
INTRODUCTION
LINE-1 elements are the largest family of human retrotransposons, mobile genetic elements that move in the human genome via an RNA intermediate. The LINE-1 family comprises about 500.000 copies, collectively accounting for as much as 17% of the human genome [1]. Each LINE-1 copy encodes a bicistronic RNA transcript which is translated into a 40 kDa RNA-binding protein (ORF-1) and a 150 kDa protein (ORF-2), the latter endowed with endonuclease and reverse transcriptase (RT) activities [2]. As such, RT is the most highly repeated protein-coding sequence in the genome of higher eukaryots and an essential component of the retrotransposition machinery, required not only for the mobilization of its own coding elements, but also for other non-autonomous retrotransposons, such as Alu and SVA [3].
At the origin of the discovery of mobile elements is their ability to produce phenotypic variations by integrating at mutliple genomic sites, hence interrupting the physical continuity and functional integrity of genes, which was historically recognized even before our understanding of gene organization and function. In the case of LINE-1, however, only a minor fraction (about 80-100) of all copies present in the human genome are full-length and retrotranspositionally competent [4], whereas the vast majority of genomic LINE-1 copies are truncated at their 5'end and thus non-mobile [5], yet still transcriptionally competent: this implies that LINE-1 elements have a higher potential for producing a proficient RT enzyme (encoded by the ORF-2 present in all copies) than for retrotransposition (of which only the full-length elements with an intact 5' end are capable). This indicates that RT production is not necessarily coupled with retroelement mobility, and highlights the notion that the transcriptional capability of the considerable high number of genomic elements provides cells with a potentially large source of RT activity.
Retroviral and retroelement-derived reverse transcriptase
The groundbreaking discovery of an RT activity encoded by infective retroviruses [6,7] has revolutionized our understanding of genome function, because it showed the existence of an unanticipated flow of genetic information, from RNA to DNA, in contrast with the central dogma of molecular biology - which considered DNA to RNA as the only possible direction. Howard Temin first predicted a functional role for RT, both in physiological differentiation, as in embryogenesis, and in its pathological loss, as in cancer [8]. Temin’s visionary prediction was fulfilled, in some way, by the discovery that non-infected cells are also endowed with an endogenous RT that can act on the genetic information stored in nuclei and provide a source of continuous genomic variability. A considerable body of evidence after Temin’s discovery has shown that the expression of endogenous RT is itself developmentally modulated and is implicated in a broad spectrum of pathological and physiological settings. Indeed, non-pathological differentiated tissues contain low levels of RT activity, if at all, while high RT activity is typically found in embryos and embryonic tissues [9].
Besides embryonic tissues, the endogenous RT is generally abundant in cells characterized by a low differentiation levels and a high proliferation rate, such as transformed cells [10], consistent with the observation that retroelements are mobilized in many pathologies, including tumors [11,12]. Thus, undifferentiated or dedifferentiated cells and tissues with a highly proliferating potential constitute permissive systems for RT expression and retrotransposition activity, while differentiated quiescent cells offer less favourable contexts [13].
While the RTs of infective retroviruses, of clear clinical relevance to infected cells, have been intensely investigated [reviewed in 14], the endogenous RT has received lower attention, in spite of the many clues that overtly suggested a potential implication in fundamental physiological and pathological processes.
