
INTRODUCTION
RAS proto-oncogenes (HRAS, KRAS and NRAS) encode a family of GDP/GTP-regulated proteins critical for signal transduction mediating cell growth and survival. The four enzymes encoded by the three RAS genes are highly homologous, sharing a high degree of identity over the first 90% of proteins [1]. The carboxyl-terminus of the proteins contains a hyper-variable region, which diverges radically in primary sequence from the remainder of these genes. This region of the gene product is susceptible to many post-translational modifications which confer major differences in trafficking and intracellular localization of the mature protein. RAS family members are frequently found in their mutated, oncogenic forms in human tumors. Mutant RAS proteins are constitutively active, owing to reduced intrinsic GTPase activity and insensitivity to GTPase-activating proteins (GAPs). Overall, activating mutations in RAS occur in approximately 20% of all human cancers [2]. Mutations in KRAS, mainly in exons 2, 3 and 4 (codons 12, 13, 59, 61, 117, 146), account for nearly 85% of all RAS mutations found in human tumors, whereas NRAS contributes to ~15%, and HRAS to less than 1% [3]. RAS somatic mutations are early drivers of tumorigenesis exclusively associated with cancer and therefore provide exquisite specificity for disease. For example, mutations in RAS are prominent drivers of colon, pancreatic and lung cancers.
Epidermal growth factor receptor (EGFR) - targeted monoclonal antibodies (MAb), such as cetuximab and panitumumab, have been used in the treatment of metastatic colorectal cancer (mCRC) since 2004. Numerous studies have demonstrated that RAS mutations are associated with resistance to anti-EGFR antibodies and that approximately 45–50% of CRCs harbor a RAS mutation [4, 5]. Based on these findings, anti-EGFR MAbs have been recommended for use in first-line therapy of metastatic CRC (mCRC) for those patients whose tumors are wild-type (WT) for RAS. The determination of RAS mutation status represents the current standard of care for determining mCRC patient eligibility for treatment with anti-EGFR therapy. KRAS is also a key biomarker in lung cancer as it is the most common alteration in NSCLC with mutations in KRAS occurring in approximately 30% of patients [6, 7]. In NSCLC, 97% of KRAS mutations occur in codons 12 and 13 [7]. Mutations in NRAS are uncommon in NSCLC (1.1%) [8] while HRAS mutations are extremely rare. KRAS is currently not targetable by any approved agents and its occurrence is largely prognostic as it is associated with poor patient outcome [6]. While remaining an elusive therapeutic target, the detection of KRAS mutations has diagnostic value as somatic alterations in NSCLC are generally considered mutually exclusive with other driver mutations such as EGFR [9, 10]. Therefore, detecting a RAS alteration may aid the clinician since it drastically decreases the chances of detecting another alteration. Testing RAS is thus of high clinical importance for mCRC and NSCLC patients, especially in mCRC in which treatment is prescribed according to RAS mutation status.
Tumor circulating-free DNA (cfDNA) are small fragments of DNA released into the bloodstream from tumor cells [11]. Mutations in cfDNA may be detected in blood using several techniques, such as: real-time PCR-based assays (Cobas® and Biocartis® assays); digital PCR (dPCR) assays such as droplet-digital PCR (ddPCR) and BEAMing, or next-generation sequencing (NGS) [12, 13]. Detecting mutations in cfDNA offers several distinct advantages when compared to traditional tissue based mutation testing: (i) the sample source for ctDNA testing is blood, which is obtained via a minimally invasive technique and is thus an easily repeatable clinical procedure; (ii) blood sampling and subsequent ctDNA survey at a specific time represent a holistic mutational assessment of the current status of a patient’s systemic disease burden, overcoming sampling bias of tissue biopsies performed at a specific disease location. Thus, cfDNA mutational analysis captures spatial and temporal tumor molecular heterogeneity providing a more complete view of the patient’s disease status. Many initiatives have emerged over the last few years to assess mutations in cfDNA across a multitude of tumor types [14–16]. The importance of defined sample collection procedures and pre-analytical conditions has been shown to be instrumental for accurate and reliable cfDNA mutation analysis [17]. Our recent work on ctDNA detection in patient samples determined specific pre-analytical considerations to enable a simplification of sample processing thereby streamlining the clinical workflow and ensuring consistency of ctDNA test results for implementation in routine clinical practice [13].
The primary goal of the present study was to compare the performance of three technologies used to detect KRAS and NRAS somatic alterations in cfDNA from mCRC and NSCLC patients. We evaluated the comparison of results between the molecular profile of FFPE tissues (considered as reference material) and cfDNA samples, and reported on the sensitivity and specificity of each of the cfDNA technologies, which included ddPCR (Biorad), BEAMing (Sysmex Inostics), and NGS using the targeted SWIFT-56G panel (Swift Biosciences). Overall, we demonstrated that the NGS technology provides broader coverage of expanded gene regions which prove useful for exploratory screening of other resistance mechanisms that may occur in addition to those with demonstrated clinical utility. The use of the OncoBEAM™ RAS-CRC assay (a BEAMing panel targeting 34 separate KRAS and NRAS mutations) provided a highly sensitive and accurate detection of RAS mutations, enabling reliable longitudinal monitoring to track the appearance and disappearance of somatic alterations by sampling only plasma-derived cfDNA.
