
INTRODUCTION
Atypical fibroxanthoma (AFX), and pleomorphic dermal sarcoma are both rare tumors that typically occur in UV-damaged skin of the elderly. Approximately 90% develop on face, scalp, ears, or neck, with a predominance of the male gender. The remaining ~10% arise on the extremities and trunk [1]. In contrast to AFX, which generally does not recur after complete excision, PDS locally reoccurs in up to 50% and metastasizes in up to 20% [2, 3]. Clinically, lesions usually suggest malignancy because they develop rapidly (over just a few weeks or months) and present as a solitary reddish pink plaque or nodule with central ulceration.
Histologically, AFX tumor cells demonstrate moderate to severe pleomorphism with spindle, epithelioid, or multinucleated forms and atypical mitotic figures. A differentiation from pleomorphic dermal sarcoma (PDS, formerly also classified as cutaneous undifferentiated pleomorphic sarcoma (UPS) or cutaneous malignant fibrous histiocytoma (MFH)) can be difficult. Helbig and colleagues compared oncogene pathways in well-defined AFX and PDS and could demonstrate that AFX and PDS present with similar oncogene expression profiles, such as PT53, CCND1, and CDK4 overexpression [2]. The authors therefore suggested that AFX is the non-infiltrating precursor lesion of PDS, which usually infiltrates into the subcutis [2].
Mild to severe solar elastosis in the AFX/PDS-adjacent normal skin is frequently present underlining the role of UV-radiation for AFX/PDS tumorigenesis. Sun exposure and radiation therapy are known risk factors for developing AFX/PDS. Early publications showed a high rate of UV-induced mutations of the TP53 gene in AFX (75%; n = 8) [4]. Using next generation sequencing, Helbig et al. could demonstrate TP53 mutations in 100% (n = 5) of PDS and 20% (n = 5) of AFX [2]. Recent findings from our own institute revealed that amplification of MYC (alias c-Myc) occurs in radiation-induced (secondary) angiosarcoma, but not in primary angiosarcoma [5]. MYC amplifications are supposed to induce tumorigeneses by increasing genomic instability [6, 7]. Since AFX and PDS primarily occur on UV-light exposed locations, it seemed reasonable to study their MYC status.
Recent genetic studies have identified similarities between AFX and PDS, such as deletions of chromosome arms 9p and 13q [8].
In contrast, other groups demonstrated distinct differences between the two entities. Sakamoto and colleagues could show that HRAS and KRAS mutations are only present in AFX, but not in PDS [9]. Mihic-Probst and colleagues used comparative genomic hybridization to demonstrate distinct chromosomal differences between AFX and PDS, such as losses of 1q, 3p, 5q, 11p, 11q and gains of 5p, 7q, 11q, 12q in PDS but not in AFX [10]. MYC is located on chromosome arm 8q. However, information about chromosome 8 alterations in AFX is sparse and fluorescence in situ hybridization (FISH) data for MYC (8q24) has not been reported so far. Therefore, the present study was undertaken to examine MYC in AFX and PDS.
RESULTS
Clinical features
A total of 51 AFX patients were included (Table 1). Patients’ age ranged from 67 to 95 years, with a median of 80 years. A predominance of the male gender (39/51 patients, 76%) was observed (Table 1). One of the patients had a recurrence of the tumor at the site of excision after one year although the tumor was resected with a safety margin of 1 cm. Development of metastases and multiple AFX were not observed.
