
INTRODUCTION
The Wnt/Wingless signaling pathway plays important roles in the regulation of various biological processes including cell proliferation/migration/fate during embryogenesis and stem cell development/differentiation. Abnormal activation of Wnt signaling is closely associated with multiple human diseases, including malignancies [1, 2]. During canonical Wnt signaling, the protein levels and activities of its downstream effector-transcriptional coactivator β-catenin is finely regulated. With the help of a protein degradation complex containing scaffold protein Axin, APC, CK1 and GSK3β, cytoplasmic β-catenin is constantly degraded via the ubiquitin-proteasome system when no Wnt ligand is present. However, if Wnt ligands bind to cell surface receptors including Frizzled (Fz) protein and LDL receptor-related protein 6 (LRP6), a signal transduction cascade would be induced to stabilize and activate β-catenin protein. Cytosolic β-catenin will accumulate and translocate into the nucleus, and further interacts with TCF/LEF to bind to their target promoters including c-Myc, CCND1 and MMP7 via a HMG (High Mobility Group) domain [1, 2].
It has been well documented that SOX (SRY-related- HMG-box) family of proteins are critical transcription factors regulating canonical Wnt/β-catenin signaling in diverse development and disease processes. More than 20 SOX proteins have been identified and classified into seven groups (group A to H) according to their highly conserved HMG sequence similarities. During transcriptional regulation, SOX proteins have to cooperate with specific partner proteins in order to bind to gene promoters in a sequence-specific manner. The human SOX10 is located at 22q13.1 and is highly conserved in vertebrates [7]. SOX10 is expressed in many different cell types and tissues and implicated in neural crest development, nervous system neurogenesis, as well as differentiation of oligodendrocyte, glia and melanocytes [8-11].
Abnormalities (over- or under- expression, or genetic mutations) of SOX factors have been shown to play critical roles in human disease pathogenesis including cancer formation and development. Studies have shown that SOX2, SOX3, SOX4, SOX9 and SOX11 are upregulated and possess oncogenic functions in different types of cancers [12-16], while SOX1, SOX7, SOX11, SOX15 and SOX17 have been identified as tumor suppressors [17-21]. SOX10 was reported to possess tumor-promoting activities in several malignancies including melanoma [22] and gliomas [23]. On the other hand, decreased expression of SOX10 was found to promote tumor cell growth and focal adhesions of Merlin-null schwannoma cells [24]. Therefore, the expression and functional role of SOX10 in cancer development needs more detailed investigation.
We previously identified SOX10 as a methylated gene in our methylome analysis of digestive cancers [25, 26]. Here, we further analyzed its epigenetic alterations, functions and in-depth mechanisms in digestive cancers including colorectal, gastric and esophageal cancers. We found that SOX10 functions as a tumor suppressor by inducing tumor cell apoptosis, inhibiting invasion, regulating cell EMT and stemness through suppressing Wnt/β-catenin signaling.
RESULTS
Epigenetic identification of SOX10 as a methylated gene
Semiquantitative RT-PCR showed wide expression of SOX10 in a series of human normal adult and fetal tissues with variable expression levels, consistent with previous observations [27] (Figure 1A and 1B). In contrast, SOX10 expression was significantly reduced or completely silenced in multiple digestive tumor cell lines of different histological origins including colorectal, gastric and esophageal cancers, but rarely silenced in melanoma cell lines which acts as a positive control (Figure 1C and Supplementary Figure S1A and S1B). SOX10 was also found to be downregulated in multiple other carcinoma cell lines including nasopharyngeal, lung, and breast (data not shown). The results were further confirmed by two more primer pairs target different regions of SOX10. These data indicate that frequent downregulation of SOX10 is involved in multiple digestive tumorigenesis.
The SOX10 contains a typical CpG island, spanning the promoter, exon 1, intron 1 and part of exon 2 (Figure 1A). We thus further examined SOX10 promoter methylation by methylation-specific PCR (MSP) and found that SOX10 was frequently methylated in multiple cell lines, correlated with expression levels (Figure 1C).
To further investigate the relationship between promoter methylation and SOX10 expression, multiple cancer cell lines with decreased SOX10 mRNA were treated with DNA-demethylating agent Aza, alone or combined with trichostatin A, a histone deacetylase inhibitor. SOX10 mRNA was significantly induced in treated cancer cells (Figure 1D). Meanwhile, the SOX10 promoter was demethylated. Interestingly, the high level of SOX10 expression in melanoma cell lines is associated with lack of promoter methylation, except for the WM852 cell line (Supplementary FigureS1B). These results demonstrate that promoter methylation mediates transcriptional silencing of SOX10 in digestive cancers.
We also found that SOX10 could be activated in the colorectal cancer cell line HCT116 which is completely methylated for this gene, by genetic demethylation through only double knockout (KO) of both DNMT1 and DNMT3B (DKO cell line), but not single KO of DNMT1 or DNMT3B alone (1KO or 3BKO cell line) (Figure 1E). Concomitantly, unmethylated SOX10 promoter alleles were detected in Aza-treated HCT116 and DKO cells, but not in DNMT1 or DNMT3B single KO cells, indicating that methylation directly mediates SOX10 silencing. These results also suggest that the maintenance of SOX10 methylation is mediated by DNMT1 and DNMT3B together.
