) from very young patients, we identified another 29 patients aged ≤35 years at diagnosis, who had data published in studies of tumor exome or genome sequencing [15–18], most of which, deposited in the COSMIC database [15–17].
In these tumors, the most frequent events were C to T transitions, representing a mean percentage of 39% of the substitutions (Supplementary Figure 2). A total of 1,617 non-synonymous variants were detected across these 29 patients, with a median number of 29 variants per patient (minimum: 9 and maximum: 546; mean: 56) (Supplementary Table 10, 10a). Some genes, that were present in our list, were also mutated in these luminal tumors, such as PIK3CA, TP53, AK8, CIITA, FLG, POC5, POLD1, SEMA6D, TTN and LCRC66.
Functional categories enriched in gene variants according to DAVID bioinformatics tool [23] included ATP binding, in five tumors and plasma membrane, in three tumors, among others less frequently represented (Supplementary Table 11).
Seven out of these 29 tumors presented SNVs in just one cancer driver, classified in the “Cancer Gene Census” database, which were: AKT1, MPL (MPL Proto-Oncogene, Thrombopoietin Receptor), TP53, GATA3 (2 samples), BCOR (BCL6 Corepressor) and KMT2C (Lysine Methyltransferase 2C), while the other 19 tumors presented SNVs in at least two cancer genes from the “Cancer Gene Census” database (Supplementary Table 12). Furthermore, three tumors did not show any variants in driver candidates from the list of “Cancer Gene Census”, but each one presented SNVs in one or two genes, already reported in the “Candidate Cancer Gene Database” category A: PDS5B (PDS5 cohesin associated factor); LPHN2/ADGRL2 (Adhesion G Protein-Coupled Receptor L2) and ETF1 (Eukaryotic translation termination factor 1); CELF2 (CUBGBD Elav-like family member 2) and NAP1L4 (Nucleosome assembly protein 1 like 4). All genes considered as causally implicated in cancer or potential cancer drivers are shown in Figure 3 and Supplementary Table 12. The score system (described in methods) identified FAT2 (FAT atypical cadherin 2) as a probable driver gene in two samples, because it is a gene ranked B in CCGD, also frequently mutated in cancers and variants were considered pathogenic in three out of four prediction models of cancer causality investigated.
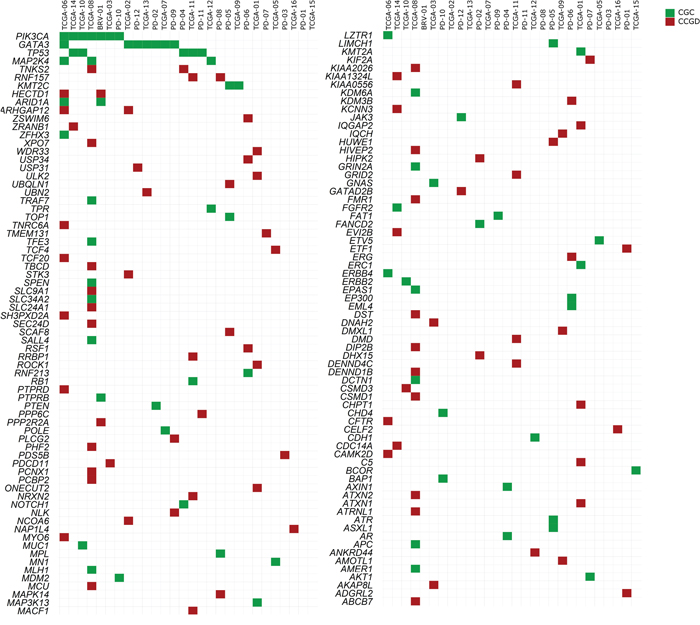
Figure 3: Distribution of mutated candidate driver genes among 28 tumor samples retrieved from the literature and COSMIC database. All cancer genes listed at “Cancer Gene Census” (CGC) database (http://cancer.sanger.ac.uk/cosmic/census) and all driver candidates listed in “Candidate Cancer Gene Database” (CCGD), ranked as A (http://ccgd-starrlab.oit.umn.edu/about.php), are shown. Note: Sample TCGA-04 is shown exclusively in Supplementary Table 10 (but not in the figure), due to a large number of somatic mutations (CGC= 30; CCGD rank A= 56). Green: CGC; Red: CCGD, rank A [18]. Causal relationship with cancer was based on a scoring system, described in Materials and Methods. All reported genes affected by SNVs appear in Supplementary Table 10.
Among the 29 tumors, six were obtained from patients whose BRCA1 and BRCA2 status was known: two wild type and four mutation carriers. Somatic SNVs in both tumors from BRCA1 and BRCA2 wild type germline patients involved GATA3; however, none of the affected genes in this pair of tumors coincided with data from our patients.
Finally, we analyzed the 37 tumors all together (29 previously reported and 8 currently evaluated). Considering only SNVs detected in the genes already included in the “Cancer Gene Census” database or the “Candidate Cancer Gene Database”, categories A or B, the median number (minimum and maximum) of driver candidates per tumor, were: 2 (0-30); 2 (0-56); 2 (0-61) respectively, totalizing a median of 6 potential drivers affected per tumor (0-147) (Supplementary Tables 12). The most frequently altered cancer causing genes according to “Cancer Gene Census” were PIK3CA (11/37: 29.7%); GATA3 (7/37; 18.9%), TP53 (6/37: 16.2%) and MAP2K4 (3/37: 8.1%). SNVs were also frequently detected in the following genes: TTN (7/37; 18.9%), CAMK1G, LYST, DALRD3 (3/29; 10.3%) and FLG (3/37; 8.1%). Among these genes, it is interesting to point out that pathogenic frameshift mutations in DALRD3 were detected in two (out of three) tumor samples. PIK3CA was concomitantly mutated with TP53 in three tumors and with GATA3 in one tumor (Figure 4; Supplementary Figure 3).
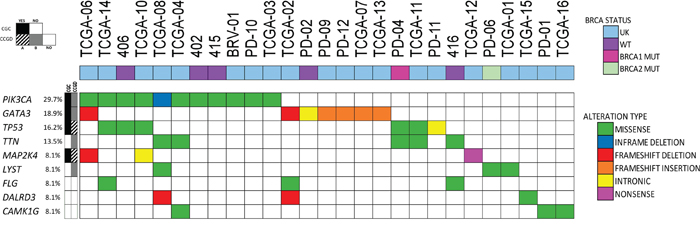
Figure 4: Most frequently mutated genes in luminal tumors. Samples (n=27) presenting SNVs in at least one of the nine most frequently mutated genes were included (current analysis, n=4; and COSMIC Database, n=23). Type of gene alteration and BRCA1/2 status are shown. Each column represents a single patient. UK: unknown.
SNVs were detected in genes involved in DNA repair mechanisms in 16 out of the 37 tumors (43.2%). In 11 samples, only one gene was altered, such as FANCD2, FANCL or BAP1, which are involved in homologous recombination repair (HRR); PARP4 (2 samples), involved in base excision repair (BER); ATR and TP53 (the latter altered in 3 samples), involved in signaling DNA damage to cell cycle checkpoints. In two tumors, SNVs uniquely affected polymerases POLD1 or POLE, which are involved in the base excision repair (BER), nucleotide excision repair (NER) and mismatch repair (MMR).
Three samples presented composite gene disturbances involving TP53 and either POLD1 or RAD51 (HRR) or POLQ (involved in translesion synthesis, TLS). The highest number of SNVs was described in two tumors, one presenting mutations in genes involved in BER (MUTYH), NER (ERCC6) and HRR (PALB2) and the other, in genes involved in MMR (MLH1) and HRR (RAD9A) [24] (Table 2).