Table 1: Clinical characteristics of patients with atypical fibroxanthoma
Patient ID |
Age at diagnosis |
Gender |
Localization |
Invasion depth (mm) |
Ratio MYC/CEN8 |
Result |
|---|---|---|---|---|---|---|
1 |
95 |
M |
Scalp |
6.25 |
1.13 |
diploid |
2 |
73 |
M |
Scalp |
6.5 |
1.13 |
diploid |
3 |
71 |
M |
Scalp |
3.25 |
1.33 |
low level gain |
4 |
87 |
M |
Hand |
4.0 |
1.11 |
diploid |
5 |
68 |
M |
Scalp |
3.75 |
1.08 |
diploid |
6 |
72 |
M |
Ear |
6.0 |
1.34 |
low level gain |
7 |
81 |
M |
Ear |
6.25 |
1.32 |
low level gain |
8 |
88 |
M |
Scalp |
17.5 |
2.06 |
amplification |
9 |
67 |
M |
Scalp |
7.5 |
1.14 |
diploid |
10 |
89 |
F |
Nose |
6.0 |
1.36 |
low level gain |
11 |
72 |
M |
Scalp |
N/A |
1.13 |
diploid |
12 |
80 |
M |
Scalp |
N/A |
1.12 |
diploid |
13 |
82 |
M |
Forehead |
5.5 |
1.25 |
low level gain |
14 |
76 |
M |
Scalp |
5.5 |
1.13 |
diploid |
15 |
84 |
M |
Ear |
5.25 |
1.28 |
low level gain |
16 |
68 |
F |
Forehead |
2.5 |
1.13 |
diploid |
17 |
80 |
M |
Ear |
3.0 |
1.67 |
low level gain |
18 |
86 |
M |
Scalp |
9.5 |
1.23 |
low level gain |
19 |
83 |
M |
Scalp |
5.75 |
1.33 |
low level gain |
20 |
75 |
M |
Cheek |
2.0 |
1.08 |
diploid |
21 |
80 |
M |
Scalp |
8.0 |
1.07 |
diploid |
22 |
76 |
M |
Scalp |
2.75 |
1.11 |
diploid |
23 |
78 |
M |
Scalp |
2.0 |
1.09 |
diploid |
24 |
79 |
M |
Forehead |
2.25 |
1.11 |
diploid |
25 |
72 |
F |
Forehead |
2.0 |
1.13 |
diploid |
26 |
77 |
M |
Scalp |
12.0 |
N/A |
N/A |
27 |
87 |
M |
Forehead |
N/A |
1.11 |
diploid |
28 |
75 |
M |
Forehead |
N/A |
1.17 |
diploid |
29 |
80 |
M |
Forehead |
2.0 |
1.12 |
diploid |
30 |
80 |
F |
Eyebrow |
2.2 |
1.37 |
low level gain |
31 |
79 |
M |
Scalp |
4.1 |
1.49 |
low level gain |
32 |
81 |
M |
Forehead |
2.2 |
N/A |
N/A |
33 |
86 |
F |
Cheek |
3.5 |
1.14 |
diploid |
34 |
76 |
M |
Eyelid |
4.15 |
1.14 |
diploid |
35 |
76 |
M |
Forehead |
10.0 |
1.13 |
diploid |
36 |
83 |
F |
Forehead |
1.45 |
1.13 |
diploid |
37 |
74 |
F |
Forehead |
3.0 |
N/A |
N/A |
38 |
87 |
M |
Cheek |
N/A |
1.11 |
diploid |
39 |
75 |
F |
Forehead |
1.7 |
1.14 |
diploid |
40 |
90 |
F |
Eyebrow |
2.0 |
1.13 |
diploid |
41 |
70 |
M |
Neck |
3.2 |
1.12 |
diploid |
42 |
85 |
M |
Scalp |
2.5 |
1.13 |
diploid |
43 |
88 |
M |
Scalp |
N/A |
N/A |
N/A |
44 |
88 |
M |
Scalp |
N/A |
1.14 |
diploid |
45 |
84 |
M |
Neck |
N/A |
N/A |
N/A |
46 |
70 |
F |
Forehead |
3.0 |
1.32 |
low level gain |
47 |
86 |
F |
Cheek |
2.0 |
1.14 |
diploid |
48 |
85 |
F |
Forehead |
1.6 |
1.13 |
diploid |
49 |
87 |
M |
Scalp |
N/A |
1.35 |
low level gain |
50 |
81 |
M |
Scalp |
1.6 |
N/A |
N/A |
51 |
73 |
M |
Scalp |
N/A |
N/A |
N/A |
Abbreviations: M: male, F: female, N/A: data not available, light grey: low level amplification, dark grey: amplification.
The single case with MYC amplification (MYC/CEP8 ratio ≥2.0) is highlighted in dark grey, the 13 cases with low level MYC copy number gain (MYC/CEN 8 ratio ≥1.2–<2.0) are accentuated in light grey.
Additionally, a cohort of 24 PDS patients was investigated (Table 2). PDS patients´ age ranged from 58 to 92, with a median of 77 years. Similar to the AFX patients, the PDS patients also demonstrated a predominance of the male gender (19/24 patients, 79%) (Table 2). Tumor thickness of the PDS cohort was not available.
