
INTRODUCTION
Brain metastases represent a frequent source of morbidity and mortality for breast cancer patients [1]. The incidence of brain metastasis in patients with metastatic breast cancer varies from 10 to 15% [2] and these rates are as high as 50% in patients with the Erb-B2 receptor tyrosine kinase 2 (ERBB2 or HER2)-positive breast cancer [3].
The central nervous system (CNS) remains a sanctuary site for HER2-positive breast cancer [4, 5]. Whilst anti-HER2 targeted therapies have resulted in better therapeutic control for systemic disease [6–8], CNS metastases often occur [9]. Several hypotheses may explain this clinical phenomenon, including the poor or non-penetration of trastuzumab, a recombinant humanized anti-HER2 monoclonal antibody, across the blood-brain barrier, better imaging methods for the diagnosis of brain metastasis and the increased life expectancy of HER2-positive breast cancer patients with newer anti-HER2-targeted therapies [10].
Yet there is a limited understanding of how brain metastases evolve from their primary HER2-positive breast cancers and how they can be effectively targeted in clinical practice. In this study we hypothesized that clinically metachronous brain metastasis from HER2-positive breast cancers would differ in their repertoire of somatic genetic alterations from their respective primary tumor, and that potentially targetable driver genetic alterations would be enriched in or restricted to the metastases, and could be employed as genetic biomarkers to guide the rational use of targeted agents. The aims of this study were i) to define the repertoire of somatic genetic alterations in primary HER2-positive breast cancers and their corresponding brain metastases in patients whose lesions metastatic to the brain were collected at rapid post-mortem examination or surgical excision of the brain metastases, ii) to analyze the temporal heterogeneity involved in the progression of HER2-positive breast cancers to brain metastasis, and iii) to identify potential clinically actionable alterations that may allow targeting brain metastasis in HER2-positive breast cancer patients.
RESULTS
The genetic diversity of a brain metastasis from a HER2-positive breast cancer patient subjected to a rapid post-mortem analysis
The index patient was a 36-year-old female with an ER-negative, HER2-positive invasive ductal carcinoma and associated liver metastasis (cT3N3M1) at presentation in 2012. The patient received first-line therapy with the anti-HER2 trastuzumab plus paclitaxel; following successive visceral and brain progression (7.5 months after diagnosis), the patient received whole brain radiotherapy and three additional lines of anti-HER2 therapies combined with cytotoxic and/or targeted agents i) capecitabine, trastuzumab, BKM120; ii) lapatinib, capecitabine, iii) vinorelbine, trastuzumab. The patient expired 18.6 months after the initial diagnosis and was subjected to a rapid post-mortem examination (Figure 1A and 1B and Table 1). In the final radiological assessment ~21 days before death, the patient had no measurable systemic extra-cranial visceral metastases (the liver metastasis was in clinical complete remission) except for the supra and infratentorial anatomically distinct brain metastases that were longitudinally followed up from the diagnosis of the CNS infiltration to the post-mortem analysis (Figure 1A and 1B).
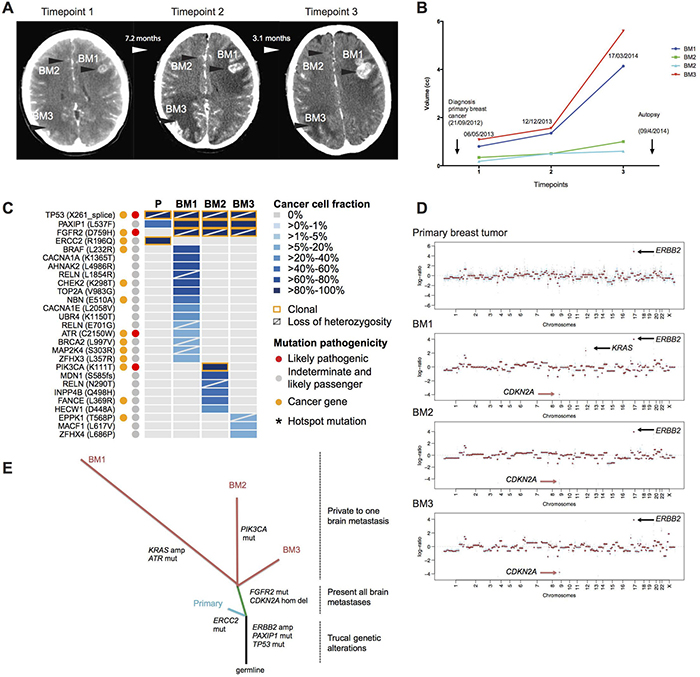
Figure 1: The somatic genetic alterations in the primary breast tumor and the three synchronous, spatially distinct brain metastatic lesions of the index case. (A) Longitudinal representation of computer tomography scans, showing coronal sections in T1W1 and the presence of 3 main lesions: BM1, left frontal lobe, BM2, basilar ganglion, BM3, right occipital lobe during the timepoint 1 (06.05.2013), timepoint 2 (12.12.2013) and timepoint 3 (17.03.2014). (B) Bar graphs representing the volumes of main brain lesions that were followed up in the clinics. Note that two small lesions in the basilar ganglion (BM2) were followed up in the clinical care of this patient. (C) Heatmap depicting the cancer cell fractions (CCF) of the mutations as defined by ABSOLUTE [11] in the primary breast cancer and the metachronous brain metastases. Color key for CCF is depicted. Red and orange dots indicate likely pathogenic mutations and mutations affecting cancer genes [12–14], respectively. Asterisk (*) indicate hotspot mutation [17]. The presence of loss of heterozygosity is represented by a diagonal bar, and clonal mutations are indicated by an orange box. (D) Genome plots of the primary tumor and the three brain metastatic lesions highlighting ERBB2 gene amplification, KRAS amplification and CDKN2A homozygous deletion. Smoothed Log2 ratios were plotted on the y-axis according to their genomic positions indicated on the x-axis. (E) Phylogenetic tree constructed from the somatic mutations, amplifications and homozygous deletions highlighting the main genetic alterations for each sample sequenced. The branch lengths are proportional to the number of genetic alterations.
