
INTRODUCTION
Diminished kidney function is commonly classified as two distinct syndromes named chronic kidney disease (CKD) and acute kidney injury (AKI) [1]. CKD is a global public health issue, and its prevalence is estimated to be 8–16% worldwide [2]. The prevalence and mortality of AKI is also increasing all over the world, especially in developing countries. Due to the different definitions of AKI, wide variation exists in the estimation of disease prevalence (1–25%) and mortality (15–60%) [3, 4]. Although massive researches have been performed in the past decades, the pathogenic mechanisms of CKD and AKI are still elusive. Moreover, to date, no specific therapies have emerged to lessen the incidence or assist the recovery of kidney diseases. Effective treatment approaches are urgently needed to be developed.
Inflammation is the established causative factor in the kidney diseases [5]. PGs are abundantly produced in the kidney (Table 1), and play an important role in triggering the inflammatory response, contributing to the occurrence and progression of kidney diseases [6–8]. PGs appear when arachidonic acid (AA) is released from the plasma membrane by phospholipases and metabolized by the peroxidase actions of COXs to PGH2 which can be subsequently converted into more stable biologically active PGs, including PGE2, PGI2, PGD2, PGF2α, and TXA2 by their respective synthases [7, 9] (Figure 1). PGs exert their functions by combining to a diverse family of membrane-spanning G protein-coupled prostanoid receptors [10, 11] (Table 2).
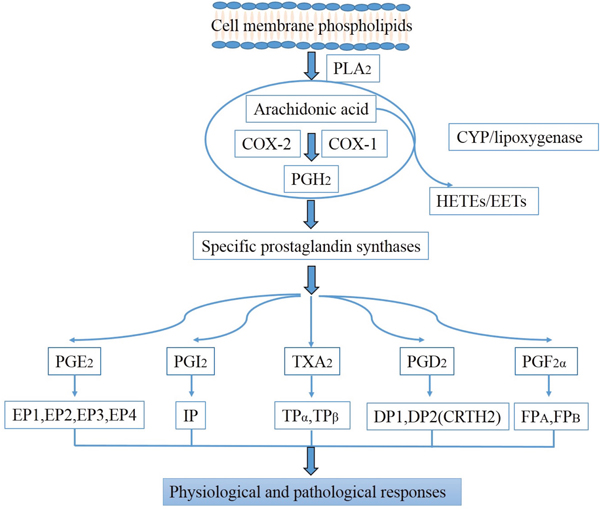
Figure 1: Biosynthesis pathways of prostaglandins.
Table 1: Renal distributions of catalytic enzymes of PGs
Enzymes |
|
Distribution |
|---|---|---|
COX-1 |
|
Collecting duct, glomeruli, medullary interstitial cells, arterial endothelial cells |
COX-2 |
|
Medullary interstitial cells, glomeruli, macula densa, thick ascending limb, |
PGES |
mPGES-1 |
Macula densa, distal convoluted tubule, collecting duct, and renal medullary interstitial cells |
mPGES-2 |
Distal convoluted tubule, collecting duct, proximal convoluted tubule, thick limbs of the loops of Henle |
|
cPGES |
All nephron segments |
|
PGIS |
|
Glomeruli, medullary collecting duct. |
TXAS |
|
Glomeruli |
PGDS |
|
Proximal convoluted tubule, thick ascending limb, distal convoluted tubule, collecting duct |
PGF2α synthase |
Not defined |
|
Table 2: Renal functions, possible signaling pathways, pharmacological modulators, and renal distributions of PG receptors
PG receptors |
Renal function |
Signaling pathways |
Agonist/Antagonist |
Expressionsites |
|
|---|---|---|---|---|---|
|
EP1 |
Haemodynamics, transport, proliferation, fibrosis, Renin release |
Gq-Ca2+ |
A: Sulprostone;17-phenyl trinor prostaglandin E2ethyl amid |
CD/MG/P/PT/V |
|
|
|
|
Ant: ONO-8711, ONO-8713 |
|
EP |
EP2 |
Haemodynamics, transport, anti-apoptosis, Renin release |
Gs-cAMP, adenylatecyclase, β-arrestin mediated signalosome |
A: CP-536, 745-01; butaprost |
IC/MD/P/V |
EP3 |
Transport, vasoconstriction |
Gi-cAMP, adenylate cyclase, G12/13-RhoA |
A: L-798106; sulprostone |
DT/CD/MD/V |
|
|
EP4 |
Haemodynamics, transport, renin release, vasodilation, proliferation, anti-apoptosis |
Gs-cAMP, Gi-cAMP, adenylate cyclase, β-arrestin mediated signalosome |
A: CP-044, 519-02; 11-deoxy-PGE1; cay10580, |
P/CD/MG/MD/DT/PT/V |
|
|
|
|
Ant: ONO-AE3-208; L-161982 |
|
|
IP |
Haemodynamics, vasodilation, transport, renin release, matrix synthesis, proliferation, anti-apoptosis |
Gs-cAMP |
A: ONO-1301; MRE-269 |
MG/MD/DT/CD/PT/P/V |
|
TP |
Vasoconstriction, haemodynamics, fibrosis, proliferation, differentiation, inflammation |
Gq-Ca2+, G12/13-RhoA |
A: U-46619 |
MG/P/V/DT/CD/PT |
|
|
|
|
Ant: S18886 |
|
|
DP |
Fibrosis, inflammation, haemodynamics, transport |
Gs-cAMP |
Ant: CAY10471 |
-- |
|
FP |
Transport and cell transformation, haemodynamics, growth |
Gq-Ca2+, Gi-cAMP, G12/13-RhoA |
A: Latanoprost |
CD/DT/P/F |
Abbreviations: CD: collecting duct; MG: mesangial cells; P: podocytes; PT: proximal tubule; DT: distal tubule; V: vasculature; F: fibroblasts; IC: interstitial cells; MD: macula densa; A: agonist; Ant: antagonist.
CD: collecting duct; MG: mesangial cells; P: podocytes; PT: proximal tubule; DT: distal tubule; V: vasculature; F: fibroblasts; IC: interstitial cells; MD: macula densa; A: agonist; Ant: antagonist.
PGs are important lipid mediators in numerous physiological and pathophysiological processes in the kidney. Under the physiological conditions, PGs exert essential functions in the regulation of renal hemodynamics, renin release, as well as water and salt balance [7, 12, 13]. Likewise, PGs also respond to distinct pathological insults. Researches on the functions of PGs in CKD and AKI have been conducted and the awareness have been drawn. In this review, we highlighted the roles of PGs, PG synthases, and the specific PG receptors in kidney diseases, with expectation to develop effective therapies targeting the PG cascades to relieve kidney injury.
COXS IN KIDNEY DISEASES
COXs are the rate-limiting enzymes that are responsible for the first two steps in the synthesis of PGs. The production of PGs is depended on the activity of COXs which are the upstream components of this process. The COXs have both COX and peroxidase activities and exist as two isoforms termed as COX-1 and COX-2 [9, 14]. COX-1 is constitutive as its abundant expression in a wide range of tissues under basal conditions. In the kidney, COX-1 highly expresses in the collecting duct, while lower level of COX-1 can be detected in interstitial cells, glomerular mesangial cells, and endothelial cells of arterioles [12, 15–17]. It is commonly recognized that COX-1 serves a constitutive housekeeping role [18]. Evidence has shown that COX-1 could regulate nephron formation via PGE2 in the zebrafish embryonic kidney [19]. COX-1 is also responsible for maintaining basic physiological functions including cytoprotection of the gastric mucosa and platelet aggregation [20, 21]. Furthermore, COX-1 functions as the major form rather than COX-2 in endothelium-dependent contraction [22, 23] or relaxation [24]. Beyond the scope of above discoveries, there are also many investigations focusing on kidney diseases [25–27]. Different from COX-1, COX-2 can be induced by inflammatory mediators and mitogens. It mediates the induction of various PGs, which play critical roles in various pathophysiological processes such as angiogenesis, inflammation and tumorigenesis [20, 28]. Whereas, several studies suggested that COX-2 also has housekeeping functions in kidney development [29, 30], ovulation, and parturition [31, 32]. In the kidney, COX-2 predominantly expresses in the macula densa, thick ascending limb, medullary interstitial cells, and glomerulus, contributing to a variety of physiological processes [12, 33]. Because of the specific distribution in the kidney, COX-2 is of vital importance in regulating renin release and water/salt metabolism [34]. More importantly, the significant role of COX-2 in kidney diseases, especially in CKD, has been documented by using COX-deficient mouse models or COX antagonists [35–42]. It is known that inhibition of COX-2 is associated with AKI incidence due to the imbalance of renal function in fluid excretion and intra-renal hemodynamics. Some investigations pointed out that suppression of COX-2 activity in AKI accounts for a depravation of renal function [43]. On the contrary, there were also some diverse results with the treatment of COX-2 inhibitor [8, 44, 45]. These diverse outcomes of COX-2 inhibition in AKI may be resulted from distinct experimental conditions such as various kinds of inhibitors and the distinction of disease models and animal species/strains. In addition, recent studies revealed that COX-2/PGE2 cascade activation mediated cisplatin-induced mesangial cell apoptosis and chronic renal failure of C57/BL6 mice with 5/6 nephrectomy [46, 47]. Apart from COXs, arachidonic acid can be metabolized via lipoxygenase and cytochrome P-450 (CYP) enzymes to some other metabolites. Early researches demonstrated the role of 20-HETE, one metabolite of arachidonic acid converted by cytochrome P-450 (CYP) enzyme, in mediating the effects on tubuloglomerular feedback [48]. 20-HETE is also consistent with the known effects of NO, both of which are the main regulators of tubuloglomerular feedback [49]. NO inhibits the production of 20-HETE, which contributes to the vasodilator effect of NO [50–52]. Strategy targeting COXs could be effective because many evidence support a significant role of COXs in the progress of kidney diseases. However, severe side effects possibly due to the nonselective antagonism on the downstream metabolites of COXs greatly limited its application. Thus, selective inhibition of the specific PG synthetic enzymes might be more promising in treating kidney diseases.