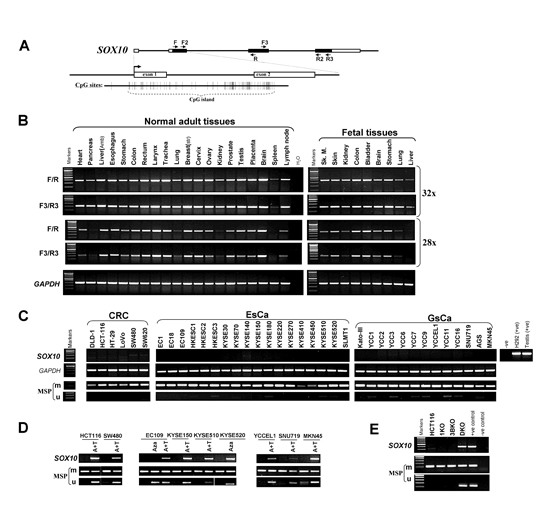
Figure 1: SOX10 is frequently silenced by promoter CpG methylation in multiple carcinomas. (A) Schematic structure of the SOX10 CpG island. Exon 1,2 (indicated with a black rectangle), CpG sites (short vertical lines). (B) SOX10 is broadly expressed in human normal adult tissues and fetal tissues. (C) SOX10 is frequently silenced and methylated in colorectal, esophageal and gastric carcinoma cell lines. Ca, carcinoma; CRC, colorectal cancer; ESCC, esophageal carcinoma; M, methylated; U, unmethylated. (D) Pharmacologic demethylation with Aza alone or combined with TSA (A+T), or (E) genetic demthylation in DKO cell line restored SOX10 expression in methylated/silenced tumor cell lines.
SOX10 expression and methylation in primary digestive tumors
We next performed immunohistochemistry (IHC) analyses to examine SOX10 expression in multiple human tissues using a monoclonal antibody described previously [22]. SOX10 expression was first examined in 16 melanomas in a tissue microarray. Clearly, strong nuclear immunostaining of SOX10 was found in all melanoma cases, while the negative control treated identically with a normal mouse IgG antibody showing no staining (Figure 2A), in agreement with previous report [22].
We next analyzed SOX10 expression using a human normal tissue microarray carrying 24 different tissue types. SOX10 was normally expressed in colon, esophagus, stomach, brain, prostate, pancreas, heart and testis and, to a lesser extent, lung tissue, in good conformity with RNA levels (Figure 2A and Supplementary Figure S2A).
SOX10 expression was further detected by IHC in multiple primary carcinomas and paired adjacent non-tumor tissue from the same patients. We found that SOX10 protein was normally expressed in paired non-cancerous colorectal, gastric and esophageal tissues, whereas it was hardly or weakly detected in tumor tissues (Figure 2B). SOX10 expression was decreased or completely silenced in multiple primary digestive cancers with variable frequencies. Downregulation of SOX10 was frequently detected in 51% (20 of 39) of colorectal, 68% (23 of 34) of gastric and 51% (20 of 39) of esophageal cancer tissues.
We further evaluated SOX10 promoter methylation in primary digestive cancers. Methylation was detected in multiple tumors, including 64% (7/11) of CRC, 77% (40/52) of gastric and 76% (13/17) of ESCC samples (Supplementary Figure S2B). These results demonstrate that SOX10 silencing by promoter methylation is a frequent event in digestive tumorigenesis.
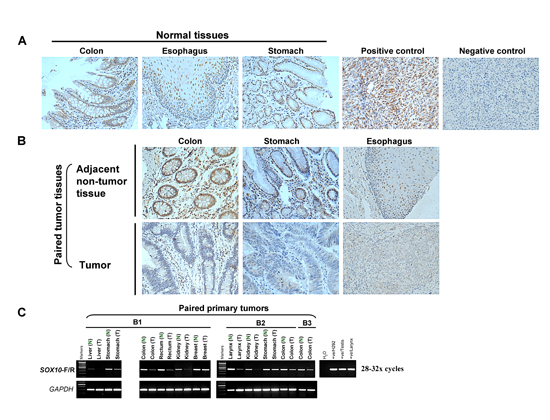
Figure 2: SOX10 expression is significantly decreased in tumor tissues. (A) Immunohistochemistry analysis (IHC) was performed with an anti-SOX10 antibody on a human normal tissue microarray (TMA). Melanoma tissue was used as a positive control and nonimmune mouse immunoglobulin G substituted for the primary antibody as negative control (original magnification ×400). (B) SOX10 expression is significantly decreased in different tumor tissues. (C) mRNA expression levels of SOX10 in different tumor tissues (T) and their paired adjacent normal tissues (N) as determined by semiquantitative RT-PCR (28 or 32 cycles).
SOX10 inhibits digestive tumor cell survival
The observations described above have implicated SOX10 as a tumor suppressor in digestive cancers. We thus explored whether tumor cells are functionally dependent on SOX10 inactivation. HCT116 (colon), KYSE150 (esophageal) and AGS (gastric) cell lines, all with fully methylated and completely silenced SOX10, were infected with lentivirus vectors encoding SOX10 or control GFP. SOX10 proteins were found localized predominantly in the nucleus (Figure 3A). Ectopic expression of SOX10 in these cells significantly suppressed cell growth, colony formation and anchorage-independent growth (Figure 3B-3D). To further confirmed the inhibitory effects of SOX10 on tumor growth in vivo, HCT116 and KYSE150 cells infected with LV-SOX10 or LV-GFP were implanted subcutaneously into the flank of nude mice. Compared to LV-GFP control group, expression of SOX10 significantly inhibited tumor growth in nude mice (Figure 3E). Together, these data demonstrate that SOX10 functions as a tumor suppressor for digestive cancers.
The inhibitory effects of SOX10 on tumor growth were likely the result of apoptosis induction as revealed by TUNEL assay of HCT116 and KYSE150 xenografted tumor tissues. When elevating the level of SOX10, the apoptotic nuclei, seen as green color excited under fluorescence microscope, were greatly increased (Figure 3F).
