
INTRODUCTION
Loss of function of tumor suppressor genes, like PTEN, can facilitate tumorigenesis and metastasis, and in nontumorigenic cell lines, can result in phenotypic changes associated with both processes [1–12]. One way to rescue loss of function of a tumor suppressor gene, is to reintroduce the normal gene, in an expression plasmid into the cells [4, 8, 13–20]. This approach shows promise, but to date has not yielded effective treatments [21, 22]. There is, however, an alternative and, possibly, less intrusive strategy that has not been explored. If the defective gene contributing to tumorigenesis has one or more functional homologs, then stimulating overexpression of one of them in mutant cells may restore the function of the defective gene. PTEN indeed has two homologs, TPTE1 and TPTE2 (TPIP), a PTEN pseudogene [23–27], a TPTE1 pseudogene [23] and seven TPTE2 (TPIP) pseudogenes [23–28] (www.ncbi.nim.nih.gov/gene; www.uniprot.org Q6XPS3; www.ensemble.org). By over-expressing a homolog of PTEN with similar catalytic and membrane-binding domains in a cell that has lost PTEN function such as TPTE2 (TPIP), one may be able to normalize the aberrant phenotype caused by the absence of PTEN. And, if expression of the homolog could be up-regulated by a surface receptor, and a chemical or monoclonal antibody could be identified that activates that receptor, one might be able to design a drug that reestablishes PTEN function in PTEN mutants and thus suppresses tumorigenesis and metastasis.
Recently, we explored this hypothesis by testing it in a model system, the simple amoeba Dictyostelium discoideum, which has been used for over 65 years as an exceptional model for human development and white blood cell function [29]. D. discoideum contains an ortholog of the human PTEN gene, ptenA, and a homolog of ptenA, lpten. Both PtenA and Lpten contain the two signature domains of human PTEN, the CDC14-protein tyrosine phosphatase domain and the lipid C2-binding domain [30]. The ptenA null mutant, ptenA-, exhibits strong behavioral defects in motility, chemotaxis, aggregation and multicellular morphogenesis [31–37]. The null mutant of the homolog, lpten-, behaves as a very weak phenocopy of ptenA- [30]. Overexpressing full length lpten rescued all developmental and behavioral defects of the ptenA- mutant [30], an observation that provided initial support for the proposed hypothesis.
Here, we have used a similar strategy to test whether overexpressing TPTE2, also known as a homolog of PTEN, in a human PTEN null mutant, PTEN–/–, generated in the nontumorigenic breast epithelial cell line, MCF-10A [10], reversed characteristics associated with the loss of PTEN function –i.e., rescued the mutant PTEN–/– phenotype. The PTEN–/– strain was generated by targeted homologous recombination, and previously demonstrated to differ from the parent strain MCF-10A in a number of phenotypic characteristics [9, 10]. The PTEN–/– -associated phenotypes explored here include 1) an increased rate of wound healing, accentuated by the removal of growth factors (GFs) from the supporting medium, 2) an increase in the rate of cell division in the presence of GFs, 3) adherence and viability on a 2D substrate in the absence of GFs, 4) viability in a 3D Matrigel model in the absence of GFs and substrate adhesion, and 5) loss of the apoptosis-associated characteristic of annexin V binding and endocytosis [38–42]. We show that overexpressing TPTE2 (TPIP) in PTEN–/– mutant cells reverses all of the phenotypic changes associated with the PTEN mutation and in the cases of wound healing and annexin V binding, actually accentuates the normal PTEN+/+ phenotype. These results add further support to the hypothesis that a receptor mediated signal that upregulates a homolog of a defective tumor suppressing gene can normalize defects in mutant cells associated with the mutation, with the potential of suppressing tumorigenesis and metastasis and stimulating apoptosis. This approach may also prove effective for other types of cancer-associated mutations, for which the mutant gene has a functional homolog that can be upregulated.