PGE2 IN KIDNEY DISEASES
It has been universally recognized that PGE2 is the most abundant renal arachidonic acid metabolite. PGE2 is produced by PGE synthases, and functions by combining to its G protein-coupled receptors: namely EP1, EP2, EP3, and EP4. Three PGE2 synthases in the kidney have been uncovered: microsomal PGE synthase 1 (mPGES-1), microsomal PGE synthase 2 (mPGES-2) and cytosolic PGE synthase (cPGES). Among these three PGESs, mPGES-1 is the best-characterized PGES. Its expression is inducible in the kidney in response to various stresses [53]. mPGES-1 expression can be detected in macula densa, distal convoluted tubule, collecting duct, and renal medullary interstitial cells. Studies revealed that mPGES-1 co-expresses with COX-1 and COX-2 in the kidney [7, 9]. Meanwhile, it has been indicated that mPGES-1 has the best PGE2 synthetic property both in vivo and vitro [5, 54, 55]. Additionally, recent studies have also shown that deletion of mPGES-2 or cPGES in mice did not reduce PGE2 levels [56]. The aggravation of AKI by downregulating mPGES-2was demonstrated to be associated with autophagy inhibition and enhanced apoptosis [57]. Multiple researches uncovered a remarkable role of mPGES-1 deletion on PGE2 production in various models including endotoxemia- and cisplatin-induced kidney injury [58], lithium-induced NDI [59], Ang II- and DOCA-salt-induced hypertension [60], aldosterone escape [61], and unilateral ureteral obstruction [62]. Different from these observations, in the diabetic kidney disease model induced by STZ, three forms of PGES were unaltered in the kidney, and deletion of mPGES-1 did not suppress renal PGE2 production [63]. These diverse results on PGE2 induction suggest that beyond three known PGESs, additional PGE synthases might be existed to exert the function in producing PGE2 under some specific pathological conditions. Due to the critical role of PGE2 in mediating the inflammatory responses, mPGES-1 is considered to contribute to chronic and acute kidney injuries, which has been confirmed in many studies from our and other groups. For instance, in the CKD model of 5/6 nephrectomy, mPGES-1 invalidation alleviated the loss of renal function characterized by the attenuated accumulation of uremic toxins in circulation and improved creatinine clearance [64]. In obese db/db mice with type-2 diabetes, the level of mPGES-1 expression in glomeruli was remarkably higher, suggesting the potential contribution of mPGES-1 in glomerular diseases associated with type-2 diabetes [65]. In kidney cells, we also found that mPGES-1-derived PGE2 could activate Stat3 signaling to promote podocyte apoptosis [66], and the proliferation of mesangial cells triggered by uric acid and uremic toxin indoxyl sulfate was attenuated by silencing mPGES-1 [67, 68]. However, in a UUO model, mPGES-1 was shown to exert a protective effect against renal fibrosis and inflammation [62]. Moreover, in the type-1 diabetic model induced by STZ, both renal PGE2 production and glomerular injury were unaffected in mPGES-1 KO mice. All these studies suggested conflict roles of mPGES-1 in mediating the pathogenesis of CKD, which indicated that the role of mPGES-1 in CKD might depend on the insults. Similarly, diverse conclusions also exist in some AKI models. When mice were challenged with cisplatin, LPS and ischemia/reperfusion, respectively, mPGES-1 deletion only played a role in cisplatin experimental setting [44]. Although above researches indicated a complexity of mPGES-1 in various kidney events, as a specific downstream enzyme of PGE2 generation, mPGES-1 is suggested to be associated with fewer adverse effects than COX inhibitors. Thus, mPGES-1 is still a promising target for the treatment of kidney diseases [69]. However, more extensive investigations on the efficacy and safety of mPGES-1 inhibition in treating various kinds of kidney diseases are required.
The role of PGE2 in the kidney has been comprehensively studied. PGE2 functions via activating four subtypes of receptors (EP1, EP2, EP3, EP4). EP1 is mostly detected in the collecting duct, and exerts its main role in natriuresis and diuresis under physiological status [70–73]. Recently, one research suggested that PGE2 regulated arginine vasopressin synthesis by hypothalamic EP1 [74]. Meanwhile, EP1 receptor, instead of EP2, was reported to increase renin expression in M-1 CD cells [73, 75]. Apart from the collecting duct, EP1 can be also detected in glomerular mesangial cells [76, 77], podocytes [78], and proximal tubule cells [79, 80], suggesting the additional functions of EP1 besides the diuretic action. In mesangial cells, PGE2-induced hypertrophy and cell cycle arrest was reproduced by EP1 agonist sulprostone [76]. It was also proved that EP1 receptor could suppress the proliferation of mouse mesangial cells to alleviate the progression of proteinuria, glomerular hypertrophy, and mesangial expansion through a selective EP1 antagonist, ONO-8713 [77, 81]. In diabetic EP1 KO mice, diabetic hyperfiltration, albuminuria, and injury markers were all suppressed possibly through modulating renin-angiotensin system. In vitro experiments with ONO-8711, an EP1 antagonist, further suggested a fundamental role of EP1 in podocyte impairment [78]. In renal tubules, EP1 deletion reduced the expressions of fibrotic markers and preserved megalin expression in diabetic mice [79, 80]. However, the controversy still exists about the role of EP1 receptor in kidney diseases. There was evidence showing that nephritic mice lacking the EP1 had increased severity of renal impairment [82].
Although PGE2 receptors have been intensively studied in the kidney, not much is known about EP2 receptor. EP2 is mainly found in vascular and interstitial compartments of the kidney [83]. Researchers once used two models including mercury chloride model of acute renal failure and 5/6 nephrectomy model of chronic renal failure to identify the effect of EP2 [84]. By use of the EP2 agonist, CP-536,745-01, reduction of glomerular sclerosis, amelioration of tubulointerstitial injury and better tubular structure were observed in the nephrotoxic mercury chloride (HgCl (2)) rat model of acute kidney failure. [85]. In a different model, EP2 receptor mediated the cystogenesis by inhibiting the apoptosis of cystic epithelial cells [86]. Another study identified that EP2 and EP4 receptors contributed to the pro-inflammatory effects of PGE2 [87]. In podocytes, COX-2/ PGE2/EP2 axis has been revealed to play a pathogenic role in hyperfiltration-mediated albuminuria, as well as the progression of CKD [88]. Meanwhile, EP2 and EP4 receptors are important in promoting the progression of chronic kidney disease induced by TGF-β, while EP1 and EP3 play the opposite role [89].
Renal EP3 expresses in thick ascending limbs and collecting duct existing as different splice variants which determine the preference for G-protein coupling. EP3 splice variants inhibit adenylate cyclase via Gi protein and increase intracellular calcium and activate the G12/G13 pathway, which later leads to the activation of Rho kinase, accounting for the action of EP3 receptor in urinary concentration. EP3 receptor may exert its role by modulating vasopressin signaling in lithium-induced and post-obstructive polyuria [16]. Similarly, EP3 could also cause vasoconstriction in rat proximal interlobular arteries, which was proved by using Misoprostol, and was suggested to protect the kidney during diabetic hyperfiltration regardless of RAAS activation [90, 91]. Beneficial effects of EP3 were also identified by EP3 antagonist, L-798106, evidenced by the retardation of normalized blood urea nitrogen, normalized glomerular cell numbers, restored synaptopodin distribution and F-actin filament arrangement in glomeruli which is mediated by PGE2 [92]. However, in a diabetic mouse model induced by streptozotocin, EP3 was shown to inhibit the water reabsorption, and contributed to polyuria [93].
EP4 receptor is the best identified subtype of the EP receptors, and is abundant in almost all types of renal cells. In addition, EP4 signaling plays a variety of roles through cAMP effectors, and is able to activate the PI3k-dependent pathway, leading to the activation of MAPK signal pathway [94]. Emerging evidence showed that it could couple to G(i)α, phosphatidylinositol 3-kinase, β-arrestin, and β-catenin [95–97]. The renal distribution of EP4 receptor involves the glomeruli, renin-secreting juxta-glomerular granular cells, glomerular epithelial cells, distal convoluted tubules, and cortical collecting ducts [98]. Lots of evidence indicates the function of EP4 in podocytes [99–102]. EP4 deletion from podocytes ameliorated kidney injury in both 5/6 nephrectomy and diabetic CKD models [99, 100]. However, diverse outcomes were seen in another kidney injury model induced by Adriamycin, showing that EP4 deletion and EP4 antagonist (L-161982) did not attenuate podocyte injury [101]. In addition, a selective EP4 agonist (11-deoxy-PGE1) promoted glomerulosclerosis and tubulointerstitial fibrosis in STZ-induced diabetic mice, possibly through IL-6 [103]. Recent evidence also showed that EP4 inhibition (ONO-AE3-208) ameliorated proteinuria and glomerular scarring in rats subjected to renal mass resection [104]. In LPS-induced renal proximal tubule cell injury, EP4 inhibition also played critical roles in anti-inflammatory and anti-apoptosis processes [105]. However, there were still lots of conflicts over EP4 receptor in kidney diseases. Several researches showed the protective role of EP4 in decreasing epithelial cells apoptosis, preventing mesangial cell injury, and preventing tubulointerstitial fibrosis [87, 102, 106]. Apart from these, EP4 is also an important component in the maintenance of body water homeostasis. EP4 expressed in the distal convoluted tubule and the cortical collecting duct and could increase AQP2 membrane trafficking and phosphorylation to enhance water reabsorption possibly via both cAMP/PKA and extracellular signal-regulated kinase (ERK) pathways [107]. Furthermore, a recent research emphasized the capability of EP4 in urine concentrating and provided a new pathway of AVP/PGE2/EP4/PRR for the regulation of AQP2 expression [108]. Overall, PGE2 couples with four EP subtypes to exert multiple functions in the pathology and physiology of kidney. With the development of novel agonists/antagonists and the researches on their application in various kidney diseases, the clinical therapies on kidney diseases by targeting EPs are becoming more feasible.
PGI2 IN KIDNEY DISEASES
Prostacyclin (PGI2) is one of the major products of COXs pathway, and is well known for its regulation on renal hemodynamics, tubular transport and renin release. Prostacyclin synthase (PGIS) is an atypical cytochrome p450 enzyme that generates prostacyclin (PGI) from prostaglandin H (PGH) derived from arachidonic acid by COXs [109]. It is a disappointment that the exact cellular localization of PGIS protein could not be identified yet, while the significant expression of PGIS mRNA was detected in the inner medullary tubules and medullary interstitial cells [110]. Similar to PGES, investigations of PGIS were carried out by use of kidney disease models and PGIS gene knockout mice. Previous reports showed that PGIS deficiency induced renal fibrosis along with the notable irregulation of renal hemodynamics, tubular atrophy, surface irregularities and cysts [111, 112]. Furthermore, overexpression of PGIS contributed to the renal protection against endotoxemia-related AKI [113]. In uremic mice, the reduction of PGI2 synthase activity could be prevented by MnTBAP, a synthetic ROS-scavenging enzyme superoxide dismutase (SOD). It was suggested that the defect of PGI2-generating pathway could be mediated by oxidative stress which was attributable to the progression of end-stage renal disease (ESRD) [114]. Besides kidney cases, PGI2 is also known for its beneficial effects during the stroke, thrombosis, atherosclerosis, and myocardial infarction [115, 116], which may indirectly affect the progression of kidney diseases. A previous review had shown the connections between PGI2 and renal growth, fibrotic response, and cell fate [112]. Recently, a lot of studies focused on Beraprost Sodium (BPS) to reveal the role of PGI2 in kidney diseases. As a prostacyclin analogue, BPS is a vasoactive substance that can expand renal vessels to increase renal blood flow, inhibit TXA2 synthesis, and prevent platelet aggregation and immune complex formation. It was also reported that BPS could prevent glomerular thrombosis to reduce proteinuria [117]. Similar results were also shown in some clinical researches [118–120]. In addition, in a diabetic kidney disease induced by STZ, BPS improved renal function possibly through the inhibition of oxidative stress and inflammation [121, 122].