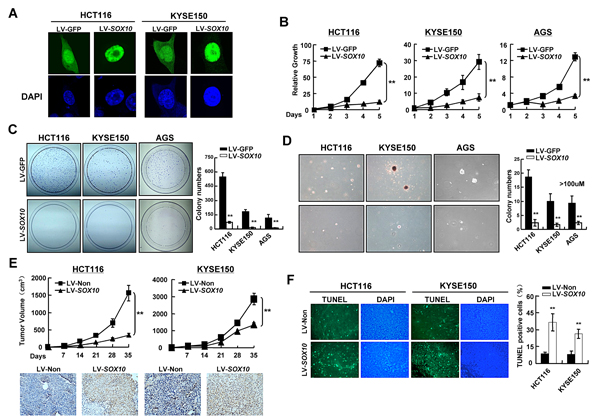
Figure 3: SOX10 represses tumor cell survival. (A) SOX10 (green) was primarily located in the nucleus in SOX10-transfeced cells by immunostaining. DAPI counterstaining (blue) was used to visualize DNA. (B) Cells were infected with LV-SOX10 or LV-GFP and seeded in 96-well plates for MTS assay. Values are mean ± SD for triplicate samples from a representative experiment. *: p<0.05. **: p<0.01. (C) Representative colony formation assays. (D) Representative anchorage-independent growth. (E) Cells infected with LV-SOX10 or LV-Non were injected subcutaneously into nude mice. Tumor volume of each group was scored every 7 days (Upper). The expresson of SOX10 in the tumors was confirmed by IHC staining (lower). (F) In situ TUNEL apoptosis analysis was performed in tumor sections derived from the same mice as in Figure 3E. The apoptotic nuclei were seen as green color excited under fluorescence microscopy. (magnification ×400).
SOX10 represses tumor cell metastasis
The effect of SOX10 on tumor cell migration and invasion was further explored. HCT116, KYSE150 and AGS cells infected with LV-SOX10 filled the wound much slower than LV-GFP infected cells (Figure 4A). Induction of SOX10 also markedly inhibited cell migration through a permeable filter and invasion through Matrigel (Figure 4B and 4C). Subsequently, we performed two sets of metastasis assays in nude mice. As shown in Figure 4D, lung metastatic clusters presented in mice subcutaneously injected with KYSE150-SOX10 cells (16.6±4.8 clusters per mice) were significantly fewer than those in the LV-GFP group (38.8±6.8 clusters per mice), as examined by HE staining. Notably, overexpression of SOX10 suppressed the establishment of colorectal cancer liver metastasis (Figure 4E). Collectively, these observations indicate that SOX10 negatively regulates metastatic properties of digestive cancer cells.
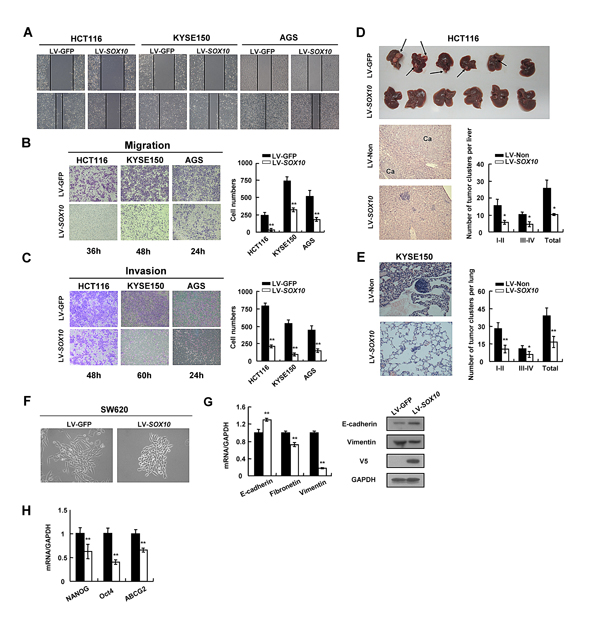
Figure 4 : SOX10 suppresses tumor metastasis and EMT phenotype. Cells were infected with LV-SOX10 or LV-GFP and subjected to wound-healing assay (A), migration assay (B) and invasion assay (C). Microscopic observations were recorded at 0 as well as 48 or 72 hours after scratching the surface of a confluent layer of cells(A). Cells that migrated (B) or invaded (C) to the lower chamber were fixed, stained, and counted using light microscopy. Values are mean ± SD for triplicate samples from a representative experiment. *: p<0.05. **: p<0.01. (D) HCT116 cells infected with LV-SOX10 or LV-GFP were inoculated into the spleen. The mice were killed and examined for the presence of hepatic metastasis 12 weeks after the intrasplenic inoculation. The upper, metastatic nodules on the surface of the liver are shown; the left, representative H&E staining of liver (original magnification, ×400); the right, the numbers of nodules were quantified and values for each group are denoted (*: p<0.05). (E) Male Balb/c nude mice were injected subcutaneously with 5x106 KYSE150 cells. Representative lung tissue sections by HE-staining from each group were shown in the left (magnification ×400). The number of lung metastatic foci in each group was calculated under microscope. (F) Morphology changes of SW620 cells infected with LV-SOX10 or empty vector by phase contrast microscopy. Original magnification, ×400. (G) qRT-PCR and western blot validation of EMT biomarkers (*: p<0.05, **: p<0.01). GAPDH was used as a control. (H) Downregulation of representative stem cell markers in SOX10-infected tumor cells.