Table 2: Clinical characteristics of patients with pleomorphic dermal sarcoma
Patient ID |
Age at diagnosis |
Gender |
Localization |
Invasion depth (mm) |
Ratio MYC/CEN8 |
Result |
|---|---|---|---|---|---|---|
1 |
68 |
M |
Scalp |
N/A |
1.13 |
diploid |
2 |
89 |
M |
Scalp |
N/A |
1.11 |
diploid |
3 |
66 |
M |
Scalp |
N/A |
1.08 |
diploid |
4 |
81 |
M |
Scalp |
N/A |
1.11 |
diploid |
5 |
58 |
M |
Forehead |
N/A |
1.08 |
diploid |
6 |
92 |
M |
Cheek |
N/A |
1.32 |
low level gain |
7 |
73 |
F |
Nose |
N/A |
1.14 |
diploid |
8 |
74 |
M |
Scalp |
N/A |
1.13 |
diploid |
9 |
80 |
F |
Cheek |
N/A |
1.14 |
diploid |
10 |
89 |
M |
Forehead |
N/A |
1.33 |
low level gain |
11 |
81 |
F |
Cheek |
N/A |
1.13 |
diploid |
12 |
88 |
M |
Forehead |
N/A |
1.12 |
diploid |
13 |
74 |
F |
Scalp |
N/A |
1.11 |
diploid |
14 |
74 |
M |
Shoulder |
N/A |
1.13 |
diploid |
15 |
77 |
M |
Scalp |
N/A |
1.28 |
low level gain |
16 |
74 |
M |
Scalp |
N/A |
1.13 |
diploid |
17 |
71 |
M |
Scalp |
N/A |
1.13 |
diploid |
18 |
75 |
M |
Scalp |
N/A |
1.23 |
low level gain |
19 |
82 |
M |
Scalp |
N/A |
1.49 |
low level gain |
20 |
84 |
M |
Scalp |
N/A |
1.08 |
diploid |
21 |
86 |
M |
Scalp |
N/A |
1.07 |
diploid |
22 |
86 |
M |
Scalp |
N/A |
1.11 |
diploid |
23 |
76 |
M |
Scalp |
N/A |
1.15 |
diploid |
24 |
59 |
F |
Shoulder |
N/A |
1.11 |
diploid |
Abbreviations: M: male, F: female, N/A: data not available, light grey: low level amplification.
The five cases with low level MYC copy number gain (MYC/CEN 8 ratio ≥1.2–<2.0) are accentuated in light grey.
Immunohistochemistry
Immunohistochemically, none of the investigated 51 AFX and 24 PDS specimen stained positive for MYC protein expression (data not shown).
Fluorescence in situ hybridization
Interphase FISH analysis for MYC was successfully performed on 44/51 AFX and 24/24 PDS cases. Seven AFX cases were not evaluable due to missing cores on the FISH TMA slide.
Interphase FISH analysis on TMA spots from each patient revealed MYC amplification (MYC/CEP8 ratio 2.06, MYC signal number 4.02) in only one AFX case (1/44 = 2.3%) (Figure 1). In 13 other AFX specimens and 5 PDS cases, low level MYC gain was detected (13/44 = 29.5% and 5/24 = 21%). The remaining 30 AFX cases (30/45 = 68.2%) and 19 PDS cases (19/24 = 79%) were found to be diploid for MYC by FISH analysis (Tables 1 and 2, Supplementary Tables 1 and 2). According to our defined criteria (Material and Methods) no polysomy was observed, however one AFX case and one PDS case fell short with an average of 2.85 and 2.8 CEP8 signals per cell (Supplementary Tables 1 and 2).
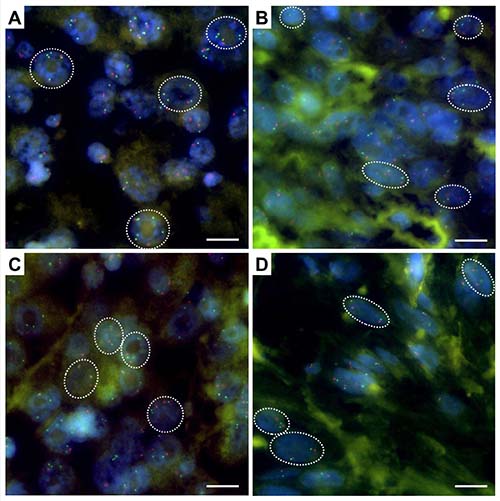
Figure 1: MYC/CEN 8 fluorescence in situ hybridization on interphase nulcei in AFX. Shown are representative immunofluorescence images of (A) MYC amplified case (patient ID 8, MYC/CEN 8 ratio 2.06), (B) case with a low level copy number gain of MYC (patient ID 6, MYC/CEN 8 ratio 1.34), (C) near-polysomic case (patient ID 17, MYC/CEN 8 ratio 1.67, on average 2.85 CEN 8 signals per cell), and (D) diploid case (patient ID 12, MYC/CEN 8 ratio 1.12). Scale bar, 10 μm. Green, MYC (8q24) signals; red, centromeric region of chromosome 8.