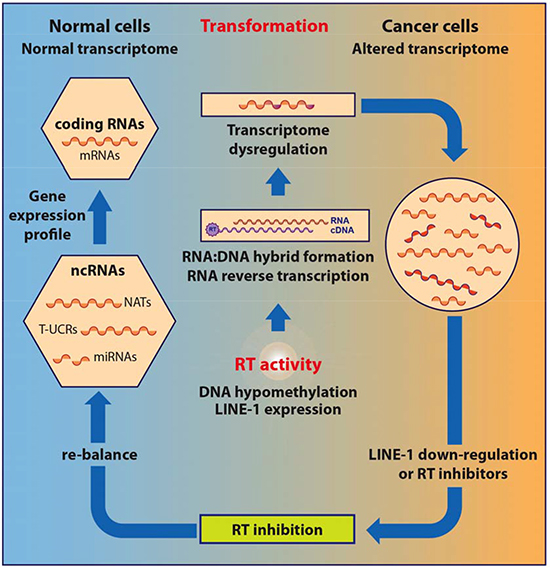
Only in the last decade have roles of the LINE-1-encoded RT been recognized, both in embryogenesis and in tumorigenesis [respectively examined in 9 and 10]. The RT has emerged as a key regulator of both these processes, in parallel with the increasingly recognized contribution of transposable elements to genome-wide regulatory networks [15]. Recent evidence indicates however that retroelement mobilization reflects only part of the roles of RT in the retrotransposition machinery. Here we review evidence linking the endogenous LINE-1-encoded RT to tumorigenesis and propose a model for a previously unrecognized regulatory role in the genesis and progression of cancer. To define the newly emerging role of RT, in the next section we will briefly recall some essential aspects of the eukaryotic transcriptome and its links with retrotransposon networks.
Genomes are pervasively transcribed on both strands: implications in cancer
The historical legacy that the eukaryotic transcriptome is constituted by messenger RNA (mRNA), transcribed from protein-coding genes, and by the non-coding ribosomal RNAs (rRNA) and transfer RNAs (tRNA), has radically changed in recent years. It is now well-established that the vast majority of eukaryotic genomes are pervasively transcribed [16].
The advent of next generation sequencing technologies led to the unexpected discovery of varieties of non-coding RNAs (ncRNAs) [reviewed in 17,18,19]. ncRNAs are grouped in two major classes, small RNAs (sncRNAs) < 200 bp, typically unstable, and long RNAs (lncRNAs) ranging from > 200 bp to 100 kb [20] more stable, transcribed on either or both of the DNA strands and classified according to distinctive sequence features [17,19,21). The discovery of microRNAs (miRNAs) and of naturally occurring small interfering RNAs (siRNAs) [reviewed in 22] provided early evidence that not only the transcriptional landscape is of higher complexity than ever thought, but also that these RNAs have regulatory roles. lncRNAs are integral components of the mammalian transcriptome [17,19,21,23] and constitute a highly heterogeneous class of thousands of polymerase II-transcribed RNA species, polyadenylated, spliced, mostly localized in the nucleus [reviewed in 24]. The evidence that the vast majority of genomic transcription is non-coding, whereas only less than 2% is transcribed in protein-coding mRNAs [16], suggests that the former cannot be dismissed as mere functionless transcriptional “noise”, but may have functional roles.
Another recently identified component of the transcriptome is composed of natural antisense non-coding RNA transcripts (NATs) from both protein-coding and non-coding genes [25, 26]. Antisense transcripts are widespreadly produced across the genome of various species [27, 28]. They represent a pervasive phenomenon, accounting for about 50-70% of annotated human coding sequences having sense partners, including genes with relevant developmental functions [29]. They are on average 10-fold less abundant than sense expression and preferentially stored in nuclei [reviewed in 30,31]. Interestingly, antisense transcription occurs nonrandomly across the genome [32] and is concentrated at preferential “hot spots” overlapping both ends of coding genes [33, 34]. Several NATs have a regulatory role on gene expression [35]. Together with sense lncRNAs, NATs are components of complex genome-wide regulatory networks that finely tune the genome expression, with roles in tumorigenesis, differentiation and development [reviewed in 29, 31, 36,37].