SOX10 suppresses the EMT phenotype and stem cell property of tumor cells
Given that SOX10 inhibits cancer metastasis, we wonder whether SOX10 plays a role in regulating epithelial-mesenchymal transition (EMT), a critical event in tumor invasion [28, 29]. To test this hypothesis, we selected the SW620 colon cancer cell line which is derived from a lymph node metastasis of a colorectal tumor. SW620 cells infected with LV-GFP showed a prominent mesenchymal-like phenotype, including a fibroblast-like morphology, loss of cell-cell contacts, expression of the mesenchymal marker vimentin as described previously [30, 31]. However, ectopic expression of SOX10 in SW620 cells led to cobblestone morphology in monolayer cultures, with tight cell-cell contacts characteristics of normal epithelial cells (Figure 4F), indicating that SOX10 most likely reversed tumor cell EMT. This was accompanied by an increase of epithelial cell marker, such as E-cadherin, a decrease of mesenchymal cell markers, such as fibronectin and vimentin and an increased migration ability (Figure 4G and Supplementary Figure S3A). Furthermore, SOX10 expression downregulated 3 stem cell markers: NANOG, ABCG2 and Oct4, at the transcriptional level (Figure 4H). Taken together, these results suggest that SOX10 inhibits both the EMT and stemness of digestive cancer cells.
SOX10 inhibits the β-catenin signaling pathway in digestive tumor cells
Previous studies have shown that SOX family members play important roles in modulating Wnt/β-catenin pathway in diverse development and disease contexts [3]. To examine whether the above observed SOX10-dependent effects were mediated by β-catenin, we performed reporter assays using both the TOPFlash construct harboring multiple TCF/LEF-binding sites and the derived FOPFlash construct with mutated TCF/LEF-binding sites as a negative control. Overexpression of SOX10 repressed the TOPFlash activity in a dose-dependent manner in KYSE150 cells, while they had no effect on the FOPFlash control (Figure 5A). The impact of SOX10 overexpression on β-catenin/TCF activity was further examined in HCT116 colon cancer cells which contain a heterozygous activating mutation in β-catenin [20]. SOX10 inhibits β-catenin promoter activities in a dose-dependent manner (Figure 5A). Similarly, increasing the amount of exogenous SOX10 progressively repressed the activities of CCND1, c-Myc and MMP7 promoter constructs as well as their expression in HCT116 cells (Figure 5B and 5C), which are typical transcriptional targets of the Wnt/β-catenin/TCF signaling pathway. Collectively, these data suggest that SOX10 can antagonize the canonical Wnt/β-catenin signaling cascade in digestive cancer cells.
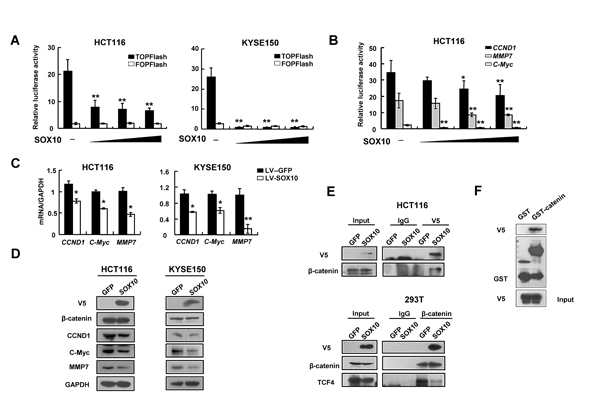
Figure 5: SOX10 suppresses Wnt signaling via compete with TCF4 to bind β-catenin. Inhibition of TCF activity (A) and CCND1, c-Myc and MMP7 promoter reporter activities (B) in SOX10-expressing tumor cells in a dose-dependent manner. *: p<0.05, **: p<0.01. qRT-PCR (C) and western blot (D) showed ectopic expression of SOX10 disrupted Wnt signaling pathway. (E) Co-immunoprecipitation (Co-IP) assays showing that exogenous V5-SOX10 binds endogenous β-catenin and vice versa. IP antibody or IgG negative control was used as indicated. (F) GST pull-down assay was performed with GST-β-catenin fusion protein or GST and V5-SOX10 as indicated.
SOX10 competes with TCF4 for interacting with β-catenin
SOX proteins regulate β-catenin/TCF activity through different mechanisms including protein-protein interactions [3]. To test whether SOX10 could interact with β-catenin, we performed co-immunoprecipitation (co-IP) analyses using cell lysates from HCT116 cells transfected with V5-tagged SOX10, and found that endogenous β-catenin could bind to SOX10 and vice versa (Figure 5D and 5E). Consistent with the findings from in vitro binding assays, V5-tagged SOX10 was able to pull down GST-tagged β-catenin (GST-β-catenin) but not GST alone, suggesting that SOX10 directly interact with β-catenin (Figure 5F).
In order to regulate gene expression, β-catenin has to interact with TCF4, then is recruited to target promoters via TCF4’s HMG domain. As SOX10 also contains a HMG domain, we tried to determine whether SOX10 and TCF4 were present in the same complex with β-catenin or compete with each other for β-catenin binding. Using anti-β-catenin antibody to pull down endogenous β-catenin in 293T cells, we detected endogenous TCF4 coprecipitated with β-catenin in the GFP group, but virtually not in the SOX10-overexpressed group, indicating that SOX10 is directly competing with TCF4 for β-catenin binding (Figure 5E).
Interaction of SOX10 with β-catenin and its DNA binding ability are required for tumor suppression
Many SOX proteins have been shown to physically interact with β-catenin through a short 9 amino acid motif (DxxEFDQYL), which shows a high degree of similarity to a loosely conserved armadillo-binding sequence found in many β-catenin-binding proteins [3, 32]. Analysis of the amino acid sequence of SOX10 revealed a match with the motif DVAELDQYL which is evolutionarily conserved (Figure 6A). To test if this conserved motif is essential for protein interaction, we mutated the conserved amino acids DQY to GGG (referred to as 3G mutant) or deleted amino acids ELDQY (referred to as DB mutant) in SOX10 (Figure 6A). Co-IP showed that endogenous β-catenin binds to V5-tagged wild-type SOX10, but not 3G or DB mutant (Figure 6B), suggesting that DVAELDQYL motif is indispensable for direct β-catenin binding.