Major renal functions of PGI2 are mediated by the cell-surface receptor termed IP. The localization of IP receptor in renal cells is considered to be controversial over species. IP was reported to be detected in mesangial cells, interstitial cells, the vasculature and the tubular epithelial cells (proximal tubule, mTAL and collecting duct) in rodent kidney [70, 123, 124]. In human kidney, the IP receptor was also detected in podocytes [125]. This potentially indicated differences in function of IP receptor in the specific parts of nephron [124]. IP receptor is found to play a significant role in maintaining renal hemodynamics, tubule transport, renin secretion, and reducing renal fibrosis and inflammation [125]. One recent research showed that prostaglandin I2 receptor agonism preserved beta-cell function and attenuated albuminuria through nephrin-dependent mechanisms [126]. ONO-1301, a novel nonprostanoid IP agonist, was used in models of type 1 diabetic nephropathy and UUO, and showed a therapeutic effect on treating diabetic nephropathy via inducing hepatocyte growth factor (HGF) to counteract TGF-β [127, 128]. ONO-1301 also ameliorated the renal lesions in type 2 diabetes by attenuating mesangial matrix accumulation, inflammation and oxidative stress through an IP receptor-mediated mechanism [129]. Interestingly, comparing with IP-knockout mice, PGIS knockout mice displayed additional glomerular, vascular and interstitial abnormalities, suggesting the contribution of other receptors in addition to IP [113]. Till now, lots of evidence indicated that PGI2 could activate peroxisome proliferator-activated receptor α (PPARα) or peroxisome proliferator-activated receptor δ (PPARδ) to protect tubular cells from apoptosis in AKI [130, 131]. In addition, recent studies lay special stress on a cyclic vasoactive peptide termed urotensin II which could induce the production of PGI2 in gentamicin-treated NRK-52E cells and protect renal cells through a PPARα-dependent mechanism [132]. Moreover, researchers figured out that L-carnitine could protect renal tubular epithelial cells in the experimental animal model induced by carboplatin by activating PGI2/ PPARα signal pathway [133]. Taken together, PGI2, as an important product of COXs, plays an important role by coupling with its receptors and the downstream signals in various types of renal diseases consisting of CKD and AKI. However, extensive studies are still required to clarify the mechanisms and additional actions of prostacyclin in various kidney disease models.
TXA2 IN KIDNEY DISEASES
TXA2 is a powerful platelet activator, and is also considered as potent smooth muscle constrictor. In addition, it also serves as a vascular smooth muscle cell mitogen. TXA2 is generated by a sequential catalyzation of phospholipase A2, COXs and TXA2 synthase (TXAS). Under physiological conditions, TXA2 derived from platelets mainly depends on the activity of COX-1. COX-1 inhibitor could inhibit the production of TXA2 and attenuate the early decrease in GFR in endotoxemia-induced acute kidney injury [134]. However, the responsible isoform of COXs for the formation of TXA2 during pathological conditions is still unknown [9]. TXAS is an endoplasmic reticulum membrane protein which is presented in different cells including smooth muscle cells, macrophages, platelets, endothelial cells, and kidney cells. TXAS expression can be regulated by a variety of factors. ONO-1301, a novel sustained-release prostacyclin analogue, was identified to inhibit TXAS expression [129].
Two subtypes of TP receptors, TPα and TPβ, have been proved to exist in various localizations of kidney and many other organs along with TXAS. In the kidney, TP receptors express in mesangial cells, podocytes, arterial vessel walls, luminal membranes of thick ascending limb of the Henle’s loop, and basolateral membranes of distal convoluted tubules and connecting tubules [135]. Generally speaking, TXA2 commonly couples with TP receptors and communicates mainly with Gq and G12/13, resulting in phospholipase C activation and Rho-GEF activation, respectively. To better understand the TXA2 on cellular signaling transduction, there is a nice review in the literature [136]. Although TPα and TPβ differ only in their C-terminal regions, several literatures had shown the differences of functions between TPα and TPβ, suggesting that the downstream signaling pathways may differ between both TP isoforms [137, 138]. TPα acts as the major TP receptor regulated by prostacyclin and NO. Moreover, RhoA signaling mediated by TPα could be directly blunted by prostacyclin and NO through protein kinase (PKA/PKG-dependent phosphorylation), while signaling of TPβ was not directly affected by prostacyclin or NO [139].
TXA2, which has been well established as efficacious activator of RhoA, was proved in detail to induce cell proliferation, differentiation, and inflammation, possibly through the mechanisms associated with AP-1, NF-KB, MRTF-A, and YAP [140]. TXA2 also played a critical role in the regulation of renal hemodynamics, which was determined by use of the TP agonist (U-46,619) [141, 142]. Patients with atrial fibrillation (AF) had a less decline in eGFR with the use of aspirin which inhibited TXA2 production [143]. One previous study reported that increased level of vascular ceramide induced by AngII contributed to the pathogenesis of renal injury possibly through TXA2-mediated vasoconstriction [144, 145]. Another study was also in line with this conclusion, showing that AngII/AT1 receptor/nSMase/ceramide-PLA2/TXA2 pathway contributed to the regulation of renal vasoconstriction [146]. In a model of type 2 diabetes, a TXA2 synthase inhibitor was beneficial in reducing glomerular lesion and proteinuria [147]. In addition to these findings, former study showed that inhibition of TXA2 synthase, pharmacological blockade of TP receptors, or genetic disruption of TP receptor could ameliorate the LPS-induced GFR decrease [148]. The reduction in GFR in response to endotoxemia was found to be resulted from the increased generation of TXA2 via COX-1 [134]. Additionally, evidence was also provided that COX-2 expressed in the macula densa regulated tubuloglomerular feedback through the generation of TXA2 and nNOS-dependent NO [149].
Beyond the role in renal vasculature, inhibition of TP receptors could rescue the kidney impairment to some degree [141]. Functions of TXA2 in mesangial cells in the regulation of growth response, matrix synthesis, glomerular thrombosis and fibrosis have been studied [147, 150]. Furthermore, in the kidney of diabetic apolipoprotein E KO mice, an antagonist of TP receptors (S18886) attenuated albuminuria, suppressed oxidative stress and inflammation, and blocked extracellular matrix deposition by activating MnSOD [151]. In accordance with this, in a CKD model of renal mass reduction, reduced renal mass led to microvascular remodeling and enhanced ET-1-induced mitochondrial ROS production and vascular contraction. All these abnormalities were related to the activation TP receptors [152]. With the treatment of puromycin amino nucleoside (PAN) or adriamycin, TXA2 was also increased in accordance with the podocyte damage. It was shown in this study that the podocyte injury was ameliorated by the TP receptor antagonist (SQ29548) or TP gene deletion [101]. In addition to this, some other studies also demonstrated the pathogenic roles of TXA2 and TP receptors in lupus nephritis [153], cyclosporine nephrotoxicity [154], and renal allograft rejection [155]. Overall, activation of TXA2/TP receptors can lead to the renal vasoconstriction, oxidative stress, and inflammation, which were involved in the onset and progression of kidney diseases.
PGD2 IN KIDNEY DISEASES
Prostaglandin D2 is known for its involvement in a variety of neurophysiological functions including the body temperature control, hormone release and the sleep-wake cycle. In the kidney, with intrarenal infusion of PGD2, renal artery flow, urine output, creatinine clearance, and sodium and potassium excretion all increased dose-dependently [156]. Similar to other types of PGs, PGD2 is converted by prostaglandin D synthase (PGDS) through the common route of the PGs synthesis. Lipocalin type prostaglandin synthase (L-PGDS), also named beta trace protein (BTP), and Hematopoietic prostaglandin D synthase (H-PGDS) are two types of PGDS. They are different from each other in terms of cellular or tissue distribution and functional relevance [157]. H-PGDS is localized mostly in inflammatory cells, especially antigen-presenting cells and mast cells. H-PGDS has both pro-inflammatory and anti-inflammatory actions under different conditions [158, 159]. Fundamentally different from H-PGDS, L-PGDS can be detected in lots of tissues including the brain, heart, kidney, and lung [160]. During the recent years, as the biomarker in large population-based study cohorts, L-PGDS has aroused wide concern [161, 162]. To date, numerous investigations have shown that L-PGDS has emerged as a novel intracellular marker of GFR better than serum creatinine level [163–167]. Besides indicating the glomerular filtration rate, L-PGDS has also been identified as a proximal tubule damage marker during kidney injures [168]. Since its sensitivity in measuring GFR reduction, L-PGDS is becoming an important indicator of the outcome in kidney diseases. It captures the risks associated with decreased kidney function or pathophysiologic processes. Thus, it may provide an opportunity for the early diagnosis and early therapy in patients with kidney diseases [169].
L-PGDS is commonly regarded as a multiple function protein, acting as the PGD2 synthase and functioning as a lipophilic ligand-binding protein after its secretion. The protective role of L-PGDS has been studied in various experiment models, especially in cardiovascular and renal systems [170–172]. Details of L-PGDS in the cardiovascular system are beyond the scope of this review. In this review, we focused on the functions of L-PGDS in the kidney. It was clearly demonstrated that L-PGDS expression in the tubular epithelium remarkably increased in obstructed kidneys. The tubulointerstitial fibrosis caused by UUO was significantly suppressed in L-PGDS-KO mice, which stressed a critical role of L-PGDS in renal fibrosis [170]. However, other investigators showed that L-PGDS KO mice developed glomerular hypertrophy, fibrosis, basement membrane thickening, and high TGF-β deposition [172]. Function of L-PGDS in early stage diabetic nephropathy in rats and adriamycin-induced nephropathy in mice further suggested a possible contribution of PGD2 in chronic kidney diseases [173, 174]. The diversity of renal consequences suggested complex roles of L-PGDS in the kidneys challenged with different insults.
PGD2 interacts with two receptors, the DP1 receptor and DP2 receptor (also termed as CRTH2). PGD2 activates the DPs which then increase the level of cAMP. DP1 receptor appears to have various functions and is more widely expressed. In contrast, DP2 receptor mostly located in the inflammatory cell types such as Th2 cells, acting as a chemo-attractant receptor-homologous molecule. Roles of DP1 have been studied in cutaneous and pulmonary venous vasodilatation [175, 176], platelet aggregation [177], and mucin secretion [178]. DP2 receptor is not structurally the same as the DP1 and belongs to the family of chemokine receptors, contributing to the production of Th2 cytokines, such as IL-4, IL-5 and IL-13 [170, 179]. PGD2 receptors have not been studied that much in the kidney as in respiratory system [175, 180, 181]. However, studies have indicated that CRTH2 may involve in the progression of tubulointerstitial fibrosis and inflammation [182]. Researchers blocked CRTH2 by generating CRTH2-KO mice or using CRTH2 antagonist (CAY10471) in UUO model, and found that interstitial collagen deposition, collagen I gene expression and soluble collagen content were significantly suppressed in CRTH2-KO UUO mice. Furthermore, they revealed that PGD2 mediated the activation of Th2 lymphocytes through combining with CRTH2, which promoted the fibrosis via the production of IL-4 and IL-13 [170]. A previous study also suggested a DP receptor-independent function of PGD2 in kidney diseases [183]. PGD2 can be metabolized to biologically active J-series cyclopentone PGs, especially the 15d-PGJ2 which is considered as a natural endogenous ligand of PPARγ. Activation of PPARγ could effectively inhibit TGF-β-induced profibrotic effects in many organs. Thus, PGD2 may inhibit fibrosis by its end product of 15d-PGJ2 to activate PPARγ, restrain AP-1 and NF-κβ transcription factors. In addition, 15d-PGJ2 could also inhibit the expressions of CXCL9, CXCL10, and CXCL11 in HK-2 cells treated with combined IFN-γ and TNF-α [184]. Together, although the roles of PGD2 in renal system has not been fully understood, above findings still implied the importance of PGD2-generating cascade in the pathophysiology of kidney diseases.