None of the evaluated clinicopathological parameters (tumor invasion thickness, preexisting diseases/malignancies, smoking behavior, location, age, gender) were associated with MYC amplification (data not shown). However, it is remarkable that the only MYC amplified AFX case showed the highest tumor thickness (17.5 mm) of all cases (17.5 mm vs. median 3.25 mm, range 1.45 to 17.5) (Figure 2).
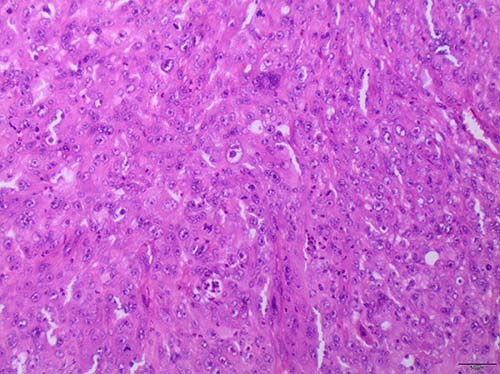
Figure 2: Histopathological examination showing distinct features of AFX, such as pronounced cell pleomorphism with spindle, epithelioid, and multinucleated forms. Numerous atypical mitotic figures were present (HE, scale bar 50 μm).
DISCUSSION
To the best of our knowledge MYC copy number alterations (CNA) have not been studied systematically in AFX or PDS. Our group could link MYC amplification to antecedent radiation treatment in angiosarcoma [5] and various other types of radiation treated sarcoma [6]. Besides, FISH analysis demonstrated that malignant melanoma, a tumor that can also be induced by UV-radiation, shows MYC copy number gain [11]. It was therefore reasonable to postulate that chronic sun exposure, the major risk factor for developing AFX or PDS, might also lead to MYC CNA. Like radiotherapy, UV-radiation can cause direct (UVB) and indirect (UVA) DNA damage [12] and plays an important role in skin cancer development [4, 13]. While UVA radiation usually causes single-strand breaks in DNA, UVB radiation can lead to double-strand DNA breakage [12]. We found that MYC amplification is a rare event in AFX and PDS; 13 of 44 (29.5%) AFX specimen showed MYC low level copy number gain and only one AFX patient (2.3%) had a MYC amplification. None of the PDS cases showed a MYC amplification and only 5 of 24 (21%) cases demonstrated MYC low level copy number gain. The one AFX case with a MYC amplification was an 88-year old male patient with an advanced AFX (17.5 mm thickness, ulcerated). However, these results are not unexpected since chronic UV radiation has a much lower energy (UVA to UVB: 3.26 to 4.43 eV) than the dosages frequently used for radiotherapy (70 Gray in a 70 kg patient: ~3.06 × 1021 eV).
The amplification of MYC did not have an impact on tumor cell morphology, a fact already known from angiosarcoma where MYC amplification did not correlate with the histological grade or other morphologic features [5]. A prognostic role for MYC could not be demonstrated since only one case in this study showed local recurrence and none of the cases presented with metastases during follow-up.
