
INTRODUCTION
Diabetic nephropathy is a major complication of diabetes mellitus, and the leading cause of end stage renal disease worldwide [1]. Twenty years after onset of diabetes, microalbuminuria –the first sign of diabetic nephropathy- is detected in up to 40% of patients [2–4]. Because of the growing number of diabetes patients in combination with an earlier onset of the disease, the incidence of diabetic nephropathy will likely rise in the coming years. With strict glycemic control and angiotensin-converting-enzyme inhibition it is possible to slow the progression of renal failure, although many patients still eventually progress towards end stage renal disease. At that stage, dialysis or kidney transplantation are the only available treatment options to date. Both dialysis and kidney transplantation have a huge social and economic impact and alternative treatment options are thus eagerly awaited for [5].
Considering the emerging role of coagulation factors in kidney disease (excellently reviewed by Madhusudhan and colleagues [6]), it is tempting to speculate that anticoagulant therapy may be a promising strategy to prevent kidney damage in diabetic patients. Many clinical trials with anticoagulants have indeed been performed [7–12], but enthusiasm for anticoagulants in the setting of diabetic nephropathy faded away due to conflicting results [13, 14]. Interestingly however, the recent notion that coagulation factor receptors, i.e. protease-activated receptors (PARs), are omnipresent in renal cells not only provides a molecular link between coagulation factors and renal cell function but also suggest that targeting PARs may hold therapeutic promise in the setting of diabetic nephropathy.
PAR-1 is a seven transmembrane domain receptor that belongs to the family of G protein-coupled receptors (GPCRs) [15]. In contrast to traditional GPCRs, PAR-1 is activated by proteolytic cleavage instead of ligand binding. Originally, PAR-1 was identified as the thrombin receptor but alternative agonists, such as activated protein C [16], plasmin [17], or metalloprotease 13 [18] have been shown to also activate this receptor - a phenomenon called biased agonism [15, 16]. In the kidney, PAR-1 expression is increased upon diabetes. Activation of PAR-1 in mesangial cells induces their proliferation and leads to the production of extracellular matrix [19]. In line with this potential profibrotic role of PAR-1 during diabetes, we recently showed that PAR-1 deficient mice develop less glomerulopathy during streptozotocin (STZ)-induced type 1 diabetes. Indeed, glomerular cell proliferation and fibronectin deposition was significantly reduced in diabetic PAR-1 deficient mice as compared to diabetic wild type mice. It is consequently tempting to speculate that PAR-1 may be a potential novel target to reduce diabetes-induced kidney damage [19].
Although transgenic mice are perfectly suited to perform mechanistic studies and to identify potential novel targets, translation of preclinical studies using knock out animals remains challenging. Before initiating clinical studies targeting PAR-1 in the setting of diabetic nephropathy it is therefore pivotal to establish whether pharmacological inhibition of PAR-1, started once diabetes has been established, also limits diabetes-induced kidney damage. Consequently, we here evaluated the potential inhibitory effect of vorapaxar (SCH530348, Zontivity), a PAR-1 antagonist used for secondary prevention of atherothrombotic events in patients with previous myocardial infarct, on STZ-induced diabetic nephropathy.
RESULTS
Vorapaxar treatment reduces diabetes-induced kidney damage
Four weeks after the induction of diabetes by streptozotocin injections, both diabetic and non-diabetic mice were randomly assigned to vorapaxar or control treatment (n = 11 in all groups). Body weight and blood glucose levels were similar in vorapaxar and control treated mice during the course of the experiment (Figure 1A and 1B). As shown in Figure 1C, urinary albumin secretion was significantly increased after streptozotocin injections in control treated mice. Despite having similar glucose levels, no increase in albuminuria was observed in diabetic mice treated with vorapaxar. Similarly, the levels of plasma cystatin C were increased in control treated diabetic mice compared to nondiabetic mice. No increase upon diabetes was observed in the vorapaxar treated mice, showing that vorapaxar limits the diabetes-induced increase in plasma cystatin C (Figure 1D). Finally, no differences on inflammation were observed between control and vorapaxar treated mice (Supplementary Figure 1).
