
INTRODUCTION
The development of novel approaches in genetics has led to the identification of 30 genetic determinants of Late Onset Alzheimer’s disease (LOAD) [1]. The most important risk locus for LOAD remains on the long arm of chromosome 19 containing the APOE locus, which was the first, and is the strongest, risk factor for Alzheimer’s disease (AD) [2]. Three APOE diplotypic alleles (Ɛ2, Ɛ3, and Ɛ4) are defined, which result from the combination of two single nucleotide polymorphisms (SNPs), rs7412:C>T and rs429358:C>T. While the Ɛ2 allele of APOE is determined by the minor allele of rs7412:C>T and is protective for LOAD [3], the minor allele of rs429358:C>T defines the Ɛ4 allele, which increases the risk of LOAD [2] by up to four-fold.
Interestingly, additional loci on chromosome 19 have been proposed, namely TOMM40 [4], EXOC3L2 [5], CD33 [6], PLD3 [7], and ABCA7 [6] [8]. While compelling genetic data have confirmed ABCA7, on the short arm of chromosome 19, as an APOE-independent AD risk factor [9, 10], inconsistent results have been reported for the other four genes, after adjustments for the effects of APOE [11, 12]. Particular interest deserves the CD33 locus which was reported in 2011 using meta-analysis of genome-wide association studies (GWAS) [6] and, its effect was nominally replicated in an additional sample [13]. However, the association signal of CD33 did not reach genome-wide significance in the International Genomics Alzheimer’s Project (IGAP) meta-analysis [9], the largest case-control study for LOAD. Thus, further investigations are required to confirm or refute the potential association of CD33 with LOAD.
Several possibilities may explain the inconsistency in the results of genetic association studies of regions around the APOE locus such as CD33, including the large contribution of the APOE allele to AD risk, the existence of long range linkage disequilibrium (LRLD) regions in different populations, the existence of hidden familial cases and population inbreeding. However, a definitive explanation for the real driving-force behind these findings is lacking.
Thus, we explored the genetic contribution of ABCA7 and CD33, including their interactions with APOE, to AD risk in a large independent AD case-control cohort composed by unrelated individuals who have at least two Spanish ancestors reported. Furthermore, we conducted LRDL analyses across several dataset to evaluate its influence on CD33 association to AD.
RESULTS
Replication results and meta-analysis
SNPs included in this study were in Hardy-Weinberg equilibrium (P < 0.05). A significant risk effect was observed for the minor allele of ABCA7 rs4147929:G>A (OR=1.15; 95% CI=1.04–1.27) in the Spanish population. ABCA7 effect survived co-variation and APOE-Ɛ4 stratification analyses (Table 1). Meta-analysis including new data generated, the IGAP results and studies fulfilling inclusion criteria (n = 182 208), re-affirmed a genome-wide significant association for ABCA7 (OR=1.15; 95% CI=1.12–1.19, Figure 1A). For CD33, we could not replicate the originally reported protective effect of rs3865444:C>A SNP. In the APOE-Ɛ4 stratified analysis, CD33 did not modulate susceptibility to AD in any stratum (Table 1). Our meta-analysis also revealed heterogeneity and a non-significant association (Figure 1B).
Table 1: Association between ABCA7 rs4147929:G>A or CD33 rs3865444:C>A and LOAD in unadjusted, adjusted and APOE-Ɛ4 stratified models
GENE marker |
Unadjusted model |
Adjusted |
Adjusted |
Stratification per APOE-ε4 |
|
|---|---|---|---|---|---|
Carriers |
Non- Carriers |
||||
ABCA7 |
OR =1.148 |
OR = 1.190 |
OR = 1.175 |
OR = 1.225 |
OR = 1.167 |
CD33 |
OR = 0.986 |
OR = 0.976 |
OR = 0.920 |
OR = 1.157 |
OR = 0.899 |
Abbreviations: LOAD, Late Onset Alzheimer’s disease; OR, Odds Ratio; CI, Confidence Interval.
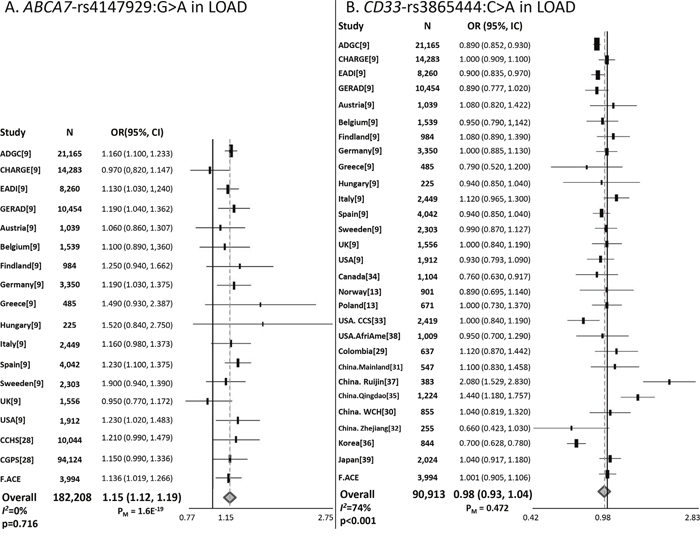
Figure 1: Forest plots for (A) ABCA7 rs4147929:G>A and (B) CD33 rs3865444:C>A. ABCA7 rs4147929:G>A and CD33 rs3865444:C>A meta-analyses comprised data from the IGAP datasets, independent replications, and the present study.