SOX10 is a transcription factor binding DNA sequence via a HMG domain. As our above data indicated that SOX10 competed with TCF4 to bind β-catenin, we next investigated whether its DNA-binding activity was required to antagonize β-catenin/TCF activity. We generated the SOX10-482ins6 mutant, which carries an insertion of 6 nucleotides between positions 482 and 483, resulting in the addition of a leucine and an arginine residue into the HMG box and disruption of the structure of SOX10 DNA-binding domain [33] (Figure 6A). This mutant maintained the same ability to interact with β-catenin but failed to repress the β-catenin/TCF reporter (Figure 6B and 6C), indicating that the repressive effect of SOX10 on β-catenin/TCF transcriptional activity depends on its DNA-binding property.
To further confirm that SOX10 was recruited to Wnt/β-catenin target gene promoters and transrepressed their expression via HMG domain instead of interacting with β-catenin and sequestering it away from target promoters, we performed a ChIP assay on c-Myc promoter. As expected, ChIP analysis demonstrated that SOX10 was recruited to c-Myc promoter in HCT116 cells, while the SOX10-DB mutant or SOX10-482ins6 failed to bind to c-Myc promoter (Figure 6E), indicating that SOX10-mediated repression of Wnt/β-catenin target genes depends on its interaction with β-catenin as well as its own DNA-binding property.
Cumulative outcomes from our above studies supported a key role of β-catenin inhibition in SOX10-induced tumor suppression of digestive tumor cells. Therefore, we next questioned whether the capacity to bind β-catenin is instrumental for SOX10 function. We first examined whether SOX10 DB or 3G mutants lose their ability to suppress Wnt/β-catenin signaling. Indeed, while ectopic SOX10 expression in HCT116 cells significantly inhibited the TOPFlash reporter as compared to control cells, transfection with SOX10-DB or SOX10-3G mutants largely reversed the suppressive effect on the reporter (Figure 6C). qRT-PCR and western blot analysis confirmed that mutations or deletion of the conserved DVAELDQYL motif as well as HMG domain mutation impaired the ability of SOX10 to attenuate Wnt signaling (Figure 6D). Furthermore, tumor cells with ectopic SOX10 expression migrated significantly slower than control cells, whereas introduction of SOX10-DB, -3G or -482ins6 mutants only partially suppressed tumor cell migration as by transwell assays (Figure 6F). These results further suggest that the interaction of SOX10 with β-catenin as well as its DNA binding ability are, at least in part, required for its tumor suppressor functions.
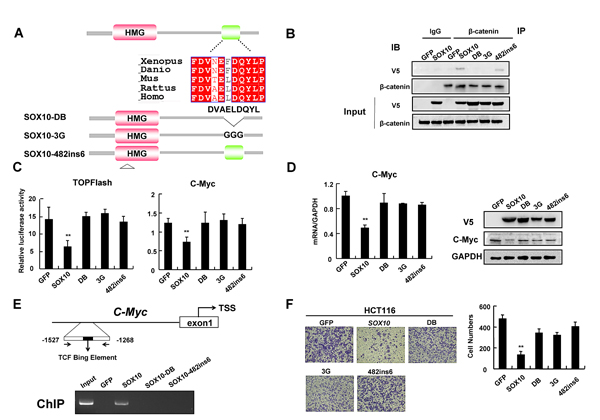
Figure 6: Interaction of SOX10 with β-catenin and its DNA binding ability are required for tumor suppression. (A) The schematic shows that the SOX10 contains a candidate β-catenin-binding region which is highly conserved. Below the schematic the ‘DB’, ‘3G’ and ‘482ins6’ mutations generated in SOX10 are shown. (B) IP assays showing that endogenous β-catenin binds exogenous V5-SOX10 and 482ins6 mutants, but not DB or 3G mutants. (C) HCT116 cells were cotransfected with V5-SOX10 or mutants plus the TOPFlash or c-Myc promoter reporter. Values are mean ± SD for triplicate samples from a representative experiment. *: p<0.05, **: p<0.01. (D) qRT-PCR and western blot showed ectopic expression of GFP, SOX10 and mutants on Wnt/β-catenin signaling pathway. (E) Schematic structure of the c-Myc promoter. TCF Binding Element (TBE) are indicated (Upper). ChIP assays on Wnt-responsive elements of c-Myc promoter gene were performed in HCT116 cells that were transfected with GFP, V5-SOX10 and mutants (lower). (F) Migration assay testing the ability of HCT116 cells expressing GFP, SOX10 and mutants to migrate through transwell chambers.
DISCUSSION
In this study, we found that while SOX10 is ubiquitously expressed in human normal adult and fetal tissues, it is frequently silenced or downregulated due to promoter methylation in digestive (colon, gastric and esophageal) cancer cell lines and primary tumors. We found that ectopic expression of SOX10 suppressed tumor cell proliferation and tumorigenicity through promoting apoptosis in vitro and in nude mice. Furthermore, we found that SOX10 inhibited the EMT and stemness of tumor cells and their migration and invasion through antagonizing Wnt/β-catenin signaling. Our study thus validated that SOX10 is a functional tumor suppressor for digestive cancers.
The canonical Wnt signaling pathway regulates multiple fundamental cellular processes including cell proliferation, migration and stemness as well as tumorigenesis, largely through modulating target gene transcription [1, 2]. Mutations in Wnt pathway components such as APC or stabilizing mutations in β-catenin itself that lead to constitutive signaling are frequently found in digestive cancers [34, 35]. Epigenetic downregulation of tumor suppressor genes, especially in the setting of key signaling pathways, have been proven to be a complementary mechanism to genetic mutation during carcinogenesis [36]. Some important Wnt signaling inhibitors such as SFRPs [37], WIF1 [38], DKKs [39] as well as SOX1, SOX7,SOX15 and SOX17 have been previously reported to be frequently methylated in multiple tumors. We reported here that SOX10, another SOX family member, functions as a TSG but is silenced by promoter CpG methylation in multiple digestive cancers. In melanomas, upregulation of SOX10 correlates well with promoter hypomethylation. These data suggest promoter methylation could be the major mechanism determining whether SOX10 is silenced or activated in tumors.