Table 2: Samples with somatic mutations in genes involved in DNA repair mechanisms
Sample |
Gene |
Mechanisms of DNA repair |
N. of variants/sample |
||||||
|---|---|---|---|---|---|---|---|---|---|
BER |
NER |
MMR |
HRR |
NHEJ |
DDC |
TLS |
|||
406 |
TP53 |
X |
7 |
||||||
413 |
POLD1 |
X |
X |
X |
6 |
||||
PD-02 |
FANCD2 |
X |
17 |
||||||
PD-04 |
POLD1 |
X |
X |
X |
55 |
||||
TP53 |
X |
||||||||
PD-05 |
ATR |
X |
X |
36 |
|||||
PD-06 |
FANCL |
X |
76 |
||||||
PD-10 |
BAP1 |
X |
17 |
||||||
PD-11 |
TP53 |
X |
16 |
||||||
TCGA-01 |
PARP4 |
X |
46 |
||||||
TCGA-04 |
MUTYH |
X |
546 |
||||||
ERCC6 |
X |
||||||||
PALB2 |
X |
||||||||
TCGA-06 |
PARP4 |
X |
48 |
||||||
TCGA-07 |
POLE |
X |
X |
X |
21 |
||||
TCGA-08 |
MLH1 |
X |
229 |
||||||
RAD9A |
X |
||||||||
TCGA-10 |
TP53 |
X |
9 |
||||||
TCGA-11 |
TP53 |
X |
84 |
||||||
RAD51 |
X |
||||||||
TCGA-14 |
POLQ |
X |
79 |
||||||
TP53 |
X |
||||||||
DNA repair genes altered and respective pathways affected per patient. Base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination repair (HRR), non-homologous end-joining (NHEJ), DNA damage signaling to cell cycle checkpoints (DDC) and translesion synthesis (TLS).
In addition, variants involving 213 genes were of nonsense or frameshift types (Supplementary Table 12). One of these genes, RBM16/SCAF8 is a driver candidate, because it is also listed in the “Candidate Cancer Gene Database”, rank A, in at least two solid tumor types. Among these genes, 42 were involved in positive regulation of gene expression [25] and one of these genes was mutated in 15 different samples and more than one gene was mutated in five other samples. Hence, 54% of the luminal samples presented at least one mutated gene involved in gene expression regulation (Table 3).
Table 3: Characterization of nonsense and frameshift variants according to CCGD A/B and biological function (Toppgene) per sample
Sample |
Gene ID |
CCGD A |
CCGD B |
Positive regulation of gene expression |
N. of variants/sample |
|---|---|---|---|---|---|
415 |
TTC21B |
- |
- |
- |
6 |
416 |
GRIN1 |
- |
- |
GRIN1 |
5 |
417 |
GRHL2 |
- |
- |
GRHL2 |
4 |
BRV-01 |
CTCFL; FAM118B; MARVELD2; VPS11 |
- |
- |
CTCFL |
28 |
PD-02 |
PTEN |
PTEN |
- |
PTEN |
17 |
PD-04 |
FSCB; IL12RB2; AKAP11 |
- |
- |
- |
55 |
PD-05 |
ATR; VIM HK1; OR7C1; RBM16/SCAF8 |
ATR; RBM16/SCAF8 |
- |
VIM |
36 |
PD-06 |
CACNA2D3; DNAH17; PEX5L MAB21L3; SYNE1; ZMYND11 |
- |
ZMYND11; DNAH17 |
- |
76 |
PD-07 |
PTRH1 |
- |
- |
- |
13 |
PD-08 |
KCNJ15; SYTL2 ENSG00000233280 |
- |
- |
- |
41 |
PD-09 |
PLCG2 SHCBP1; GATA3 |
PLCG2 |
GATA3 |
GATA3 |
15 |
PD-10 |
NEMF |
NEMF |
- |
17 |
|
PD-11 |
KRTAP2-1; SLC2A3; NARG2/ICE2 COL22A1 |
- |
- |
NARG2/ICE2 |
16 |
PD-12 |
GATA3; PDE7A |
- |
GATA3 |
GATA3 |
22 |
TCGA-01 |
ABCA10; NTRK2; MAP3K6_ENST00000374040; CX3CR1; KBTBD4; KMT2A |
- |
- |
KMT2A; NTRK2 |
46 |
TCGA-02 |
GATA3; DALRD3; RASGRP2; SALL3; TNFSF9 |
- |
GATA3 |
GATA3 |
18 |
TCGA-03 |
C1orf187; NR1I3; FAM155A; GNAS; PCDHA2; SSC5D; SEC14L5; WDR81_ENST00000409644 |
- |
- |
NR1I3 |
21 |
TCGA-04 |
41 |
UBR5 |
BTBD7; ITGB1; KLHDC2; MTA2; ODF2; PCCA; PPFIA3; RASGRF1 |
ITGA8; MTA2; NFKBIA; ATF7IP; SPAG8; TARBP2; TLR3 |
546 |
TCGA-05 |
NMS; FAM111B; DYNC2H1_ENST00000398093 |
- |
- |
- |
31 |
TCGA-06 |
ARID1A; CFTR; SYNM; GATA3; CCDC61; CDK18; IRF7; TCF20; KIAA0430/MARF1; LZTR1; MAP2K4; SH3PXD2A |
ARID1A; TCF20; CFTR; MAP2K4; SH3PXD2A; |
GATA3 |
ARID1A; GATA3; IRF7; TCF20 |
48 |
TCGA-07 |
GATA3 |
GATA3 |
GATA3 |
21 |
|
TCGA-08 |
73 |
ATXN2; DIP2B; KIAA2026_E NST0000039 9933; PCNX1; PHF2; TNKS2; SLC9A1 |
CLMN; KIAA0947_ ENST0000 0296564/ICE1; OSBPL1A; RAB11A; ARHGAP2 9; SLTM; |
17 |
229 |
TCGA-09 |
DYNC1H1; IGSF3; MAST1; AKAP12; ASB10_ENST00000422024; NASP; TENM1/ODZ1; THOC5; ZNF799 |
- |
THOC5 |
THOC5 |
35 |
TCGA-10 |
C9orf66 |
- |
- |
- |
9 |
TCGA-11 |
A2M; CHKB; NBR1; RB1; SYT3; ARR3; KIFC3; PPP1R3C; ZBTB24 |
RB1 |
NBR1 |
RB1 |
84 |
TCGA-12 |
EFEMP1; MAP2K4; C1orf35; KIF26A |
MAP2K4 |
- |
- |
15 |
TCGA-13 |
SNUPN_ENST00000371091; GATA3; GRM6 |
- |
GATA3 |
GATA3 |
13 |
TCGA-14 |
PRDM5; COL14A1; POLA1; SLC22A25 |
- |
PRDM5 |
- |
79 |
TCGA-15 |
IGSF1; NFYB; SCN2A; TRAF5 |
- |
NFYB |
NFYB; TRAF5 |
34 |
TCGA-16 |
JHDM1D/KDM7A |
- |
- |
JHDM1D/KDM7A |
30 |
Samples with genes affected by nonsense or frameshift variants were searched for candidate cancer genes (CCGD database ranks A or B) and involvement in positive regulation of gene expression (GO: biological process).
DISCUSSION
Our goal was to characterize BRCA1 and BRCA2 germline mutations in a group of very young Brazilian patients and to identify somatic mutations in luminal HER2 negative breast cancer.