Linkage disequilibrium analysis
Next, we sought to explore potential reasons for the lack of replication of the association of CD33 rs3865444:C>A with AD in some populations. Specifically, patterns of LD were studied in five datasets, comprising data from the present study and four non-imputed GWAS datasets.
We detected significant LRLD between APOE rs7412:C>T and CD33 rs3865444:C>A in two of the five datasets analysed (D’ ≥ 0.2) (Table 2). The probability of these results being spurious was 0.011 for the Murcia dataset and 0.002 for the NIA data (Supplementary Table 1). However, the other datasets did not display LRLD: ADNI, D’ = 0.17 (P = 0.11); GenADA, D’ = 0.14 (P = 0.004); and F.ACE, D’ = 0.01 (P = 0.86) (Table 2). In contrast, only one dataset exhibited significant LD between APOE rs429358:C>T and CD33 rs3865444:C>A; however it did not reach the lower threshold for LD (D’ ≥ 0.2).
Table 2: Linkage disequilibrium results and test of significance for LD between APOE rs7412:C>T or rs429358:C>T and CD33 rs3865444:C>A
Dataset |
LD |
||||
|---|---|---|---|---|---|
N |
D’ |
r2 |
Chi sq |
P value |
|
APOE rs429358:C>T – CD33 rs3865444:C>A |
|||||
ADNI |
358 |
- 0.087 |
0.001 |
0.86 |
0.350 |
GenADA |
1555 |
- 0.105 |
0.001 |
5.32 |
0.020 |
Murcia |
1088 |
+ 0.036 |
5.00e-04 |
1.16 |
0.280 |
NIA |
1789 |
+ 0.015 |
2.00e-04 |
0.75 |
0.380 |
F_ACE |
4402 |
+ 0.013 |
8.00e-05 |
0.70 |
0.400 |
APOE rs7412:C>T – CD33 rs3865444:C>A |
|||||
ADNI |
358 |
+ 0.174 |
0.003 |
2.62 |
0.105 |
GenADA |
1555 |
+ 0.138 |
0.002 |
8.00 |
0.004 |
Murcia |
1088 |
- 0.312 |
0.002 |
5.75 |
0.016 |
NIA |
1789 |
- 0.228 |
0.001 |
4.70 |
0.030 |
F_ACE |
4402 |
+ 0.005 |
3.25e-06 |
0.03 |
0.860 |
APOE rs429358:C>T – APOE rs7412:C>T |
|||||
ADNI |
358 |
- 1 |
0.020 |
13.79 |
2.00e-04 |
GenADA |
1555 |
- 1 |
0.020 |
63.78 |
1.44e-15 |
Murcia |
1088 |
- 1 |
0.010 |
23.42 |
1.30e-06 |
NIA |
1789 |
- 1 |
0.030 |
93.22 |
4.68e-22 |
F_ACE |
4402 |
- 1 |
0.010 |
91.06 |
1.39e-21 |
We next tested the LRLD hypothesis by assessing the pattern of LD along chromosome 19. This alternative strategy highlighted common areas of high LD in all datasets, particularly at positions 10.7–13.5 Mb, 42–45.1 Mb, and 47.8–50 Mb (Figure 2A). In addition, the Murcia and ADNI dataset exhibited the largest segments of LD at positions 9.7–10.7Mb and 9.8–13.8 Mb, respectively. ADNI dataset also presents the largest LD segment at positions 41.8–51.1Mb, the latter of which contained the APOE gene at position 50.1 Mb (Figure 2B). However, this strategy did not reveal a general LD between APOE and CD33. This observation is supporting the notion that long range LD between CD33 and APOE, if exist, would be affecting to a discrete fraction of chromosomes in some human populations.