Based on the detected low frequency of MYC amplification in AFX and PDS it is difficult to speculate about its role for AFX/PDS tumorigenesis. Apparently it is a rare genetic event and not specific for these entities. Furthermore, sun exposure does not seem to lead to the same genetic alterations that are known from radiation induced angiosarcoma, in which MYC amplification was present in 55% of the cases [5]. Besides of secondary angiosarcoma [5], MYC amplification has also been reported in malignant fibrous histiocytoma [14], high grade chondrosarcoma [15], epitheloid sarcoma of the proximal type [16], and in high grade myxoid liposarcomas [17]. For leiomyosarcoma, MYC amplification was shown to have an adverse prognostic impact [18]. In total, it seems that in certain sarcoma entities MYC amplification is associated with higher tumor grades and worse prognosis. MYC CNA in high grade adult soft tissue sarcomas have been reported as rare events (7 of 207, 3.4%) [19]. In an own study, we observed a MYC amplification frequency of 6% for non-radiated PDS (vs. 29% for radiation induced sarcoma) [6]. Helbig and colleagues found amplifications and deletions in 6 of 27 PDS cases but not in AFX [20]. Becerikli and colleagues could not detect any MYC CNA in 19 PDS samples [21]. Furthermore, it has been reported that AFX and PDS may share distinct genetic events (e.g., 9p & 13q losses) [8]. Thus, after acquiring additional genetic alteration, AFX could progress into PDS and may therefore present a superficial form of PDS. However, our findings do rule out that MYC amplification is a major genetic driver for the development of AFX or PDS since it was a very rare event, the only AFX case with MYC amplification did not show histological signs for transition into PDS, even though it was the most advanced lesion and none of the PDS cases demonstrated a MYC amplification. Immunhistochemically, none of the investigated cases was positive for MYC protein expression. Apparently, even in the single amplified AFX case, the level of MYC amplification was not sufficient to result into immunohistochemically detectable MYC protein expression. It still has to be determined by which pathway chronic sun exposure leads to the formation of AFX/PDS, most certainly MYC amplification can be excluded as the major genetic driver. However, our data indicate that MYC amplification occurs as a late event during the tumorigenesis and might only be detectable in advanced AFX lesions.
MATERIALS AND METHODS
Patients and tissue microarray
In total, 51 AFX formalin-fixed paraffin-embedded (FFPE) tumor specimens were retrospectively collected from the archive of the Institute of Pathology and Dermatohistological Laboratory Prof. Kind. The 24 PDS FFPE specimens were provided from the archive of the Institute for Pathology of the University Hospital Cologne. Additionally, demographic and clinical characteristics, such as age, gender, location, tumor specific data, in some cases co-morbidities and smoking behavior, were collected. All lesions were reassessed by two independent pathologists (TG, AQ) to reassure the AFX/PDS diagnosis. The study was approved by the local board of ethics of the University Medical Center Mannheim (2014-835R-MA) and the University Hospital Cologne (Registration No. 15-307).
Tumor areas were marked on whole sections and each tumor sample was assembled on tissue microarrays (TMA) in triplicates (Mannheim TMA 1) and duplicates (Cologne TMA 1 and Mannheim TMA 2) using TissueMax Automated and Personal Tissue Microarrayers according to the manufacturer’s protocol [22].
Immunohistochemistry
Tissue sections were stained for MYC (clone N262, Santa Cruz, 1:50). Sections were subjected to heat-induced EDTA-based antigen retrieval. Antibody binding was visualized using the EnVision Detection System, Peroxidase/DAB, Rabbit/Mouse (cat# K5007, Dako) according to the manufacturer’s instructions.
Fluorescence in situ hybridization
FISH for quantitation of MYC and chromosome 8 centromere (CEP8) was performed on 5 μm FFPE tissue sections using the ZytoLight SPEC MYC/CEN 8 Dual Color Probe (Zytomed Systems GmbH, Berlin, Germany). Deparaffinization, denaturation and hybridization were done according to the manufacturer’s instructions. For denaturation and hybridization a StatSpin Hybridizer instrument (cat # S2450, Dako, Hamburg, Germany) was used. Hybridized slides were counterstained with Vectashield Antifade Mounting Medium with DAPI (H-1200, Vector Laboratories, Inc., Burlingame, CA, USA) and then analyzed using an Olympus BX41 fluorescence microscope (Olympus Deutschland GmbH, Hamburg, Germany) with optical filters for DAPI, SpectrumGreen and SpectrumOrange (Olympus) with a UPlanSApo 60x objective (oil, numerical aperture 1.35; Olympus). The microscope was connected to an F-View II CCD-Camera (Soft Imaging System GmbH, Muenster, Germany). Cell^F software (Olympus Soft Imaging Solutions GmbH, Muenster, Germany) was used to acquire representative images with each filter. Sixty interphase nuclei were counted per sample. Following previous publications [23–25] the following signal patterns were defined:
• MYC amplification: MYC/CEP8 ratio ≥2.0
• MYC low level gain: MYC/CEP8 ratio ≥1.2–<2.0
• Polysomy: CEP8 (≥3.0)
Author contributions
TG, DH and MRG wrote the manuscript text. TG, AM, AQ and MRG designed the experiments. DH and AQ performed the experiments. TG, DH, JU, AM and MRG analyzed the data. DH, AO, MB, PK and MRG provided technical support, collected the paraffin samples and clinical data. All authors reviewed the manuscript.