Our data indicates that in very young Brazilian patients, BRCA1 and BRCA2 mutation frequency is 16%, similar to that already reported in comparable groups of patients from Brazil [7], as well as from other countries [4–8]. However, there is still a lack of information regarding the spectrum of mutations and VUS in the average Brazilian population, that harbors peculiar characteristics of miscegenation, comprehending a mixture of 70% European, 15% African and 15% Amerindian ancestry genes [26]. In our patients we could detect a new mutation in the BRCA2 gene, as well as another 13 variants of unknown significance.
Somatic mutation in the group of eight luminal samples (HER2 negative) from BRCA1 and BRCA2 wild type carriers were then investigated. The overall mutation rate in these tumor samples was 1.93 per Mbp, as compared with 1.18 per Mbp and 1.66 per Mbp reported in luminal samples from post-menopausal women [27] and other breast cancer samples in general, irrespective of subtype or age [18], respectively. We have also detected a predominance of C>T substitutions, a signature previously associated with advancing age, indicating that these alterations are also the most prevalent in early onset breast cancer [28]. In accordance, the same signature was also the most frequent among other luminal tumors from very young patients deposited in COSMIC [15–17].
In the present series, somatic SNVs affected, among others, five known cancer causing genes, PIK3CA, TP53, PRKAR1A, POLD1 and CIITA [19]. PIK3CA was the only recurrent finding, which was detected in three different tumors. Other cancer causing candidates were SMURF2, PIK3AP1, RSBN1, TTN and SEMA6D, which were ranked in the top 25% potential drivers in transposon insertional mutagenesis studies in mice [21, 29]. These genes variants were also considered pathogenic/deleterious/not tolerated in at least two out of five mutation function assessment algorithms. In addition, SNVs were detected in genes that were previously associated with cancer, such as CACNA1E, PRKD1, NDST4, and were also considered pathogenic/deleterious/not tolerated in at least three mutation function models. Moreover, nonsense mutations were detected in GRHL2, GRIN1, NOL9 and TTC21B, however only GRHL2 and GRIN1 were previously shown to be involved in cancer.
GRHL2 (grainyhead-like transcription factor 2), is a transcription factor that mainly suppresses epithelial mesenchymal transition (EMT) process. It is considered a potential tumor suppressor gene in breast cancer [30]. GRIN1 or NMDAR1 (N-Methyl-D-Aspartate Receptor Subunit NR1) was shown to be expressed in breast cancer specimens, but not in normal breast and to be involved in tumor growth [31], being thus, a potential oncogene. SMURF2 (SMAD specific E3 ubiquitin protein ligase 2) is a tumor suppressor involved in the maintenance of genomic stability and suppression of breast cancer cells invasiveness [32, 33]. PIK3AP1 (phosphoinositide-3-kinase adaptor protein 1), also known as BCAP, is involved in the phosphatidylinositol 3-kinase (PI3K) pathway and genome wide association studies suggest that the PIK3AP1 gene region might be involved in breast cancer predisposition [34]. RSBN1 encodes a round spermatid basic protein 1, which function is not well established. In breast cancer lineages RSBN1 expression is induced by hypoxia and the gene is a potential HIF target [35]. Besides, in luminal breast cancer, RSBN1 high expression is associated with a better prognosis in luminal breast cancer [20].
CACNA1E, calcium voltage-gated channel subunit alpha-1 E, was shown to be underexpressed in breast cancer compared with normal tissue and was hypothesized to be a tumor suppressor gene in some types of cancer [36]. In the current study, CACNA1E mutation occurred in a hot spot site already reported as altered in at least five different types of cancers. PRKD1 codes for a serine-threonine kinase and mutations all over the gene were described in various types of cancer. A recurrent activating mutation in the kinase domain described in polymorphous low grade adenocarcinoma of salivary glands, was associated with improved metastasis free survival in a transfection cell model [37]. In breast cancer cells however, PRKD1 may display a dual function as an oncogene, stimulating drug resistance in breast cancer stemness [38] or as a tumor suppressor, blocking invasion and metastasis. In our patient, PRKD1 mutation was located in the distal region in the kinase domain.
POLD1 codes for the catalytic subunit of DNA polymerase delta, which plays a role in DNA replication and DNA repair [39]. Both germline and somatic gene mutations may cause an ultra-mutated phenotype, and mutations affecting the exonuclease domain are associated with high risk of colorectal and endometrial carcinomas [40]. In our patient, POLD1 amino-acid change occurred in the exonuclease domain. In addition, POLD1 was also mutated in another luminal sample from a very young patient present in COSMIC database [16]. Although infrequent in breast cancer, five of ten POLD1 somatic mutations reported in the COSMIC database were of frameshift nature, therefore, potentially pathogenic (http://cancer.sanger.ac.uk/cosmic) (July, 2017).
NDST4 (N-deacetylase/N-sulfotransferase-4), is involved in heparan sulfate (HS) biosynthesis and may be implicated in positive or negative aspects of tumor progression. In colorectal cancer, NDST4 loss of function was implicated in tumor progression and the gene was considered a candidate tumor suppressor [41].
ELMO3 (Engulfment and Motility 3) is involved in induction of cell proliferation, invasion and metastasis in colorectal cancer cells [42]. In addition, ELMO3 positive/higher expression is associated with poor overall survival in non-small cell lung cancer and head and neck cancer, as well as in breast cancer, corroborating its role as an oncogene [43, 44]. MTHFD2 (methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 2) is a source of carbon units for purine synthesis in rapidly growing cancer cells and has been associated with poor prognosis in patients with breast cancer [45, 46]. SEMA6D (Semaphorin 6D) encodes a transmembrane protein and its overexpression increases proliferation and tumor formation, playing an oncogene role in osteosarcoma [47]. SEMA6D high expression is also associated with better patient survival, especially among triple negative breast cancer [48].
The results, considering all the 37 tumors (29 previously analyzed and the eight currently analyzed), suggest that the median number of driver candidates per tumor is six, however, this number is quite variable. Moreover, in luminal tumors from very young patients the most frequent cancer drivers are PIK3CA; GATA3, and TP53. In accordance with a recent analysis that included some of these very young patients (≤ 35 years) but mainly older patients, with ages up to 45 years, the most prevalent mutated genes were also PIK3CA, TP53, GATA3 and TTN [49].
Other genes frequently mutated were CAMK1G (Calcium/Calmodulin Dependent Protein Kinase IG), DALRD3 (DALR Anticodon Binding Domain Containing 3), LYST (Lysosomal Trafficking Regulator) and MAP2K4 (3/37: 8.1%). DALRD3 contains two microRNA (miRNA) precursors (miR-191 and miR-425) in one of its introns and the expression of both microRNAs is higher in estrogen receptor alpha (ER) positive cells. However, estrogen regulation of miR191/425-DALRD3 transcriptional unit is complex and may be unparalleled. Although the exact function DALRD3 is not known, in estrogen receptor positive cells, miR-191/425 works as oncogenes by inducing proliferation. Interestingly, SNVs in DALRD3 detected in two out of three samples from young patients were of the frameshift kind [50]. LYST gene silencing may inhibit cell proliferation and induce apoptosis in myeloma cells [51].
It is worth mentioning that somatic mutations in genes involved in DNA repair mechanisms were quite common and any of these pathways might be altered: base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination repair (HRR) as well as signaling DNA damage to cell cycle checkpoints. The highest number of SNVs was described in two tumors presenting mutations in genes involved in HRR, as well as in other DNA repair mechanisms concomitantly [24]. In accordance, an association between younger age at diagnosis and risk genotypes for genes involved in DNA repair, such as NER, MMR and NHEJ (Non-homologous end-joining) have been already reported [52].