Table 1: Clinico-pathologic characteristics and outcome of the six patients included in the study
Patient ID |
Breast cancer subtype |
Histologic subtype |
Age at diagnosis (years) |
Anti-HER2 therapy administered between the diagnosis of the primary breast cancer and the collection of the brain metastases |
Time to brain metastasis diagnosis (months) |
Extra-cranial disease |
Overall survival (months) |
|---|---|---|---|---|---|---|---|
Index |
ER-negative/HER2-positive |
IDC |
36 |
Trastuzumab plus taxanes |
7.5 |
No extra-cranial disease at the last clinical assessment before death |
18.6 |
Case 1 |
ER-negative/HER2-positive |
ILC |
44 |
Neo (adjuvant): anthracycline and taxanes-based plus trastuzumab |
26.3 |
Bone metastasis in lumbar vertebrae (L4) synchronous to brain metastasis |
67 |
Case 2 |
ER-positive/HER2-positive |
IDC |
40 |
Adjuvant: anthracycline and taxanes-based plus endocrine therapy. No adjuvant trastuzumab due to low ventricular ejection fraction |
26.1 |
Axillary lymph node and leptomeningeal carcinomatosis a posteriori to the diagnosis of brain metastasis |
58.3 |
Case 6 |
ER-positive/HER2-positive |
IDC |
30 |
Adjuvant: anthracycline and taxanes-based plus trastuzumab and endocrine therapy. Metastatic setting: Capecitabine plus vinorelbine |
39 |
Bone lesion resected, then irradiated |
52 |
Case 12 |
ER-positive/HER2-positive |
IDC |
40 |
Neo (adjuvant): anthracycline and taxanes-based plus adjuvant chemotherapy followed by trastuzumab and endocrine therapy |
18.1* |
No |
62.6 |
Case 14 |
ER-negative/HER2-positive |
IDC |
52 |
Neoadjuvant: anthracycline and taxanes-based plus trastuzumab |
19.5 |
No |
27.6 |
ER, estrogen receptor; IDC, invasive ductal carcinoma; ILC, invasive lobular carcinoma.
*Time to the first brain metastasis.
The primary tumor and three spatially separated brain metastases were subjected to high-depth targeted sequencing of 254 genes frequently mutated in breast cancer and/or related to DNA repair (Supplementary Table 1) to 238× in the primary tumor, a median depth of 1,267× (range: 1,056×–1,503×) in the metastases and to 1,109× in the matched normal sample (Supplementary Table 2). All mutations found by targeted sequencing where genomic DNA was available were subjected to orthogonal validation using amplicon resequencing (Supplementary Tables 3 and 4).
We sought to define whether intra-tumor genetic heterogeneity would be present between the primary breast cancer and the brain metastatic deposits, and whether the progression from HER2-positive primary breast cancer metastatic to the brain following anti-HER2 therapy was associated with shifts in clonal composition. Overall, the primary breast cancer and the three synchronous and spatially distinct brain metastatic lesions, in addition to displaying a clonal ERBB2 (HER2) gene amplification, harbored 27 somatic mutations (Figure 1C). The likely pathogenic TP53 X261_splice mutation was clonal (as defined by ABSOLUTE [11]) and ubiquitously present in the primary tumor and in the three brain metastatic deposits. The PAXIP1 L537F mutation was subclonal in the primary tumor and expanded to become clonal in the brain metastases. Among the 27 somatic mutations, 24 were restricted to the brain metastases, of which 4 affected cancer-related genes (i.e., FGFR2, MAP2K4, ATR and PIK3CA) (Figure 1C) [12–16]. The likely pathogenic FGFR2 D759H missense mutation was clonal in the three metastatic deposits found in the patient. Most of the somatic mutations identified were private to one of the metastatic deposits and the likely pathogenic hotspot PIK3CA K111T mutation, a recurrently mutated residue in breast cancer [16], was private to and clonal in the brain metastasis #2 (BM2).
Analysis of the CNAs demonstrated that ERBB2 amplification was present both in the primary tumor and all brain metastases; reinforcing its role as an early ‘truncal’ event (Figure 1D). By contrast, we observed a homozygous deletion on chromosome 9p21.3 in the three brain metastases but not in the primary breast cancer. This locus encompasses the CDKN2A (p16) gene, an important tumor suppressor gene with a central role in cell cycle regulation. Furthermore, the brain metastasis #1 (BM1) showed a private (i.e., not present in the primary tumor or in the other synchronous brain metastases) amplification on chromosome 12p12.1, encompassing the KRAS gene.
A phylogenetic analysis of the somatic mutations and CNAs of the index patient suggested that the brain metastatic lesions diverged from the primary tumor and acquired additional likely pathogenic mutations in the cancer genes FGFR2, PIK3CA and ATR, homozygous deletion in CDKN2A and amplification in KRAS (Figure 1E). This led us to expand our cohort to investigate temporal heterogeneity in pairs of HER2-positive primary breast tumor and brain metastases.
Characterization of somatic genetic alterations of primary breast cancers and their metachronous brain metastases in five additional patients
Sequencing of an additional five primary HER2-positive breast cancers and corresponding six brain metastases was performed to median depths of 447× (range: 84×–645×), 392× (range: 139×–1,148×) and 467× (range: 196×–1,790×) in the primary tumors, brain metastases and matched normal counterparts, respectively (Table 1, Supplementary Table 2).
Across the five cases, the primary tumors and the metastases harbored a median of 3 (range: 1–24) and 10 (range: 1–38) somatic mutations, respectively (Supplementary Table 4), with a higher number of somatic mutations in the brain metastases than the corresponding primary tumors (P = 0.02, paired Mann-Whitney U-test, Figures 1C and 2). Overall, 111 somatic mutations targeting 73 genes were detected (median 16.5 per case, range 1–49), including 18 likely pathogenic mutations in 11 cancer genes (AKAP9, ATM, ATR, BRAF, CDH1, ERBB2, FGFR2, MLH1, PIK3CA, RAF1 and TP53, Figures 1C and 2). A pairwise comparison revealed that a median of 17% (range: 0%–100%) of the mutations were shared by the primary tumors and corresponding brain metastases, while a median of 15% (range: 0%–22%) and 60% (range: 0%–88%) were restricted to the primary tumors and metastases, respectively (Figures 1C and 2). Hotspot mutations [17] present in both the primary tumor and brain metastasis were identified in Cases 1 (PIK3CA E542K) and 12 (TP53 H214R and PIK3CA H1047R, Figure 2). Most CNAs were present in both the primary tumor and the brain metastases (Supplementary Figure 2), consistent with the notion that CNAs are early events in breast cancer tumorigenesis, likely constituting punctuated evolutionary bursts [18]. Importantly, ERBB2 amplification was present and inferred by FACETS [19] to be clonal in all primary tumors and their respective brain metastases, therefore likely represented a ‘truncal’ genetic event. Further focal amplifications present in both the primary tumors and their respective brain metastases affected 19q13.33 (encompassing NR1H2 and POLD1, Case 2) and 12q13.2–15 (ERBB3, CDK4 and MDM2, Case 6).