RESULTS
CfDNA input
In the two cohorts (NSCLC and mCRC), we observed a correlation between Qubit quantification of cfDNA quantity and the number of WT haploid GE for KRAS among samples obtained from mCRC patients (R 2 = 0.91) and NSCLC patients (R 2 = 0.86) (Supplementary Figure 1). In these cohorts, the average haploid GE numbers of cfDNA from patients within the CIRCAN cohort was approximately 1200 WT haploid GE/ddPCR reaction (150 WT GE/μL of cfDNA). The basic requirements to appropriately compare the different detection platforms were as follows: (i) 123 μL of cfDNA for the OncoBEAM-TM-RAS-CRC method; (ii) 8 μL of cfDNA/per reaction for the ddPCR Biorad; (iii) and 10 μL of cfDNA for sequencing library preparation (Swift-56G Biosciences) with a cfDNA input quantity ranging between 1 ng and 25 ng. To compare the three assays with the same cfDNA sample and to avoid bias generated by the extraction step, cfDNA was extracted from 4.5 mL of plasma that was eluted in 210 μL of AVE elution buffer. Under these conditions, the concentrations of cfDNA ranged from 0.1 to 9.1 ng/μL (200 to 16,000 WT haploid GE/8 μL) for colon cancer samples, and from 0.1 to 6.2 ng/μL (500 to 1,1000 WT haploid GE/8 μL) for lung cancer samples. These concentrations were sufficient to enable us to carry out the somatic detections for all patients of the cohort. The specificity and sensitivity evaluations are presented in supplementary data (Supplementary Tables 3 and 4). First, we used commercial gold standard from Horizon Diagnostics. secondly, we used expected KRAS wild-type patients samples. Since EGFR and KRAS mutations are mostly mutually exclusive [9], we selected EGFR mutated patients to confirm that the BEAMing and NGS assays did not give rise to false-positives cases. The signals are largely below the threshold of positivity (set up at 50 mutated positive signals/reaction for BEAMing and 0.5% for NGS) (Supplementary Table 1).
Head-to-head performance comparisons of all cfDNA assays with respect to mutational analysis of FFPE samples
Supplementary Tables 2 (mCRC) and 3 (NSCLC) show KRAS/NRAS molecular profiles in FFPE reference biopsies (sampled at diagnosis) and in cfDNA using the three different assays. These tables report the: cfDNA concentration per sample (used to assess impact on assay performance); absolute number of mutated and wild-type KRAS/NRAS haploid GE for each sample (positive tests are highlighted in red and additional mutations are presented); corresponding mutated allelic fraction (AF); and the KRAS and NRAS mutation status. Finally, we calculated the concordance rate between each assay compared to FFPE results. For few patients, we found 2 different KRAS/NRAS mutations in cfDNA.
CfDNA and FFPE biopsies of mCRC were both carried out at diagnosis, enabling paired mutation profile comparisons. In mCRC patients, we found two, four and three mismatches in the ddPCR, the OncoBEAM-TM-RAS-CRC and the NGS assays respectively, compared with FFPE samples (Supplementary Table 2). CIRCAN-colon samples #3 and #23 displayed very low KRAS MT-positive signal (59 and 54 absolute count respectively) with the OncoBEAM-TM-RAS-CRC assay and were not detected using the other two assays or in the FFPE samples. CIRCAN-Colon #16 harbored a p.K117D mutation detected with the NGS assay (low level: 1.1%). This mutation is not assessed in the ddPCR and BEAMing panels. The CIRCAN-colon #15 plasma sample harbored a KRAS p.G13D mutation and was detected with both ddPCR (22MT haploid GE) and BEAMing (1113 MT-positive signals). NGS assessment also detected this same mutation but at a low allelic fraction (0.8%). FFPE assessment of this sample resulted in a WT determination. In CIRCAN-Colon #21 FFPE, a KRAS p.G12V mutation (above positivity threshold) was detected with all cfDNA assays (Supplementary Table 2), though analysis of FFPE was determined to be WT. Using these results, sensitivity was higher for BEAMing (93% vs. 47% and 67% for ddPCR and NGS, respectively), while specificity was lower (69% vs. 76% in both NGS and ddPCR).
In the NSCLC cohort, FFPE biopsy profiling was performed at diagnosis and the cfDNA sampling was performed during disease progression (several months or years after initial diagnosis in some cases). Thus, not all patients had FFPE blocks available for mutation analysis. Among 6 patients with a known WT FFPE sample, 1 was determined to have a p.G12C KRAS mutation with BEAMing and NGS (insufficient quantity of cfDNA was available to perform the ddPCR assay). In FFPE samples with known KRAS mutations, 6 carried insufficient quantity to perform ddPCR (Supplementary Table 3, ddPCR green box). Finally, 4 KRAS mutations were accurately found by ddPCR while 4 samples remained WT (Supplementary Table 3, ddPCR green box).
Fourteen (14) patients were classified as KRAS mutant via FFPE. Six (6) of these patients lacked sufficient sample volume to perform ddPCR assessment (Supplementary Table 3, ddPCR green box). Of the 8 patients tested by ddPCR, 4 patients were KRAS positive while the other 4 were KRAS WT (Supplementary Table 3, ddPCR green box). In a WT FFPE specimen, the OncoBEAM-TM-RAS-CRC assay detected one p.G12C KRAS mutation (216 mutated signals) in cfDNA that was confirmed by NGS at a low 0.6% allelic frequency (Supplementary Table 3, BEAMing blue box and NGS orange box). There was insufficient cfDNA for ddPCR analysis in this patient. In KRAS MT FFPE samples, OncoBEAM-TM-RAS-CRC assay detected a mutation in 8 samples (Supplementary Table 3, BEAMing blue box); while 3 samples remained negative (mutated signal found but under the threshold); and 3 were not covered by the OncoBEAM-TM-RAS-CRC panel. The NGS assay detected mutations in 9 samples (Supplementary Table 3, BEAMing blue box); while 5 samples did not display any mutations (Supplementary Table 3, NGS orange box).