The weaknesses and the strengths of our study involve the number of exomes analyzed, though small, add around 20% of samples to the available data thus far.
In summary, in luminal tumors (HER2 negative) from very young patients, the most frequent events were C to T transitions. SNVs were detected in a median number of six potential driver genes per sample, and 43% of the tumors presented mutations in DNA repair genes and 54% of the tumors presented at least one pathogenic mutation in a gene involved in positive regulation of gene transcription. The most frequent somatic mutations involved cancer driver genes, such as PIK3CA, TP53 and GATA3. Other potential driver candidates currently identified were GRHL2, PIK3AP1, CACNA1E and SEMA6D.
MATERIALS AND METHODS
Patients
This study was approved by the Institutional Ethics Committee of Instituto Brasileiro de Controle do Câncer (IBCC) and Instituto do Câncer do Estado de São Paulo (ICESP)/Faculdade de Medicina da Universidade de São Paulo (FMUSP). All patients were informed and signed an informed consent.
Early onset breast cancer was defined as a disease diagnosed in very young women aged ≤35 years. No patients received previous medical treatment for their breast cancer before the tumor collection through biopsy or mastectomy procedures.
Patients were interviewed for family history suggestive of Hereditary Breast and Ovarian Cancer Syndrome (HBOCS) in close relatives, such as first, second, and third degree family members. Family history was considered informative if the patient could report on at least two first or second degree female relatives having lived beyond age 45 in both parental lineages, otherwise it was considered unknown or limited (National Comprehensive Cancer Network, NCCN, https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf, (February 2012). Genetic/Familial high-risk assessment: breast and ovarian. Patients were also asked about their ancestry, to obtain information of country or continent where their parents and grandparents (at least) were born.
The median age of the 79 patients at diagnosis was 32 years, most of whom diagnosed with invasive ductal carcinoma (91.1%), high histological grade (48%), Ki67 >14% (90.4%), luminal subtype (65.8%; ER and/or PR positive and HER2 negative), and advanced stage disease (clinical stages III/IV; 47.1%) (Supplementary Table 1). HER2 positivity was defined as immunohistochemistry 3+ or 2+, the latter, associated with Fluorescence in situ hybridization (FISH)-amplification. HER2 immunohistochemistry and FISH were scored according to ASCO/CAP guidelines [53].
All women had a blood sample collected for BRCA1 and BRCA2 whole gene sequencing (see below).
Among the 79 women, 12 had fresh-frozen tumor samples collected during breast surgery. Among the latter, eight patients, who were BRCA1 and BRCA2 wild type carriers bearing luminal HER2 negative tumors, had their samples subsequently analyzed through whole exome sequencing (see below) (Supplementary Figure 4).
DNA extraction from blood and tumor tissue
DNA was extracted from 8mL of whole blood using the Kit Illustra Blood GenomicPrep Mini Spin Kit (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA/28-9042-64); and from cancer cells enriched areas from fresh-frozen or FFPE samples, using the QIAamp DNA Mini Kit - Qiagen (Qiagen, Valencia, CA, USA/51304) and QIAamp® DNA FFPE Tissue (Qiagen/56404), respectively, following instructions of the manufacturer.
Direct sequencing of BRCA1 and BRCA2 genes
Polymerase chain reaction (PCR) amplification and sanger sequencing
Briefly, the complete coding region of BRCA1 (U14680 or NM_7294.2) and BRCA2 (U43746 or NM_000059.1) genes were amplified and sequenced in both forward and reverse directions. Primers and conditions are described in Supplementary Table 13 for BRCA1 [54, 55] and Supplementary Table 14 for BRCA2 [56]. Sequences obtained were visualized by Chromas (v2.33; Technelysium Pty, Ltd Eden Prairie, MN, USA) and by Mutation Surveyor software (v3.20, SoftGenetics LLC, State College, PA, USA). If a pathogenic mutation was identified, a new DNA sample derived from a second venipuncture was resequenced for confirmation. Full details of methods are given in the Supplementary Methods.
Multiplex ligation-dependent probe amplification (MLPA) of BRCA1 and BRCA2 genes
Samples from patients, who were negative for BRCA1 and BRCA2 pathogenic mutations were investigated for large deletions and duplications, using the MLPA commercial kits SALSA® MLPA® P002 BRCA1 probemix (P002 - 100R) and SALSA® MLPA® P045 BRCA2/CHEK2 probemix (P045 - 100R) (MRC-Holland, Amsterdam, The Netherlands), as described in Supplementary Methods. Sequencing to detect the presence of CHEK2 hot spot (c.1100delC) was also performed.
BRCA1 and BRCA2 sequencing analysis and reporting criteria
All sequence variants were named according to nomenclature used by The Human Gene Mutation Database, HGMD (http://www.hgmd.cf.ac.uk/ac/index.php). The variants were searched for their classification in five publicly accessible databases: Breast Cancer Information Core (BIC) [57], Leiden Open Variation Database (LOVD v3.0 build 13), [58], Leiden Open Variation Database - International Agency for Research on Cancer (LOVD-IARC v.2.0 Build 22), Universal Mutation Database (UMD), [59, 60], and ClinVar [61], this search was performed on the months of April - June 2017.
Gene variants were submitted to the following in silico prediction models: Polymorphism Phenotyping (PolyPhen; v2.2.2) [62], Sorting Intolerant From Tolerant (SIFT; v1.0.3) [63], Align-GVGD [64, 65], for missense variants; Protein Variation Effect Analyzer (Provean; v1.1) [66] for in-frame deletions, and Human Splicing Finder [67] to check for intronic and exonic variants leading to potential splicing defects.
Minor allele frequency was checked in the 1000 Genomes Project database [68], the Exome Aggregation Consortium (ExAC) [69, 70], the Global MAF dbSNP [71], and the Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP) [72].
The variants were classified according to recommendations of the American College of Medical Genetics and Genomics in: pathogenic, likely pathogenic, benign, likely benign and variant of uncertain significance (VUS) [73]. Variants for BRCA1 were also checked for co-occurrence with known pathogenic mutations in the same patient. If VUS were classified in two of the five databases, and categorized as benign (BIC and ClinVar), no known pathogenicity (LOVD), 1-not pathogenic (LOVD-IARC), or 1-neutral (UMD), they were reclassified as benign.
Exome sequencing
DNA extracted from mononuclear cells and fresh tumor samples (containing at least 70% malignant cells) from eight patients was used to prepare a DNA library with the Illumina Nextera Rapid Capture Expanded kit (Illumina, Inc., San Diego, CA, USA/FC-140-1004), as detailed in Supplementary Methods. Shortly, genomic DNA (gDNA) was enzymatically fragmented while tags were simultaneously added. After purification, a limited-cycle PCR program was performed to ligate adapters and amplify libraries. Once gDNA libraries were prepared, exon-specific capture probes attached to streptavidin beads were used to enrich fragments containing only regions of interest, comprising 201,121 exons, totaling 62 mega base pairs (Mbp) of the genome. Exome libraries were then evaluated on a DNA 1000 Agilent 2100 Bioanalyzer chip (Agilent Technologies, Santa Clara, CA, USA) and quantified using KAPA SYBR FAST qPCR Kits (Kapa Biosystems, Wilmington, MA, USA, part #KK4602) prior to cluster generation. Pooled libraries were loaded on six lanes of one flow cell and sequenced on HiSeq 1000 platform (Illumina, Inc.) using 2 x 100bp paired-end reads, with a median of 95.3% of targeted bases covered at least 30-fold across the sample set.