PGF2α IN KIDNEY DISEASES
PGF2α is also one of the major colooxygenase-mediated arachidonate metabolite in the kidney. PGF2α can be derived from either PGH2 via the PGF synthase or enzymatic conversion of PGE2 to PGF2α by PGE9 ketoreductase [185]. The cellular effects of PGF2α are mediated by G protein-coupled transmembrane receptors, namely FPA and FPB. FP receptors are found to be linked to the increase of intracellular Ca2+ in response to PGF2α in renal cells, participating in the transformation of kidney fibroblasts [186]. Commonly, the FP receptors can be detected in the kidney. FPs were found to have high expression in the principal cells of the collecting duct, distal convoluted tubule, and podocytes of the glomeruli with less level in the thick ascending limb [187, 188]. Unlike other PGs, little is known about PGF2α in the renal diseases. Previous studies have shown that PGF2α is associated with natriuresis and diuresis through the activation of FP receptors in the cortical collecting duct [189, 190]. PGF2α inhibited the basolateral 40 pS K channels at high concentrations and stimulated these channels at low concentrations because of the different activation of FPA and FPB in PKC and Rho pathway, respectively [191]. FP activation could also inhibit arginine vasopressin-stimulated water permeability without increasing intracellular Ca2+ in cortical collecting ducts [189]. In animals, it was shown that FP KO mice exhibited modest polyuria and polydipsia with a mild defect in regulating renal medullary osmolality during water deprivation. This research also indicated that FP deletion reduced blood pressure through the inactivation of RAS, despite a compensatory enhancement of AT1 receptors and an augmented hypertensive response to AngII infusion [190]. In a renovascular hypertensive rat (RHR) model, ROS was considered as an initiator that activates endothelial COX-2 to form PGF2α to participate in endothelial dysfunction in the rabbit arteries. Treatment with celecoxib or tempol reduced blood pressure, increased renal blood flow, and restored endothelial function in renovascular hypertension rats, providing a new insight into understanding renal-related hypertension [192]. With respect to the role of PGF2α in oxidative stress, the production level of PGF2α has been shown to be changed dramatically during inflammatory response [193]. In a study carried out in autosomal dominant polycystic kidney disease (ADPKD) patients, increased serum concentrations of the oxidative stress markers of 8-isoprostane and PGF2α were associated with the GFR and kidney volume, suggesting a relationship between PGF2α and renal function during the growth of cystic kidney. Meanwhile, PGF2α was also confirmed to increase serum levels of MMP-1 and MMP-9 in this study [194]. Due to the abundance of PGF2α and its receptors in the kidney, there must be more functions in the renal incidences that have not been figured out. Thus, further investigation has to be conducted to fill the vacancy.
CONCLUSIONS
In summary, PGs exert various functions in the pathology and physiology of kidney. The levels of PGs can be regulated at multiple steps. Among these steps, COXs and prostanoid synthases are of importance in the control of PGs’ production. PGs act by combining to their specific receptors or crosstalk with other receptors. Studies have shown that PGs exert multiple physiological functions in the kidney, such as maintaining glomerular filtration, modulating water and sodium excretion. Moreover, PGs are also involved in the pathology of various renal diseases including CKD and AKI. Strategies targeting any step of the COXs/PG synthases/PGs/receptors cascade might be potentially effective for the treatment of kidney diseases. Although there are still many problems and controversies in understanding the PGs system in kidney pathology, a number of current studies already provided valuable insights into the in-depth investigation of this field in the future.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by Grants from the National Natural Science Foundation of China (nos. 81370802, 81770740, 81300591, 81670647, 81600557, 81600352, 81600532, 81770690 and 81570616), the National Key Research and Development Program (no. 2016YFC0906103).
CONFLICTS OF INTEREST
There is no conflicts of interest to disclose.
REFERENCES
1. Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. 2014; 371:58–66.
2. Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, Saran R, Wang AY, Yang CW. Chronic kidney disease: global dimension and perspectives. Lancet. 2013; 382:260–72.
3. Lameire NH, Bagga A, Cruz D, De Maeseneer J, Endre Z, Kellum JA, Liu KD, Mehta RL, Pannu N, Van Biesen W, Vanholder R. Acute kidney injury: an increasing global concern. Lancet. 2013; 382:170–79.
4. Li PK, Burdmann EA, Mehta RL, and World Kidney Day Steering Committee 2013. Acute kidney injury: global health alert. Kidney Int. 2013; 83:372–76.
5. Jia Z, Zhang Y, Ding G, Heiney KM, Huang S, Zhang A. Role of COX-2/mPGES-1/prostaglandin E2 cascade in kidney injury. Mediators Inflamm. 2015; 2015:147894.
6. Aoki T, Narumiya S. Prostaglandins and chronic inflammation. Trends Pharmacol Sci. 2012; 33:304–11.
7. Hao CM, Breyer MD. Physiological regulation of prostaglandins in the kidney. Annu Rev Physiol. 2008; 70:357–77.
8. Ranganathan PV, Jayakumar C, Mohamed R, Dong Z, Ramesh G. Netrin-1 regulates the inflammatory response of neutrophils and macrophages, and suppresses ischemic acute kidney injury by inhibiting COX-2-mediated PGE2 production. Kidney Int. 2013; 83:1087–98.
9. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011; 31:986–1000.
10. Hsu YH, Chen TH, Chen YC, Cheng CY, Sue YM, Chen JR, Chen CH. Urotensin II exerts antiapoptotic effect on NRK-52E cells through prostacyclin-mediated peroxisome proliferator-activated receptor alpha and Akt activation. Mol Cell Endocrinol. 2013; 381:168–74.
11. Ward JE, Gould H, Harris T, Bonacci JV, Stewart AG. PPAR gamma ligands, 15-deoxy-delta12,14-prostaglandin J2 and rosiglitazone regulate human cultured airway smooth muscle proliferation through different mechanisms. Br J Pharmacol. 2004; 141:517–25.
12. Nørregaard R, Kwon TH, Frøkiær J. Physiology and pathophysiology of cyclooxygenase-2 and prostaglandin E2 in the kidney. Kidney Res Clin Pract. 2015; 34:194–200.
13. Breyer MD, Harris RC. Cyclooxygenase 2 and the kidney. Curr Opin Nephrol Hypertens. 2001; 10:89–98.
14. Green T, Gonzalez AA, Mitchell KD, Navar LG. The complex interplay between cyclooxygenase-2 and angiotensin II in regulating kidney function. Curr Opin Nephrol Hypertens. 2012; 21:7–14.
15. Câmpean V, Theilig F, Paliege A, Breyer M, Bachmann S. Key enzymes for renal prostaglandin synthesis: site-specific expression in rodent kidney (rat, mouse). Am J Physiol Renal Physiol. 2003; 285:F19–32.
16. Olesen ET, Fenton RA. Is there a role for PGE2 in urinary concentration? J Am Soc Nephrol. 2013; 24:169–78.
17. Smith WL, Langenbach R. Why there are two cyclooxygenase isozymes. J Clin Invest. 2001; 107:1491–95.
18. Warford-Woolgar L, Peng CY, Shuhyta J, Wakefield A, Sankaran D, Ogborn M, Aukema HM. Selectivity of cyclooxygenase isoform activity and prostanoid production in normal and diseased Han:SPRD-cy rat kidneys. Am J Physiol Renal Physiol. 2006; 290:F897–904.
19. Poureetezadi SJ, Cheng CN, Chambers JM, Drummond BE, Wingert RA. Prostaglandin signaling regulates nephron segment patterning of renal progenitors during zebrafish kidney development. eLife. 2016; 5:5.
20. Smith WL. Prostanoid biosynthesis and mechanisms of action. Am J Physiol. 1992; 263:F181–91.
21. Wallace JL. Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn’t the stomach digest itself? Physiol Rev. 2008; 88:1547–65.
22. Liu B, Luo W, Zhang Y, Li H, Zhu N, Huang D, Zhou Y. Involvement of cyclo-oxygenase-1-mediated prostacyclin synthesis in the vasoconstrictor activity evoked by ACh in mouse arteries. Exp Physiol. 2012; 97:277–89.
23. Zhu N, Liu B, Luo W, Zhang Y, Li H, Li S, Zhou Y. Vasoconstrictor role of cyclooxygenase-1-mediated prostacyclin synthesis in non-insulin-dependent diabetic mice induced by high-fat diet and streptozotocin. Am J Physiol Heart Circ Physiol. 2014; 307:H319–27.
24. Liu B, Luo W, Zhang Y, Li H, Zhang J, Tan XR, Zhou Y. Concomitant activation of functionally opposing prostacyclin and thromboxane prostanoid receptors by cyclo-oxygenase-1-mediated prostacyclin synthesis in mouse arteries. Exp Physiol. 2012; 97:895–904.
25. Chen CH, Lin H, Hsu YH, Sue YM, Cheng TH, Chan P, Chen TH. The protective effect of prostacyclin on adriamycin-induced apoptosis in rat renal tubular cells. Eur J Pharmacol. 2006; 529:8–15.
26. Cheng CY, Sue YM, Chen CH, Hou CC, Chan P, Chu YL, Chen TH, Hsu YH. Tetramethylpyrazine attenuates adriamycin-induced apoptotic injury in rat renal tubular cells NRK-52E. Planta Med. 2006; 72:888–93.
27. Lin H, Hou CC, Cheng CF, Chiu TH, Hsu YH, Sue YM, Chen TH, Hou HH, Chao YC, Cheng TH, Chen CH. Peroxisomal proliferator-activated receptor-alpha protects renal tubular cells from doxorubicin-induced apoptosis. Mol Pharmacol. 2007; 72:1238–45.
28. Hao CM, Breyer MD. Physiologic and pathophysiologic roles of lipid mediators in the kidney. Kidney Int. 2007; 71:1105–15.
29. Kusakabe T, Maeda M, Hoshi N, Sugino T, Watanabe K, Fukuda T, Suzuki T. Fatty acid synthase is expressed mainly in adult hormone-sensitive cells or cells with high lipid metabolism and in proliferating fetal cells. J Histochem Cytochem. 2000; 48:613–22.
30. Yang T, Huang YG, Ye W, Hansen P, Schnermann JB, Briggs JP. Influence of genetic background and gender on hypertension and renal failure in COX-2-deficient mice. Am J Physiol Renal Physiol. 2005; 288:F1125–32.
31. Fang L, Cheng JC, Chang HM, Sun YP, Leung PC. EGF-like growth factors induce COX-2-derived PGE2 production through ERK1/2 in human granulosa cells. J Clin Endocrinol Metab. 2013; 98:4932–41.
32. Lyons TR, O’Brien J, Borges VF, Conklin MW, Keely PJ, Eliceiri KW, Marusyk A, Tan AC, Schedin P. Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and COX-2. Nat Med. 2011; 17:1109–15.
33. Nørregaard R, Jensen BL, Li C, Wang W, Knepper MA, Nielsen S, Frøkiaer J. COX-2 inhibition prevents downregulation of key renal water and sodium transport proteins in response to bilateral ureteral obstruction. Am J Physiol Renal Physiol. 2005; 289:F322–33.