Figure 2: The repertoire of somatic genetic alterations in primary breast cancers and their respective brain metastases. Heatmap depicting the cancer cell fractions (CCF) of the mutations as defined by ABSOLUTE [11] in the primary breast tumors and the metachronous brain metastases. CCFs are depicted according to the color key. Red and orange dots indicate likely pathogenic mutations and mutations affecting cancer genes [12–14], respectively. Asterisks (*) indicate hotspot mutations [17]. The presence of loss of heterozygosity (LOH) of the wild-type allele is represented by a diagonal bar, and mutations considered clonal by ABSOLUTE [11] are indicated by an orange box.
Of particular interest are the 61 genes affected by mutations selected for in the brain metastases, including those mutations private to, enriched in, or associated with secondary LOH event in the brain metastasis. These genes included 54 genes affected by 73 mutations that were private to the brain metastases, 31 of which that were clonal in at least one of the brain metastases analyzed, including two hotspot mutations in TP53 (Case 1, Y236C and Case 2, R248W), as well as seven likely pathogenic mutations in ATR, FGFR2, PIK3CA, RAF1, TP53, and WDFY3 (Figures 1C and 2). We further identified 11 genes affected by 12 mutations that were enriched in the brain metastasis, including pathogenic hotspot mutations in PIK3CA (Case 12, H1047R) and TP53 (Case 12, H214R). Two genes (TOP2A and TP53) were affected by three shared mutations that were associated with a secondary LOH event in the metastases but not in the primary tumor in Case 14, including a TP53 Q331* truncating mutation. Collectively, the mutations private to, enriched in, or associated with a secondary LOH event in the brain metastasis affected 61 genes. Additionally, the index case harbored copy number alterations restricted to the brain metastases (KRAS amplification in brain metastasis #1, CDKN2A homozygous deletion in all three brain metastases) (Figure 1D).
Mutational processes that shape the human genome can be deduced from the pattern of somatic mutations detected in cancer cells [20]. For Case 14, the high mutation burden allowed us to identify signatures associated with increased APOBEC cytidine deaminase activity (2 and 13 [20]) as the predominant signatures among the mutations shared between the primary tumor and the brain metastasis and among the mutations private to the metastasis (Supplementary Figure 1). Mutations private to the primary breast cancer were associated with signature 2 (APOBEC) and 3 (homologous recombination deficiency), although the number of mutations private to the primary was small and the mutations were not associated with an increased burden of small insertions and deletions. In the remaining cases, this analysis was not possible, as the tumors harbored < 20 somatic mutations.
Taken together, these results suggest that primary HER2-positive breast cancers and their respective metachronous brain metastases displayed important genetic differences that affected cancer genes. Of note, four of the six cases harbored TP53 likely pathogenic mutations (three hotspot mutations and one truncating mutation) that were private to or enriched in the brain lesions, all of which were associated with LOH of the wild-type allele in the metastatic deposit.
Tumor progression from the breast cancer to the brain site is underpinned by temporal genetic heterogeneity
The tissue samples in Case 12 were longitudinally collected, thus we sought to define whether shifts in clonal composition would take place in the progression from the primary tumor to the brain metastases in this HER2-positive breast cancer following anti-HER2 therapy (Table 1).
Case 12 was a 40-year-old woman, diagnosed in 2005 with locally advanced ER-positive/HER2-positive breast cancer (Figure 3A). The patient was treated with neoadjuvant therapy (anthracycline and taxane-based therapy), mastectomy and adjuvant chemotherapy, followed by adjuvant radiotherapy and trastuzumab plus tamoxifen. The patient developed a single cerebellum metastasis (brain metastasis # 1) 18.1 months from after the diagnosis of primary breast cancer. This patient had a complete surgical excision of the brain metastasis in 2007 (ER- and PR-negative, HER2 not tested during clinical care), followed by radiotherapy. The patient recurred after 17 months and was treated with radiosurgery and anti-HER2 and taxane-based therapies achieving partial response with an incomplete surgical excision in 2009. The residual lesion increased in size five months after surgery. The patient started treatment with lapatinib and capecitabine. The brain metastasis # 2 was surgically treated in 2010 after six months on chemotherapy (ER- and PR-negative, HER2-positive) (Supplementary Figure 3). After a new CNS progression, the patient was treated with anti-HER2 systemic therapy, and expired in 2011.
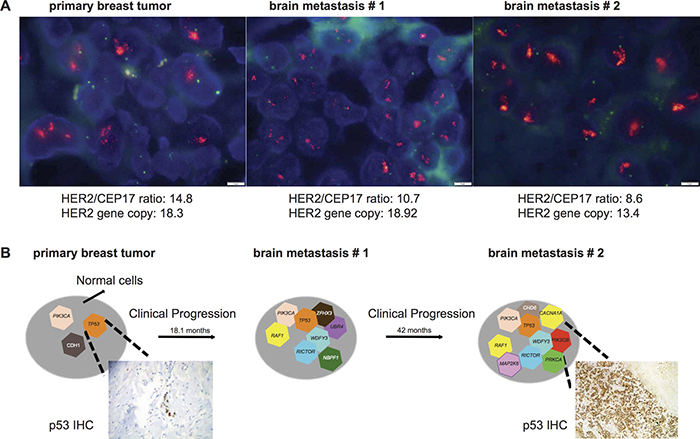
Figure 3: Progression and associated shift in clonal composition of Case 12. (A) Fluorescent in situ hybridization representative micrographs characterize the HER2 status of the primary breast cancer, brain metastasis #1, brain metastasis #2 as HER2-amplified. HER2/CEP17 ratio and HER2 gene copy number are shown for each sample. Scale bar: 5 microns. (B) Schematic shows shifts in clonal composition from the diagnosis of primary breast cancer to the development of two sequential brain metastases over a period of 60.8 months. Genes affected by likely pathogenic mutations are illustrated. P53 expression has a focal pattern in the primary tumor and is enriched in the brain metastasis (more than 80% of the cells) as depicted in the representative micrograph of immunohistochemistry with antibodies against p53 (See Materials and Methods, original magnifications 100X).
The analysis of the sequential brain metastases revealed that two likely pathogenic hotspot mutations TP53 H214R and PIK3CA H1047R were enriched in the brain metastasis as compared to the primary tumor (Figure 2 and Figure 3B). In particular, the enrichment of the TP53 H214R mutation coupled with LOH of the wild-type allele in the brain metastases was confirmed by p53 IHC demonstrating the evident enrichment of p53 expression in more than 80% of the cells in brain metastasis #2 compared to the primary breast cancer (Figure 3B). We identified six mutations that were present in both brain metastases but not in the primary tumor, including three likely pathogenic clonal mutations affecting RAF1 and WDFY3. Additionally, private mutations were identified in each of the two brain metastases, including a likely pathogenic mutation in ZNF703 in brain metastasis #1 and likely pathogenic mutations affecting MAP2K6, PRKCA, CACNA1A and CHD6 in brain metastasis #2 (Figure 3B). These results suggest that progression from primary tumor to brain metastasis after several lines of chemo and targeted therapy was associated with clonal shifts and the acquisition of additional mutations.