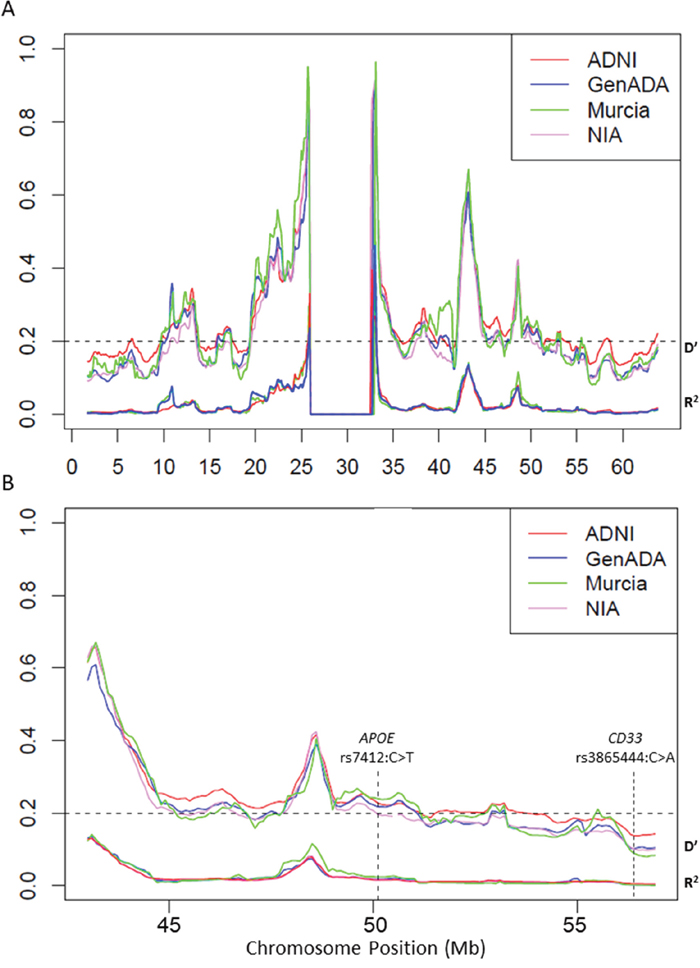
Figure 2: Linkage disequilibrium patterns across (A) chromosome 19 and (B) APOE region. Average D’ (top groups) and r2 coefficients (bottom groups) plotted in sliding windows containing all common polymorphisms separated by 50 and 500 kb in successive 1.7-Mb segments (1.6-Mb overlap). Genome assembly NCBI36/hg18.
Finally, an association analysis approach according to the presence or absence of the APOE Ɛ2 genotype also supported that long range association between APOE and CD33 is not very common in studied populations. After Bonferroni correction, the majority of significant signals mapped to a region of chromosome 19, 231 kb upstream of APOE rs7412C>T (Supplementary Table 2).
APOE stratification analysis and meta-analysis of GWAS data
Stratification analysis revealed a significant protective association for CD33 rs3865444:C>A only in Ɛ4 allele carriers in the GenADA dataset (OR = 0.69; P = 0.009) (Table 3). This protective trend for CD33 rs3865444:C>A was also observed in Ɛ2 carriers of Spanish origin (Murcia study: OR = 0.49, 95% CI, 0.19–1.25; F.ACE: OR = 0.68, 95% CI 0.45–1.03). Interestingly, the minor allele of CD33 rs3865444:C>A was associated with increased susceptibility to AD in Ɛ4 carriers in the same Spanish datasets (Murcia study: OR = 1.13, 95% CI, 0.79–1.63; F.ACE: OR = 1.15, 95% CI, 0.97–1.37). Opposite effect directions were observed in the NIA and ADNI datasets. After meta-analysis, homozygous Ɛ3Ɛ3 carriers were identified as the most homogeneous stratum for CD33 effects, while Ɛ4 allele carriers were the most heterogeneous (Table 3). Finally, we excluded hidden relatedness and population stratification as a potential cause for LRLD by analysing inbreeding (Supplementary Table 3) and conducting principal components (PCs) analysis across the datasets (Supplementary Figure 1).
Table 3: Stratification per Ɛ2, Ɛ4 APOE alleles and Ɛ3Ɛ3 APOE genotype for five studied dataset and meta- analysis results per stratums
Unadjusted model |
Ɛ2 allele carriers |
Ɛ4 allele carriers |
Ɛ3Ɛ3 genotype carriers |
|
|---|---|---|---|---|
ADNI |
OR = 0.865 |
OR = 2.2 |
OR = 0.979 |
OR = 0.706 |
GenADA |
OR = 0.864 |
OR = 0.863 |
OR = 0.697 |
OR = 1.085 |
Murcia |
OR = 0.938 |
OR = 0.492 |
OR = 1.133 |
OR = 0.849 |
NIA |
OR = 0.956 |
OR = 1.356 |
OR = 0.873 |
OR = 0.908 |
F.ACE |
OR = 0.986 |
OR = 0.682 |
OR = 1.153 |
OR = 0.928 |
Meta-Analysis |
OR = 0.947 |
OR = 0.850 |
OR = 0.946 |
OR = 0.932 |
Heterogeneity |
I2 = 0% |
I2 = 32% |
I2 = 66% |
I2 = 0% |
DISCUSSION
Confirmatory data on AD associations with loci neighbouring APOE are still lacking, despite several replication efforts. In our work, we clearly replicated the ABCA7 signal (P = 1.60 x 10-19), which is located far from the APOE locus. However, we failed to replicate the CD33 association in the Spanish population. Both observations are independent and consistent with previous IGAP observations [9].