ACKNOWLEDGMENTS
We thank Yonca Ceribas and Alexandra Eichhorn for excellent technical assistance. This work was supported in part by grants of the German research foundation to MRG (Deutsche Forschungsgemeinschaft) GRK2099).
CONFLICTS OF INTEREST
All authors have no conflicts of interest to declare.
REFERENCES
1. Beer TW, Drury P, Heenan PJ. Atypical fibroxanthoma: a histological and immunohistochemical review of 171 cases. Am J Dermatopathol. 2010; 32:533–40. https://doi.org/10.1097/DAD.0b013e3181c80b97.
2. Helbig D, Ihle MA, Putz K, Tantcheva-Poor I, Mauch C, Buttner R, Quaas A. Oncogene and therapeutic target analyses in atypical fibroxanthomas and pleomorphic dermal sarcomas. Oncotarget. 2016; 7:21763–74. https://doi.org/10.18632/oncotarget.7845.
3. Tardio JC, Pinedo F, Aramburu JA, Suarez-Massa D, Pampin A, Requena L, Santonja C. Pleomorphic dermal sarcoma: a more aggressive neoplasm than previously estimated. J Cutan Pathol. 2016; 43:101–12. https://doi.org/10.1111/cup.12603.
4. Dei Tos AP, Maestro R, Doglioni C, Gasparotto D, Boiocchi M, Laurino L, Fletcher CD. Ultraviolet-induced p53 mutations in atypical fibroxanthoma. Am J Pathol. 1994; 145:11–7.
5. Manner J, Radlwimmer B, Hohenberger P, Mossinger K, Kuffer S, Sauer C, Belharazem D, Zettl A, Coindre JM, Hallermann C, Hartmann JT, Katenkamp D, Katenkamp K, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010; 176:34–9. https://doi.org/10.2353/ajpath.2010.090637.
6. Kacker C, Marx A, Mossinger K, Svehla F, Schneider U, Hogendoorn PC, Nielsen OS, Kuffer S, Sauer C, Fisher C, Hallermann C, Hartmann JT, Blay JY, et al. High frequency of MYC gene amplification is a common feature of radiation-induced sarcomas. Further results from EORTC STBSG TL 01/01. Genes Chromosomes Cancer. 2013; 52:93–8. https://doi.org/10.1002/gcc.22009.
7. Felsher DW, Bishop JM. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci U S A. 1999; 96:3940–4.
8. Lopez L, Velez R. Atypical Fibroxanthoma. Arch Pathol Lab Med. 2016; 140:376–9. https://doi.org/10.5858/arpa.2014-0495-RS.
9. Sakamoto A, Oda Y, Itakura E, Oshiro Y, Tamiya S, Honda Y, Ishihara A, Iwamoto Y, Tsuneyoshi M. H-, K-, and N-ras gene mutation in atypical fibroxanthoma and malignant fibrous histiocytoma. Hum Pathol. 2001; 32:1225–31. https://doi.org/10.1053/hupa.2001.28953.
10. Mihic-Probst D, Zhao J, Saremaslani P, Baer A, Oehlschlegel C, Paredes B, Komminoth P, Heitz PU. CGH analysis shows genetic similarities and differences in atypical fibroxanthoma and undifferentiated high grade pleomorphic sarcoma. Anticancer Res. 2004; 24:19–26.
11. Kraehn GM, Utikal J, Udart M, Greulich KM, Bezold G, Kaskel P, Leiter U, Peter RU. Extra c-myc oncogene copies in high risk cutaneous malignant melanoma and melanoma metastases. Br J Cancer. 2001; 84:72–9. https://doi.org/10.1054/bjoc.2000.1535.
12. Svobodova AR, Galandakova A, Sianska J, Dolezal D, Lichnovska R, Ulrichova J, Vostalova J. DNA damage after acute exposure of mice skin to physiological doses of UVB and UVA light. Arch Dermatol Res. 2012; 304:407–12. https://doi.org/10.1007/s00403-012-1212-x.
13. Halliday GM, Byrne SN, Damian DL. Ultraviolet A radiation: its role in immunosuppression and carcinogenesis. Semin Cutan Med Surg. 2011; 30:214–21. https://doi.org/10.1016/j.sder.2011.08.002.