Clinical ‘actionability’ of genetic alterations present in HER2-positive brain metastases
Given that the progression of the breast cancers to the brain metastasis (median 22.75 months, range 7.5–39, Table 1) showed evidence of spatial and temporal genetic heterogeneity, we interrogated the 402-gene list derived from DGIdb [21, 22] and the 373-variant list from OncoKB [23] (Supplementary Tables 5 and 6) to determine the actionability of the likely pathogenic mutations that were private to, enriched in or associated with secondary LOH events in the brain lesions, and the genes affected by CNAs private to the brain lesions.
Among the 54 genes affected by 73 mutations, as well as KRAS and CDKN2A (affected by amplification and homozygous deletion, respectively, restricted to the brain metastases in the Index case) private to, enriched in or associated with secondary LOH events in the brain lesions, 11 genes were considered actionable based on DGIdb (Table 2). In particular, both mutations in PIK3CA and all four mutations in TP53 affected hotspot residues (Table 2). Considering the actionability of specific alterations based on OncoKB, CDKN2A homozygous deletion (level 4 evidence), and both oncogenic mutations in PIK3CA were considered clinically actionable (level 3A evidence for breast cancer, Table 2). Taken together, our results suggest that a small but important subset of genetic alterations private to, enriched in or associated with secondary LOH events in the brain lesions may be clinically targetable.
Table 2: Potentially targetable genes and systemic therapies for genes harboring likely pathogenic mutations that were private to, enriched in or associated with secondary LOH events in the brain lesions, and the genes affected by CNAs private to the brain lesions
Actionable genes (DGIdb) |
Private to the brain metastases |
Hotspot mutation |
Actionable alteration (OncoKB) |
Selected systemic therapies |
|---|---|---|---|---|
ATR |
Index case (C2150W)* |
- |
- |
Nirapanib, BMN673, Olaparib, Rucaparib, |
Case 14 (E1878Q)* |
- |
- |
Veliparib |
|
BRAF |
Case 14 (E703D)* |
- |
- |
Vemurafenib, Dabrafenib, Dabrafenib + Trametinib, Vemurafenib + Cobimetinib, Trametinib (Level 1) |
CDKN2A |
Index case (homozygous deletion)* |
- |
Yes |
Palbociclib + Letrozole (Level 4) |
FGFR2 |
Index case (D759H)* |
- |
- |
JNJ-42756493, Debio1347 (Level 3A) |
KRAS |
Index case (amplification)* |
- |
# |
Docetaxel + Trametinib, Abemaciclib, Erlotinib + Binimetinib, Selumetinib, Binimetinib, Ribociclib + Trametinib, Palbociclib, Ribociclib, Palbociclib + PD0325901, Trametinib Binimetinib + Alpelisib, Cobimetinib + GDC-0994 (Level 4) |
MLH1 |
Case 14 (R100Q)* |
- |
- |
- |
PIK3CA |
Case 12 (H1047R)** |
Yes |
Yes |
Buparlisib, Serabelisib, Alpelisib + Fulvestrant, Copanlisib, Fulvestrant + Taselisib, GDC-0077, |
Index case (K111T)** |
Yes |
Yes |
Alpelisib, Buparlisib + Fulvestrant, Taselisib (Level 3A) |
|
RAF1 |
Case 12 (D19Y and D23H)* |
- |
- |
Sorafenib (Level 4) |
TOP2A |
Case 14 (D524N and D526N)*** |
- |
- |
Doxorrubicin, Teniposide, Valrubicin, Idarubicin, etoposide |
TP53 |
Case 1 (Y236C)* |
Yes |
- |
- |
Case 2 (R248W)* |
Yes |
- |
||
Case 12 (H214R)** |
Yes |
- |
||
Case 14 (Q331*)*** |
Yes |
- |
||
ZNF703 |
Case 12 (S389W)* |
- |
- |
- |
*Private to the brain metastasis
**Enriched in the brain metastasis
***Associated with LOH in the brain metastasis
#as per OncoKB, oncogenic mutations in KRAS are actionable (level 4 evidence).
DISCUSSION
The development of brain metastases is a major limitation to life expectancy and contributes to the poor outcomes to patients with HER2-positive breast cancer. Patients with HER2-positive breast cancer have a higher risk of developing brain metastases [24]. This may be a direct result of the inability for trastuzumab to efficiently overcome the blood-brain barrier. Although traditional modes of administration of trastuzumab improve the outcome of HER2-positive breast cancer patients with brain metastases [25, 26], intrathecal administration might represent an alternative to overcome blood-brain barrier [27]. Studies comparing the genetic alterations in paired primary breast cancer and distant metastases have been performed [28–31]. However, the unique biology and challenging clinical course of HER2-positive breast cancers metastasized to the brain suggest that they warrant further and specific characterization to identify candidate molecular targets.
To define the genetic alterations associated with the development and progression of brain metastasis from patients with HER2-positive breast cancers, we assessed the somatic genetic alterations in the primary breast cancer and in up to three brain metastatic deposits. Recent studies of paired primary breast cancer and distant metastases (including but not limited to the brain) revealed that mutations in TP53, PTEN, KRAS and SMAD4 were frequently restricted to the metastases [28–31]. In addition, ESR1, PALB2, FSIP2, AGRN, FRAS1, IGFN1, EDC4, OSBPL3 were found to be significantly more frequent in prospectively accrued metastatic breast cancers than in primary breast cancers from The Cancer Genome Atlas (TCGA) [32]. These observations, however, largely stem from the analysis of ER-positive/HER2-negative metastatic breast cancers [32]. In our series of six HER2-positive breast cancers with brain metastasis, four cases harbored TP53 likely pathogenic mutations private to or enriched in the brain metastases. Indeed, our results here, as well as those from previous studies [33–36], suggest that breast cancers may undergo clonal shifts and acquire additional somatic genetic alterations in the progression from primary to metachronous brain metastasis after systemic therapy. HER2 amplification as a truncal alteration is likely to be true for the majority of HER2-positive breast cancers. A small subset of HER2-positive breast cancers, however, exhibit non-uniform patterns of HER2 overexpression and HER2 gene amplification [37]. In these cases, it is uncertain whether HER2 amplification is an early alteration that is lost during tumor development or is gained later in the evolution of the tumor. In fact, loss of HER2-positive status can occur in metastatic tumors from patients with primary HER2-positive breast cancer [38], probably due to eradication of the HER2-positive major clone and secondary expansion of a minor clone lacking HER2 gene amplification. An important clinical implication of this observation is that sequencing of primary biopsies alone may miss a substantial number of opportunities for targeted therapy in this cohort of patients [34].