ABCA7 marker was, firstly, identified such an AD locus by Hollingworth et. al. [6] and Naj et al. [8], in a large cohort of individuals (n=29,544). Following, this association was supported by IGAP consortium [9] and confirmed in non-European populations [10]. Our finding is in accordance with previously studies. It reinforces the role of ABCA7 as a candidate gene for AD and warrants its functional characterisation. ABCA7 is involved in lipid metabolism [14] and apoptotic cell clearance [15]. Its loss has been related to deficient macrophage clearance of amyloid plaques [16] and to accelerated amyloid-beta production [14].
CD33 locus appeared such as a promising signal [6] especially as a candidate target for immune-related therapies [17]. Despite that, the signal disappeared after the IGAP meta-analysis [9], casting doubts on its real contribution to AD. In that scenario, we believed that the CD33 locus appears to be affected by the “APOE curse”; i.e., the impossibility of determining whether additional AD loci truly exist around APOE. Similar to CD33, conflicting data exist for TOMM40’523, EXOC3L2-597668, and PLD3 (V232M) [11, 12]. Several strategies have been attempted to tackle this issue; for example, using phylogenetic analysis, the TOMM40’523 poly-T marker was shown to be associated with age-at-onset in the APOE Ɛ3 subgroup [4]. However, this finding has not been extensively replicated [18].
The lack of association between AD and CD33 prompted us to search for an underlying explanation. Our data suggested significant, weak and non-universal, LRLD between the APOE-Ɛ2 allele and CD33 rs3865444:C>A. Of course, there are limitations on these observations. First of all, very low levels of r2 were detected, however the intrinsic properties of r2 prompts to consider D’ such as more informative measure for assessing historical recombination in a given population [19]. Furthermore, we are dealing with relatively weak D’ values, roughly 0.3, which means that 30% of the chromosomes will be carrying the long LD tract in specific populations. This could be compromising the capacity to detect LRLD when a direct measure of LD between studied markers is not determined.
Second, LRLD patterns differ across populations and are dependent on many factors, including admixture or migration, genetic drift, chromosome inversions, epistatic selection and hitchhiking effects [20]. Furthermore, differential natural selection pressures across genomic regions, depending on specific geographical or environmental conditions, can lead to differential patterns of allele micro-heterogeneity. Although differential population structure was not identified in our GWAS datasets analysis, there is compelling evidence that micro-stratification cannot be detected by standard methods [21] and, therefore, this remains a potential limitation. Thus, the existence of undetected population sub-structure, with different LRLD patterns, could act as confounding factor, explaining the divergent observations and lack of replication between CD33 and AD across studies [9]. Of note, the genuine or spurious character of the association would remain masked under LRLD patterns, being its appearance highly dependent of the population structure.
An additional explanation for the lack of association between CD33 and AD could be that the original CD33 signals were simply chance findings. We feel that this possibility is less likely because under the assumption of a random association between CD33 and AD, the chance of observing effects in opposite directions in independent studies would have the same probability. This latter observation is clearly not the situation reported in the literature to date.
Importantly, we detected LRLD upstream of the APOE locus using a previously described method [22]. Confirmation of LRLD around APOE is relevant for several reasons. First, the data will assist in clarifying whether or not reported AD signals are genuine. Second, differential LRLD across populations may reveal the existence of structural variations, such as large inversions, insertions, or deletions, unequally affecting human populations. Structural variants have been implicated in the aetiology of the majority of multifactorial diseases [23].
In summary, we confirm ABCA7 is associated with LOAD. However, we could not confirm the association between CD33 and AD. Our data suggest that LRLD between CD33 markers and the APOE alleles might explain the observed lack of consistency of CD33 signal. Further studies using independent populations are required to clarify whether LRLD interferes with real associations between loci around APOE and AD.
MATERIALS AND METHODS
Replication study in the spanish population: subjects and genotyping
The Spanish sample comprised 1796 unrelated sporadic AD patients (mean age, 82.1 ± 7.9 years, 70.2% women) and 2642 healthy controls (mean age, 54.1 ± 11.6 years, 64.3% women) recruited at Fundació ACE, Institut Català de Neurociències Aplicades (Barcelona, Spain); Unidad de Memoria, Hospital Universitario La Paz-Cantoblanco (Madrid, Spain); Hospital Clínico Sán Carlos Unidad de Demencias, Hospital Universitario Virgen de la Arrixaca (Murcia, Spain) and Neocodex S.L. (Supplementary Table 4). Sample characteristics were previously described by Antunez et al. [24]. Briefly, all AD patients fulfilled Diagnostic and Statistical Manual of Mental Disorders IV criteria for dementia and were diagnosed according to National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s disease and Related Disorders Association criteria for possible and probable AD. Ethics committees from each referral centre approved the research protocol. All participants provided written informed consent.