Exome sequencing analysis
BWA (v0.5.7) [74] software was used to align 8 paired tumor/blood exome samples, using hg19 as the reference genome and Picard (v1.92) to mark duplicates. Paired tumor-normal samples were processed together using GATK (v2.4.9) [75] for local realignment and for base quality recalibration. SAMtools (v0.1.9) and Picard (v1.107) were then used to process the bam headers and to index the samples, respectively [76].
To detect somatic single nucleotide variants (SNVs), SomaticSniper (v1.0.2) [77] was utilized. Default parameters were used to call SNVs, except for the mapping quality threshold, which was set to 1, as recommended by the developer. Standard, LOH, bam-readcount, false positive and lastly high confidence filters were applied using SAMtools (v0.1.6) and scripts provided by the SomaticSniper package. The final VCF file, containing high-confidence somatic SNVs, was used in downstream analyses.
An in-house perl- and R- based pipeline was used to identify recurrent mutations. Parameters were set to find genes that were mutated in at least 2 samples. This pipeline uses lists of SNPs compiled from various studies to filter out likely false positive SNPs from the samples, unless they are found in the Catalogue of Somatic Mutations in Cancer (COSMIC v71) database for coding and non-coding mutations [78]. After somatic SNVs were called using SomaticSniper, the SNPs were annotated by ANNOVAR (v2014-07-14) [79], using the RefGene database. Nonsynonymous, stop-loss, stop-gain and splice-site SNVs (based on RefGene annotations) were considered functional. SNVs were filtered using tabixpp (3b299cc) [80], removing SNVs found in any of the following databases: Fuentes, 2012 [81], dbSNP142 [82], 1000 Genomes Project (v3) [68], AccuSNP blacklist (invalidated SNVs from 68 human colorectal cancer exomes (in preparation), generated from GATK (v2.4.9 UG) and AccuSNP platform (Roche NimbleGen) analyses), and ENCODE DAC and Duke [83]. SeqSig (v3.6.4)[84] was used to identify likely driver non-synonymous mutations. This test assumes that for each patient, mutations are independent among nucleotides and homogeneous across all positions on coding regions and compute the background mutation rate for non-synonymous mutations. It uses the convolution law and may be used in situations where samples are not abundant. Discrepancies between the number of genes found in Supplementary Table 15 and that plotted in Figure 2, are due to the collapsing of variants into genes. SnpEff (v4) [85] was then used to predict amino acid changes. Data visualization used the BPG package (v5.2.1) in R [86].
Analysis of somatic variants to identification of candidate driver genes
Genes candidates were then searched for in the “Cancer Gene Census” (CGC) database (http://cancer.sanger.ac.uk/census/) [19] to identify genes causally implicated in cancer, as well as in the “Candidate Cancer Genes Database” (CCGD) (http://ccgd-starrlab.oit.umn.edu/search.php) [21], to identify potential cancer drivers, detected in mouse insertional mutagenesis experiments. In this model, candidate genes were associated with common insertion sites (CIS), which were ranked based either on the number of insertions or the p-value: A for the top 10%; B for the top 11-25%, C for the top 26-50% and D for the bottom 50%. CISs identified in screens that did not include insertion numbers or p-values are denoted as Not Ranked [21]. Afterwards, gene mutations were analyzed through mutation function assessment algorithms: PolyPhen, SIFT, Align GV/GD [62–65], Functional analysis through Hidden Markov Models (FATHMM; v2.3) (http://fathmm.biocompute.org.uk/) [87], and Cancer-Related Analysis of Variants Toolkit (CRAVAT), [88]. This search was performed between April and June 2017, and the latter three algorithms were reviewed in December 2017.
We have then developed a scoring system in order to identify potential cancer drivers. The genes found in CGC were scored 3 points; CCGD was scored according to the highest rank for each sample: “A”: 2 points, “B”: 1.5 points, “C”: 1.0 point, “D”: 0.5 point; “Not Ranked” variants were not scored; mutation domain, frequency of the variant in other cancers and/or in breast cancer (≥1%) were scored 0.5 point each; mutation consequence when nonsense or frameshift was scored 1.5 points; mutation function assessment algorithms FATHMM, PolyPhen, SIFT, GV/GD and CRAVAT-CHASM (3.0) were scored by 1 point, if the variant was considered pathogenic at least in 3 of them.
Analysis of somatic variants identified in other published manuscripts and COSMIC database
For this analysis, publicly available data about 29 patients, aged 35 years or younger, was obtained.
Most patients (n=28) had data for tumor exome or genome sequencing deposited in the COSMIC database [15–17] (TCGA, 2012, n=16; Nik-Zainal et al., 2016, n=9; Stephens et al., 2012, n=3). Additionally, data for one patient was recovered from a published manuscript [18], which was not available in COSMIC. Only HER2 negative tumors were included. One and four of these patients were BRCA1 and BRCA2 mutation carriers, respectively. BRCA mutation status of the remaining patients was unknown [16, 17].
For the present analysis, among the total number of mutations per patient, repeated substitutions detected in the same chromosomal position were considered only once. In addition, only non-synonymous mutations were contemplated.
The list of nonsynonymous variants derived from each tumor was then clustered using the DAVID v6.7 bioinformatics tool (The Database for Annotation, Visualization, and Integrated Discovery) [23], in order to explore its biological meaning. Only one Gene Ontology category (p ≤0.05) or Interpro process (in the absence of GO category) was selected for each tumor sample. If more than one GO category was enriched, the one containing the largest number of genes was chosen.
To identify potential cancer driver genes a scoring system has been developed. The genes found in CGC were scored 3.0 points; CCGD was scored according to the highest rank for each sample: “A”: 2 points, “B”: 1.5 points, “C”: 1.0 point, “D”: 0.5 point; “Not Ranked” variants were not scored; mutation domain, frequency of the variant in other cancers and/or in breast cancer (≥1%) were scored 0.5 point each; mutation consequence when nonsense or frameshift was scored 1.5 points; mutation function assessment algorithms FATHMM, PolyPhen, SIFT and CRAVAT-CHASM (3.0) were scored by 0.5 point, if the variant was considered pathogenic at least in 2 of them; were scored by 1 point, if the variant was considered pathogenic in 3 or 4 of them. Gene variants scoring ≥3.5 were considered as candidates for cancer drivers (Supplementary Table 10, 10a).
The search in the referred databases and prediction tools was performed for this analysis until December, 2017.
Toppgene was used to identify biological processes enriched in the list of genes affected by pathogenic mutations (nonsense and frame shift). (https://toppgene.cchmc.org/enrichment.jsp). Gene ID followed by ENST number was searched using the gene ID without ENST number.
Ten functions (biological process) presented more than 10 affected genes and had a p value, Bonferroni and FDR <0.05, including positive regulation of gene expression. Analysis was performed in March 2018.
Author contributions
GE, ECL, and MAAKF conceived of the study. GE, ECL, ACRCG, MDPED, and JCGSG included patients and collected clinical data. GE, SM, MLHK, PWMVV, and FSP performed the genomic experiments. FCG, GE, GFLP, LMR, MAAKF, PAMPS, PCB, RAB and VYS analyzed data. GE, VYS, SM, MLHK, LMR, GFLP, PAMPS, MMB, PCB, RC, and MAAKF interpreted data. GE, VYS, PCB and MAAKF drafted the manuscript. All authors revised and approved the final manuscript.