Currently, there are increasing efforts to match potential clinically actionable genetic alterations with targeted therapy to accomplish the goals of precision medicine in the context of brain metastases [34]. A recent sequencing analysis of a series of 86 pairs of primary solid tumors and matched brain metastasis, including 21 cases with breast cancer primary, demonstrated that in 53% of cases, at least one clinically actionable genetic alteration was restricted to the brain metastasis, in particular alterations that may confer sensitivity to PI3K/AKT/mTOR, CDK, HER2/EGFR and MAPK pathway inhibitors [34]. In the six patients analyzed in this study, we found alterations that may confer sensitivity to PI3K/AKT/mTOR, CDK and MAPK pathway inhibitors in two (Index case and Case 12, PIK3CA K111T and H1047R, respectively), one (Index Case, CDKN2A homozygous deletion), and two cases (Case 14, BRAF E703D and Case 12, RAF1 D19Y and D23H), respectively. Additionally, we identified likely pathogenic mutations private to or enriched in the brain metastases that are potentially targetable with PARP inhibitors and/or drugs that target replication stress in two cases (likely pathogenic mutations in ATR in Index case and Case 14), or with multi-target angiokinase inhibitors in one case (FGFR2 D759H in Index case). Taken together, four of six cases (67%) were found to harbor at least one likely pathogenic genetic alteration private to or enriched in the brain metastases that may be clinically targetable (Table 2). Of note, most of these genetic alterations can be targeted with small molecule inhibitors that can potentially cross the blood-brain barrier. Moreover, the majority of brain macrometastases (> 1 mm diameter) show variable extent of disturbance of the blood-brain barrier [39, 40], potentially allowing the small molecule inhibitors and other agents to reach the brain metastases. Our results suggest that different subtypes of HER2-positive breast cancer patients may be defined based on their repertoire of genetic alterations in the brain metastasis and potential targeted drugs directed against these alterations tested in the context of clinical trials.
In Case 14 (ER-negative/HER2-positive) we identified an enrichment in the signatures related to APOBEC-mediated mutagenesis (signatures 2 and 13) [20], which was previously described as enriched in metastatic tumors compared to primary tumors, in the context of HER2-negative breast cancers [32]. Our observation provides evidence to suggest that APOBEC-mediated mutagenesis may contribute to the genetic heterogeneity between the primary tumor and the brain metastasis in HER2-positive breast cancer. Our findings suggest that further studies investigating shifts in mutational signatures in the progression of breast cancers from patients receiving specific modalities of systemic therapies are warranted.
Our study has several limitations. First, due to the challenges posed in tissue procurement of brain metastasis, our cohort size was small. Our patient population comprises patients with HER2-positive breast cancers that developed brain metastasis as a unique or main metastatic site and underwent brain metastasis excision or rapid autopsy. Despite the small sample size, however, we identified potentially actionable genetic alterations private to, enriched in or associated with secondary LOH events in the brain lesions in ATR, BRAF, FGFR2, MAP2K4, PIK3CA, RAF1 and TP53. It should be noted that some of the TP53 and PI3KCA mutations were private mutations (that is, not found in matched primary tumors – TP53: cases 1 and 2; PI3KCA: case index). These variants are usually clonal in primary breast tumors, although they have also been reported to be subclonal in a subset of primary breast cancers [41]. In this cohort, these alterations were clonal in the brain metastasis of cases index and 1. We cannot exclude the possibility that these mutations would have been detected in the primary tumors with deeper sequencing or as a minor clone in spatially distinct areas of the primary tumor. It is unlikely, however, that these alterations would not have been detected in the primary tumor if they were present in the dominant clone of the primary tumor. In addition, caution should be exercised in the interpretation the clinical impact of these somatic genetic alterations, given that these databases are evolving. For example, TP53 mutations are not considered therapeutically actionable by OncoKB [23] but TP53 was considered an actionable gene by DGIdb [21, 22]. Second, we performed a targeted sequencing analysis for the 254 genes frequently mutated in breast cancer and/or involved in DNA repair. Whole-exome/genome sequencing may reveal even more potentially actionable or targetable genetic alterations restricted to or enriched in the metastatic lesions or primary breast cancers [42]. Third, in the expansion cohort, the brain metastases were not amenable to punches or biopsies in spatially separated areas of the tumors. In clinical practice, sampling brain metastases as the sole site of disease or in the context of disseminated extra-cranial breast disease is practically unfeasible [8, 43, 44]. Cell-free DNA from cerebro-spinal fluid [43] may be more appropriate in capturing the genetic heterogeneity and private mutations within the brain metastases, but unlike the current study, we would not have been able to attribute the mutations to the brain metastasis.
Despite these limitations, this study provides evidence that genetic alterations that are private to or enriched in the brain metastases of HER2-positive breast cancers may represent potential targets for pharmacological inhibition. We would contend that studies of larger patient cohorts and of larger collections of genes are warranted to expand the characterization of potentially targetable genetic alterations in HER2-positive breast cancer patients with brain metastasis.
MATERIALS AND METHODS
Patients and samples
An index HER2-positive breast cancer patient with multiple anatomically distinct brain metastatic deposits and complete clinical remission of the systemic disease at the time of death was subjected to a rapid autopsy. Five additional HER2-positive breast cancer patients, who had minimal or absent extra-cranial disease, were subjected to brain metastases excision were also included (Table 1, Supplementary Methods). Tumor and matched normal tissue were subjected to microdissection and DNA extraction as previously reported [43].
Immunohistochemistry and fluorescence in situ hybridization
HER2 status was assessed in all primary breast tumor tissues by either immunohistochemistry (IHC) and/or fluorescence in situ hybridization (FISH) according to the American Society of Clinical Oncologists (ASCO)/College of American Pathologists (CAP) guidelines [45] (Supplementary Methods). p53 IHC was performed using the clone DO7 antibody (Cell Marque, Rocklin, CA). Tumors were classified as p53-positive if > 10% of morphologically unequivocal neoplastic cells displayed strong nuclear expression as previously described [46].
Targeted capture massively parallel sequencing
Tumor and matched normal DNA samples were subjected to targeted sequencing using a previously described customized panel targeting all exons of 254 genes recurrently mutated in breast cancer and/or related to DNA repair (Supplementary Table 1 and Supplementary Methods) [47, 48]. Bioinformatics analyses were performed as previously described [46–51]. Allele-specific copy number alterations (CNAs) and loss of heterozygosity (LOH) of the wild-type allele in genes harboring a somatic mutation were inferred using FACETS [19]. Sequence reads have been deposited to the NCBI Sequence Read Archive (SRP070781).