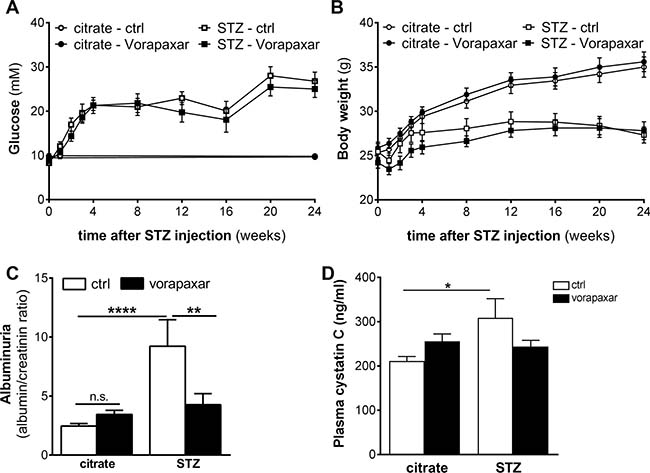
Figure 1: Reduced nephropathy after vorapaxar treatment in streptozotocin-induced diabetic mice. (A) Glucose levels of control (citrate) and streptozotocin-induced (STZ) diabetic mice during control (ctrl) or vorapaxar treatment (B) Body weight of control (citrate) and streptozotocin-induced (STZ) diabetic mice during control (ctrl) or vorapaxar treatment (C) Albumin-to-creatinine ratio in urine and (D) plasma cystatin C levels in control and vorapaxar treated mice 24 weeks after streptozotocin (STZ) or citrate injections. Indicated is the mean ± SEM. Citrate ctrl, n = 11; citrate vorapaxar, n = 11; STZ ctrl, n = 6; STZ vorapaxar, n = 8. One-way ANOVA with Bonferroni post-hoc analysis and unpaired t-test were used, *p < 0.05; **p < 0.01; ****p < 0.0001.
Vorapaxar treatment reduces diabetes-induced mesangial expansion and glomerular fibronectin deposition
We previously showed that diminished albuminuria in PAR-1 deficient mice was accompanied by a reduction of mesangial expansion [19]. Consequently, we next evaluated the effect of vorapaxar on mesangial expansion in diabetic mice. An increase in mesangial expansion was observed in control treated diabetic mice as evident from an increase in the number of deviated glomeruli (Figure 2). Compared to control treatment, vorapaxar significantly reduced the number of glomeruli with mesangial expansion.
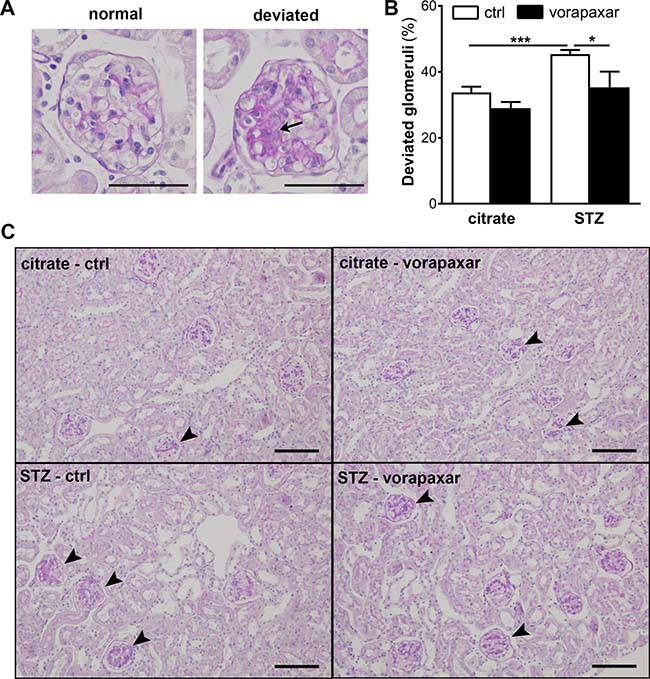
Figure 2: Limited mesangial expansion upon vorapaxar treatment in STZ-induced diabetic mice. (A) Micrograph showing a PAS-D stained glomerulus scored ‘normal’ and ‘deviated’. The arrow pinpoints area of mesangial expansion. Scale bars: 50 μm (B) Quantification of mesangial expansion in glomeruli of diabetic (STZ) and non-diabetic (citrate) mice, upon control or vorapaxar treatment. Glomeruli were scored either normal or deviated as described in the materials and methods section. (C) Representative pictures of deviated glomeruli in control and vorapaxar treated diabetic mice. The arrowheads pinpoint glomeruli that were scored ‘deviated’. Scale bars: 100 μm. Citrate ctrl, n = 11; citrate vorapaxar, n = 10; STZ ctrl, n = 5; STZ vorapaxar, n = 8. One-way ANOVA with Bonferroni post-hoc analysis was used, *p < 0.05; ***p < 0.005.