34. Kömhoff M, Grone HJ, Klein T, Seyberth HW, Nüsing RM. Localization of cyclooxygenase-1 and -2 in adult and fetal human kidney: implication for renal function. Am J Physiol. 1997; 272:F460–68.
35. Cheng H, Wang S, Jo YI, Hao CM, Zhang M, Fan X, Kennedy C, Breyer MD, Moeckel GW, Harris RC. Overexpression of cyclooxygenase-2 predisposes to podocyte injury. J Am Soc Nephrol. 2007; 18:551–59.
36. Dey A, Maric C, Kaesemeyer WH, Zaharis CZ, Stewart J, Pollock JS, Imig JD. Rofecoxib decreases renal injury in obese Zucker rats. Clin Sci (Lond). 2004; 107:561–70.
37. Cheng H, Fan X, Moeckel GW, Harris RC. Podocyte COX-2 exacerbates diabetic nephropathy by increasing podocyte (pro)renin receptor expression. J Am Soc Nephrol. 2011; 22:1240–51.
38. Agrawal S, Guess AJ, Chanley MA, Smoyer WE. Albumin-induced podocyte injury and protection are associated with regulation of COX-2. Kidney Int. 2014; 86:1150–60.
39. Sankaran D, Bankovic-Calic N, Ogborn MR, Crow G, Aukema HM. Selective COX-2 inhibition markedly slows disease progression and attenuates altered prostanoid production in Han:SPRD-cy rats with inherited kidney disease. Am J Physiol Renal Physiol. 2007; 293:F821–30.
40. Wang L, Sha Y, Bai J, Eisner W, Sparks MA, Buckley AF, Spurney RF. Podocyte-specific knockout of cyclooxygenase 2 exacerbates diabetic kidney disease. Am J Physiol Renal Physiol. 2017; 313:F430–39.
41. Wang X, Yao B, Wang Y, Fan X, Wang S, Niu A, Yang H, Fogo A, Zhang MZ, Harris RC. Macrophage Cyclooxygenase-2 Protects Against Development of Diabetic Nephropathy. Diabetes. 2017; 66:494–504.
42. Li C, Mo Z, Xie J, Xu L, Tan L, Luo D, Chen H, Yang H, Li Y, Su Z, Su Z. Chongcao-Shencha Attenuates Liver and Kidney Injury through Attenuating Oxidative Stress and Inflammatory Response in D-Galactose-Treated Mice. Evid Based Complement Alternat Med. 2016; 2016:3878740.
43. Patel NS, Cuzzocrea S, Collino M, Chaterjee PK, Mazzon E, Britti D, Yaqoob MM, Thiemermann C. The role of cycloxygenase-2 in the rodent kidney following ischaemia/reperfusion injury in vivo. Eur J Pharmacol. 2007; 562:148–54.
44. Jia Z, Wang N, Aoyagi T, Wang H, Liu H, Yang T. Amelioration of cisplatin nephrotoxicity by genetic or pharmacologic blockade of prostaglandin synthesis. Kidney Int. 2011; 79:77–88.
45. Feitoza CQ, Câmara NO, Pinheiro HS, Gonçalves GM, Cenedeze MA, Pacheco-Silva A, Santos OF. Cyclooxygenase 1 and/or 2 blockade ameliorates the renal tissue damage triggered by ischemia and reperfusion injury. Int Immunopharmacol. 2005; 5:79–84.
46. Yu X, Yang Y, Yuan H, Wu M, Li S, Gong W, Yu J, Xia W, Zhang Y, Ding G, Huang S, Jia Z, Zhang A. Inhibition of COX-2/PGE2 cascade ameliorates cisplatin-induced mesangial cell apoptosis. Am J Transl Res. 2017; 9:1222–29.
47. Jin J, Tang Q, Li Z, Zhao Z, Zhang Z, Lu L, Zhu T, Vanhoutte PM, Leung SW, Tu R, Shi Y. Prostaglandin E2 regulates renal function in C57/BL6 mouse with 5/6 nephrectomy. Life Sci. 2017; 174:68–76.
48. Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002; 82:131–85.
49. Schnermann J. Concurrent activation of multiple vasoactive signaling pathways in vasoconstriction caused by tubuloglomerular feedback: a quantitative assessment. Annu Rev Physiol. 2015; 77:301–22.
50. Dordea AC, Vandenwijngaert S, Garcia V, Tainsh RE, Nathan DI, Allen K, Raher MJ, Tainsh LT, Zhang F, Lieb WS, Mikelman S, Kirby A, Stevens C, et al. Androgen-sensitive hypertension associated with soluble guanylate cyclase-α1 deficiency is mediated by 20-HETE. Am J Physiol Heart Circ Physiol. 2016; 310:H1790–800.
51. Tunctan B, Sari AN, Kacan M, Unsal D, Buharalioglu CK, Sahan-Firat S, Korkmaz B, Falck JR, Malik KU. NS-398 reverses hypotension in endotoxemic rats: contribution of eicosanoids, NO, and peroxynitrite. Prostaglandins Other Lipid Mediat. 2013; 104-105:93–108.
52. Tunctan B, Korkmaz B, Cuez T, Kemal Buharalioglu C, Sahan-Firat S, Falck J, Malik KU. Contribution of vasoactive eicosanoids and nitric oxide production to the effect of selective cyclooxygenase-2 inhibitor, NS-398, on endotoxin-induced hypotension in rats. Basic Clin Pharmacol Toxicol. 2010; 107:877–82.
53. Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Ikeda T, Fueki M, Ueno A, Oh S, Kudo I. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000; 275:32783–92.
54. Jia Z, Zhang A, Zhang H, Dong Z, Yang T. Deletion of microsomal prostaglandin E synthase-1 increases sensitivity to salt loading and angiotensin II infusion. Circ Res. 2006; 99:1243–51.
55. Soodvilai S, Jia Z, Yang T. Hydrogen peroxide stimulates chloride secretion in primary inner medullary collecting duct cells via mPGES-1-derived PGE2. Am J Physiol Renal Physiol. 2007; 293:F1571–76.
56. Jia Z, Sun Y, Liu S, Liu Y, Yang T. COX-2 but not mPGES-1 contributes to renal PGE2 induction and diabetic proteinuria in mice with type-1 diabetes. PLoS One. 2014; 9:e93182.
57. Li T, Liu Y, Zhao J, Miao S, Xu Y, Liu K, Liu M, Wang G, Xiao X. Aggravation of acute kidney injury by mPGES-2 down regulation is associated with autophagy inhibition and enhanced apoptosis. Sci Rep. 2017; 7:10247.
58. Salazar F, Vazquez ML, Masferrer JL, Mbalaviele G, Llinas MT, Saez F, Arhancet G, Salazar FJ. Renal effects induced by prolonged mPGES1 inhibition. Am J Physiol Renal Physiol. 2014; 306:F68–74.
59. Jia Z, Wang H, Yang T. Mice lacking mPGES-1 are resistant to lithium-induced polyuria. Am J Physiol Renal Physiol. 2009; 297:F1689–96.
60. Jia Z, Aoyagi T, Yang T. mPGES-1 protects against DOCA-salt hypertension via inhibition of oxidative stress or stimulation of NO/cGMP. Hypertension. 2010; 55:539–46.
61. Jia Z, Aoyagi T, Kohan DE, Yang T. mPGES-1 deletion impairs aldosterone escape and enhances sodium appetite. Am J Physiol Renal Physiol. 2010; 299:F155–66.
62. Luo R, Kakizoe Y, Wang F, Fan X, Hu S, Yang T, Wang W, Li C. Deficiency of mPGES-1 exacerbates renal fibrosis and inflammation in mice with unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2017; 312:F121–33.
63. Jia Z, Liu G, Downton M, Dong Z, Zhang A, Yang T. mPGES-1 deletion potentiates urine concentrating capability after water deprivation. Am J Physiol Renal Physiol. 2012; 302:F1005–12.
64. Jia Z, Wang H, Yang T. Microsomal prostaglandin E synthase 1 deletion retards renal disease progression but exacerbates anemia in mice with renal mass reduction. Hypertension. 2012; 59:122–28.
65. Sun Y, Jia Z, Liu G, Zhou L, Liu M, Yang B, Yang T. PPARγ Agonist Rosiglitazone Suppresses Renal mPGES-1/PGE2 Pathway in db/db Mice. PPAR Res. 2013; 2013:612971.
66. Yu J, Wu Y, Wang L, Zhang W, Xu M, Song J, Fu Y, Cui Y, Gong W, Li S, Xia W, Huang S, Zhang A, Jia Z. mPGES-1-derived prostaglandin E2 stimulates Stat3 to promote podocyte apoptosis. Apoptosis. 2017; 22:1431–40.
67. Li S, Cheng S, Sun Z, Mungun HK, Gong W, Yu J, Xia W, Zhang Y, Huang S, Zhang A, Jia Z. Indoxyl Sulfate Induces Mesangial Cell Proliferation via the Induction of COX-2. Mediators Inflamm. 2016; 2016:5802973.
68. Li S, Sun Z, Zhang Y, Ruan Y, Chen Q, Gong W, Yu J, Xia W, He JC, Huang S, Zhang A, Ding G, Jia Z. COX-2/mPGES-1/PGE2 cascade activation mediates uric acid-induced mesangial cell proliferation. Oncotarget. 2017; 8:10185-10198. https://doi.org/10.18632/oncotarget.14363
69. Soodvilai S, Jia Z, Wang MH, Dong Z, Yang T. mPGES-1 deletion impairs diuretic response to acute water loading. Am J Physiol Renal Physiol. 2009; 296:F1129–35.
70. Hébert RL, O’Connor T, Neville C, Burns KD, Laneuville O, Peterson LN. Prostanoid signaling, localization, and expression of IP receptors in rat thick ascending limb cells. Am J Physiol. 1998; 275:F904–14.
71. Guan Y, Zhang Y, Breyer RM, Fowler B, Davis L, Hébert RL, Breyer MD. Prostaglandin E2 inhibits renal collecting duct Na+ absorption by activating the EP1 receptor. J Clin Invest. 1998; 102:194–201.
72. Gonzalez AA, Salinas-Parra N, Leach D, Navar LG, Prieto MC. PGE2 upregulates renin through E-prostanoid receptor 1 via PKC/cAMP/CREB pathway in M-1 cells. Am J Physiol Renal Physiol. 2017; 313:F1038–49.
73. Srivastava T, Dai H, Heruth DP, Alon US, Garola RE, Zhou J, Duncan RS, El-Meanawy A, McCarthy ET, Sharma R, Johnson ML, Savin VJ, Sharma M. Mechanotransduction signaling in podocytes from fluid flow shear stress. Am J Physiol Renal Physiol. 2018; 314:F22–34.
74. Kennedy CR, Xiong H, Rahal S, Vanderluit J, Slack RS, Zhang Y, Guan Y, Breyer MD, Hébert RL. Urine concentrating defect in prostaglandin EP1-deficient mice. Am J Physiol Renal Physiol. 2007; 292:F868–75.