Validation of mutations by amplicon sequencing
Selected mutations found by targeted sequencing (n = 108, consisting of 104 unique mutations) were subjected to orthogonal validation using amplicon resequencing in all samples for a given patient, where sufficient genomic DNA was available (Supplementary Table 3 and Supplementary Methods). The validation rate of the somatic mutations with sufficient coverage was 93% (100/108).
Identification of mutations private to or enriched in the brain metastatic lesion
ABSOLUTE (v1.0.6) [11, 52] was used to infer the cancer cell fraction (CCF) of mutations and the mutations were classified as clonal or subclonal as previously described [46–48, 53] (Supplementary Methods). Mutations were defined as ‘private to the metastatic lesion’ if they were absent in the primary tumor but present in the brain metastasis. Mutations were defined as ‘enriched in the brain metastasis’ if their CCFs increased by at least 20% in the brain metastases compared to the respective primary tumors. Mutations associated with LOH of the wild-type allele in the metastasis but not in the corresponding primary tumor were defined as ‘LOH in metastasis’.
Identification of potentially pathogenic and/or actionable somatic genetic alterations
MutationTaster, CHASM (breast) and FATHMM [54–56] were used to define the potential functional effect of missense single nucleotide variants as previously described [46–48, 53, 57] (Supplementary Methods). Frameshift, splice-site and nonsense mutations were considered likely pathogenic if they were targeted by loss of the wild-type allele or affected haploinsufficient genes [58]. The Drug-Gene Interaction database (DGIdb) [21, 22] and the OncoKB platform [23] were used to investigate the clinical actionability of mutated genes (Supplementary Methods).
Phylogenetic tree construction
A maximum parsimony tree was built for the index case using binary presence/absence matrix based on the somatic non-synonymous and synonymous mutations, gene amplifications and homozygous deletions in the biopsies of the primary tumor and the metastatic lesions, as previously described [59, 60] and Supplementary Methods.
Mutational signatures
Decomposition of the mutational signature was performed using deconstructSigs [61], based on the set of 30 mutational signatures (“signature.cosmic”, based on the signatures at http://cancer.sanger.ac.uk/cosmic/signatures [15, 20]), for the case with at least 20 somatic mutations.
Statistical analysis
All statistical analyses were performed in R v3.1.2. Comparisons of continuous variables were performed using Mann-Whitney U tests. Association was performed using the Spearman rank-correlation test. All statistical tests were two-tailed and P < 0.05 was considered statistically significant.
Author contributions
LDM-A and JSR-F conceived and designed the study. LDM-A, MG-C, SV, VP, JS, JC provided the samples. NF, VP, FT performed sample microdissection and histological review. LDM-A, CKYN, SP, CT, NF, AMS, RSL, FTC, SRC, GMS, BW, JSR-F developed methodology. LDM-A, CKYN, SP, MRDF, NF, AMS, GMS, BW, JS, JSR-F analyzed and interpreted the data. BW and JSR-F supervised the study. LDM-A wrote the first draft of the manuscript, which was initially revised by CKYN and JSR-F. All authors subsequently revised and approved the manuscript.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
FUNDING
J.S. Reis-Filho is funded in part by the Breast Cancer Research Foundation. L. De Mattos-Arruda was partly funded by Rafael de Pino and Asociacion Espanola Contra el Cancer. The authors also acknowledge CELLEX Foundation and ESMO Fellowship (for LDM-A). S. Piscuoglio is currently funded in part by the Swiss National Foundation (Ambizione grant number PZ00P3_168165). A. M. Schultheis was funded by a stipend from the German Cancer Aid (Dr. Mildred Scheel Stiftung). Research reported in this publication was supported in part by a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (Grant No. P30CA008748). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
1. De Ieso PB, Schick U, Rosenfelder N, Mohammed K, Ross GM. Breast cancer brain metastases - A 12 year review of treatment outcomes. Breast. 2015; 24:426–33. https://doi.org/10.1016/j.breast.2015.03.007.
2. Gonzalez-Angulo AM, Hortobagyi GN. Brain metastases and breast cancer subtypes. Onkologie. 2010; 33:143–44. https://doi.org/10.1159/000296307.
3. Hess KR, Esteva FJ. Effect of HER2 status on distant recurrence in early stage breast cancer. Breast Cancer Res Treat. 2013; 137:449–55. https://doi.org/10.1007/s10549-012-2366-0.
4. Lin NU, Winer EP. Brain metastases: the HER2 paradigm. Clin Cancer Res. 2007; 13:1648–55. https://doi.org/10.1158/1078-0432.CCR-06-2478.
5. Singh JC, Jhaveri K, Esteva FJ. HER2-positive advanced breast cancer: optimizing patient outcomes and opportunities for drug development. Br J Cancer. 2014; 111:1888–98. https://doi.org/10.1038/bjc.2014.388.
6. Mendes D, Alves C, Afonso N, Cardoso F, Passos-Coelho JL, Costa L, Andrade S, Batel-Marques F. The benefit of HER2-targeted therapies on overall survival of patients with metastatic HER2-positive breast cancer—a systematic review. Breast Cancer Res. 2015; 17:140. https://doi.org/10.1186/s13058-015-0648-2.
7. Swain SM, Baselga J, Miles D, Im YH, Quah C, Lee LF, Cortés J. Incidence of central nervous system metastases in patients with HER2-positive metastatic breast cancer treated with pertuzumab, trastuzumab, and docetaxel: results from the randomized phase III study CLEOPATRA. Ann Oncol. 2014; 25:1116–21. https://doi.org/10.1093/annonc/mdu133.
8. Lin NU, Amiri-Kordestani L, Palmieri D, Liewehr DJ, Steeg PS. CNS metastases in breast cancer: old challenge, new frontiers. Clin Cancer Res. 2013; 19:6404–18. https://doi.org/10.1158/1078-0432.CCR-13-0790.
9. Weil RJ, Palmieri DC, Bronder JL, Stark AM, Steeg PS. Breast cancer metastasis to the central nervous system. Am J Pathol. 2005; 167:913–20. https://doi.org/10.1016/S0002-9440(10)61180-7.
10. Zhu X, Verma S. Targeted therapy in her2-positive metastatic breast cancer: a review of the literature. Curr Oncol. 2015; 22:S19–28. https://doi.org/10.3747/co.22.2363.
11. Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, Beroukhim R, Pellman D, Levine DA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012; 30:413–21. https://doi.org/10.1038/nbt.2203.
12. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013; 502:333–39. https://doi.org/10.1038/nature12634.
13. Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004; 4:177–83. https://doi.org/10.1038/nrc1299.
14. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014; 505:495–501. https://doi.org/10.1038/nature12912.
15. Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, Van Loo P, Ju YS, Smid M, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016; 534:47–54. https://doi.org/10.1038/nature17676.
16. Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, Tsui DW, Liu B, Dawson SJ, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016; 7:11479. https://doi.org/10.1038/ncomms11479.
17. Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, Gao J, Socci ND, Solit DB, Olshen AB, Schultz N, Taylor BS. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016; 34:155–63. https://doi.org/10.1038/nbt.3391.
18. Gao R, Davis A, McDonald TO, Sei E, Shi X, Wang Y, Tsai PC, Casasent A, Waters J, Zhang H, Meric-Bernstam F, Michor F, Navin NE. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat Genet. 2016; 48:1119–30. https://doi.org/10.1038/ng.3641.
19. Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016; 44:e131. https://doi.org/10.1093/nar/gkw520.
20. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al, and Australian Pancreatic Cancer Genome Initiative, and ICGC Breast Cancer Consortium, and ICGC MMML-Seq Consortium, and ICGC PedBrain. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–21. https://doi.org/10.1038/nature12477. Erratum in: Nature. 2013; 502:258. Imielinsk, Marcin [corrected to Imielinski, Marcin].
21. Griffith M, Griffith OL, Coffman AC, Weible JV, McMichael JF, Spies NC, Koval J, Das I, Callaway MB, Eldred JM, Miller CA, Subramanian J, Govindan R, et al. DGIdb: mining the druggable genome. Nat Methods. 2013; 10:1209–10. https://doi.org/10.1038/nmeth.2689.
22. Wagner AH, Coffman AC, Ainscough BJ, Spies NC, Skidmore ZL, Campbell KM, Krysiak K, Pan D, McMichael JF, Eldred JM, Walker JR, Wilson RK, Mardis ER, et al. DGIdb 2.0: mining clinically relevant drug-gene interactions. Nucleic Acids Res. 2016; 44:D1036–44. https://doi.org/10.1093/nar/gkv1165.
23. Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH, Chang MT, Chandarlapaty S, Traina TA, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol. 2017; 1–16. https://doi.org/10.1200/PO.17.00011.
24. Leyland-Jones B. Human epidermal growth factor receptor 2-positive breast cancer and central nervous system metastases. J Clin Oncol. 2009; 27:5278–86. https://doi.org/10.1200/JCO.2008.19.8481.
25. Dawood S, Gonzalez-Angulo AM, Albarracin C, Yu TK, Hortobagyi GN, Buchholz TA, Woodward WA. Prognostic factors of survival in the trastuzumab era among women with breast cancer and brain metastases who receive whole brain radiotherapy: a single-institution review. Cancer. 2010; 116:3084–92. https://doi.org/10.1002/cncr.25115.
26. Le Scodan R, Jouanneau L, Massard C, Gutierrez M, Kirova Y, Cherel P, Gachet J, Labib A, Mouret-Fourme E. Brain metastases from breast cancer: prognostic significance of HER-2 overexpression, effect of trastuzumab and cause of death. BMC Cancer. 2011; 11:395. https://doi.org/10.1186/1471-2407-11-395.
27. Bousquet G, Darrouzain F, de Bazelaire C, Ternant D, Barranger E, Winterman S, Madelaine-Chambin I, Thiebaut JB, Polivka M, Paintaud G, Culine S, Janin A. Intrathecal Trastuzumab Halts Progression of CNS Metastases in Breast Cancer. J Clin Oncol. 2016; 34:e151–55. https://doi.org/10.1200/JCO.2012.44.8894.
28. Bertucci F, Finetti P, Guille A, Adélaïde J, Garnier S, Carbuccia N, Monneur A, Charafe-Jauffret E, Goncalves A, Viens P, Birnbaum D, Chaffanet M. Comparative genomic analysis of primary tumors and metastases in breast cancer. Oncotarget. 2016; 7:27208–19. https://doi.org/10.18632/oncotarget.8349.
29. Goswami RS, Patel KP, Singh RR, Meric-Bernstam F, Kopetz ES, Subbiah V, Alvarez RH, Davies MA, Jabbar KJ, Roy-Chowdhuri S, Lazar AJ, Medeiros LJ, Broaddus RR, et al. Hotspot mutation panel testing reveals clonal evolution in a study of 265 paired primary and metastatic tumors. Clin Cancer Res. 2015; 21:2644–51. https://doi.org/10.1158/1078-0432.CCR-14-2391.
30. Brannon AR, Vakiani E, Sylvester BE, Scott SN, McDermott G, Shah RH, Kania K, Viale A, Oschwald DM, Vacic V, Emde AK, Cercek A, Yaeger R, et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014; 15:454. https://doi.org/10.1186/s13059-014-0454-7.
31. Kovaleva V, Geissler AL, Lutz L, Fritsch R, Makowiec F, Wiesemann S, Hopt UT, Passlick B, Werner M, Lassmann S. Spatio-temporal mutation profiles of case-matched colorectal carcinomas and their metastases reveal unique de novo mutations in metachronous lung metastases by targeted next generation sequencing. Mol Cancer. 2016; 15:63. https://doi.org/10.1186/s12943-016-0549-8.
32. Lefebvre C, Bachelot T, Filleron T, Pedrero M, Campone M, Soria JC, Massard C, Lévy C, Arnedos M, Lacroix-Triki M, Garrabey J, Boursin Y, Deloger M, et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 2016; 13:e1002201. https://doi.org/10.1371/journal.pmed.1002201.
33. Thomson AH, McGrane J, Mathew J, Palmer J, Hilton DA, Purvis G, Jenkins R. Changing molecular profile of brain metastases compared with matched breast primary cancers and impact on clinical outcomes. Br J Cancer. 2016; 114:793–800. https://doi.org/10.1038/bjc.2016.34.
34. Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, Van Allen EM, Lawrence MS, Horowitz PM, Cibulskis K, Ligon KL, Tabernero J, Seoane J, et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015; 5:1164–77. https://doi.org/10.1158/2159-8290.CD-15-0369.
35. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012; 486:395–99.
36. Savas P, Teo ZL, Lefevre C, Flensburg C, Caramia F, Alsop K, Mansour M, Francis PA, Thorne HA, Silva MJ, Kanu N, Dietzen M, Rowan A, et al. The Subclonal Architecture of Metastatic Breast Cancer: Results from a Prospective Community-Based Rapid Autopsy Program “CASCADE”. PLoS Med. 2016; 13:e1002204. https://doi.org/10.1371/journal.pmed.1002204. Erratum in: PLoS Med. 2017; 14:e1002302.
37. Ng CK, Martelotto LG, Gauthier A, Wen HC, Piscuoglio S, Lim RS, Cowell CF, Wilkerson PM, Wai P, Rodrigues DN, Arnould L, Geyer FC, Bromberg SE, et al. Intra-tumor genetic heterogeneity and alternative driver genetic alterations in breast cancers with heterogeneous HER2 gene amplification. Genome Biol. 2015; 16:107. https://doi.org/10.1186/s13059-015-0657-6.