In the lung cancer cohort, we observed 67%, 57% and 64% sensitivity and 100%, 83% and 83% specificity for the ddPCR, BEAMing and NGS assays, respectively. We carried out both OncoBEAM-TM-RAS-CRC and NGS assays on 14 additional samples with unknown FFPE statuses. We found 3 positive cases with the OncoBEAM-TM-RAS-CRC assay, for which only two were confirmed by NGS assay (Supplementary Table 3).
Additional results assessing internal performance and sensitivity of each assay are provided in Supplementary File 2.
Exploration of concomitant variants in negative and positive KRAS/NRAS groups in colon and lung cancer
Since the coverage of mutations by NGS sequencing is larger than that provided by digital PCR assays, we analyzed the concomitant presence of additional alterations in our cohort (Supplementary Table 4). We analyzed the Variant Caller Files generated by bioinformatics analyses and we retained all non-synonymous and splice variants found at 1% with at least 50 mutated reads both for KRAS/NRAS-positive (Supplementary Table 4A) and negative (Supplementary Table 4B) colon and lung cancer biopsies. In the first group, in addition to the presence of KRAS/NRAS mutations, we detected 9 alterations of TP53, PIK3CA and APC at a frequency exceeding 15%. Other genes, including HRAS and PTEN, yielded alterations at lower frequencies (Supplementary Table 4A). For the second group, we listed 14 genes with somatic alterations. The three main mutated genes with mutational rates exceeding 10% were EGFR, PIK3CA and TP53. We also observed mutations in tyrosine-kinases, such as ERBB2 and ERRB4, belonging to the same family as EGFR (Supplementary Table 4A).
DISCUSSION
Prior investigations have shown that cfDNA is detected in almost all patients with advanced cancer [18]. However the low abundance of this tumor-specific genetic material requires highly sensitive techniques to ensure reliable detection for routine practice. The main benefits of utilizing cfDNA as a source of tumor genetic material are based on the safety and convenience associated with minimally-invasive procedures as well as ease of accessibility at any time point. This approach enables clinicians to monitor tumor dynamics and evolution unaffected by sample selection bias. However, accuracy and concordance with tumor tissue mutation testing techniques have not been fully elucidated in patients followed in clinical practices. In this paper, we evaluated the performance of three independent assays to determine RAS status in cfDNA using tissue biopsy as reference standard.
Although somatic mutation testing in cfDNA is an area of intense interest, there are still some concerns regarding which assay delivers optimal results for use in the setting of a routine clinical laboratory. Only a handful of studies have approached this issue by comparing several analytical techniques using matched and identical samples [19]. Recently, Bartels et al. compared digital PCR (QuantStudio 3D Digital PCR System,ThermoFisher Scientific), NGS (IonTorent, ThermoFisher) and quantitative PCR in 55 cfDNA samples from lung cancer patients and found a 96% concordance when testing for EGFR T790M. The limit of detection for identification of mutations in that study was set at > 0.1% for ddPCR and > 0.2% for NGS. In comparison, we have previously determined that for the analysis of EGFR T790M in cfDNA by ddPCR, the threshold of 0.1% can only be achieved for samples with at least 5000 haploid GE cfDNA input. Below this level, the achieved threshold is only of 1% [13]. In the present study, we determined that a 0.5% allelic fraction represents an adequate threshold for the NGS assay. A lower threshold of positivity can be determined, when the mutation detection of a sample for a specific alteration is carried out by two independent assays (using replicates). Nevertheless, in routine testing generally relying on a single assay to determine mutational status, the threshold of positivity must be increased to avoid a greater risk of obtaining false positives.
Recently, Beije et al. [20] and Iwama et al.[21] showed that ddPCR performance was more accurate for the detection of EGFR mutations in lung cancer and colon cancer than NGS [21]. Interestingly, Beije et al. also found that detection of only certain alterations, namely KRAS, PIK3CA and TP53 somatic mutations, were consistently more detectable than others also detected in the larger panel (including also APC, ATM, CREBBP, FBXW7, and KMT2D genes). The concordance between cfDNA and tissue biopsy was higher in patients presenting liver metastasis (55%) than in patients undergoing cfDNA analysis when only the primary tumor was present (39%). This finding may reflect a more robust performance given a higher level of spatial tumor heterogeneity in metastatic cancer patients and underscores the fact that cfDNA represents a more comprehensive surveying of cancer clones that co-exist within the metastatic patient [20]. By contrast, Goldstein et al. showed that cfDNA mutation detection in prostate cancers by NGS in the androgen receptor axis may lead to false positive cases that are not observed with ddPCR assays [22]. This highlighted the importance of setting the proper clinical cut-off values specific for the platform being used to detect mutations in cfDNA with particular attention to establishing margins of security that are assay-specific. To our knowledge, only one study performed a cross-comparison of BEAMing with another cfDNA mutation detection method in this setting. In cfDNA samples from lung cancer, both platforms were used to detect EGFR sensitizing mutations as well as the EGFR T790M resistance mutation. In this study, the detection sensitivity of BEAMing was slightly higher than that of the Cobas® EGFR mutation test, Therascreen™ EGFR ARMS-PCR and ddPCR™ for all three types of EGFR mutations that are clinically-actionable (deletion 19, L858R and T790M). Conversely, specificity was lower with BEAMing compared to the three other platforms [23]. Thus, the present study is the first to cross-compare three different platforms, in two different cancers, using external controls as well as paired samples from blood and tissue. Our results are also in line with those previously reported, namely that BEAMing demonstrates a higher sensitivity than either ddPCR or NGS. Indeed, without increasing background noise (false positive cases), PCR pre-amplification enabled us to detect very low mutant allelic fractions [23]. The enhanced BEAMing sensitivity was further evidenced by the fact that the OncoBEAM-TM-RAS-CRC assay confirmed 93% of patients with a FFPE RAS+ mutation status (we used tissue biopsy samples as previously reported as reference standards for evaluating cfDNA [23, 19, 20]) and even identified positive cases classified as WT through FFPE testing (i.e. resulted in mismatches). These mismatches or “false-positive” cases are defined as somatic alterations detected in cfDNA that are not detected in FFPE analyses. This is common in the liquid biopsy setting and may be attributed to spatial and temporal tumor heterogeneity. With regards to the spatial heterogeneity, tissue biopsy samples originate from single (and often small) biopsies while a cfDNA sample may capture the patient’s mutational status more comprehensively by examining the entire tumor burden [20].