A vast body of data implicate ncRNA classes, both sense and antisense transcripts, in tumorigenesis [reviewed by 38,27], a context in which the transcriptome is profoundly affected by altered genome methylation [39,40]. miRNAs were the first class of ncRNAs to be implicated in cancer formation and spreading: miRNAs can act as oncogenes or tumor suppressors [41,42], are frequently located within cancer-associated genomic regions [43] and show significantly altered expression profiles in human cancers, a dysregulation caused by and [38], often sufficient to induce oncogenesis [44,45]. Long non-coding RNAs (lncRNAs) expression [46,47] is highly tissue-specific compared with coding genes, a finding consistent with the hypothesis that lncRNAs contribute to confer target specificity to regulatory networks [48, 49]. Serial analysis of gene expression libraries (SAGE) indicate that the tissue-specificity of lncRNA expression is altered in many cancer types [50], suggesting roles in tumorigenesis. T-UCRs are lncRNAs transcribed from ultra-conserved regions (UCRs) [51] acting as possible developmental enhancers in mammalian genomes [52] and aberrantly expressed in a variety of human cancers [53,54,55].
ncRNAs and transposable elements: a long-lasting relationship
Remarkably, ncRNAs and transposable elements (TEs) share many biogenetic, functional and structural aspects [56,57, reviewed in 58]:
First, a high proportion of miRNAs originates from TE families, including DNA transposons, LTR-containing retrotransposons, LINE-1 and SINE elements [58, 59].
Second, TE sequences are embedded in about three-quarters of all mature long non-coding (lnc) RNA transcripts, while being virtually absent from protein-coding exons, and account for about 30-42% of total human lncRNA sequences [56,57]. Interestingly, TEs - particularly LTR-containing ERVs - target preferential positions and orientations within lncRNAs; they are frequently associated with transcription starting sites (TSS), and hence may have roles in regulation of lncRNA transcription. Importantly, a relevant regulatory role has been assigned to HERV-H-containing lncRNAs expressed in embryonic stem cells (ESCs) [56,57] and to other lncRNAs enriched in LTRs, the expression of which is implicated in pluripotency of ESCs [60,61].
Additionally, retrotransposition events can generate thousands of pseudogenes, which also have global regulatory roles [62]. The “competing endogenous RNA” (ceRNA) hypothesis [63] highlights the regulatory role played, among others, by pseudogenes generated via mRNA retrotransposition. In the ceRNA hypothesis, which takes into account the variety of targets for each miRNA and the variety of miRNAs capable of acting on a common target, cross-talks are generated between distinct regulatory RNAs; pseudogenes may “sequester” specific miRNAs and hence modulate their actual availability as functional regulatory molecules. In this framework, pseudogene transcripts, mRNAs and lncRNAs constitute regulatory networks, the “communication” of which is mediated by a limited pool of miRNAs [62].
Overall, the phenomena briefly outlined above entail different levels of control (e.g., transcriptional in the case of some inserted retrocopies, post-transcriptional in cases in which retrotransposons originate regulatory RNAs or, on the contrary, block their function); their common reverse transcription-dependent origin indicates the many ways through which RT can globally shape genome functions. Collectively therefore these data indicate a broad reach of TEs in shaping the transcriptome of ncRNAs and influencing their regulatory role and tissue specificity.
Retrotransposons and the endogenous RT in tumorigenesis
The notion that expression of retroelements increases in tumors, while being low in normal tissues, is consistent with recent findings that proteins encoded by the LINE-1 bicistronic open reading frames, i.e. ORF1p and ORF2p, are abundant in a variety of cancers [64], breast [65; 66], gastric [67] and pediatric germ cell tumors [68], but not in their healthy tissue counterparts.
In agreement with these studies, using a specific RT-targeted monoclonal antibody we have depicted a quantitative increase of LINE-1-ORF2p proteins in progressive breast cancer stages in a cancer-prone transgenic mouse model [69]. Exemplifying immunofluorescence panels in Fig. 1A illustrate this increase in progressively advanced stages of breast cancer. Cancer samples withdrawn from mice at regular intervals after birth were staged (1 to 6 ) according to several parameters (e.g. expression of epidermal growth factor receptor (ERB2), down-regulation of the estrogen receptor (ER) and others; see [69] for detailed description). We found that both LINE-1 and SINEB1 retroelements undergo progressive copy number amplification in advancing cancer stages, indicating that the activation of the retrotransposon machinery yields not only increased expression, but also an increase in the content of retroelements in the cancer cell genome [69].