Standard methods were used to isolate DNA. The SNP rs4147929:G>A, this is hg19 chr19:g.1063444A>G located in the ABCA7 gene and the SNP rs3865444:C>A, this is hg19 chr19:g.51727962C>A located in CD33 gene, were genotyped using Sequenom technology (Sequenom, California, USA), as previously described [25]. Primer sequences and assay conditions for the genotyped SNPs are available upon request.
APOE rs7412C>T and rs429358:C>T markers were genotyped using real-time PCR. Primers design was previously described by Calero et al [26]. Briefly, PCR reactions were performed in a final volume of 5μl, using 11 ng of genomic DNA, 0.3 μM of each amplification primer and 2.65μl of 2X SYBR Fast Master Mix (Kapa Biosystems). We used an initial denaturation step of 95 °C for 2 min, followed by 33 cycles of 95 °C for 10 s, and 69 °C for 30 s. Melting curves were 95 °C for 15 s (ramping rate 5.5 °C s), 45 °C for 15 s (ramping rate of 5.5°C s−1) and 95 °C for 15 s (ramping rate of 5.5°C s−1). In the last step of each melting curve, a continuous fluorimetric register was performed by the system at one acquisition register per each degree Celsius. Melting peaks and genotype calls were obtained by using the Eco Real-Time PCR system (Illumina).
Statistical analysis and meta-analysis
Comparisons of allele frequencies between cases and controls were performed using Chi-square tests. Logistic regression analysis (additive model) was used to adjust for: 1) sex and APOE Ɛ4, and 2) age, sex, and APOE Ɛ4. Stratification was also conducted according to the presence or absence of the APOE Ɛ4 allele. All statistical analyses were performed using PLINK 1.9 software (http://www.cog-genomics.org/plink2/) [27].
Meta-analysis techniques were used to estimate: ABCA7 rs4147929:G>A and CD33 rs3865444:C>A effects across studies. Meta-analysis datasets comprise new data generated, samples overlapping with IGAP were excluded (n = 3,994), data from IGAP [9] and available studies published (Supplementary Table 5). Briefly, we carried out a literature search in Pubmed for studies published before March 2018. The search terms were: ABCA7 and Alzheimer’s disease; and CD33 and Alzheimer’s disease, respectively. Only studies meeting the following criteria were included: (1) case/control studies evaluating the effect of rs4147929:G>A or rs3865444:C>A markers in AD’s risk; (2) studies that provided an odds ratio with 95% confidence interval as well as the p-value or provide sufficient data to calculate them. Reviews were excluded. We included 1 article for ABCA7 [28] (32 articles were excluded due to rs4147929:G>A was not genotyped; 1 presented sample overlapping; 66 were not case/control studies and 3 presented limited access). Thus, the meta-analysis sample size for ABCA7 rs4147929:G>A comprises 182,208 individuals. In case of CD33, we included 12 articles [13, 29–39] (6 studies did not genotype rs3865444:C>A; 5 presented sample overlapping; and 43 were not case/control studies). Thus, the meta-analysis sample size for CD33 rs3865444:C>A comprises 90,913 individuals. Meta-analysis was conducted using the inverse variant method (fixed-effects model) but in the case of heterogeneity, the DerSimonian and Liard method (random-effects model) was used. Heterogeneity was considered significant when I2 > 50% and p < 0.05. Meta-analysis results and forest plots were generated using OpenMeta (http://www.cebm.brown.edu/openmeta/).
Linkage disequilibrium analysis using GWAS datasets
Patterns of LD were studied in data from the present study and four non-imputed GWAS datasets: the Alzheimer’s Disease Neuroimaging Initiative (ADNI) longitudinal study [40]; the Genotype-Phenotype Alzheimer Disease Association (GenADA) study [41]; the National Institute of Aging (NIA) Genetic Consortium for Late Onset Alzheimer’s disease study [42]; and the Murcia study [24] (Supplementary Table 6). The genome assembly for the four non-imputed GWAS datasets was NCBI36/hg18.
LD measures (D’ and r2) and tests of the significance of LD were calculated between the APOE rs429358:C>T or rs7412:C>T markers and CD33 rs3865444:C>A using Plink 1.9 software (http://www.cog-genomics.org/plink2/) [27] and the R statistics package. LRLD was accepted where D’ ≥ 0.2 and P < 0.05. With the objective of discarding LD results generated by chance, we performed bootstrapping analyses; we calculated LD between APOE rs7412:C>T or rs429358:C>T and 10,000 random markers, which presented minor allele frequencies (MAF) ≥ 0.20 and ≤ 0.40 (since CD33 rs3865444:C>A, MAF = 0.30) and that did not localize to chromosome 19.
To further investigate these results, two additional methods were employed. First, the pattern of disequilibrium across the whole of chromosome 19 was calculated according to the methods of Dawson and colleagues [22]; including only markers with MAFs ≥ 0.2. Briefly, we considered 1.7Mb window (1.6Mb overlap) and calculated average values of D' and r2 for all marker pairs, which were separated by at least 50kb and at most 500kb. D’ and r2 were calculated using Plink software 1.9 [27]. Second, association analysis was conducted in Plink 1.9 software [27] according to the presence or absence of the Ɛ2 allele. In this second approach, markers in strong LD with the Ɛ2 allele will exhibit stronger associations. P values were adjusted using the Bonferroni correction method.