SOX familiy members regulate canonical Wnt signaling through different mechanisms, including protein-protein interactions, binding of SOX factors to Wnt-target gene promoters, the recruitment of co-repressors or co-activators, modulation of protein stability, and nuclear translocation [3]. Many SOX proteins can physically interact with β-catenin via a conserved 9 amino acid motif (DXXEFDQYL), for example, SOX7 and SOX17 [3]. Sequence analysis indicate that SOX10 also contains a similar highly conserved motif, DVAELDQYL, which is essential to the intercation of SOX10 with β-catenin, also to the abilities of SOX10 to bind to target promoters and exert its transcriptional regulator and tumor suppressor functions.
We used digestive cancer cell lines with mutant (HCT116 [40] and AGS [41] ) or wild-type β-catenin (KYSE150), all of which have complete silenced expression of SOX10. Ectopic expression of SOX10 could effectively suppress tumor proliferation and metastasis in cell lines harboring wild-type as well as mutant β-catenin. Consistent with this, SOX10 could interact with mutant β-catenin and repress target gene transcription. These data suggest that SOX10 functions as a tumor suppressor probably independent of β-catenin mutation and is another checkpoint for aberrant β-catenin activation in cancers.
As no DNA binding domain existed in β-catenin, other DNA binding partners are needed to assist β-catenin to its target promoters. Here, we discovered that SOX10 could compete with TCF4 to form a complex with β-catenin and be recruited to target promoters to suppress their expression. This transrepresson ability is dependent on the DNA-binding activity of SOX10’s HMG domain, because a SOX10 mutant that is incapable of DNA binding can still interact with β-catenin, but fails to suppress the TOPFlash reporter as well as Wnt-target gene expression. Together, these findings demonstrated the unexpected role of SOX10 as an important DNA binding transcription factor that recruits β-catenin to repress Wnt/β-catenin signaling in cancer progression.
Since disrupting SOX10-β-catenin interaction could only partially abolished tumor suppressor functions of SOX10, there may be other signaling pathways participated in the complex mechanisms of tumor suppression by SOX10. This issue remains to be explored in further depth in future.
Although emerging evidences together with ours support that SOX10 functions as a tumor suppressor in certain tumors, overexpression and oncogenic property of SOX10 have been reported in melanoma and glioma, indicating that SOX10 plays a complex role in tumorigenesis. The SOX family of transcription factors regulate a wide range of biological events by cooperating with specific partner factors to select specific target genes. When a SOX-partner is changed, the set of activated genes might be dramatically altered. For example, during Schwann cell development, SOX10 cooperates with OCT6/BRN2 to upregulate the Krox20 gene. However, during melanocyte development, SOX10 is partnered with PAX3 and activates the gene encoding MITF [5, 42]. In melanoma, SOX10 haploinsufficiency abrogates NrasQ61K-driven tumor initiation as well as progression [22]. SOX10 increases KROX20 and myelin protein zero (MPZ) expression, and promotes NFATC4 nucleus translocation, further suppressing Merlin-null cell proliferation [24]. These results suggested that SOX10 function in a context- and likely cell-type dependent manner. The molecular mechanisms and biological functions of SOX10 in other tumors need to be determined in future studies.
In summary, our study identifies SOX10 as a functional tumor suppressor and an important regulator of the Wnt/β-catenin signaling pathway, with frequent epigenetic inactivation in certain digestive tumor. These findings improves our understanding of the molecular mechanisms underlying β-catenin activation and tumor progression and provide a potential target for cancer therapy.
MATERIALS and METHODS
Cell lines and culture condition
Tumor cell lines were obtained from American Type Culture Collection. Colon cancer cell line HCT116 with genetic knock-out (KO) of DNA methyltransferases (DNMT): HCT116 DNMT1-/- (1KO), HCT116 DNMT3B-/- (3BKO) and HCT116 DNMT1-/- DNMT3B-/- (DKO) cells (gifts of Bert Vogelstein, Johns Hopkins University Medical Institutes) were grown with either 0.4 mg/ml genecitin or 0.05 mg/ml hygromycin or both.
Tissue samples and immunohistochemistry (IHC)
Normal adult and fetal tissue RNA samples were purchased commercially (Stratagene, La Jolla, CA, USA or Millipore-Chemicon, Billerica, MA,). DNA samples of primary carcinomas (T) and matched surgical margin normal tissues (N) have been described previously. For IHC staning, human normal tissue TMA composed of 24 different tissue types (Superchip Co., Ltd., Shanghai, China) and Human melanoma tissue TMA (US Biomax, Rockville, MD) was utilized. Paired tumor tissues were collected in Luoyang 150 Hospital and Chinese PLA General Hospital in China. Tissue samples were collected with informed consent and the procedure was approved by the hospital ethics committees. Further details can be found in the Supplementary information.
DNA bisulfite treatment, methylation analysis, 5-Aza-2’-deoxycytidine(Aza) and trichostatin A treatment
See Supplementary information.
Plasmid construction and lentivirus production
See Supplementary information.
qRT-PCR, semi-quantitative RT–PCR , western blot and immunofluorescence staining, dual-luciferase reporter assay
See Supplementary information.
MTS Assay, colony formation assay, anchorage-independent growth assay, TUNEL Assay, wound-Healing, transwell migration and invasion assay
See Supplementary information.
Glutathione-S-transferase pull-Down assay
Glutathione-S-transferase (GST) and GST-tagged β-catenin were expressed in Escherichia coli and purified by glutathione-sepharose beads (GE Healthcare). We incubated SOX10 protein extracted from 293T cells transfected with pcSOX10 with GST or the GST-catenin for 1 hour at 4°C. After washing, we resolved the adsorbed proteins by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and analyzed them by western blotting. Further details are available in the Supplementary information.