The heterogeneity of CRC has been underscored and demonstrated in numerous studies as researchers have highlighted significant differences in the KRAS mutational status between primary vs metastatic tumors [24–26]. Intra-tumor heterogeneity may lead to the underestimation of the tumor genomic landscape that is provided by testing of single-site tumor biopsy samples. Because plasma receives cfDNA from the various heterogeneous tumor clones in the body, NGS sequencing of cfDNA allows a readily available and non-invasive method for studying and monitoring tumoral heterogeneity and the total tumor burden in the body [27]. In lung cancer, temporal heterogeneity of the tumor may affect the efficacy of particular cancer treatments. Thus, the molecular pattern of tissues sampled at diagnosis may differ from patterns observed in cfDNA using a highly sensitive technique when the cfDNA sample is evaluated in a patient undergoing disease progression.
In our study, the false-positive rate of cfDNA results vs. the RAS status reported for lung cancer patient cohort whose tumors were reported as WT using a tissue method ranged from 0% to 17% (Supplementary Table 3) depending on the assay used. The false-positive rate in the colon cancer cohort was higher and ranged from 8% to 31% (Supplementary Table 2). Overall, we observed a very low real rate of false-positive (high specificity) in all three assays. For all assays, specific thresholds of positivity were defined. For the Biorad ddPCR assay, the threshold of positivity was defined as detection of 5 mutated haploid GE/reaction, which translates to acute-off threshold of 0.08% using commercial control DNA. For the OncoBEAM-TM-RAS-CRC assay, the threshold of positivity was 50 mutated positive signals/reaction (according to the manufacturer’s determine cut-off threshold) using Horizon’s wild-type standard DNA. Finally, for the NGS assay, the threshold of positivity was set up at 0.5% mutant allelic frequency.
Interestingly, the pre-amplification of starting cfDNA material targeting KRAS/NRAS exons 2, 3 and 4 by multiplex PCR in the OncoBEAM-TM-RAS-CRC assay did not increase the risk of obtaining a false-positive background. Moreover, this assay was able to expose 4 colon cancer and 1 lung cancer patients as true-positives when their FFPE profile was WT, as evidenced by the consistently lower rates of concordance obtained (Supplementary Tables 2 and 3). Secondly, we observed “false-negative” cases (meaning an alteration found in tissue biopsy but not in plasma). This may originate from the limited sensitivity of conventional methods used. Conventional quantitative PCR has a threshold at 1% allelic frequency. This was demonstrated in the Aura study [23], for the T790M mutation, the sensitivity and specificity were 73% and 67%, respectively, with the cobas® EGFR Mutation Test, and 81% and 58%, respectively, with BEAMing dPCR. The lower specificity of BEAMing assay was likely due to the detection of additional positive cases in WT reference samples.
In the present study, 49% of samples examined had less than 1000 haploid GE cfDNA input (Supplementary Figure 1), explaining the necessity for using a highly sensitive method to compensate for the low cfDNA input and low ctDNA release encountered in certain patients. Notably, in case of low input of cfDNA, BIORAD digital PCR assay trended towards to overestimating the allelic mutated frequencies in some cases (CIRCAN-COLON #12 and #16, for example), while the qualitative aspect of positivity of results were in agreement with the other assays. Allelic factions in ddPCR was moderately correlated to those in NGS or BEAMing (R2 coefficient 0.64 and 0.68 respectively), but mutated allelic fractions in BEAMing correlated strongly to mutated allelic fractions in NGS (R2 coefficient 0.99). Here, we found that the BEAMing assay was the best to detect somatic mutations and thus limit the rate of true false-negative results. Indeed, another explanation of false-negative are the panels used; i.e. a targeted assay would not detect an alteration not covered by the panel but is considered here as a false-negative if the panel used in tissue was wider. The detection panels between assays are very distinct, since the OncoBEAM-TM-RAS-CRC includes 34 frequent RAS (KRAS and NRAS) DNA alterations, Biorad’s assay is restricted to alterations of KRAS on exon 2, and the 56G oncology panel provides the overall molecular profile of the patient in one assay [28, 29].
Another explanation for false-negative cases is the copy-number threshold defined to consider a positive sample. Indeed, using our data, we set this threshold at 0.5% for NGS and > 4 haploid GE and > 0.08% of MAF for ddPCR from Biorad and 50 positive mutation signals for the OncoBEAM-TM-RAS-CRC assay (See OncoBEAM RAS CRC IVD instructions for use) [30]. Most mismatches detected via the OncoBEAM-TM-RAS-CRC assay had low numbers of mutated signal, which remained undetected using the other two assays, emphasizing the sensitivity of this ddPCR technology. Overall the best matches were obtained with the paired colon cancer samples (FFPE molecular profile and cfDNA sampling conducted at diagnosis) for all three assays, while lung cancer cfDNA samples were collected during disease progression.