Unadjusted, stratified, and meta-analysis models were explored (using the methods described above). In the stratification analysis, subjects were classified into three groups: carriers of Ɛ2 or Ɛ4 alleles, and carriers of the Ɛ3Ɛ3 genotype. Ɛ2Ɛ4 genotype carriers were excluded from this analysis.
Estimation of inbreeding and population structure
Wright’s population inbreeding coefficient (F) was calculated according to heterozygote reduction, with regard to Hardy-Weinberg expectations, according to the formulae: F = 1 – Ho/He, where Ho is the frequency of heterozygotes observed in the sample population and He is the frequency of heterozygotes expected under Hardy Weinberg. The final inbreeding calculation responds to the mean of F per number of markers included in the analysis. Markers were included in this analysis if they were common markers for all the dataset, had MAF > 0.2 [43], and HWE > 0.001. Individuals with less than 99% of available genotypes were excluded. Inbreeding calculations for F.ACE dataset are very imprecise compared to other datasets available, due to it comprises a small number of SNPs. Thus, inbreeding calculation with a higher number of SNPs was also performed excluding F.ACE dataset.
PCs analysis was conducted to discard LRLD was caused by differential population structure. Plink 1.9 software was used to perform the analysis. PCs analysis was conducted in 19,979 markers, which were common between studies, and which presents low linkage disequilibrium (LD) (r2<0.3). In addition, long range LD regions were excluded to the analysis.
Availability of the data and material
Fundacio ACE datasets used during the current study are available from the corresponding author on reasonable request. Murcia study data analysed are available upon request to Carmen Antunez, Manuel Serrano-Rios and Agustin Ruiz. The GENADA and NIA datasets that support the findings of this study are available from dbGaP but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. The ADNI datasets analysed during the current study are available in the ADNI repository, http://adni.loni.usc.edu/.
Author contribution
SMG and ARu contributed to the study design and analysed data. SMG prepared the first draft of the manuscript. IH, SR, MR, AM, LV, ORG, MA, AE, GO, MT, CA, DS and JLA contributed to phenotyping, database curation and patients selection. CA, MSR and ARu carried out Murcia study. SMG and OSG contributed to data processing methods. SHH and ARa carried out genotyping procedures. WM, LT, MB and ARu participated in funds procurement. ARa and ARu were responsible for the critic revision of the manuscript. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
We would like to thank patients and controls who participated in this project. We are indebted to Trinitat Port-Carbó and her family for their support of Fundació ACE research programs. Fundació ACE collaborates with the Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED, Spain) and is one of the participating centers of the Dementia Genetics Spanish Consortium (DEGESCO). The genotypic and associated phenotypic data used in the study, 'Multi-Site Collaborative Study for Genotype-Phenotype Associations in Alzheimer's Disease (GenADA)', were provided by GlaxoSmithKline, R&D Limited. The datasets used for analyses described in this manuscript were obtained from dbGaP through dbGaP accession numbers phs000219.v1.p1 and phs000168.v1.p1. Funding support for the 'Genetic Consortium for Late Onset Alzheimer's Disease' was provided through the Division of Neuroscience, NIA. The Genetic Consortium for Late Onset Alzheimer's Disease included a GWAS, funded as part of the Division of Neuroscience, NIA. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by the Genetic Consortium for Late Onset Alzheimer's Disease. Furthermore, part of the data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbV ie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company, Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research provides funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. The present work was performed as part of the Biochemistry, Molecular Biology and Biomedicine doctoral program of S. Moreno-Grau at Universitat Autònoma de Barcelona (Barcelona, Spain).
CONFLICTS OF INTEREST
None. The authors declare that they have no competing interest.
FUNDING
The results were generated with the assistance of financial support from the Innovative Medicines Initiative 2 Joint Undertaking which receives support from the European Union’s Horizon 2020 research and innovation programme (ADAPTED Grant No. 115975). A.R. research is also supported by grants PI13/02434 and PI16/01861. Acción Estratégica en Salud, integrated in the Spanish National R + D + I Plan and financed by ISCIII (Instituto de Salud Carlos III)-Subdirección General de Evaluación and the Fondo Europeo de Desarrollo Regional (FEDER- “Una manera de Hacer Europa”), by Fundación bancaria “La Caixa” and Grifols SA (GR@ACE project). Part of the data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbV ie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company, Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research provides funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. The present work was performed as part of the Biochemistry, Molecular Biology and Biomedicine doctoral program of S. Moreno-Grau at Universitat Autònoma de Barcelona (Barcelona, Spain). Funding sources were not involved in the design of the study, the analysis and interpretation of data and in writing the manuscript.