Co-immunoprecipitation (IP)
Whole-cell extracts of HCT116 or 293T cells expressing V5-SOX10 or its mutants were immunoprecipitated with anti-V5 or anti-β-catenin, followed by western blot analysis using anti-V5, anti-β-catenin, or anti-TCF4 antibodies. Further details are available in the Supplementary information.
ChIP assay
ChIP was performed using the EZ ChIP™ Chromatin Immunoprecipitation Kit (Millipore, Billerica, MA) according to the manufacturer’s protocol. Sonicated chromatin was immunoprecipitated with anti-V5 antibody and normal mouse IgG as the negative controls. Immunoprecipitated DNA was then reverse cross-linked, purified and subjected to PCR. Primers used in this study are listed in Supplementary information, Table S1.
Animal studies
Male BALB/c nude mice at six week-old were housed under standard conditions and cared for according to the institutional guidelines for animal care. All animal experiments were approved by Institutional Animal Care and Use Committee (IACUC) of PLA General Hospital. Further details can be found in the Supplementary information.
Statistic analysis
The analyses were carried out using SPSS 16.0 for Windows software (SPSS, Chicago, IL, USA). Results are shown as values of mean ± SD. Statistical analyses were performed using Student’s t test to determine P values.
Competing interest statement
The authors declare that they have no competing financial interests.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (81172582, 81330061 and 81000941), HK RGC (#475009), Group Research Schemes of CUHK and Shandong Provincial Natural Science Foundation (ZR2013HZ002).
Abbreviations
APC, adenomatous polyposis coli; ChIP, chromatin-immunoprecipitation; CK1, casein kinase 1; CRC, colorectal cancer; EMT, epithelial-mesenchymal transition; DAPI, 4′,6-diamidino-2-phenylindol; ESCC, esophageal cancer; GSK3β, glycogen synthase kinase 3β; GST, Glutathione-S-transferase; IF, Immunofluorescence; IHC, Immunohistochemisty; IP, Immunoprecipitation; qRT-PCR, quantitative reverse-transcription polymerase chain reaction; TCF/LEF, T cell factor/lymphoid enhancer factors; TMA, tissue microarray; TSG, tumor suppressor gene; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
REFERENCES
1. Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer 2008;8:387-98.
2. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell 2012;149:1192-205.
3. Kormish JD, Sinner D, Zorn AM. Interactions Between SOX Factors and Wnt/beta-Catenin Signaling in Development and Disease. Developmental Dynamics 2010;239:56-68.
4. Kiefer JC. Back to basics: Sox genes. Developmental Dynamics 2007;236:2356-2366.
5. Koopman P. HMG Domain Superfamily of DNA-bending Proteins: HMG, UBF, TCF, LEF, SOX, SRY and Related Proteins.
6. Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell 2013;12:15-30.
7. Pusch C, Hustert E, Pfeifer D, Sudbeck P, Kist R, Roe B, Wang Z, Balling R, Blin N, Scherer G. The SOX10/Sox10 gene from human and mouse: sequence, expression, and transactivation by the encoded HMG domain transcription factor. Hum Genet 1998;103:115-23.
8. Hong CS, Saint-Jeannet JP. Sox proteins and neural crest development. Semin Cell Dev Biol 2005;16:694-703.
9. Harris ML, Baxter LL, Loftus SK, Pavan WJ. Sox proteins in melanocyte development and melanoma. Pigment Cell Melanoma Res 2010;23:496-513.
10. Haldin CE, LaBonne C. SoxE factors as multifunctional neural crest regulatory factors. International Journal of Biochemistry & Cell Biology 2010;42:441-444.
11. Stolt CC, Wegner M. SoxE function in vertebrate nervous system development. International Journal of Biochemistry & Cell Biology 2010;42:437-440.
12. Chen YP, Shi L, Zhang LR, Li RF, Liang J, Yu WH, Sun LY, Yang XH, Wang Y, Zhang Y, Shang YF. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. Journal of Biological Chemistry 2008;283:17969-17978.
13. Liu PB, Ramachandran S, Ali Seyed M, Scharer CD, Laycock N, Dalton WB, Williams H, Karanam S, Datta MW, Jaye DL, Moreno CS. Sex-determining region Y box 4 is a transforming oncogene in human prostate cancer cells. Cancer Research 2006;66:4011-4019.
14. Weigle B, Ebner R, Temme A, Schwind S, Schmitz M, Kiessling A, Rieger MA, Schackert G, Schackert HK, Rieber EP. Highly specific overexpression of the transcription factor SOX11 in human malignant gliomas. Oncology Reports 2005;13:139-144.
15. Li K, Wang RW, Jiang YG, Zou YB, Guo W. Overexpression of Sox3 is Associated with Diminished Prognosis in Esophageal Squamous Cell Carcinoma. Annals of Surgical Oncology 2013;20:S459-S466.
16. Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JP, Pan FC, Akiyama H, Wright CVE, Jensen K, Hebrok M, Sander M. Identification of Sox9-Dependent Acinar-to-Ductal Reprogramming as the Principal Mechanism for Initiation of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012;22:737-750.
17. Thu KL, Radulovich N, Becker-Santos DD, Pikor LA, Pusic A, Lockwood WW, Lam WL, Tsao MS. SOX15 is a candidate tumor suppressor in pancreatic cancer with a potential role in Wnt/beta-catenin signaling. Oncogene 2014;33:279-288.
18. Gustavsson E, Sernbo S, Andersson E, Brennan DJ, Dictor M, Jerkeman M, Borrebaeck CAK, Ek S. SOX11 expression correlates to promoter methylation and regulates tumor growth in hematopoietic malignancies. Molecular Cancer 2010;9.