75. Nüsing RM, Treude A, Weissenberger C, Jensen B, Bek M, Wagner C, Narumiya S, Seyberth HW. Dominant role of prostaglandin E2 EP4 receptor in furosemide-induced salt-losing tubulopathy: a model for hyperprostaglandin E syndrome/antenatal Bartter syndrome. J Am Soc Nephrol. 2005; 16:2354–62.
76. Qian Q, Kassem KM, Beierwaltes WH, Harding P. PGE2 causes mesangial cell hypertrophy and decreases expression of cyclin D3. Nephron, Physiol. 2009; 113:7–p14.
77. Makino H, Tanaka I, Mukoyama M, Sugawara A, Mori K, Muro S, Suganami T, Yahata K, Ishibashi R, Ohuchida S, Maruyama T, Narumiya S, Nakao K. Prevention of diabetic nephropathy in rats by prostaglandin E receptor EP1-selective antagonist. J Am Soc Nephrol. 2002; 13:1757–65.
78. Srivastava T, McCarthy ET, Sharma R, Kats A, Carlton CG, Alon US, Cudmore PA, El-Meanawy A, Sharma M. Fluid flow shear stress upregulates prostanoid receptor EP2 but not EP4 in murine podocytes. Prostaglandins Other Lipid Mediat. 2013; 104-105:49–57.
79. Nasrallah R, Hassouneh R, Zimpelmann J, Karam AJ, Thibodeau JF, Burger D, Burns KD, Kennedy CR, Hébert RL. Prostaglandin E2 increases proximal tubule fluid reabsorption, and modulates cultured proximal tubule cell responses via EP1 and EP4 receptors. Lab Invest. 2015; 95:1044–55.
80. Thibodeau JF, Nasrallah R, Carter A, He Y, Touyz R, Hébert RL, Kennedy CR. PTGER1 deletion attenuates renal injury in diabetic mouse models. Am J Pathol. 2013; 183:1789–802.
81. Ni WJ, Tang LQ, Zhou H, Ding HH, Qiu YY. Renoprotective effect of berberine via regulating the PGE2 -EP1-Gαq-Ca(2+) signalling pathway in glomerular mesangial cells of diabetic rats. J Cell Mol Med. 2016; 20:1491–502.
82. Rahal S, McVeigh LI, Zhang Y, Guan Y, Breyer MD, Kennedy CR. Increased severity of renal impairment in nephritic mice lacking the EP1 receptor. Can J Physiol Pharmacol. 2006; 84:877–85.
83. Kennedy CR, Zhang Y, Brandon S, Guan Y, Coffee K, Funk CD, Magnuson MA, Oates JA, Breyer MD, Breyer RM. Salt-sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat Med. 1999; 5:217–20.
84. Chun KS, Shim M. EP2 Induces p38 Phosphorylation via the Activation of Src in HEK 293 Cells. Biomol Ther (Seoul). 2015; 23:539–48.
85. Vukicevic S, Simic P, Borovecki F, Grgurevic L, Rogic D, Orlic I, Grasser WA, Thompson DD, Paralkar VM. Role of EP2 and EP4 receptor-selective agonists of prostaglandin E(2) in acute and chronic kidney failure. Kidney Int. 2006; 70:1099–106.
86. Elberg G, Elberg D, Lewis TV, Guruswamy S, Chen L, Logan CJ, Chan MD, Turman MA. EP2 receptor mediates PGE2-induced cystogenesis of human renal epithelial cells. Am J Physiol Renal Physiol. 2007; 293:F1622–32.
87. Zahner G, Schaper M, Panzer U, Kluger M, Stahl RA, Thaiss F, Schneider A. Prostaglandin EP2 and EP4 receptors modulate expression of the chemokine CCL2 (MCP-1) in response to LPS-induced renal glomerular inflammation. Biochem J. 2009; 422:563–70.
88. Srivastava T, Alon US, Cudmore PA, Tarakji B, Kats A, Garola RE, Duncan RS, McCarthy ET, Sharma R, Johnson ML, Bonewald LF, El-Meanawy A, Savin VJ, Sharma M. Cyclooxygenase-2, prostaglandin E2, and prostanoid receptor EP2 in fluid flow shear stress-mediated injury in the solitary kidney. Am J Physiol Renal Physiol. 2014; 307:F1323–33.
89. Xi PP, Xu YY, Chen XL, Fan YP, Wu JH. Role of the prostaglandin E2 receptor agonists in TGF-β1-induced mesangial cell damage. Biosci Rep. 2016; 36:e00383.
90. van Rodijnen WF, Korstjens IJ, Legerstee N, Ter Wee PM, Tangelder GJ. Direct vasoconstrictor effect of prostaglandin E2 on renal interlobular arteries: role of the EP3 receptor. Am J Physiol Renal Physiol. 2007; 292:F1094–101.
91. Badzynska B, Sadowski J. Opposed effects of prostaglandin E2 on perfusion of rat renal cortex and medulla: interactions with the renin-angiotensin system. Exp Physiol. 2008; 93:1292–302.
92. Kvirkvelia N, McMenamin M, Chaudhary K, Bartoli M, Madaio MP. Prostaglandin E2 promotes cellular recovery from established nephrotoxic serum nephritis in mice, prosurvival, and regenerative effects on glomerular cells. Am J Physiol Renal Physiol. 2013; 304:F463–70.
93. Hassouneh R, Nasrallah R, Zimpelmann J, Gutsol A, Eckert D, Ghossein J, Burns KD, Hébert RL. PGE2 receptor EP3 inhibits water reabsorption and contributes to polyuria and kidney injury in a streptozotocin-induced mouse model of diabetes. Diabetologia. 2016; 59:1318–28.
94. Fujino H, Xu W, Regan JW. Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J Biol Chem. 2003; 278:12151–56.
95. Jang MW, Yun SP, Park JH, Ryu JM, Lee JH, Han HJ. Cooperation of Epac1/Rap1/Akt and PKA in prostaglandin E(2) -induced proliferation of human umbilical cord blood derived mesenchymal stem cells: involvement of c-Myc and VEGF expression. J Cell Physiol. 2012; 227:3756–67.
96. Leduc M, Breton B, Galés C, Le Gouill C, Bouvier M, Chemtob S, Heveker N. Functional selectivity of natural and synthetic prostaglandin EP4 receptor ligands. J Pharmacol Exp Ther. 2009; 331:297–307.
97. Yang T, Xu C. Physiology and Pathophysiology of the Intrarenal Renin-Angiotensin System: an Update. J Am Soc Nephrol. 2017; 28:1040–49.
98. Yokoyama U, Iwatsubo K, Umemura M, Fujita T, Ishikawa Y. The prostanoid EP4 receptor and its signaling pathway. Pharmacol Rev. 2013; 65:1010–52.
99. Stitt-Cavanagh EM, Faour WH, Takami K, Carter A, Vanderhyden B, Guan Y, Schneider A, Breyer MD, Kennedy CR. A maladaptive role for EP4 receptors in podocytes. J Am Soc Nephrol. 2010; 21:1678–90.
100. Faour WH, Thibodeau JF, Kennedy CR. Mechanical stretch and prostaglandin E2 modulate critical signaling pathways in mouse podocytes. Cell Signal. 2010; 22:1222–30.
101. Cheng H, Fan X, Guan Y, Moeckel GW, Zent R, Harris RC. Distinct roles for basal and induced COX-2 in podocyte injury. J Am Soc Nephrol. 2009; 20:1953–62.
102. Yamamoto E, Izawa T, Juniantito V, Kuwamura M, Sugiura K, Takeuchi T, Yamate J. Involvement of endogenous prostaglandin E2 in tubular epithelial regeneration through inhibition of apoptosis and epithelial-mesenchymal transition in cisplatin-induced rat renal lesions. Histol Histopathol. 2010; 25:995–1007.
103. Mohamed R, Jayakumar C, Ramesh G. Chronic administration of EP4-selective agonist exacerbates albuminuria and fibrosis of the kidney in streptozotocininduced diabetic mice through IL-6. Lab Invest. 2013; 93:933–45.
104. Thieme K, Majumder S, Brijmohan AS, Batchu SN, Bowskill BB, Alghamdi TA, Advani SL, Kabir MG, Liu Y, Advani A. EP4 inhibition attenuates the development of diabetic and non-diabetic experimental kidney disease. Sci Rep. 2017; 7:3442.
105. Hong YA, Yang KJ, Jung SY, Chang YK, Park CW, Yang CW, Kim SY, Hwang HS. Paricalcitol attenuates lipopolysaccharide-induced inflammation and apoptosis in proximal tubular cells through the prostaglandin E2 receptor EP4. Kidney Res Clin Pract. 2017; 36:145–58.
106. Nakagawa N, Yuhki K, Kawabe J, Fujino T, Takahata O, Kabara M, Abe K, Kojima F, Kashiwagi H, Hasebe N, Kikuchi K, Sugimoto Y, Narumiya S, Ushikubi F. The intrinsic prostaglandin E2-EP4 system of the renal tubular epithelium limits the development of tubulointerstitial fibrosis in mice. Kidney Int. 2012; 82:158–71.
107. Gao M, Cao R, Du S, Jia X, Zheng S, Huang S, Han Q, Liu J, Zhang X, Miao Y, Kang J, Gustafsson JÅ, Guan Y. Disruption of prostaglandin E2 receptor EP4 impairs urinary concentration via decreasing aquaporin 2 in renal collecting ducts. Proc Natl Acad Sci USA. 2015; 112:8397–402.
108. Wang F, Lu X, Peng K, Fang H, Zhou L, Su J, Nau A, Yang KT, Ichihara A, Lu A, Zhou SF, Yang T. Antidiuretic Action of Collecting Duct (Pro)Renin Receptor Downstream of Vasopressin and PGE2 Receptor EP4. J Am Soc Nephrol. 2016; 27:3022–34.
109. Klein T, Klaus G, Kömhoff M. Prostacyclin synthase: upregulation during renal development and in glomerular disease as well as its constitutive expression in cultured human mesangial cells. Mediators Inflamm. 2015; 2015:654151.
110. Tone Y, Inoue H, Hara S, Yokoyama C, Hatae T, Oida H, Narumiya S, Shigemoto R, Yukawa S, Tanabe T. The regional distribution and cellular localization of mRNA encoding rat prostacyclin synthase. Eur J Cell Biol. 1997; 72:268–77.
111. Yokoyama C, Yabuki T, Shimonishi M, Wada M, Hatae T, Ohkawara S, Takeda J, Kinoshita T, Okabe M, Tanabe T. Prostacyclin-deficient mice develop ischemic renal disorders, including nephrosclerosis and renal infarction. Circulation. 2002; 106:2397–403.
112. Nasrallah R, Hébert RL. Prostacyclin signaling in the kidney: implications for health and disease. Am J Physiol Renal Physiol. 2005; 289:F235–46.
113. Lim H, Dey SK. A novel pathway of prostacyclin signaling-hanging out with nuclear receptors. Endocrinology. 2002; 143:3207–10.
114. D’Apolito M, Du X, Pisanelli D, Pettoello-Mantovani M, Campanozzi A, Giacco F, Maffione AB, Colia AL, Brownlee M, Giardino I. Urea-induced ROS cause endothelial dysfunction in chronic renal failure. Atherosclerosis. 2015; 239:393–400.