REFERENCES
1. Moreno-Grau S, Ruiz A. Genome research in pre-dementia stages of Alzheimer’s disease. Expert Rev Mol Med. 2016; 18: e11.
2. Corder E, Saunders A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993; 8: 41–3.
3. Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, Rimmler JB, Locke PA, Conneally PM, Schmader KE. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994; 7: 180–4.
4. Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, Huentelman MJ, Welsh-Bohmer KA, Reiman EM. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 2010; 10: 375–84.
5. Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, Bis JC, Smith AV, Carassquillo MM, Lambert JC, Harold D, Schrijvers EM, Ramirez-Lorca R, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010; 303: 1832–40.
6. Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011; 43: 429–35.
7. Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, Harari O, Norton J, Budde J, Bertelsen S, Jeng AT, Cooper B, Skorupa T, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer ’ s disease. Nature. 2014; 505: 550–4.
8. Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011; 43: 436–41.
9. Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013 ; 45: 1452–8.
10. Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang LS, Valladares O, Lin CF, Larson EB, Graff-Radford NR, Evans D, De Jager PL, Crane PK, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ϵ4,and the risk of late-onset Alzheimer disease in African Americans. JAMA. 2013; 309: 1483–92.
11. Chouraki V, Seshadri S. Genetics of Alzheimer’s disease. Adv Genet. 2014; 87: 245–94.
12. Heilmann S, Drichel D, Clarimon J, Fernández V, Lacour A, Wagner H, Thelen M, Hernández I, Fortea J, Alegret M, Blesa R, Mauleón A, Roca MR, et al. PLD3 in non-familial Alzheimer’s disease. Nature. 2015; 520: E3–5.
13. Carrasquillo MM, Belbin O, Hunter TA, Ma L, Bisceglio GD, Zou F, Crook JE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, et al. Replication of EPHA1 and CD33 associations with late-onset Alzheimer’s disease: a multi-centre case-control study. Mol Neurodegener. 2011; 6: 54.
14. Sakae N, Liu CC, Shinohara M, Frisch-Daiello J, Ma L, Yamazaki Y, Tachibana M, Younkin L, Kurti A, Carrasquillo MM, Zou F, Sevlever D, Bisceglio G, et al. ABCA7 Deficiency Accelerates Amyloid-β Generation and Alzheimer’s Neuronal Pathology. J Neurosci. 2016; 36: 3848–59.
15. Jehle AW, Gardai SJ, Li S, Linsel-Nitschke P, Morimoto K, Janssen WJ, Vandivier RW, Wang N, Greenberg S, Dale BM, Qin C, Henson PM, Tall AR. ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J Cell Biol. 2006; 174: 547–56.
16. Kim WS, Li H, Ruberu K, Chan S, Elliott DA, Low JK, Cheng D, Karl T, Garner B. Deletion of Abca7 increases cerebral amyloid-β accumulation in the J20 mouse model of Alzheimer’s disease. J Neurosci. 2013; 33: 4387–94.
17. Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, Morris MC, Evans DA, Johnson K, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013; 16: 848–50.
18. Cruchaga C, Nowotny P, Kauwe JSK, Ridge PG, Mayo K, Bertelsen S, Hinrichs A, Fagan AM, Holtzman DM, Morris JC, Goate AM. Association and expression analyses with single-nucleotide polymorphisms in TOMM40 in Alzheimer disease. Arch Neurol. 2011; 68: 1013–9.
19. Mueller JC. Linkage disequilibrium for different scales and applications. Brief Bioinform. 2004; 5: 355–64.
20. Ardlie KG, Kruglyak L, Seielstad M. Patterns of Linkage Disequilibrium in the Human Genome. Nat Rev Genet. 2002; 3: 299–309.
21. Campbell CD, Ogburn EL, Lunetta KL, Lyon HN, Freedman ML, Groop LC, Altshuler D, Ardlie KG, Hirschhorn JN. Demonstrating stratification in a European American population. Nat Genet. 2005; 37: 868–72.
22. Dawson E, Abecasis GR, Bumpstead S, Chen Y, Hunt S, Beare DM, Pabial J, Dibling T, Tinsley E, Kirby S, Carter D, Papaspyridonos M, Livingstone S, et al. A first-generation linkage disequilibrium map of human chromosome 22. Nature. 2002; 418: 544–8.
23. Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet. 2007; 8: 639–46.
24. Antúnez C, Boada M, González-Pérez A, Gayán J, Ramírez-Lorca R, Marín J, Hernández I, Moreno-Rey C, Morón FJ, López-Arrieta J, Mauleón A, Rosende-Roca M, Noguera-Perea F, et al. The membrane-spanning 4-domains, subfamily A (MS4A) gene cluster contains a common variant associated with Alzheimer’s disease. Genome Med. 2011; 3: 33.
25. Ruiz A, Heilmann S, Becker T, Hernández I, Wagner H, Thelen M, Mauleón A, Rosende-Roca M, Bellenguez C, Bis JC, Harold D, Gerrish A, Sims R, et al. Follow-up of loci from the International Genomics of Alzheimer’s Disease Project identifies TRIP4 as a novel susceptibility gene. Transl Psychiatry. 2014; 4: e358.