The activation of the retrotransposition machinery can yield extensive genomic insertions, typical of human cancers: indeed, hundreds of novel cancer-specific retrotranspositions have been mapped in genomes from lung [70], colon [71,72], prostate [71], ovarian [71] and liver [73] carcinomas. While these data confirm that tumors offer a highly permissive environment for retrotransposition, they do not indicate whether these insertions are “passenger” (irrelevant to the onset of oncogenesis) or “driver” mutations with a causative potential [74], favouring the emergence of a typically altered “cancer genome” [75].
Compelling evidence indicate that retrotransposition events exert a profound impact on genome function and expression, with a crucial role of the endogenous RT. LINE-1-encoded RT is an essential mechanistic component for the mobility of retrotransposons, usually not thought to play any other role beyond retrotransposition. Intrigued by the evidence implicating retrotransposition in the physiological state of cells and tissues, we sought to directly assess the role of RT in tumorigenesis using two experimental approaches. In the first one, RT was pharmacologically inhibited in cancer cell lines using nonnucleoside inhibitors widely employed in AIDS treatment, e.g. nevirapine or efavirenz (EFV) [76,77,78]; in the second one, we used the RNA interference (RNAi) methodology to down-regulate the expression of full-length RT-encoding LINE-1 elements, the major source of RT activity in human cells [4], in cancer cell lines [78,79]. Both approaches yielded consistent responses: first, RT inhibition caused a reduced cell proliferation rate in cancer cells transformed but not in normal cells (e.g. WI38 fibroblasts) (Fig. 1B); second, cells assumed a differentiated phenotype (exemplifying panels from A375 melanoma cultures are depicted in Fig.1C). The changes in cell functional morphology (such as seen in Fig. 1C), including the appearance of dendritic-like extensions and increased adhesion to the plate surface, distinctive of differentiating melanoma (see [78] for an in-depth characterization), are accompanied by global alterations of the transcriptome of coding and non-coding sequences, as will be seen below. Several laboratories have independently confirmed these conclusions in studies of various human tumorigenic cell lines treated with RT inhibitors, both of the nonnucleoside [80,81, 82] and the nucleoside type [83, 84, 85].
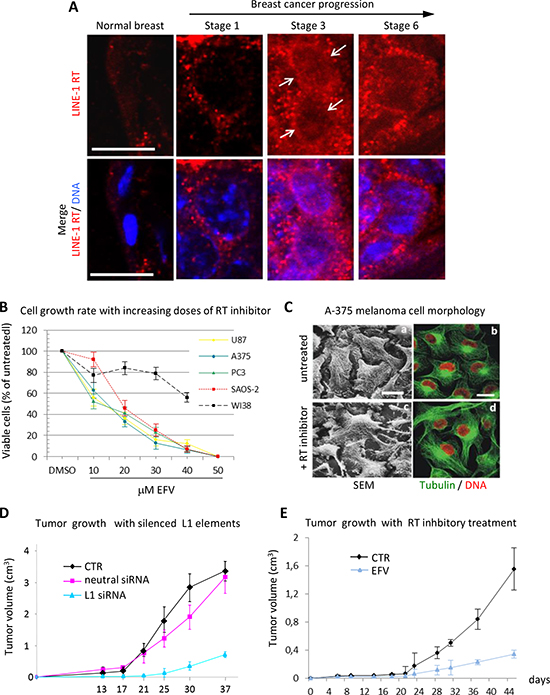
Figure 1: RT inhibition recapitulates the global reprogramming of cancer cell phenotypes observed with LINE-1 element silencing. (A)
“”
“”
“”
?&
ò””“”“”“”
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
ü–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
—–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
ć–
–
–
'’–
–
–
–
–
ñ–
–
–
–
à–
–
–
–
–
–
–
ä–
ü–
ò–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
ö–
–
–
–
–
š