Finally, glomerular fibronectin expression was assessed as PAR-1 directly induces fibronectin production by mesangial cells and fibronectin deposition was reduced in diabetic PAR-1 deficient mice as compared to control diabetic mice [19]. In line with our previous experiments, glomerular fibronectin expression was induced in diabetic control treated mice. Importantly however, no induction of fibronectin deposition was observed in vorapaxar treatment diabetic mice (Figure 3).
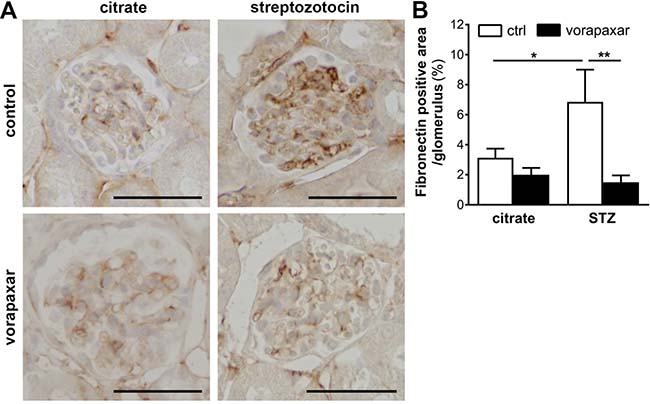
Figure 3: Limited glomerular fibronectin deposition upon vorapaxar treatment in STZ-induced diabetic mice. (A) Representative pictures of fibronectin stained paraffin sections of kidneys of control and vorapaxar treated mice, 24 weeks after streptozotocin or citrate injections. Scale bars: 50 μm (B) Quantification of fibronectin deposition. Indicated is the mean ± SEM. Citrate ctrl, n = 10; citrate vorapaxar, n = 11; STZ ctrl, n = 5; STZ vorapaxar, n = 7. One-way ANOVA with Bonferroni post-hoc analysis was used, *p < 0.05; **p < 0.01.
DISCUSSION
Diabetic nephropathy is a major complication of diabetes which ultimately requires renal replacement therapy. Treatment options to delay or prevent the development of diabetic nephropathy are therefore eagerly awaited. We have recently shown that genetic ablation of PAR-1 limits the development of diabetic nephropathy in streptozotocin-induced diabetic mice by diminishing mesangial cell proliferation and extracellular matrix production. To evaluate the clinical potential of PAR-1 inhibition, we here treated diabetic mice with the PAR-1 antagonist vorapaxar. We show that inhibition of PAR-1 by vorapaxar treatment, started after the onset of diabetes, limits the development of diabetic nephropathy. In line with PAR-1 deficiency, vorapaxar treated mice developed less albuminuria, mesangial expansion and fibronectin deposition, as compared to control treated mice. Our data thus confirm the key role of PAR-1 in the pathogenesis of diabetic nephropathy and point to the potential clinical significance of vorapaxar treatment in diabetic patients.
Vorapaxar (SCH530349, Zontivity) is a PAR-1 antagonist, used for secondary prevention of atherothrombotic events in patients with previous myocardial infarction. In the TRA-CER trial [20] and the TRA 2° P-TIMI trial [21], treatment with vorapaxar led to reduced cardiovascular death, myocardial infarction, or stroke both in patients at high risk for cardiovascular events and in patients with a history of myocardial infarction, ischemic stroke, or peripheral arterial disease [22, 23]. Interestingly, treatment with vorapaxar appeared especially beneficial in patients with diabetes, with an absolute risk reduction for cardiovascular events of 3,5% (compared to 1,36% absolute risk reduction in non-diabetic patients) [24, 25].