38. Niikura N, Liu J, Hayashi N, Mittendorf EA, Gong Y, Palla SL, Tokuda Y, Gonzalez-Angulo AM, Hortobagyi GN, Ueno NT. Loss of human epidermal growth factor receptor 2 (HER2) expression in metastatic sites of HER2-overexpressing primary breast tumors. J Clin Oncol. 2012; 30:593–99. https://doi.org/10.1200/JCO.2010.33.8889.
39. Lockman PR, Mittapalli RK, Taskar KS, Rudraraju V, Gril B, Bohn KA, Adkins CE, Roberts A, Thorsheim HR, Gaasch JA, Huang S, Palmieri D, Steeg PS, Smith QR. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin Cancer Res. 2010; 16:5664–78. https://doi.org/10.1158/1078-0432.CCR-10-1564.
40. Percy DB, Ribot EJ, Chen Y, McFadden C, Simedrea C, Steeg PS, Chambers AF, Foster PJ. In vivo characterization of changing blood-tumor barrier permeability in a mouse model of breast cancer metastasis: a complementary magnetic resonance imaging approach. Invest Radiol. 2011; 46:718–25.
41. McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015; 7:283ra54. https://doi.org/10.1126/scitranslmed.aaa1408.
42. Hoadley KA, Siegel MB, Kanchi KL, Miller CA, Ding L, Zhao W, He X, Parker JS, Wendl MC, Fulton RS, Demeter RT, Wilson RK, Carey LA, et al. Tumor Evolution in Two Patients with Basal-like Breast Cancer: A Retrospective Genomics Study of Multiple Metastases. PLoS Med. 2016; 13:e1002174. https://doi.org/10.1371/journal.pmed.1002174. Erratum in: PLoS Med. 2017; 14:e1002222.
43. De Mattos-Arruda L, Mayor R, Ng CK, Weigelt B, Martínez-Ricarte F, Torrejon D, Oliveira M, Arias A, Raventos C, Tang J, Guerini-Rocco E, Martínez-Sáez E, Lois S, et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun. 2015; 6:8839. https://doi.org/10.1038/ncomms9839.
44. Chien AJ, Rugo HS. Emerging treatment options for the management of brain metastases in patients with HER2-positive metastatic breast cancer. Breast Cancer Res Treat. 2013; 137:1–12. https://doi.org/10.1007/s10549-012-2328-6.
45. Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, Allred DC, Bartlett JM, Bilous M, Fitzgibbons P, Hanna W, Jenkins RB, Mangu PB, et al, and American Society of Clinical Oncology, and College of American Pathologists. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013; 31:3997–4013. https://doi.org/10.1200/JCO.2013.50.9984.
46. Guerini-Rocco E, Hodi Z, Piscuoglio S, Ng CK, Rakha EA, Schultheis AM, Marchiò C, da Cruz Paula A, De Filippo MR, Martelotto LG, De Mattos-Arruda L, Edelweiss M, Jungbluth AA, et al. The repertoire of somatic genetic alterations of acinic cell carcinomas of the breast: an exploratory, hypothesis-generating study. J Pathol. 2015; 237:166–78. https://doi.org/10.1002/path.4566.
47. Piscuoglio S, Ng CK, Murray M, Burke KA, Edelweiss M, Geyer FC, Macedo GS, Inagaki A, Papanastasiou AD, Martelotto LG, Marchio C, Lim RS, Ioris RA, et al. Massively parallel sequencing of phyllodes tumours of the breast reveals actionable mutations, and TERT promoter hotspot mutations and TERT gene amplification as likely drivers of progression. J Pathol. 2016; 238:508–18. https://doi.org/10.1002/path.4672.
48. Piscuoglio S, Geyer FC, Burke KA, Murray MP, Ng CK, Mota A, Marchio C, Berman SH, Norton L, Brogi E, Weigelt B, Reis-Filho JS. Massively parallel sequencing analysis of synchronous fibroepithelial lesions supports the concept of progression from fibroadenoma to phyllodes tumor. NPJ Breast Cancer. 2016; 2:16035. https://doi.org/10.1038/npjbcancer.2016.35.
49. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–60. https://doi.org/10.1093/bioinformatics/btp324.
50. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–303. https://doi.org/10.1101/gr.107524.110.
51. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–79. https://doi.org/10.1093/bioinformatics/btp352.
52. Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, Wan Y, Zhang W, Shukla SA, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013; 152:714–26. https://doi.org/10.1016/j.cell.2013.01.019.
53. Ng CK, Bidard FC, Piscuoglio S, Geyer FC, Lim RS, de Bruijn I, Shen R, Pareja F, Berman SH, Wang L, Pierga JY, Vincent-Salomon A, Viale A, et al. Genetic Heterogeneity in Therapy-Naïve Synchronous Primary Breast Cancers and Their Metastases. Clin Cancer Res. 2017; 23:4402–15. https://doi.org/10.1158/1078-0432.CCR-16-3115.
54. Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010; 7:575–76. https://doi.org/10.1038/nmeth0810-575.
55. Carter H, Chen S, Isik L, Tyekucheva S, Velculescu VE, Kinzler KW, Vogelstein B, Karchin R. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 2009; 69:6660–67. https://doi.org/10.1158/0008-5472.CAN-09-1133.
56. Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, Day IN, Gaunt TR. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013; 34:57–65. https://doi.org/10.1002/humu.22225.
57. Martelotto LG, Ng CK, De Filippo MR, Zhang Y, Piscuoglio S, Lim RS, Shen R, Norton L, Reis-Filho JS, Weigelt B. Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome Biol. 2014; 15:484. https://doi.org/10.1186/s13059-014-0484-1.
58. Dang VT, Kassahn KS, Marcos AE, Ragan MA. Identification of human haploinsufficient genes and their genomic proximity to segmental duplications. Eur J Hum Genet. 2008; 16:1350–57. https://doi.org/10.1038/ejhg.2008.111.
59. Schliep KP. phangorn: phylogenetic analysis in R. Bioinformatics. 2011; 27:592–93. https://doi.org/10.1093/bioinformatics/btq706.
60. Murugaesu N, Wilson GA, Birkbak NJ, Watkins T, McGranahan N, Kumar S, Abbassi-Ghadi N, Salm M, Mitter R, Horswell S, Rowan A, Phillimore B, Biggs J, et al. Tracking the genomic evolution of esophageal adenocarcinoma through neoadjuvant chemotherapy. Cancer Discov. 2015; 5:821–31. https://doi.org/10.1158/2159-8290.CD-15-0412.
61. Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016; 17:31. https://doi.org/10.1186/s13059-016-0893-4.