115. Kawabe J, Ushikubi F, Hasebe N. Prostacyclin in vascular diseases. - Recent insights and future perspectives -. Circ J. 2010; 74:836–43.
116. Stitham J, Midgett C, Martin KA, Hwa J. Prostacyclin: an inflammatory paradox. Front Pharmacol. 2011; 2:24.
117. Yamada M, Sasaki R, Sato N, Suzuki M, Tamura M, Matsushita T, Kurumatani H. Amelioration by beraprost sodium, a prostacyclin analogue, of established renal dysfunction in rat glomerulonephritis model. Eur J Pharmacol. 2002; 449:167–76.
118. Chen S, Xie S, He W, Wei D, Li S, Chen W. Beneficial Effect of Beraprost Sodium Plus Aspirin in the Treatment of Acute Ischemic Stroke. Med Sci Monit. 2017; 23:4401–07.
119. Okaka EI, Ojeh-Oziegbe OE, Unuigbe EI. Factors associated with blood pressure control in predialysis chronic kidney disease patients: short-term experience from a single center in Southern Nigeria. Niger J Clin Pract. 2017; 20:537–41.
120. Koyama A, Fujita T, Gejyo F, Origasa H, Isono M, Kurumatani H, Okada K, Kanoh H, Kiriyama T, Yamada S. Orally active prostacyclin analogue beraprost sodium in patients with chronic kidney disease: a randomized, double-blind, placebo-controlled, phase II dose finding trial. BMC Nephrol. 2015; 16:165.
121. Guan J, Long L, Chen YQ, Yin Y, Li L, Zhang CX, Deng L, Tian LH. Effects of beraprost sodium on renal function and inflammatory factors of rats with diabetic nephropathy. Genet Mol Res. 2014; 13:4154–58.
122. Peng L, Li J, Xu Y, Wang Y, Du H, Shao J, Liu Z. The Protective Effect of Beraprost Sodium on Diabetic Nephropathy by Inhibiting Inflammation and p38 MAPK Signaling Pathway in High-Fat Diet/Streptozotocin-Induced Diabetic Rats. Int J Endocrinol. 2016; 2016:1690474.
123. Nasrallah R, Nusing RM, Hébert RL. Localization of IP in rabbit kidney and functional role of the PGI(2)/IP system in cortical collecting duct. Am J Physiol Renal Physiol. 2002; 283:F689–98.
124. Reid HM, Kinsella BT. Prostacyclin receptors: Transcriptional regulation and novel signalling mechanisms. Prostaglandins Other Lipid Mediat. 2015; 121:70–82. https://doi.org/10.1016/j.prostaglandins.2015.04.008.
125. Nasrallah R, Clark J, Hébert RL. Prostaglandins in the kidney: developments since Y2K. Clin Sci (Lond). 2007; 113:297–311.
126. Batchu SN, Majumder S, Bowskill BB, White KE, Advani SL, Brijmohan AS, Liu Y, Thai K, Azizi PM, Lee WL, Advani A. Prostaglandin I2 Receptor Agonism Preserves β-Cell Function and Attenuates Albuminuria Through Nephrin-Dependent Mechanisms. Diabetes. 2016; 65:1398–409.
127. Yamasaki H, Maeshima Y, Nasu T, Saito D, Tanabe K, Hirokoshi-Kawahara K, Sugiyama H, Sakai Y, Makino H. Intermittent administration of a sustained-release prostacyclin analog ONO-1301 ameliorates renal alterations in a rat type 1 diabetes model. Prostaglandins Leukot Essent Fatty Acids. 2011; 84:99–107.
128. Nasu T, Kinomura M, Tanabe K, Yamasaki H, Htay SL, Saito D, Hinamoto N, Watatani H, Ujike H, Suzuki Y, Sugaya T, Sugiyama H, Sakai Y, et al. Sustained-release prostacyclin analog ONO-1301 ameliorates tubulointerstitial alterations in a mouse obstructive nephropathy model. Am J Physiol Renal Physiol. 2012; 302:F1616–29.
129. Watatani H, Yamasaki H, Maeshima Y, Nasu T, Hinamoto N, Ujike H, Sugiyama H, Sakai Y, Tanabe K, Makino H. ONO-1301, a sustained-release prostacyclin analog, ameliorates the renal alterations in a mouse type 2 diabetes model possibly through its protective effects on mesangial cells. Acta Med Okayama. 2015; 69:1–15.
130. Wang L, Li SJ, Sidhu A, Zhu L, Liang Y, Freedman RB, Wang CC. Reconstitution of human Ero1-Lalpha/proteindisulfide isomerase oxidative folding pathway in vitro. Position-dependent differences in role between the a and a′ domains of protein-disulfide isomerase. J Biol Chem. 2009; 284:199–206.
131. Chen HH, Sue YM, Chen CH, Hsu YH, Hou CC, Cheng CY, Lin SL, Tsai WL, Chen TW, Chen TH. Peroxisome proliferator-activated receptor alpha plays a crucial role in L-carnitine anti-apoptosis effect in renal tubular cells. Nephrol Dial Transplant. 2009; 24:3042–49.
132. Chen HH, Chen TW, Lin H. Prostacyclin-induced peroxisome proliferator-activated receptor-alpha translocation attenuates NF-kappaB and TNF-alpha activation after renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2009; 297:F1109–18.
133. Sue YM, Chou HC, Chang CC, Yang NJ, Chou Y, Juan SH. L-carnitine protects against carboplatin-mediated renal injury: AMPK- and PPARα-dependent inactivation of NFAT3. PLoS One. 2014; 9:e104079.
134. Mederle K, Meurer M, Castrop H, Höcherl K. Inhibition of COX-1 attenuates the formation of thromboxane A2 and ameliorates the acute decrease in glomerular filtration rate in endotoxemic mice. Am J Physiol Renal Physiol. 2015; 309:F332–40.
135. Sakariassen KS, Alberts P, Fontana P, Mann J, Bounameaux H, Sorensen AS. Effect of pharmaceutical interventions targeting thromboxane receptors and thromboxane synthase in cardiovascular and renal diseases. Future Cardiol. 2009; 5:479–93.
136. Nakahata N. Thromboxane A2: physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol Ther. 2008; 118:18–35.
137. Miyosawa K, Sasaki M, Ohkubo S, Nakahata N. Different pathways for activation of extracellular signal-regulated kinase through thromboxane A2 receptor isoforms. Biol Pharm Bull. 2006; 29:719–24.
138. Ashton AW, Ware JA. Thromboxane A2 receptor signaling inhibits vascular endothelial growth factor-induced endothelial cell differentiation and migration. Circ Res. 2004; 95:372–79.
139. Wikström K, Kavanagh DJ, Reid HM, Kinsella BT. Differential regulation of RhoA-mediated signaling by the TPalpha and TPbeta isoforms of the human thromboxane A2 receptor: independent modulation of TPalpha signaling by prostacyclin and nitric oxide. Cell Signal. 2008; 20:1497–512.
140. Yu OM, Brown JH. G Protein-Coupled Receptor and RhoA-Stimulated Transcriptional Responses: Links to Inflammation, Differentiation, and Cell Proliferation. Mol Pharmacol. 2015; 88:171–80.
141. Chien CT, Fan SC, Lin SC, Kuo CC, Yang CH, Yu TY, Lee SP, Cheng DY, Li PC. Glucagon-like peptide-1 receptor agonist activation ameliorates venous thrombosis-induced arteriovenous fistula failure in chronic kidney disease. Thromb Haemost. 2014; 112:1051–64.
142. Welch WJ. Effects of isoprostane on tubuloglomerular feedback: roles of TP receptors, NOS, and salt intake. Am J Physiol Renal Physiol. 2005; 288:F757–62.
143. Pastori D, Pignatelli P, Perticone F, Sciacqua A, Carnevale R, Farcomeni A, Basili S, Corazza GR, Davì G, Lip GY, Violi F, and ARAPACIS (Atrial Fibrillation Registry for Ankle-Brachial Index Prevalence Assessment-Collaborative Italian Study) study group. Aspirin and renal insufficiency progression in patients with atrial fibrillation and chronic kidney disease. Int J Cardiol. 2016; 223:619–24.
144. Boini KM, Xia M, Li C, Zhang C, Payne LP, Abais JM, Poklis JL, Hylemon PB, Li PL. Acid sphingomyelinase gene deficiency ameliorates the hyperhomocysteinemiainduced glomerular injury in mice. Am J Pathol. 2011; 179:2210–19.
145. Smith AR, Visioli F, Frei B, Hagen TM. Age-related changes in endothelial nitric oxide synthase phosphorylation and nitric oxide dependent vasodilation: evidence for a novel mechanism involving sphingomyelinase and ceramideactivated phosphatase 2A. Aging Cell. 2006; 5:391–400.
146. Bautista-Pérez R, del Valle-Mondragón L, Cano-Martínez A, Pérez-Méndez O, Escalante B, Franco M. Involvement of neutral sphingomyelinase in the angiotensin II signaling pathway. Am J Physiol Renal Physiol. 2015; 308:F1178–87.
147. Okumura M, Imanishi M, Okamura M, Hosoi M, Okada N, Konishi Y, Morikawa T, Miura K, Nakatani T, Fujii S. Role for thromboxane A2 from glomerular thrombi in nephropathy with type 2 diabetic rats. Life Sci. 2003; 72:2695–705.
148. Boffa JJ, Just A, Coffman TM, Arendshorst WJ. Thromboxane receptor mediates renal vasoconstriction and contributes to acute renal failure in endotoxemic mice. J Am Soc Nephrol. 2004; 15:2358–65.
149. Araujo M, Welch WJ. Cyclooxygenase 2 inhibition suppresses tubuloglomerular feedback: roles of thromboxane receptors and nitric oxide. Am J Physiol Renal Physiol. 2009; 296:F790–94.
150. Schmitz PG, Zhang K, Dalal R. Eicosapentaenoic acid suppresses PDGF-induced DNA synthesis in rat mesangial cells: involvement of thromboxane A2. Kidney Int. 2000; 57:1041–51.
151. Xu S, Jiang B, Maitland KA, Bayat H, Gu J, Nadler JL, Corda S, Lavielle G, Verbeuren TJ, Zuccollo A, Cohen RA. The thromboxane receptor antagonist S18886 attenuates renal oxidant stress and proteinuria in diabetic apolipoprotein E-deficient mice. Diabetes. 2006; 55:110–19.
152. Wang C, Luo Z, Kohan D, Wellstein A, Jose PA, Welch WJ, Wilcox CS, Wang D. Thromboxane prostanoid receptors enhance contractions, endothelin-1, and oxidative stress in microvessels from mice with chronic kidney disease. Hypertension. 2015; 65:1055–63.
153. Salvati P, Lamberti E, Ferrario R, Ferrario RG, Scampini G, Pugliese F, Barsotti P, Patrono C. Long-term thromboxanesynthase inhibition prolongs survival in murine lupus nephritis. Kidney Int. 1995; 47:1168–75.
154. Smith SR, Creech EA, Schaffer AV, Martin LL, Rakhit A, Douglas FL, Klotman PE, Coffman TM. Effects of thromboxane synthase inhibition with CGS 13080 in human cyclosporine nephrotoxicity. Kidney Int. 1992; 41:199–205.