14. Tarkkanen M, Larramendy ML, Bohling T, Serra M, Hattinger CM, Kivioja A, Elomaa I, Picci P, Knuutila S. Malignant fibrous histiocytoma of bone: analysis of genomic imbalances by comparative genomic hybridisation and C-MYC expression by immunohistochemistry. Eur J Cancer. 2006; 42:1172–80. https://doi.org/10.1016/j.ejca.2006.01.035.
15. Morrison C, Radmacher M, Mohammed N, Suster D, Auer H, Jones S, Riggenbach J, Kelbick N, Bos G, Mayerson J. MYC amplification and polysomy 8 in chondrosarcoma: array comparative genomic hybridization, fluorescent in situ hybridization, and association with outcome. J Clin Oncol. 2005; 23:9369–76. https://doi.org/10.1200/JCO.2005.03.7127.
16. Lualdi E, Modena P, Debiec-Rychter M, Pedeutour F, Teixeira MR, Facchinetti F, Dagrada GP, Pilotti S, Sozzi G. Molecular cytogenetic characterization of proximal-type epithelioid sarcoma. Genes Chromosomes Cancer. 2004; 41:283–90. https://doi.org/10.1002/gcc.20086.
17. Schneider-Stock R, Boltze C, Jager V, Epplen J, Landt O, Peters B, Rys J, Roessner A. Elevated telomerase activity, c-MYC-, and hTERT mRNA expression: association with tumour progression in malignant lipomatous tumours. J Pathol. 2003; 199:517–25. https://doi.org/10.1002/path.1315.
18. Tsiatis AC, Herceg ME, Keedy VL, Halpern JL, Holt GE, Schwartz HS, Cates JM. Prognostic significance of c-Myc expression in soft tissue leiomyosarcoma. Mod Pathol. 2009; 22:1432–8. https://doi.org/10.1038/modpathol.2009.113.
19. Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, Decarolis PL, Shah K, Socci ND, Weir BA, Ho A, Chiang DY, Reva B, Mermel CH, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010; 42:715–21. https://doi.org/10.1038/ng.619.
20. Helbig D, Quaas A, Mauch C, Merkelbach-Bruse S, Buttner R, Emberger M, Wobser M, Russeler V, Putz K, Binot E, Rehker J, Budczies J, Ihle MA. Copy number variations in atypical fibroxanthomas and pleomorphic dermal sarcomas. Oncotarget. 2017; 8:109457–67. https://doi.org/10.18632/oncotarget.22691.
21. Becerikli M, Wieczorek S, Stricker I, Nambiar S, Rittig A, Epplen JT, Tannapfel A, Lehnhardt M, Steinstraesser L, Jacobsen F. Numerical and structural chromosomal anomalies in undifferentiated pleomorphic sarcoma. Anticancer Res. 2014; 34:7119–27.
22. Wagner NB, Weide B, Reith M, Tarnanidis K, Kehrel C, Lichtenberger R, Pflugfelder A, Herpel E, Eubel J, Ikenberg K, Busch C, Holland-Letz T, Naeher H, et al. Diminished levels of the soluble form of RAGE are related to poor survival in malignant melanoma. Int J Cancer. 2015; 137:2607–17. https://doi.org/10.1002/ijc.29619.
23. Hofmann M, Stoss O, Gaiser T, Kneitz H, Heinmoller P, Gutjahr T, Kaufmann M, Henkel T, Ruschoff J. Central HER2 IHC and FISH analysis in a trastuzumab (Herceptin) phase II monotherapy study: assessment of test sensitivity and impact of chromosome 17 polysomy. J Clin Pathol. 2008; 61:89–94. https://doi.org/10.1136/jcp.2006.043562.
24. Shon W, Sukov WR, Jenkins SM, Folpe AL. MYC amplification and overexpression in primary cutaneous angiosarcoma: a fluorescence in-situ hybridization and immunohistochemical study. Mod Pathol. 2014; 27:509–15. https://doi.org/10.1038/modpathol.2013.163.
25. Gaiser T, Berroa-Garcia L, Kemmerling R, Dutta A, Ried T, Heselmeyer-Haddad K. Automated analysis of protein expression and gene amplification within the same cells of paraffin-embedded tumour tissue. Cell Oncol (Dordr). 2011; 34:337–42. https://doi.org/10.1007/s13402-011-0032-x.