26. Calero O, Hortigüela R, Bullido M, Calero M. Apolipoprotein E genotyping method by Real Time PCR, a fast and cost-effective alternative to the TaqMan® and FRET assays. J Neurosci Methods. 2009; 183: 238–40.
27. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007; 81: 559–75.
28. Kjeldsen EW, Tybjærg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. ABCA7and risk of dementia and vascular disease in the Danish population. Ann Clin Transl Neurol. 2018; 5: 41–51.
29. Moreno DJ, Ruiz S, Ríos Á, Lopera F, Ostos H, Via M, Bedoya G. Association of GWAS Top Genes With Late-Onset Alzheimer’s Disease in Colombian Population. Am J Alzheimers Dis Other Demen. 2017; 32: 27–35.
30. Zhang DF, Li J, Wu H, Cui Y, Bi R, Zhou HJ, Wang HZ, Zhang C, Wang D, Kong QP, Li T, Fang Y, Jiang T, et al. CFH Variants Affect Structural and Functional Brain Changes and Genetic Risk of Alzheimer’s Disease. Neuropsychopharmacology. 2015; 41:1034–1035.
31. Jiao B, Liu X, Zhou L, Wang MH, Zhou Y, Xiao T, Zhang W, Sun R, Waye MM, Tang B, Shen L. Polygenic Analysis of Late-Onset Alzheimer’s Disease from Mainland China. PLoS One. 2015; 10: e0144898.
32. Mao YF, Guo ZY, Pu JL, Chen YX, Zhang BR. Association of CD33 and MS4A cluster variants with Alzheimer’s disease in East Asian Populations. Neurosci Lett. 2015; 609: 235–239.
33. Ebbert MT, Ridge PG, Wilson AR, Sharp AR, Bailey M, Norton MC, Tschanz JT, Munger RG, Corcoran CD, Kauwe JSK. Population-based Analysis of Alzheimer’s Disease Risk Alleles Implicates Genetic Interactions. Biol Psychiatry. 2014; 75: 732–7.
34. Omoumi A, Fok A, Greenwood T, Sadovnick AD, Feldman HH, Hsiung GY. Evaluation of late-onset Alzheimer disease genetic susceptibility risks in a Canadian population. Neurobiol Aging. 2014; 35: 936.e5-12.
35. Tan L, Yu JT, Zhang W, Wu ZC, Zhang Q, Liu QY, Wang W, Wang HF, Ma XY, Cui WZ. Association of GWAS-linked loci with late-onset Alzheimer’s disease in a northern Han Chinese population. Alzheimers Dement. 2013; 9: 546–53.
36. Chung SJ, Lee JH, Kim SY, You S, Kim MJ, Lee JY, Koh J. Association of GWAS top hits with late-onset Alzheimer disease in Korean population. Alzheimer Dis Assoc Disord. 2013; 27: 250–7.
37. Deng YL, Liu LH, Wang Y, Tang HD, Ren RJ, Xu W, Ma JF, Wang LL, Zhuang JP, Wang G, Chen SD. The prevalence of CD33 and MS4A6A variant in Chinese Han population with Alzheimer’s disease. Hum Genet. 2012; 131: 1245–9.
38. Logue MW. A Comprehensive Genetic Association Study of Alzheimer Disease in African Americans. Arch Neurol. 2011; 68: 1569.
39. Miyashita A, Koike A, Jun G, Wang LS, Takahashi S, Matsubara E, Kawarabayashi T, Shoji M, Tomita N, Arai H, Asada T, Harigaya Y, Ikeda M, et al. SORL1 is genetically associated with late-onset Alzheimer’s disease in Japanese, Koreans and Caucasians. PLoS One. 2013; 8: e58618.
40. Weiner M, Aisen P, Jack C Jr, Jaugust W, Trojanowski J, Shaw L, Saykin AJ, Morris JC, Cairns N, Laurel A, Toga A, Green R, Walter S, et al. The Alzheimer’s disease neuroinmaging iniciative: Progress report and future plans. Alzheimers Dement. 2010; 6: 202–11.
41. Li H, Wetten S, Li L, St Jean PL, Upmanyu R, Surh L, Hosford D, Barnes MR, Briley JD, Borrie M, Coletta N, Delisle R, Dhalla D, et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol. 2008; 65: 45–53.
42. Wijsman EM, Pankratz ND, Choi Y, Rothstein JH, Faber KM, Cheng R, Lee JH, Bird TD, Bennett DA, Diaz-Arrastia R, Goate AM, Farlow M, Ghetti B, et al. Genome-wide association of familial late-onset Alzheimer’s disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet. 2011; 7: e1001308.
43. Zhang Q, Calus MP, Guldbrandtsen B, Lund MS, Sahana G. Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. BMC Genet. 2015; 16: 88.