Our data show that vorapaxar limits diabetic nephropathy in mice and suggest that vorapaxar may be pursued in patients. Importantly however, vorapaxar not only acts upon mesangial cells but (amongst others) also blocks PAR-1-dependent platelet aggregation which may lead to bleeding complications. Indeed, clinical trials using vorapaxar note an increased risk in bleeding and the TRA-CER trial was even terminated early due to excessive bleeding in patients on vorapaxar treatment (on top of aspirin and P2Y12 inhibitor) [22]. In a clinical setting, it should be critically evaluated whether kidney preservation by vorapaxar would outweigh the bleeding risk in individual patients. Ultimately, finding the PAR-1 agonist responsible for initiating the PAR-1 response in diabetic nephropathy, may lead to the development of a PAR-1 based treatment strategy avoiding the increased risk of bleeding.
In conclusion, vorapaxar treatment prevents the development of diabetic nephropathy in streptozotocin-induced diabetic mice, pinpointing PAR-1 as a novel therapeutic target to pursue in the setting of diabetic nephropathy.
MATERIALS AND METHODS
Mice
Wild type C57BL/6 mice were purchased from Charles River (Maastricht, the Netherlands). All experiments were approved by the Institutional Animal Care and Use Committee of the University of Amsterdam. All mice were maintained according to institutional guidelines. Animal procedures were carried out in compliance with the Institutional Standards for Humane Care and Use of Laboratory Animals of the Academic Medical Center. The Animal Care and Use Committee of the Academic Medical Center approved all experiments.
Experimental diabetic nephropathy model
Eight to twelve week-old male wild type C57Bl/6 mice were injected intraperitoneally with streptozotocin (50 mg/kg body weight) or citrate for 5 consecutive days to induce diabetes. Four weeks after the induction of diabetes, mice were randomly assigned to control (n = 11) or vorapaxar (n = 11) treatment. Vorapaxar (1,75 mg/kg) was administered orally twice a week at 100 μl/10 g, whereas a similar amount of vehicle solution (0,02% hydroxyproline-β-cyclodextrin in 0,1% DMSO) was administered as control treatment. After 20 weeks of treatment, 24 h urine samples were collected using metabolic cages, after which the mice were sacrificed and blood and kidneys were harvested for further analysis. During the experiment, blood glucose levels were measured from tail vein blood using an Accu-Chek Aviva glucose meter (Roche). Five control treated mice and three vorapaxar treated mice did not develop diabetes (i.e. glucose levels remained below 15 mM) and were therefore excluded from further analysis. Plasma cystatin C (R&D systems) and urine albumin (Bethyl laboratories) levels were determined by ELISA according to the manufacturer’s instructions. Urine creatinine levels were determined using an enzymatic mouse creatinine assay kit (CrystalChem), according to the manufacturer’s instructions. Glomerular injury was determined by histological analysis and fibronectin deposition by immunohistochemical staining analysis.
(Immuno)histopathology
Formalin-fixed, paraffin embedded, kidney slides were periodic acid–Schiff–diastase (PAS-D) stained following routine procedures. The extent of glomerular injury was determined by two independent observers in a blinded fashion. To quantify glomerular injury, 50 glomeruli per mouse, were scored as either normal or deviated. Glomeruli were scored as deviated when mesangial expansion was apparent as clusters of >3 mesangial cells.