ACKNOWLEDGMENTS
The authors would like to acknowledge Dr. Gustavo Gasparin for helpful assistance on exome sequencing, Dr. Fernando Nalesso Aguiar for reviewing immunohistochemistry analysis, Tauana Rodrigues Nagy for assistance in the analysis, Mrs. Cristina Pineiro Grandal for figure edition. The authors thank Richard de Borja, Takafumi Yamaguchi and Nicholas Harding for help in creating bioinformatics pipelines.
CONFLICTS OF INTEREST
All authors declare that they have no conflicts of interest.
FINANCIAL SUPPORT
This work was supported by São Paulo Research Foundation (FAPESP, grant #2012/12306-4). Maria Aparecida Azevedo Koike Folgueira and Roger Chammas received a research productivity grant by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). Giselly Encinas received a PhD scholarship grant by FAPESP (grant #2011/09572-1) and Simone Maistro received a postdoctoral scholarship by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (grant #029/2012). Gláucia Fernanda de Lima Pereira, Pedro Wilson Mompean de Vasconcellos Valle and Pedro Adolpho de Menezes Pacheco Serio received a grant from Professional Improvement Program by Secretaria de Saúde do Estado de São Paulo.
This study was conducted with the support of the Ontario Institute for Cancer Research to PCB through funding provided by the Government of Ontario. Dr. Boutros was supported by a Terry Fox Research Institute New Investigator Award and a CIHR New Investigator Award.
REFERENCES
1. FOSP - Fundação Oncocentro de São Paulo. 2015. http://www.fosp.saude.sp.gov.br/htmL/tabnet.htmL.
2. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67:7–30. https://doi.org/10.3322/caac.21387.
3. Bleyer A, Barr R, Hayes-Lattin B, Thomas D, Ellis C, Anderson B, and Biology and Clinical Trials Subgroups of the US National Cancer Institute Progress Review Group in Adolescent and Young Adult Oncology. The distinctive biology of cancer in adolescents and young adults. Nat Rev Cancer. 2008; 8:288–98. https://doi.org/10.1038/nrc2349.
4. Lalloo F, Varley J, Moran A, Ellis D, O’Dair L, Pharoah P, Antoniou A, Hartley R, Shenton A, Seal S, Bulman B, Howell A, Evans DG. BRCA1, BRCA2 and TP53 mutations in very early-onset breast cancer with associated risks to relatives. Eur J Cancer. 2006; 42:1143–50. https://doi.org/10.1016/j.ejca.2005.11.032.
5. Loizidou M, Marcou Y, Anastasiadou V, Newbold R, Hadjisavvas A, Kyriacou K. Contribution of BRCA1 and BRCA2 germline mutations to the incidence of early-onset breast cancer in Cyprus. Clin Genet. 2007; 71:165–70. https://doi.org/10.1111/j.1399-0004.2007.00747.x.
6. Haffty BG, Choi DH, Goyal S, Silber A, Ranieri K, Matloff E, Lee MH, Nissenblatt M, Toppmeyer D, Moran MS. Breast cancer in young women (YBC): prevalence of BRCA1/2 mutations and risk of secondary malignancies across diverse racial groups. Ann Oncol. 2009; 20:1653–59. https://doi.org/10.1093/annonc/mdp051.
7. Carraro DM, Koike Folgueira MA, Garcia Lisboa BC, Ribeiro Olivieri EH, Vitorino Krepischi AC, de Carvalho AF, de Carvalho Mota LD, Puga RD, do Socorro Maciel M, Michelli RA, de Lyra EC, Grosso SH, Soares FA, et al. Comprehensive Analysis of BRCA1, BRCA2 and TP53 Germline Mutation and Tumor Characterization: A Portrait of Early-Onset Breast Cancer in Brazil. PLoS One. 2013; 8:e57581. https://doi.org/10.1371/journal.pone.0057581.
8. Kurian AW. BRCA1 and BRCA2 mutations across race and ethnicity: distribution and clinical implications. Curr Opin Obstet Gynecol. 2010; 22:72–78. https://doi.org/10.1097/GCO.0b013e328332dca3.
9. Churpek JE, Walsh T, Zheng Y, Moton Z, Thornton AM, Lee MK, Casadei S, Watts A, Neistadt B, Churpek MM, Huo D, Zvosec C, Liu F, et al. Inherited predisposition to breast cancer among African American women. Breast Cancer Res Treat. 2015; 149:31–39. https://doi.org/10.1007/s10549-014-3195-0.
10. Cancello G, Maisonneuve P, Rotmensz N, Viale G, Mastropasqua MG, Pruneri G, Veronesi P, Torrisi R, Montagna E, Luini A, Intra M, Gentilini O, Ghisini R, et al. Prognosis and adjuvant treatment effects in selected breast cancer subtypes of very young women (<35 years) with operable breast cancer. Ann Oncol. 2010; 21:1974–81. https://doi.org/10.1093/annonc/mdq072.
11. Azim HA Jr, Michiels S, Bedard PL, Singhal SK, Criscitiello C, Ignatiadis M, Haibe-Kains B, Piccart MJ, Sotiriou C, Loi S. Elucidating prognosis and biology of breast cancer arising in young women using gene expression profiling. Clin Cancer Res. 2012; 18:1341–51. https://doi.org/10.1158/1078-0432.CCR-11-2599.
12. Loibl S, Jackisch C, Lederer B, Untch M, Paepke S, Kümmel S, Schneeweiss A, Huober J, Hilfrich J, Hanusch C, Gerber B, Eidtmann H, Denkert C, et al. Outcome after neoadjuvant chemotherapy in young breast cancer patients: a pooled analysis of individual patient data from eight prospectively randomized controlled trials. Breast Cancer Res Treat. 2015; 152:377–87. https://doi.org/10.1007/s10549-015-3479-z.
13. Cancello G, Maisonneuve P, Mazza M, Montagna E, Rotmensz N, Viale G, Pruneri G, Veronesi P, Luini A, Gentilini O, Goldhirsch A, Colleoni M. Pathological features and survival outcomes of very young patients with early breast cancer: how much is “very young”? Breast. 2013; 22:1046–51. https://doi.org/10.1016/j.breast.2013.08.006.
14. Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, Greenman CD, Jia M, Latimer C, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009; 462:1005–10. https://doi.org/10.1038/nature08645.
15. Network TC, and Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70. https://doi.org/10.1038/nature11412.
16. Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, Van Loo P, Ju YS, Smid M, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016; 534:47–54. https://doi.org/10.1038/nature17676.
17. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, et al, and Oslo Breast Cancer Consortium (OSBREAC). The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012; 486:400–04. https://doi.org/10.1038/nature11017.
18. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012; 486:405–09. https://doi.org/10.1038/nature11154.
19. Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004; 4:177–83. https://doi.org/10.1038/nrc1299.
20. Lánczky A, Nagy Á, Bottai G, Munkácsy G, Szabó A, Santarpia L, Győrffy B. miRpower: a web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res Treat. 2016; 160:439–46. https://doi.org/10.1007/s10549-016-4013-7.
21. Abbott KL, Nyre ET, Abrahante J, Ho YY, Isaksson Vogel R, Starr TK. The Candidate Cancer Gene Database: a database of cancer driver genes from forward genetic screens in mice. Nucleic Acids Res. 2015; 43:D844–48. https://doi.org/10.1093/nar/gku770.