Interestingly, when considering BEAMing as the reference standard, the sensitivity of cfDNA NGS was 71%, specificity 88%, PPV 84% and NPV 76%. At this stage of the discussion, this may explain some of the mismatches observed since the profile of patients may have evolved in the case of RAS-negative patients or chemotherapy may decrease the proliferation and survival of the mutated KRAS/NRAS subclones in RAS-positive patients. Furthermore, mismatches could also be due to (i) the weak release of mutated cfDNA by the tumor (lowering assay sensitivity), (ii) the presence of low proportion of mutated tumoral cells in the samples, (iii) tumor heterogeneity (the tissues used for FFPE molecular profiling may not be representative of the entire tumor), or (iv) the presence of mutated clones in the brain (the release of mutated cfDNA into the blood being restricted in this localization).
Hence, our findings have several strengths but also show some limitations. The main shortcoming is that we did not assess clinical relevance of our molecular results. Indeed, this study aimed only at cross-comparing three different platforms for RAS mutations. Obviously, the next step will be to assess whether detection of certain mutations at low allelic frequencies are clinically meaningful. Another limitation is that our study only included a relatively small cohort of patients (59 in all). Nevertheless, this relatively small cohort is one of the largest groups of patients to be analyzed for cfDNA RAS mutation in cross-platform comparisons [19]. Another drawback of our study arises from the kinetics of blood sampling in the lung cancer cohort (at diagnosis vs during progression). Indeed, this difference in sampling may have generated a risk of bias regarding temporal tumor heterogeneity. However, the strengths of our study reside in the comparison of three platforms for cfDNA analysis based on paired samples. To our knowledge, a head-to-head performance comparison of these three cfDNA mutation detection platforms (BEAMing, ddPCR, and NGS) has never been undertaken. Furthermore, most blood samples were compared to their paired tissue samples.
Costs and turnaround time, in routine practice, of these assays are not similar. Thus, these outcomes may be considered. With respect to the two most complementary assays examined here, the BEAMing assay had the quickest turnaround time (2 days) versus the NGS assay (1 week), which required more time-consuming steps from library preparation until the time of bio-informatics analysis of the results. The digital droplet PCR assay, although less informative than the two other assays, had the benefit of the shortest turnaround time (8 hours) to complete mutational analysis of KRAS exon 2. Concerning the cost, the BEAMing and NGS assays were comparable, whereas the ddPCR assay required only half the cost to perform the analysis versus that of the other two methods.
In conclusion, our findings argue in favor of the use of the Sysmex Inostics BEAMing technology for the detection of somatic KRAS and NRAS mutations, as a highly specific and sensitive alternative to conventional ddPCR (Biorad). Though, we also highlighted the complementarity between the OncoBEAM-TM-RAS-CRC and NGS combined with the 56G oncology panel, which often confirmed dPCR results and provided a larger overview of the major targetable alterations of 56 genes in one run at diagnosis with a 0.5% threshold. The low detection threshold of the OncoBEAM-TM-RAS-CRC assay (0.03%) is also notable as it reduces the amount of detectable ctDNA (owing to the pre-amplification step) and thus enables clinicians to work from smaller cfDNA concentrations. This technique would thus be interesting to monitor the kinetics of mutated haploid GE numbers for cancer patients under treatment in a minimally-invasive manner, since longitudinal testing of serial tissue biopsy samples is both practical and feasible.
MATERIALS AND METHODS
Patients
Samples were collected within the framework of the CIRCAN (“CIRculating CANcer”) study, which is a prospective program established to comprehensively evaluate tumor biomarkers in cfDNA at the Lyon University Hospital. In the present study, we analyzed cfDNA in plasma samples derived from blood from mCRC patients at diagnosis (n = 25), as well as metastatic NSCLC patients during disease progression (n = 34). All tumor cases were histologically or cytologically confirmed on FFPE biopsy specimens. RAS mutation testing was performed on FFPE for 45 patients (25 mCRC and 20 NSCLC). However, in 14 NSCLC cases, the quantity and/or quality of the FFPE specimen were insufficient for RAS mutation analysis. Patient outcome and demographic data were collected during the course of the study. The extraction, amplification and mutation analyses of cfDNA were performed by investigators who did not have access to or prior knowledge of clinical data and were also blinded to patient outcome following therapy.
Sample collection
FFPE tumor samples (n = 25 for mCRC and n = 20 for NSCLC) were microdissected (microdissector LMD2000, Leica, Germany, EU) and DNA was purified from the areas of the samples with the highest percentage of tumor cells using the QIAamp DNA FFPE Tissue kit (Qiagen, Cat No./ID: 56404, Valencia, CA, USA) as per manufacturer’s instructions. These samples were then analyzed using a customized Ampliseq library and next-generation sequencing (NGS) on the Ion PGM (Life Technologies, Carlsbad, CA, USA).
Plasma was prepared from 30 mL of blood collected in K2 EDTA tubes (BD, 367525, 18 mg). All blood samples were delivered to the laboratory within 24 hours after collection. Detailed pre-analytical considerations have been previously published [13]. The haploid GE corresponds to the haploid Genome Equivalent (330 GE blood DNA for 1 ng/μL cfDNA).