Glomerular extracellular matrix accumulation was determined using goat-anti-fibronectin (1:500; sc-6953; Santa Cruz Biotechnology) antibody as described before [26]. In short, paraffin embedded slides were deparaffinized, and endogenous peroxidases were inhibited by 15 minutes incubation in 0,3% H2O2 at room temperature. Slides were boiled in citrate buffer (pH6.0) for 10 min, blocked with normal goat serum or Ultra V block (Thermo Scientific, Runcorn, UK) for 30 min, and incubated overnight with the primary antibody. Slides were incubated with HRP conjugated rabbit-anti-goat IgG (P0160; Dako) for 30 min at room temperature, visualized with DAB (BS04-999; Immunologic) and counterstained using haematoxylin. Slides incubated without the primary antibody were used as negative controls to exclude nonspecific binding of the secondary antibody. Pictures were taken at 20 times magnification using a Leica DM5000B microscope equipped with a Leica DFC500 camera and Image Pro Plus software (vs 5.02; Media Cybernatics). Fibronectin positive areas (expressed as a percentage) was determined, per glomerulus (expressing the density of ECM) using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA) in 25 glomeruli per mouse.
RNA isolation and RT-qPCR
For gene expression analysis, mRNA was isolated from kidney homogenates using TriReagent (#11667165001; Roche Diagnostics) according to the manufacturers recommendations. All mRNA samples were quantified by spectrophotometry and stored at −80° C until further analysis. One μg of mRNA was treated with RQ1 DNAse (M6101, Promega, Madison, WI, USA) and subsequently converted to cDNA using M-MLV reverse transcriptase (M1705, Promega, Madison, WI, USA) and random hexamer primers (#SO142, Fisher scientific, Landsmeer, the Netherlands) according to the manufacturers recommendations. qPCR and subsequent analysis were performed using sensiFAST No-ROX PCR master mix (GC Biotech) on a Lightcycler 480 machine and corresponding software (Software release 1.5.0 (1.5.0.39), Roche, Almere, the Netherlands). Expression levels were normalized using the average expression levels of HPRT1, GAPDH and TBP. The following primer sequences were used:
mIL6 forward: 5′- GCTACCAAACTGGATATA ATCAGGA-3′ reverse: 5′- CCAGGTAGCTATGGTACTC CAGAA-3′; mTNFα forward: 5′- CTGTAGCCCACGTC GTAGC-3′ reverse: 5′- TTGAGATCCATGCCGTTG-3′; mMCP1 forward: 5′- CATCCACGTGTTGGCTCA-3′ reverse: 5′- GATCATCTTGCTGGTGAATGAGT-3′; mIL1β forward: 5′- TGAGCACCTTCTTTTCCTTCA-3′ reverse: 5′- TTGTCTAATGGGAACGTCACAC-3′; mTBP forward: 5′- CCTTGTACCCTTCACCAATGAC-3′ reverse: 5′- ACAGCCAAGATTCACGGTAGA-3′; mGAPDH forward: 5′- CTCATGACCACAGTCCATGC-3′ reverse: 5′- CACATTGGGGGTAGGAACAC-3′; mHPRT1 forward: 5′- CAGGCCAGACTTTGTTGGAT-3′ reverse: 5′- GACACAAACGTGATTCAAATCC-3′.
Statistics
All values are expressed as mean ± SEM. All groups were tested for normality using the D’Agostino-Pearson omnibus normality test. Detected outliers were excluded from analysis. Differences between two groups were analysed using a t-test if data were normally distributed, or a Mann-Whitney U-test for non-parametric data. Multiple comparisons were analysed using one-way-ANOVA analysis or Kruksal-Wallis test (for nonparametric values), followed by Bonferroni’s or Dunns multiple comparison tests, respectively. All analyses were performed using GraphPad Prism version 5.01.
Abbreviations
PAR: Protease-activated receptor; GPCR: G protein-coupled receptor; STZ: streptozotocin; TNF-α: Tumor necrosis factor-a; IL: Interleukin; HPRT1: Hypoxanthine-guanine phosphoribosyltransferase; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; TBP: TATA-binding protein.
Author contributions
M. Waasdorp wrote the manuscript, designed experiments and researched data. JW. Duitman designed experiments, researched data and critically reviewed the manuscript. S. Florquin contributed to conception, discussion and critically reviewed the manuscript. C.A. Spek contributed to conception, design of experiments, discussion and critically reviewed the manuscript. All authors approved the final version for publication. M. Waasdorp is the guarantor of this work and takes responsibility for the integrity of the data and the accuracy of the data analysis.