155. Coffman TM, Ruiz P, Sanfilippo F, Klotman PE. Chronic thromboxane inhibition preserves function of rejecting rat renal allografts. Kidney Int. 1989; 35:24–30.
156. Rao PS, Cavanagh D, Dietz JR, Marsden KA, O’Brien WF, Spaziani E. Dose-dependent effects of prostaglandin D2 on hemodynamics, renal function, and blood gas analyses. Am J Obstet Gynecol. 1987; 156:843–51.
157. Helliwell RJ, Adams LF, Mitchell MD. Prostaglandin synthases: recent developments and a novel hypothesis. Prostaglandins Leukot Essent Fatty Acids. 2004; 70:101–13.
158. Christ AN, Labzin L, Bourne GT, Fukunishi H, Weber JE, Sweet MJ, Smythe ML, Flanagan JU. Development and characterization of new inhibitors of the human and mouse hematopoietic prostaglandin D(2) synthases. J Med Chem. 2010; 53:5536–48.
159. Trivedi SG, Newson J, Rajakariar R, Jacques TS, Hannon R, Kanaoka Y, Eguchi N, Colville-Nash P, Gilroy DW. Essential role for hematopoietic prostaglandin D2 synthase in the control of delayed type hypersensitivity. Proc Natl Acad Sci USA. 2006; 103:5179–84.
160. Joo M, Sadikot RT. PGD synthase and PGD2 in immune resposne. Mediators Inflamm. 2012; 2012:503128.
161. Foster MC, Inker LA, Levey AS, Selvin E, Eckfeldt J, Juraschek SP, Coresh J, and CKD Biomarkers Consortium. Novel filtration markers as predictors of all-cause and cardiovascular mortality in US adults. Am J Kidney Dis. 2013; 62:42–51.
162. Astor BC, Shafi T, Hoogeveen RC, Matsushita K, Ballantyne CM, Inker LA, Coresh J. Novel markers of kidney function as predictors of ESRD, cardiovascular disease, and mortality in the general population. Am J Kidney Dis. 2012; 59:653–62.
163. Beuckmann CT, Aoyagi M, Okazaki I, Hiroike T, Toh H, Hayaishi O, Urade Y. Binding of biliverdin, bilirubin, and thyroid hormones to lipocalin-type prostaglandin D synthase. Biochemistry. 1999; 38:8006–13.
164. White CA, Ghazan-Shahi S, Adams MA. β-Trace protein: a marker of GFR and other biological pathways. Am J Kidney Dis. 2015; 65:131–46.
165. Bhavsar NA, Appel LJ, Kusek JW, Contreras G, Bakris G, Coresh J, Astor BC, and AASK Study Group. Comparison of measured GFR, serum creatinine, cystatin C, and beta-trace protein to predict ESRD in African Americans with hypertensive CKD. Am J Kidney Dis. 2011; 58:886–93.
166. Rebholz CM, Inker LA, Chen Y, Liang M, Foster MC, Eckfeldt JH, Kimmel PL, Vasan RS, Feldman HI, Sarnak MJ, Hsu CY, Levey AS, Coresh J, and Chronic Kidney Disease Biomarkers Consortium. Risk of ESRD and Mortality Associated With Change in Filtration Markers. Am J Kidney Dis. 2017; 70:551–60.
167. Inker LA, Coresh J, Sang Y, Hsu CY, Foster MC, Eckfeldt JH, Karger AB, Nelson RG, Liu X, Sarnak M, Appel LJ, Grams M, Xie D, et al, and CKD Biomarkers Consortium. Filtration Markers as Predictors of ESRD and Mortality: Individual Participant Data Meta-Analysis. Clin J Am Soc Nephrol. 2017; 12:69–78.
168. Dajak M, Ignjatović S, Stojimirović B, Gajić S, Majkić-Singh N. Urinary beta-trace protein as a tubular marker of renal dysfunction in patients with chronic kidney disease. Clin Chim Acta. 2010; 411:1154–55.
169. Uehara Y, Makino H, Seiki K, Urade Y, and L-PGDS Clinical Research Group of Kidney. Urinary excretions of lipocalin-type prostaglandin D synthase predict renal injury in type-2 diabetes: a cross-sectional and prospective multicentre study. Nephrol Dial Transplant. 2009; 24:475–82.
170. Ito H, Yan X, Nagata N, Aritake K, Katsumata Y, Matsuhashi T, Nakamura M, Hirai H, Urade Y, Asano K, Kubo M, Utsunomiya Y, Hosoya T, et al. PGD2-CRTH2 pathway promotes tubulointerstitial fibrosis. J Am Soc Nephrol. 2012; 23:1797–809.
171. Orenes-Piñero E, Manzano-Fernández S, López-Cuenca Á, Marín F, Valdés M, Januzzi JL. β-Trace protein: from GFR marker to cardiovascular risk predictor. Clin J Am Soc Nephrol. 2013; 8:873–81.
172. Ragolia L, Palaia T, Hall CE, Maesaka JK, Eguchi N, Urade Y. Accelerated glucose intolerance, nephropathy, and atherosclerosis in prostaglandin D2 synthase knock-out mice. J Biol Chem. 2005; 280:29946–55.
173. Tsuchida T, Eguchi N, Eguchi Y, Numabe A, Nakajima H, Oda H, Seiki K, Hakamada-Taguchi R, Urade Y, Uehara Y. Lipocalin-type prostaglandin D synthase in urine in adriamycin-induced nephropathy of mice. Nephron Physiol. 2004; 96:p42–51.
174. Hirawa N, Uehara Y, Ikeda T, Gomi T, Hamano K, Totsuka Y, Yamakado M, Takagi M, Eguchi N, Oda H, Seiki K, Nakajima H, Urade Y. Urinary prostaglandin D synthase (beta-trace) excretion increases in the early stage of diabetes mellitus. Nephron. 2001; 87:321–27.
175. Kupczyk M, Kuna P. Targeting the PGD2/CRTH2/DP1 Signaling Pathway in Asthma and Allergic Disease: Current Status and Future Perspectives. Drugs. 2017; 77:1281–94.
176. Hearon CM Jr, Richards JC, Racine ML, Luckasen GJ, Larson DG, Joyner MJ, Dinenno FA. Sympatholytic effect of intravascular ATP is independent of nitric oxide, prostaglandins, Na+ /K+ -ATPase and KIR channels in humans. J Physiol. 2017; 595:5175–90.
177. Hubertus K, Mischnik M, Timmer J, Herterich S, Mark R, Moulard M, Walter U, Geiger J. Reciprocal regulation of human platelet function by endogenous prostanoids and through multiple prostanoid receptors. Eur J Pharmacol. 2014; 740:15–27.
178. Wright DH, Ford-Hutchinson AW, Chadee K, Metters KM. The human prostanoid DP receptor stimulates mucin secretion in LS174T cells. Br J Pharmacol. 2000; 131:1537–45.
179. Schuligoi R, Sturm E, Luschnig P, Konya V, Philipose S, Sedej M, Waldhoer M, Peskar BA, Heinemann A. CRTH2 and D-type prostanoid receptor antagonists as novel therapeutic agents for inflammatory diseases. Pharmacology. 2010; 85:372–82.
180. Takahashi G, Asanuma F, Suzuki N, Hattori M, Sakamoto S, Kugimiya A, Tomita Y, Kuwajima G, Abraham WM, Deguchi M, Arimura A, Shichijo M. Effect of the potent and selective DP1 receptor antagonist, asapiprant (S-555739), in animal models of allergic rhinitis and allergic asthma. Eur J Pharmacol. 2015; 765:15–23.
181. Oguma T, Asano K, Ishizaka A. Role of prostaglandin D(2) and its receptors in the pathophysiology of asthma. Allergol Int. 2008; 57:307–12.
182. Jandl K, Heinemann A. The therapeutic potential of CRTH2/DP2 beyond allergy and asthma. Prostaglandins Other Lipid Mediat. 2017; 133:42–48.
183. Zhang A, Dong Z, Yang T. Prostaglandin D2 inhibits TGF-beta1-induced epithelial-to-mesenchymal transition in MDCK cells. Am J Physiol Renal Physiol. 2006; 291:F1332–42.
184. Deng YL, Xiong XZ, Cheng NS. Organ fibrosis inhibited by blocking transforming growth factor-β signaling via peroxisome proliferator-activated receptor γ agonists. Hepatobiliary Pancreat Dis Int. 2012; 11:467–78.
185. Schieber A, Frank RW, Ghisla S. Purification and properties of prostaglandin 9-ketoreductase from pig and human kidney. Identity with human carbonyl reductase. Eur J Biochem. 1992; 206:491–502.
186. Almirza WH, Peters PH, van Zoelen EJ, Theuvenet AP. Role of Trpc channels, Stim1 and Orai1 in PGF(2α)-induced calcium signaling in NRK fibroblasts. Cell Calcium. 2012; 51:12–21.
187. Bek M, Nüsing R, Kowark P, Henger A, Mundel P, Pavenstädt H. Characterization of prostanoid receptors in podocytes. J Am Soc Nephrol. 1999; 10:2084–93.
188. Saito O, Guan Y, Qi Z, Davis LS, Kömhoff M, Sugimoto Y, Narumiya S, Breyer RM, Breyer MD. Expression of the prostaglandin F receptor (FP) gene along the mouse genitourinary tract. Am J Physiol Renal Physiol. 2003; 284:F1164–70.
189. Hébert RL, Carmosino M, Saito O, Yang G, Jackson CA, Qi Z, Breyer RM, Natarajan C, Hata AN, Zhang Y, Guan Y, Breyer MD. Characterization of a rabbit kidney prostaglandin F(2{alpha}) receptor exhibiting G(i)-restricted signaling that inhibits water absorption in the collecting duct. J Biol Chem. 2005; 280:35028–37.
190. Yu Y, Lucitt MB, Stubbe J, Cheng Y, Friis UG, Hansen PB, Jensen BL, Smyth EM, FitzGerald GA. Prostaglandin F2alpha elevates blood pressure and promotes atherosclerosis. Proc Natl Acad Sci USA. 2009; 106:7985–90.
191. Wang L, Zhang C, Su XT, Lin DH, Wu P, Schwartzman ML, Wang WH. PGF2α regulates the basolateral K channels in the distal convoluted tubule. Am J Physiol Renal Physiol. 2017; 313:F254–61.
192. Tian XY, Wong WT, Leung FP, Zhang Y, Wang YX, Lee HK, Ng CF, Chen ZY, Yao X, Au CL, Lau CW, Vanhoutte PM, Cooke JP, Huang Y. Oxidative stress-dependent cyclooxygenase-2-derived prostaglandin f(2α) impairs endothelial function in renovascular hypertensive rats. Antioxid Redox Signal. 2012; 16:363–73.
193. Basu S. Bioactive eicosanoids: role of prostaglandin F(2α) and F2-isoprostanes in inflammation and oxidative stress related pathology. Mol Cells. 2010; 30:383–91.
194. Klawitter J, Reed-Gitomer BY, McFann K, Pennington A, Klawitter J, Abebe KZ, Klepacki J, Cadnapaphornchai MA, Brosnahan G, Chonchol M, Christians U, Schrier RW. Endothelial dysfunction and oxidative stress in polycystic kidney disease. Am J Physiol Renal Physiol. 2014; 307:F1198–206.