Droplet-digital PCR for detection of KRAS mutations
The sensitive and quantitative QX100 droplet digital PCR system from Biorad (ddPCR, Biorad, Hercules, CA, USA) combines a water-oil emulsion droplet technology with microfluidics (Biorad, 186-3005). All reactions were prepared using the ddPCR Supermix for probes concentrated 2× (Biorad, 186-3024). KRAS somatic alterations were detected using a commercial KRAS Screening Multiplex Kit provided by Biorad (ref 186-3506). This multiplex ready-to-use detection assay is designed to screen 7 mutations in KRAS codons 12 and 13 (Figure 1, labelled in green). Detailed analytical considerations for this assay have been previously published ([13], Supplementary File 1).
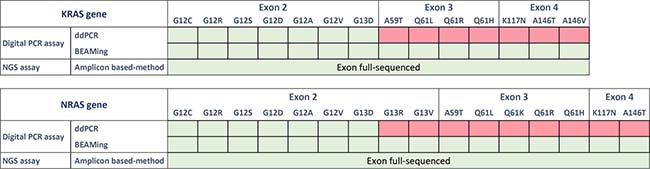
Figure 1: List of the panels provided by Biorad (ddPCR), Sysmex Inostics (OncoBEAM-TM-RAS-CRC) and Swift Biosciences (56G oncology library panel). The “KRAS multiplex kit” screens seven KRAS mutations in a single well by ddPCR. NRAS mutations are not included in the panel. The OncoBEAM-TM-RAS-CRC panel enables the detection of 16 KRAS mutations and 18 NRAS mutations in exons 2, 3 and 4. Panel of the genes included in the “56G Oncology Panel”. In total, 56 genes (263 amplicons) are amplified and sequenced by NGS using the NextSeq 500 of Illumina. Here, cfDNA samples were analyzed using the three assays while FFPE samples were only tested by NGS.
Digital BEAMing for detection of KRAS and NRAS mutations
OncoBEAM is a highly sensitive and quantitative digital PCR platform utilizing Beads, Emulsion, Amplification and Magnetics (BEAMing). This platform is CE-IVD labelled and produced by Sysmex Inostics (Hamburg, Germany, EU) [31–33]. This assay is based on multiplex PCR targeting somatic alterations which are then followed by a massively parallel second PCR amplification performed on magnetic beads compartmentalized in millions of oil emulsions. Finally, a hybridization step utilizing fluorescent probes specific to wild-type (WT) and mutant (MT) signals is performed with flow cytometry in order to discriminate WT and MT bead populations. Here, we used the OncoBEAM-TM-RAS-CRC™ kit (Sysmex Inostics, Hamburg Germany) which enables the screening of 34 somatic genomic alterations in KRAS and NRAS genes and 30 somatic non-synonymous protein alterations in one run (Figure 1, labelled in green). All experiments were performed according to the supplier’s IVD recommendations for clinical application (Instructions for Use, IfU). The pre-specified positivity threshold for each codon was established in a clinical study of 238 patients [30], and on average, the clinical cut-offs provided by the manufacturer were determined to be ~50 mutant beads detected (according to the OncoBEAM RAS CRC kit IfU).
Targeted next-generation sequencing library preparation
CfDNA libraries were created using the multiple targeted amplicon technology provided by Swift Biosciences according to the manufacturer’s instructions (56G Oncology Panel Kit, Swift Biosciences, Ann Arbor, MI, Cat. No AL-56248). Detailed analytical considerations are fully described in the Supplementary File 1. Fastq files, obtained by the demultiplexing of base-call files (BCL), were aligned against the human genome reference Hg19 (GRCh37.p5). Bioinformatics analyses were conducted by Sophia Genetics (Saint-Sulpice). The resulting BAM files were re-aligned for soft-clipping regions to recover potential indels. Variant calls were conducted by comparing the non-reference base against the averaged mean error of the corresponding averaged base quality for a given position. The subsequent Variant Call Files (VCF) were subjected to cross sample background filtering with potential artefacts removed below 3 standard deviations of the mean background noise for each position. Filter criteria for variant calling for absolute mutated allele read counts was set to = > 50 and the depth > 500.
Ethical considerations
The CIRCAN_ALL study is a prospective observational study conducted at the Lyon University Hospital since December 2015 and intended to comprehensively perform cfDNA mutational analysis in in patients with various types of malignancies. This study was approved by the regional ethics committee Lyon Sud Est IV (CPP L15-188 11/04/2015; amended by L16-160 09/21/2016) and French National committee in Informatics (CNIL 15-131 01.12.2015). Written informed consent for total blood sampling was obtained from all patients included in the study. The study was carried out in accordance with international guidelines and French regulations. All samples and medical data used in this study were anonymized.
Statistical analysis
Statistics were performed using the latest version of GraphPad InStat software (GraphPad Software, Inc., La Jolla, CA, USA). Normal distribution of continuous variables was assessed with the Shapiro-Wilk normality test. We used the Mann–Whitney test for non-normally distributed variables, and the Student t-test for normalized data. A 2-sided P-value < 0.05 was considered statistically significant.
Abbreviations
cfDNA: cell-free DNA; FFPE: Formalin-Fixed Paraffin-Embedded; RT: room temperature; ddPCR: digital droplet polymerase chain reaction; WT: wild-type; EGFR: epidermal growth-factor; NGS: next generation sequencing.
Author contributions
Conceptualization and design: JF, TW, SC, LP and JG; Development of methodology: TW, LP, JG and SC; Material support: JG, JF, ASW, FG, ED, PM, PF, CFP, FJ, VC, CB, GV, ZX, PPB, MG, MC, BM, MB, CRL, VH, SC, TW and LP; Analysis and interpretation of data: LP, JG and SC; Writing: JG, LP, BM, TW, JF and SC; Critical revision: All.