ACKNOWLEDGMENTS
We thank Joost Daalhuisen and Marieke ten Brink for their technical assistance during the animal experiments.
CONFLICTS OF INTEREST
The authors declare that there is no conflicts of interest associated with this manuscript.
FUNDING
This work was supported by the Dutch Diabetes Foundation (2012.00.1471 and 2009.11.001).
REFERENCES
1. Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001; 414:782–7. https://doi.org/10.1038/414782a.
2. Hovind P, Tarnow L, Rossing P, Jensen BR, Graae M, Torp I, Binder C, Parving HH. Predictors for the development of microalbuminuria and macroalbuminuria in patients with type 1 diabetes: inception cohort study. BMJ. 2004; 328:1105. https://doi.org/10.1136/bmj.38070.450891.FE.
3. Adler AI, Stevens RJ, Manley SE, Bilous RW, Cull CA, Holman RR, UKPDS GROUP. Development and progression of nephropathy in type 2 diabetes: the United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int. 2003; 63: 225–32. https://doi.org/10.1046/j.1523-1755.2003.00712.x.
4. Martinez-Castelao A, Navarro-Gonzalez JF, Gorriz JL, de Alvaro F. The Concept and the Epidemiology of Diabetic Nephropathy Have Changed in Recent Years. J Clin Med. 2015; 4:1207–16. https://doi.org/10.3390/jcm4061207.
5. Foley RN, Collins AJ. The growing economic burden of diabetic kidney disease. Curr Diab Rep. 2009; 9:460–5.
6. Madhusudhan T, Kerlin BA, Isermann B. The emerging role of coagulation proteases in kidney disease. Nat Rev Nephrol. 2016; 12:94–109. https://doi.org/10.1038/nrneph.2015.177.
7. Packham DK, Wolfe R, Reutens AT, Berl T, Heerspink HL, Rohde R, Ivory S, Lewis J, Raz I, Wiegmann TB, Chan JC, de Zeeuw D, Lewis EJ, et al. Sulodexide fails to demonstrate renoprotection in overt type 2 diabetic nephropathy. J Am Soc Nephrol. 2012; 23:123–30. https://doi.org/10.1681/ASN.2011040378.
8. Lewis EJ, Lewis JB, Greene T, Hunsicker LG, Berl T, Pohl MA, de Zeeuw D, Heerspink HL, Rohde RD, Atkins RC, Reutens AT, Packham DK, Raz I, et al. Sulodexide for kidney protection in type 2 diabetes patients with microalbuminuria: a randomized controlled trial. Am J Kidney Dis. 2011; 58:729–36. https://doi.org/10.1053/j.ajkd.2011.06.020.
9. Heerspink HL, Greene T, Lewis JB, Raz I, Rohde RD, Hunsicker LG, Schwartz SL, Aronoff S, Katz MA, Eisner GM, Mersey JH, Wiegmann TB; Collaborative Study Group. Effects of sulodexide in patients with type 2 diabetes and persistent albuminuria. Nephrol Dial Transplant. 2008; 23:1946–54. https://doi.org/10.1093/ndt/gfm893.
10. Sulikowska B, Olejniczak H, Muszynska M, Odrowaz-Sypniewska G, Gaddi A, Savini C, Cicero AF, Laghi L, Manitius J. Effect of sulodexide on albuminuria, NAG excretion and glomerular filtration response to dopamine in diabetic patients. Am J Nephrol. 2006; 26:621–8. https://doi.org/10.1159/000098195.
11. Gambaro G, Kinalska I, Oksa A, Pont’uch P, Hertlova M, Olsovsky J, Manitius J, Fedele D, Czekalski S, Perusicova J, Skrha J, Taton J, Grzeszczak W, et al. Oral sulodexide reduces albuminuria in microalbuminuric and macroalbuminuric type 1 and type 2 diabetic patients: the Di.N.A.S. randomized trial. J Am Soc Nephrol. 2002; 13:1615–25.
12. Dedov I, Shestakova M, Vorontzov A, Palazzini E. A randomized, controlled study of sulodexide therapy for the treatment of diabetic nephropathy. Nephrol Dial Transplant. 1997; 12:2295–300.