22. Chen HJ, Wei Z, Sun J, Bhattacharya A, Savage DJ, Serda R, Mackeyev Y, Curley SA, Bu P, Wang L, Chen S, Cohen-Gould L, Huang E, et al. A recellularized human colon model identifies cancer driver genes. Nat Biotechnol. 2016; 34:845–51. https://doi.org/10.1038/nbt.3586.
23. Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003; 4:3. https://doi.org/10.1186/gb-2003-4-5-p3.
24. Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012; 12:801–17. https://doi.org/10.1038/nrc3399.
25. Chen J, Xu H, Aronow BJ, Jegga AG. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinformatics. 2007; 8:392. https://doi.org/10.1186/1471-2105-8-392.
26. Pena SD, Di Pietro G, Fuchshuber-Moraes M, Genro JP, Hutz MH, Kehdy Fde S, Kohlrausch F, Magno LA, Montenegro RC, Moraes MO, de Moraes ME, de Moraes MR, Ojopi EB, et al. The Genomic Ancestry of Individuals from Different Geographical Regions of Brazil Is More Uniform Than Expected. PLoS One. 2011; 6:e17063. https://doi.org/10.1371/journal.pone.0017063.
27. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012; 486:353–60. https://doi.org/10.1038/nature11143.
28. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al, and Australian Pancreatic Cancer Genome Initiative, and ICGC Breast Cancer Consortium, and ICGC MMML-Seq Consortium, and ICGC PedBrain. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–21. https://doi.org/10.1038/nature12477.
29. Guo Y, Updegraff BL, Park S, Durakoglugil D, Cruz VH, Maddux S, Hwang TH, O’Donnell KA. Comprehensive Ex Vivo Transposon Mutagenesis Identifies Genes That Promote Growth Factor Independence and Leukemogenesis. Cancer Res. 2016; 76:773–86. https://doi.org/10.1158/0008-5472.CAN-15-1697.
30. Cieply B, Farris J, Denvir J, Ford HL, Frisch SM. Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res. 2013; 73:6299–309. https://doi.org/10.1158/0008-5472.CAN-12-4082.
31. North WG, Gao G, Memoli VA, Pang RH, Lynch L. Breast cancer expresses functional NMDA receptors. Breast Cancer Res Treat. 2010; 122:307–14. https://doi.org/10.1007/s10549-009-0556-1.
32. Blank M, Tang Y, Yamashita M, Burkett SS, Cheng SY, Zhang YE. A tumor suppressor function of Smurf2 associated with controlling chromatin landscape and genome stability through RNF20. Nat Med. 2012; 18:227–34. https://doi.org/10.1038/nm.2596.
33. Chandhoke AS, Chanda A, Karve K, Deng L, Bonni S. The PIAS3-Smurf2 sumoylation pathway suppresses breast cancer organoid invasiveness. Oncotarget. 2017; 8:21001–14. https://doi.org/10.18632/oncotarget.15471.
34. Politopoulos I, Gibson J, Tapper W, Ennis S, Eccles D, Collins A. Composite likelihood-based meta-analysis of breast cancer association studies. J Hum Genet. 2011; 56:377–82. https://doi.org/10.1038/jhg.2011.23.
35. Abu-Jamous B, Buffa FM, Harris AL, Nandi AK. In vitro downregulated hypoxia transcriptome is associated with poor prognosis in breast cancer. Mol Cancer. 2017; 16:105. https://doi.org/10.1186/s12943-017-0673-0.
36. Phan NN, Wang CY, Chen CF, Sun Z, Lai MD, Lin YC. Voltage-gated calcium channels: novel targets for cancer therapy. Oncol Lett. 2017; 14:2059–74. https://doi.org/10.3892/ol.2017.6457.
37. Weinreb I, Piscuoglio S, Martelotto LG, Waggott D, Ng CK, Perez-Ordonez B, Harding NJ, Alfaro J, Chu KC, Viale A, Fusco N, da Cruz Paula A, Marchio C, et al. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat Genet. 2014; 46:1166–69. https://doi.org/10.1038/ng.3096.
38. Kim Y, Park EY, Chang E, Kang HG, Koo Y, Lee EJ, Ko JY, Kong HK, Chun KH, Park JH. A novel miR-34a target, protein kinase D1, stimulates cancer stemness and drug resistance through GSK3/β-catenin signaling in breast cancer. Oncotarget. 2016; 7:14791–802. https://doi.org/10.18632/oncotarget.7443.
39. {POLD}1 - {GENE} - {NCBI} []. 2017. https://www.ncbi.nlm.nih.gov/gene/5424.
40. Briggs S, Tomlinson I. Germline and somatic polymerase ε and δ mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol. 2013; 230:148–53. https://doi.org/10.1002/path.4185.
41. Tzeng ST, Tsai MH, Chen CL, Lee JX, Jao TM, Yu SL, Yen SJ, Yang YC. NDST4 Is a Novel Candidate Tumor Suppressor Gene at Chromosome 4q26 and Its Genetic Loss Predicts Adverse Prognosis in Colorectal Cancer. PLoS One. 2013; 8:e67040. https://doi.org/10.1371/journal.pone.0067040.
42. Peng HY, Yu QF, Shen W, Guo CM, Li Z, Zhou XY, Zhou NJ, Min WP, Gao D. Knockdown of ELMO3 Suppresses Growth, Invasion and Metastasis of Colorectal Cancer. Int J Mol Sci. 2016; 17:2119. https://doi.org/10.3390/ijms17122119.
43. Kadletz L, Heiduschka G, Wiebringhaus R, Gurnhofer E, Kotowski U, Haymerle G, Brunner M, Barry C, Kenner L. ELMO3 expression indicates a poor prognosis in head and neck squamous cell carcinoma - a short report. Cell Oncol (Dordr). 2017; 40:193–98. https://doi.org/10.1007/s13402-016-0310-8.
44. Fan W, Yang H, Xue H, Sun Y, Zhang J. ELMO3 is a novel biomarker for diagnosis and prognosis of non-small cell lung cancer. Int J Clin Exp Pathol. 2015; 8:5503–8. http://www.ncbi.nlm.nih.gov/pubmed/26191257.
45. Tedeschi PM, Scotto KW, Kerrigan J, Bertino JR. MTHFD2--a new twist? Oncotarget. 2016; 7:7368–9. https://doi.org/10.18632/oncotarget.7147.
46. Liu F, Liu Y, He C, Tao L, He X, Song H, Zhang G. Increased MTHFD2 expression is associated with poor prognosis in breast cancer. Tumour Biol. 2014; 35:8685–90. https://doi.org/10.1007/s13277-014-2111-x.
47. Moriarity BS, Otto GM, Rahrmann EP, Rathe SK, Wolf NK, Weg MT, Manlove LA, LaRue RS, Temiz NA, Molyneux SD, Choi K, Holly KJ, Sarver AL, et al. A Sleeping Beauty forward genetic screen identifies new genes and pathways driving osteosarcoma development and metastasis. Nat Genet. 2015; 47:615–24. https://doi.org/10.1038/ng.3293.
48. Chen D, Li Y, Wang L, Jiao K. SEMA6D Expression and Patient Survival in Breast Invasive Carcinoma. Int J Breast Cancer. 2015; 2015:539721. https://doi.org/10.1155/2015/539721.
49. Azim HA Jr, Nguyen B, Brohée S, Zoppoli G, Sotiriou C. Genomic aberrations in young and elderly breast cancer patients. BMC Med. 2015; 13:266. https://doi.org/10.1186/s12916-015-0504-3.