CONFLICTS OF INTEREST
Dr. Cheynet and Dr. Brengel-Pesce are employees of BioMérieux SA. Daniel Edelstein and Frederick Jones are employees of Sysmex Inostics, Inc. Dr. Xu reports to be employed by Sophia Genetics. Dr. Forestier reports personal fees from AMGEN, personal fees from SANOFI, personal fees from MERCK, personal fees from CELGENE, personal fees from ROCHE, personal fees from IPSEN, outside the submitted work. Dr. Bringuier reports personal fees from Astra Zeneca, outside the submitted work. Dr. Brevet reports grants from Astra Zeneca, personal fees from MSD, personal fees from BMS, grants from Pfizer, outside the submitted work. Dr. Garcia and Dr Payen report grants from Sysmex-innostics during the conduct of the study. Dr. Couraud reports grants and non-financial support from Sysmex Innostics, grants and personal fees from Astra Zeneca, grants from Merck, grants and personal fees from BMS, during the conduct of the study; grants and personal fees from Astra Zeneca, grants, personal fees and other from Pfizer, grants, personal fees and other from Roche, grants, personal fees and other from Chugai, grants, personal fees and other from MSD, grants, personal fees and other from Boehringher Ingelheim, grants and personal fees from Lilly, grants and personal fees from Novartis, grants and personal fees from Laidet Medical, grants from Amgen, other from Eformed, outside the submitted work. Other authors have nothing to disclose.
FUNDING
Our team is accredited by the “Ligue Nationale contre le Cancer” of Saône et Loire Comity. JG was recipient of fellowships from Astra-Zeneca.
REFERENCES
1. Cox AD, Der CJ. Ras history: The saga continues. Small GTPases. 2010; 1:2–27.
2. Němeček R, Svoboda M, Slabý O. Current Possibilities for Predicting Responses to EGFR Blockade in Metastatic Colorectal Cancer. [Article in Czech] Klin Onkol. 2016;29:187–95.
3. Yao Y, Liu J, Li L, Yuan Y, Nan K, Wu X, Zhang Z, Wu Y, Li X, Zhu J, Meng X, Wei L, Chen J, Jiang Z. Detection of circulating tumor DNA in patients with advanced non-small cell lung cancer. Oncotarget. 2017; 8:2130–2140. https://doi.org/10.18632/oncotarget.12883.
4. Garde Noguera J, Jantus-Lewintre E, Gil-Raga M, Evgenyeva E, Macia Escalante S, Llombart-Cussac A, Camps Herrero C. Role of RAS mutation status as a prognostic factor for patients with advanced colorectal cancer treated with first-line chemotherapy based on fluoropyrimidines and oxaliplatin, with or without bevavizumab: A retrospective analysis. Mol Clin Oncol. 2017; 6:403–408.
5. Fiala O, Buchler T, Mohelnikova-Duchonova B, Melichar B, Matejka VM, Holubec L, Kulhankova J, Bortlicek Z, Bartouskova M, Liska V, Topolcan O, Sedivcova M, Finek J. G12V and G12A KRAS mutations are associated with poor outcome in patients with metastatic colorectal cancer treated with bevacizumab. Tumour Biol. 2016; 37:6823–6830.
6. Barlesi F, Mazieres J, Merlio JP, Debieuvre D, Mosser J, Lena H, Ouafik L, Besse B, Rouquette I, Westeel V, Escande F, Monnet I, Lemoine A, et al, and Biomarkers France contributors. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet. 2016; 387:1415–1426.
7. Chapman AM, Sun KY, Ruestow P, Cowan DM, Madl AK. Lung cancer mutation profile of EGFR, ALK, and KRAS: Meta-analysis and comparison of never and ever smokers. Lung Cancer. 2016; 102:122–134.
8. Illei PB, Belchis D, Tseng LH, Nguyen D, De Marchi F, Haley L, Riel S, Beierl K, Zheng G, Brahmer JR, Askin FB, Gocke CD, Eshleman JR, et al. Clinical mutational profiling of 1006 lung cancers by next generation sequencing. Oncotarget. 2017; 8:96684–96. https://doi.org/10.18632/oncotarget.18042.
9. Mounawar M, Mukeria A, Le Calvez F, Hung RJ, Renard H, Cortot A, Bollart C, Zaridze D, Brennan P, Boffetta P, Brambilla E, Hainaut P. Patterns of EGFR, HER2, TP53, and KRAS mutations of p14arf expression in non-small cell lung cancers in relation to smoking history. Cancer Res. 2007; 67:5667–5672.
10. Guibert N, Barlesi F, Descourt R, Lena H, Besse B, Beau-Faller M, Mosser J, Pichon E, Merlio JP, Ouafik L, Guichard F, Mastroianni B, Moreau L, et al. Characteristics and Outcomes of Patients with Lung Cancer Harboring Multiple Molecular Alterations: Results from the IFCT Study Biomarkers France. J Thorac Oncol. 2017; 12:963–973.
11. Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011; 11:426–437.
12. Couraud S, Vaca-Paniagua F, Villar S, Oliver J, Schuster T, Blanché H, Girard N, Trédaniel J, Guilleminault L, Gervais R, Prim N, Vincent M, Margery J, et al, and BioCAST/IFCT-1002 investigators. Noninvasive diagnosis of actionable mutations by deep sequencing of circulating free DNA in lung cancer from never-smokers: a proof-of-concept study from BioCAST/IFCT-1002. Clin Cancer Res. 2014; 20:4613–4624.