13. Coccheri S. Game not over for sulodexide. Am J Kidney Dis. 2012; 59: 467. https://doi.org/10.1053/j.ajkd.2011.12.010.
14. House AA, Weir MA. Sulodexide for diabetic nephropathy: another one bites the dust. Am J Kidney Dis. 2011; 58:692–4. https://doi.org/10.1053/j.ajkd.2011.08.011.
15. Coughlin SR. Protease-activated receptors in vascular biology. Thromb Haemost. 2001; 86:298–307.
16. Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood. 2012; 120:5237–46. https://doi.org/10.1182/blood-2012-08-452169.
17. Pendurthi UR, Ngyuen M, Andrade-Gordon P, Petersen LC, Rao LV. Plasmin induces Cyr61 gene expression in fibroblasts via protease-activated receptor-1 and p44/42 mitogen-activated protein kinase-dependent signaling pathway. Arterioscler Thromb Vasc Biol. 2002; 22:1421–6.
18. Jaffre F, Friedman AE, Hu Z, Mackman N, Blaxall BC. Beta-adrenergic receptor stimulation transactivates protease-activated receptor 1 via matrix metalloproteinase 13 in cardiac cells. Circulation. 2012; 125:2993–3003. https://doi.org/10.1161/CIRCULATIONAHA.111.066787.
19. Waasdorp M, Duitman J, Florquin S, Spek CA. Protease-activated receptor-1 deficiency protects against streptozotocin-induced diabetic nephropathy in mice. Sci Rep. 2016; 6:33030. https://doi.org/10.1038/srep33030.
20. TRA*CER Executive and Steering Committees. The Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome (TRA*CER) trial: study design and rationale. Am Heart J. 2009; 158:327–34 e4. https://doi.org/10.1016/j.ahj.2009.07.001.
21. Morrow DA, Scirica BM, Fox KA, Berman G, Strony J, Veltri E, Bonaca MP, Fish P, McCabe CH, Braunwald E; TRA 2(o)P-TIMI 50 Investigators. Evaluation of a novel antiplatelet agent for secondary prevention in patients with a history of atherosclerotic disease: design and rationale for the Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events (TRA 2 degrees P)-TIMI 50 trial. Am Heart J. 2009; 158:335–41.e3. https://doi.org/10.1016/j.ahj.2009.06.027.
22. Tricoci P, Huang Z, Held C, Moliterno DJ, Armstrong PW, Van de Werf F, White HD, Aylward PE, Wallentin L, Chen E, Lokhnygina Y, Pei J, Leonardi S, et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012; 366:20–33. https://doi.org/10.1056/NEJMoa1109719.
23. Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, Fox KA, Lipka LJ, Liu X, Nicolau JC, Ophuis AJ, Paolasso E, Scirica BM, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012; 366:1404–13. https://doi.org/10.1056/NEJMoa1200933.
24. Cavender MA, Scirica BM, Bonaca MP, Angiolillo DJ, Dalby AJ, Dellborg M, Morais J, Murphy SA, Ophuis TO, Tendera M, Braunwald E, Morrow DA. Vorapaxar in patients with diabetes mellitus and previous myocardial infarction: findings from the thrombin receptor antagonist in secondary prevention of atherothrombotic ischemic events-TIMI 50 trial. Circulation. 2015; 131:1047–53. https://doi.org/10.1161/CIRCULATIONAHA.114.013774.
25. Moschonas IC, Tselepis AD. Increased Benefit With Vorapaxar Use in Patients With a History of Myocardial Infarction and Diabetes Mellitus: What the Data Show Us. J Cardiovasc Pharmacol Ther. 2017; 22:133–141. https://doi.org/10.1177/1074248416662347.
26. Duitman J, Ruela-de-Sousa RR, Shi K, de Boer OJ, Borensztajn KS, Florquin S, Peppelenbosch MP, Spek CA. Protease activated receptor-1 deficiency diminishes bleomycin-induced skin fibrosis. Mol Med. 2014; 20:410–6. https://doi.org/10.2119/molmed.2014.00027.