50. Di Leva G, Piovan C, Gasparini P, Ngankeu A, Taccioli C, Briskin D, Cheung DG, Bolon B, Anderlucci L, Alder H, Nuovo G, Li M, Iorio MV, et al. Estrogen Mediated-Activation of miR-191/425 Cluster Modulates Tumorigenicity of Breast Cancer Cells Depending on Estrogen Receptor Status. Clurman BE, editor. PLoS Genet. 2013; 9: e1003311. https://doi.org/10.1371/journal.pgen.1003311.
51. Bong IP, Ng CC, Fakiruddin SK, Lim MN, Zakaria Z. Small interfering RNA-mediated silencing of nicotinamide phosphoribosyltransferase (NAMPT) and lysosomal trafficking regulator (LYST) induce growth inhibition and apoptosis in human multiple myeloma cells: A preliminary study. Bosn J Basic Med Sci. 2016; 16:268–75. https://doi.org/10.17305/bjbms.2016.1568.
52. Smith TR, Liu-Mares W, Van Emburgh BO, Levine EA, Allen GO, Hill JW, Reis IM, Kresty LA, Pegram MD, Miller MS, Hu JJ. Genetic polymorphisms of multiple DNA repair pathways impact age at diagnosis and TP53 mutations in breast cancer. Carcinogenesis. 2011; 32:1354–60. https://doi.org/10.1093/carcin/bgr117.
53. Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, Allred DC, Bartlett JM, Bilous M, Fitzgibbons P, Hanna W, Jenkins RB, Mangu PB, et al, and American Society of Clinical Oncology, and College of American Pathologists. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013; 31:3997–4013. https://doi.org/10.1200/JCO.2013.50.9984.
54. Arnold N, Gross E, Schwarz-Boeger U, Pfisterer J, Jonat W, Kiechle M. A highly sensitive, fast, and economical technique for mutation analysis in hereditary breast and ovarian cancers. Hum Mutat. 1999; 14:333–39. https://doi.org/10.1002/(SICI)1098-1004(199910)14:4<333::AID-HUMU9>3.0.CO;2-C.
55. Friedman LS, Ostermeyer EA, Szabo CI, Dowd P, Lynch ED, Rowell SE, King MC. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat Genet. 1994; 8:399–404. https://doi.org/10.1038/ng1294-399.
56. Wagner TM, Hirtenlehner K, Shen P, Moeslinger R, Muhr D, Fleischmann E, Concin H, Doeller W, Haid A, Lang AH, Mayer P, Petru E, Ropp E, et al. Global sequence diversity of BRCA2: analysis of 71 breast cancer families and 95 control individuals of worldwide populations. Hum Mol Genet. 1999; 8:413–23. http://www.ncbi.nlm.nih.gov/pubmed/9971877 https://doi.org/10.1093/hmg/8.3.413.
57. Szabo C, Masiello A, Ryan JF, Brody LC. The breast cancer information core: database design, structure, and scope. Hum Mutat. 2000; 16:123–31. https://doi.org/10.1002/1098-1004(200008)16:2<123::AID-HUMU4>3.0.CO;2-Y.
58. Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011; 32:557–63. https://doi.org/10.1002/humu.21438.
59. Béroud C, Collod-Béroud G, Boileau C, Soussi T, Junien C. UMD (Universal mutation database): a generic software to build and analyze locus-specific databases. Hum Mutat. 2000; 15:86–94. https://doi.org/10.1002/(SICI)1098-1004(200001)15:1<86::AID-HUMU16>3.0.CO;2-4.
60. Caputo S, Benboudjema L, Sinilnikova O, Rouleau E, Béroud C, Lidereau R, and French BRCA GGC Consortium. Description and analysis of genetic variants in French hereditary breast and ovarian cancer families recorded in the UMD-BRCA1/BRCA2 databases. Nucleic Acids Res. 2012; 40:D992–1002. https://doi.org/10.1093/nar/gkr1160.
61. Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014; 42:D980–85. https://doi.org/10.1093/nar/gkt1113.
62. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010; 7:248–49. https://doi.org/10.1038/nmeth0410-248.
63. Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001; 11:863–74. https://doi.org/10.1101/gr.176601.
64. Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, de Silva D, Zharkikh A, Thomas A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006; 43:295–305. https://doi.org/10.1136/jmg.2005.033878.
65. Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006; 34:1317–25. https://doi.org/10.1093/nar/gkj518.
66. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the Functional Effect of Amino Acid Substitutions and Indels. de Brevern AG, editor. PLoS One. 2012; 7:e46688. https://doi.org/10.1371/journal.pone.0046688.
67. Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009; 37:e67–67. https://doi.org/10.1093/nar/gkp215.
68. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA, 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012; 491:56–65. https://doi.org/10.1038/nature11632.
69. Exome Aggregate Consortium. ExAC Browser, 2016. http://exac.broadinstitute.org/variant/9-139413097-T-G.
70. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, et al, and Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–91. https://doi.org/10.1038/nature19057.
71. PubChem. National Center for Biotechnology Information, U.S. National Library of Medicine, database, 2004: https://pubchem.ncbi.nlm.nih.gov/.
72. EVS. Exome Variant Server. NHLBI GO Exome Sequencing Project (ESP). 2014. http://evs.gs.washington.edu/EVS/.
73. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, and ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–24. https://doi.org/10.1038/gim.2015.30.
74. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–60. https://doi.org/10.1093/bioinformatics/btp324.
75. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–303. https://doi.org/10.1101/gr.107524.110.
76. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–79. https://doi.org/10.1093/bioinformatics/btp352.
77. Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, Ding L. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012; 28:311–17. https://doi.org/10.1093/bioinformatics/btr665.
78. Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, Stefancsik R, Harsha B, Kok CY, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017; 45:D777–83. https://doi.org/10.1093/nar/gkw1121.
79. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164–164. https://doi.org/10.1093/nar/gkq603.
80. Li H. Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics. 2011; 27:718–19. https://doi.org/10.1093/bioinformatics/btq671.
81. Fuentes Fajardo KV, Adams D, Mason CE, Sincan M, Tifft C, Toro C, Boerkoel CF, Gahl W, Markello T, and NISC Comparative Sequencing Program. Detecting false-positive signals in exome sequencing. Hum Mutat. 2012; 33:609–13. https://doi.org/10.1002/humu.22033.
82. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001; 29:308–11. http://www.ncbi.nlm.nih.gov/pubmed/11125122 https://doi.org/10.1093/nar/29.1.308.
83. Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, Min R, Alves P, Abyzov A, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012; 489:91–100. https://doi.org/10.1038/nature11245.
84. Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, Shiah YJ, Yousif F, Lin X, Masella AP, Fox NS, Xie M, Prokopec SD, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature. 2017; 541:359–64. https://doi.org/10.1038/nature20788.
85. Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012; 6:80–92. https://doi.org/10.4161/fly.19695.
86. P'ng C, Green J, Chong LC, Waggott D, Prokopec SD, Shamsi M, Nguyen F, Mak DYF, Lam F, Albuquerque MA, Wu Y, Jung EH, Starmans MHW, et al. BPG: Seamless, Automated and Interactive Visualization of Scientific Data. bioRxiv. 2017: 156067. https://doi.org/10.1101/156067.
87. Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, Day IN, Gaunt TR. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013; 34:57–65. https://doi.org/10.1002/humu.22225.
88. Douville C, Carter H, Kim R, Niknafs N, Diekhans M, Stenson PD, Cooper DN, Ryan M, Karchin R. CRAVAT: cancer-related analysis of variants toolkit. Bioinformatics. 2013; 29:647–48. https://doi.org/10.1093/bioinformatics/btt017.