13. Garcia J, Dusserre E, Cheynet V, Bringuier PP, Brengle-Pesce K, Wozny AS, Rodriguez-Lafrasse C, Freyer G, Brevet M, Payen L, Couraud S. Evaluation of pre-analytical conditions and comparison of the performance of several digital PCR assays for the detection of majorEGFRmutations in circulating DNA from non-small cell lung cancers: the CIRCAN_0 study. Oncotarget. 2017; 8:87980–96. https://doi.org/10.18632/oncotarget.21256.
14. Olmedillas-Lopez S, Garcia-Arranz M, Garcia-Olmo D. Current and Emerging Applications of Droplet Digital PCR in Oncology. Mol Diagn Ther. 2017; 21:493–510. https://doi.org/10.1007/s40291-017-0278-8.
15. Perdigones N, Murtaza M. Capturing tumor heterogeneity and clonal evolution in solid cancers using circulating tumor DNA analysis. Pharmacol Ther. 2017; 174:22–26.
16. Gao J, Wang H, Zang W, Li B, Rao G, Li L, Yu Y, Li Z, Dong B, Lu Z, Jiang Z, Shen L. Circulating tumor DNA functions as an alternative for tissue to overcome tumor heterogeneity in advanced gastric cancer. Cancer Sci. 2017; 108:1881–87.
17. Medina Diaz I, Nocon A, Mehnert DH, Fredebohm J, Diehl F, Holtrup F. Performance of Streck cfDNA Blood Collection Tubes for Liquid Biopsy Testing. PLoS One. 2016; 11:e0166354.
18. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014; 6:224ra224.
19. Bartels S, Persing S, Hasemeier B, Schipper E, Kreipe H, Lehmann U. Molecular Analysis of Circulating Free DNA from Lung Cancer Patients in Routine Laboratory Practice: A Cross-Platform Comparison of Three Different Molecular Methods for Mutation Detection. J Mol Diagn. 2017; 19:722–32.
20. Beije N, Helmijr JC, Weerts MJ, Beaufort CM, Wiggin M, Marziali A, Verhoef C, Sleijfer S, Jansen MP, Martens JW. Somatic mutation detection using various targeted detection assays in paired samples of circulating tumor DNA, primary tumor and metastases from patients undergoing resection of colorectal liver metastases. Mol Oncol. 2016; 10:1575–84. https://doi.org/10.1016/j.molonc.2016.10.001.
21. Iwama E, Sakai K, Azuma K, Harada T, Harada D, Nosaki K, Hotta K, Ohyanagi F, Kurata T, Fukuhara T, Akamatsu H, Goto K, Shimose T, et al. Monitoring of somatic mutations in circulating cell-free DNA by digital PCR and next-generation sequencing during afatinib treatment in patients with lung adenocarcinoma positive for EGFR activating mutations. Ann Oncol. 2017; 28:136–141.
22. Goldstein A, Toro PV, Lee J, Silberstein JL, Nakazawa M, Waters I, Cravero K, Chu D, Cochran RL, Kim M, Shinn D, Torquato S, Hughes RM, et al. Detection fidelity of AR mutations in plasma derived cell-free DNA. Oncotarget. 2017; 8:15651–62. https://doi.org/10.18632/oncotarget.14926.
23. Thress KS, Brant R, Carr TH, Dearden S, Jenkins S, Brown H, Hammett T, Cantarini M, Barrett JC. EGFR mutation detection in ctDNA from NSCLC patient plasma: A cross-platform comparison of leading technologies to support the clinical development of AZD9291. Lung Cancer. 2015; 90:509–515.
24. Larki P, Gharib E, Yaghoob Taleghani M, Khorshidi F, Nazemalhosseini-Mojarad E, Asadzadeh Aghdaei H. Coexistence of KRAS and BRAF Mutations in Colorectal Cancer: A Case Report Supporting The Concept of Tumoral Heterogeneity. Cell J. 2017; 19:113–117.
25. Baldus SE, Schaefer KL, Engers R, Hartleb D, Stoecklein NH, Gabbert HE. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res. 2010; 16:790–799.
26. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366:883–892.
27. Chan KC, Jiang P, Zheng YW, Liao GJ, Sun H, Wong J, Siu SS, Chan WC, Chan SL, Chan AT, Lai PB, Chiu RW, Lo YM. Cancer genome scanning in plasma: detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin Chem. 2013; 59:211–224.
28. Shu Y, Wu X, Tong X, Wang X, Chang Z, Mao Y, Chen X, Sun J, Wang Z, Hong Z, Zhu L, Zhu C, Chen J, et al. Circulating Tumor DNA Mutation Profiling by Targeted Next Generation Sequencing Provides Guidance for Personalized Treatments in Multiple Cancer Types. Sci Rep. 2017; 7:583.
29. Chevrier S, Arnould L, Ghiringhelli F, Coudert B, Fumoleau P, Boidot R. Next-generation sequencing analysis of lung and colon carcinomas reveals a variety of genetic alterations. Int J Oncol. 2014; 45:1167–1174.
30. Jones F, Edelstein D, Wichner K, Ross C, Stieler K, Boehm V, Lenfert E, Bien J, Lukas A, Macioszek J. Performance of standardized BEAMing platform for detecting RAS mutations in the blood of metastatic colorectal cancer (mCRC) patients. J Clin Oncol. 2016; ASCO 2016 Poster presentation 11538:11538–11538.
31. Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A. 2003; 100:8817–8822.
32. Vidal J, Muinelo L, Dalmases A, Jones F, Edelstein D, Iglesias M, Orrillo M, Abalo A, Rodriguez C, Brozos E, Vidal Y, Candamio S, Vazquez F, et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann Oncol. 2017; 28:1325–1332.
33. Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz LA Jr, Goodman SN, David KA, Juhl H, Kinzler KW, Vogelstein B. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005; 102:16368–16373.