19. Tsao CM, Yan MD, Shih YL, Yu PN, Kuo CC, Lin WC, Li HJ, Lin YW. SOX1 Functions as a Tumor Suppressor by Antagonizing the WNT/beta-Catenin Signaling Pathway in Hepatocellular Carcinoma. Hepatology 2012;56:2277-2287.
20. Zhang W, Glockner SC, Guo MZ, Machida EO, Wang DH, Easwaran H, Van Neste L, Herman JG, Schuebel KE, Watkins DN, Ahuja N, Baylin SB. Epigenetic inactivation of the canonical Wnt antagonist SRY-Box containing gene 17 in colorectal cancer. Cancer Research 2008;68:2764-2772.
21. Guo LH, Zhong DS, Lau S, Liu XJ, Dong XY, Sun XD, Yang VW, Vertino PM, Moreno CS, Varma V, Dong JT, Zhou W. Sox7 is an independent checkpoint for beta-catenin function in prostate and colon epithelial cells. Molecular Cancer Research 2008;6:1421-1430.
22. Shakhova O, Zingg D, Schaefer SM, Hari L, Civenni G, Blunschi J, Claudinot S, Okoniewski M, Beermann F, Mihic-Probst D, Moch H, Wegner M, Dummer R, et al. Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nat Cell Biol 2012;14:882-90.
23. Ferletta M, Uhrbom L, Olosson T, Ponten F, Westermark B. Sox10 has a broad expression pattern in gliomas and enhances platelet-derived growth factor-B-induced gliomagenesis. Molecular Cancer Research 2007;5:891-897.
24. Doddrell RDS, Dun XP, Shivane A, Feltri ML, Wrabetz L, Wegner M, Sock E, Hanemann CO, Parkinson DB. Loss of SOX10 function contributes to the phenotype of human Merlin-null schwannoma cells. Brain 2013;136:549-563.
25. Wang JW, Xia YD, Li LL, Gong DS, Yao Y, Luo HJ, Lu HL, Yi N, Wu HL, Zhang XQ, Tao Q, Gao F. Double restriction-enzyme digestion improves the coverage and accuracy of genome-wide CpG methylation profiling by reduced representation bisulfite sequencing. Bmc Genomics 2013;14.
26. Ying JM, Li HY, Yu J, Ng KM, Poon FF, Wong SCC, Chan ATC, Sung JJY, Tao Q. WNT5A exhibits tumor-suppressive activity through antagonizing the Wnt/beta-catenin signaling, and is frequently methylated in colorectal cancer. Clinical Cancer Research 2008;14:55-61.
27. Bondurand N, Kobetz A, Pingault V, Lemort N, Encha-Razavi F, Couly G, Goerich DE, Wegner M, Abitbol M, Goossens M. Expression of the SOX10 gene during human development. Febs Letters 1998;432:168-172.
28. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell 2011;144:646-674.
29. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009;139:871-890.
30. Buck E, Eyzaguirre A, Barr S, Thompson S, Sennello R, Young D, Iwata KK, Gibson NW, Cagnoni P, Haley JD. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Molecular Cancer Therapeutics 2007;6:532-541.
31. Dhawan P, Singh AB, Deane NG, No Y, Shiou SR, Schmidt C, Neff J, Washington MK, Beauchamp RD. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. Journal of Clinical Investigation 2005;115:1765-1776.
32. Sinner D, Rankin S, Lee M, Zorn AM. Sox17 and beta-catenin cooperate to regulate the transcription of endodermal genes. Development 2004;131:3069-80.
33. Kuhlbrodt K, Schmidt C, Sock E, Pingault V, Bondurand N, Goossens M, Wegner M. Functional analysis of Sox10 mutations found in human Waardenburg-Hirschsprung patients. J Biol Chem 1998;273:23033-8.
34. Choi YW, Heath EI, Heitmiller R, Forastiere AA, Wu TT. Mutations in beta-catenin and APC genes are uncommon in esophageal and esophagogastric junction adenocarcinomas. Mod Pathol 2000;13:1055-9.
35. Ogasawara N, Tsukamoto T, Mizoshita T, Inada K, Cao X, Takenaka Y, Joh T, Tatematsu M. Mutations and nuclear accumulation of beta-catenin correlate with intestinal phenotypic expression in human gastric cancer. Histopathology 2006;49:612-21.
36. Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nature Reviews Cancer 2006;6:107-116.
37. Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, Pretlow TP, Bin Y. a. n. g. , Akiyama Y, van Engeland M, Toyota M, Tokino T, Hinoda Y, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nature Genetics 2004;36:417-422.
38. Zou HZ, Molina JR, Harrington JJ, Osborn NK, Klatt KK, Romero Y, Burgart LJ, Ahlquist DA. Aberrant methylation of secreted frizzled-related protein genes in esophageal adenocarcinoma and Barrett’s esophagus. International Journal of Cancer 2005;116:584-591.
39. Aguilera O, Fraga MF, Ballestar E, Paz MF, Herranz M, Espada J, Garcia JM, Munoz A, Esteller M, Gonzalez-Sancho JM. Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene 2006;25:4116-4121.
40. Sekine S, Shibata T, Sakamoto M, Hirohashi S. Target disruption of the mutant beta-catenin gene in colon cancer cell line HCT116: preservation of its malignant phenotype. Oncogene 2002;21:5906-5911.
41. Oguma K, Oshima H, Aoki M, Uchio R, Naka K, Nakamura S, Hirao A, Saya H, Taketo MM, Oshima M. Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. Embo Journal 2008;27:1671-1681.
42. Kondoh H, Kamachi Y. SOX-partner code for cell specification: Regulatory target selection and underlying molecular mechanisms. International Journal of Biochemistry & Cell Biology 2010;42:391-399.