RESULTS
Generating PTEN–/–TPTE2OE strains
The human genome contains one copy of PTEN [3, 5, 43], one each of the two PTEN homologs TPTE1 and TPTE2 (TPIP) [23, 24, 26, 27], and a pseudogene [25]. PTEN produces two transcripts, PTEN and PTEN long (Figure 1A) [43, 44]. Here, we have focused on the effects of overexpressing TPTE2 (TPIP), which will be referred to hence as TPTE2, in the mutant strain PTEN–/–, the latter generated from the breast epithelial cell line MCF-10A [10]. PTEN–/– was previously shown to differ phenotypically from MCF-10A (PTEN+/+). The differences included independence of growth on growth factors, resistance to apoptosis, a decrease in susceptibility to doxorubicin and shape changes in suspension [9, 10]. TPTE2 was selected over TPTE1 (TPTE) because the former was predicted to have phosphatase activity [25, 27, 45, 46] whereas TPTE1 was predicted, based on amino acid sequence, not to have activity [26]. Amplification and sequencing of TPTE2 from cDNA preparations of MCF-10A revealed three TPTE2 transcripts, TPTE2-1, TPTE2-2 and TPTE2-3, diagrammed in Figure 1B. The variants differed at both the N- and C- terminal regions (Figure 1B). All three, however, contained the CDC14 protein tyrosine phosphatase domain involved in the conversion of PIP3 to PIP2, and the PTEN-C2 domain, a lipid-binding domain involved in localization of PTEN to the inner face of the plasma membrane (Figure 1B) [3, 25]. TPTE2-3 corresponded to TPIPγ, identified by Walker et al. [27]. All three TPTE2 variants contained four transmembrane (TM) domains in the N-terminal half of the transcript (Figure 1B). These TMs are absent in PTEN (Figure 1A). TPTE2-1 (Figure 1B) was selected for overexpression because of its high sequence homology to PTEN, including similar relative positions of the CDC14 and C2 domains (Figure 1A). To generate TPTE2-1 overexpression strains in the PTEN mutant PTEN–/–-1, a transformation plasmid was generated that contained the TPTE2-1 cDNA fused in frame to GFP and under the control of the cytomegalovirus (cmv) promoter (Figure 1C). The TPTE2 overexpression plasmid integrated ectopically into the genome of the mutant strain PTEN–/–-1. Two independent clones of TPTE2-1 overexpressors, TPTE2oe-1 and TPTE2oe-2, were employed in subsequent analyses.
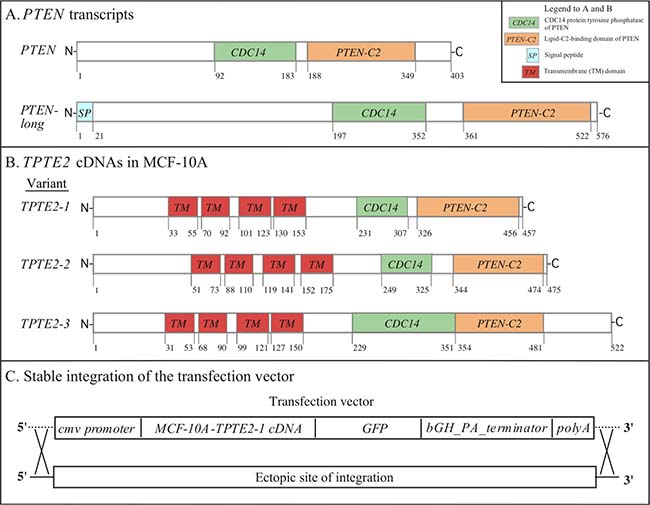
Figure 1: The PTEN and TPTE2 transcripts, and integration of the TPTE2 overexpression vector. (A) The PTEN and PTEN long transcripts. (B) The three TPTE2 transcript variants, TPTE2-1, TPTE2-2 and TPTE2-3. (C) The ectopic, random integration of the construct containing the cmv promoter, TPTE2 variant 1 (TPTE2-1) open reading frame, GFP tag, bGH-PA-terminator sequence and poly A tail. Box in upper right-hand corner of panel A shows color-coded domains in panels A and B.
Proof of overexpression
TPTE2 has been found to be highly expressed in the testes, and at lower levels in spermatocytes, the brain and stomach [26, 27, 47, 48]. It is expressed at negligible levels in other tissues [47, 48]. To demonstrate that TPTE2 was actually overexpressed in strains TPTE2oe-1 and TPTE2oe-2, growth cultures were analyzed by RT-PCR. MCF-10A cells and the two PTEN–/– derivative mutants expressed TPTE2, but the PTEN–/– derivative mutants did so at approximately 60% and 40% that of MCF-10A cells (Figure 2A, 2B). TPTE2-1oe and TPTE2oe-2, expressed TPTE2-1 at 3 fold and 2.5 fold, respectively the level of the PTEN–/– mutants (Figure 2A, 2B). To demonstrate that the fusion protein TPTE2-GFP was expressed, we employed indirect immunostaining, in which cells were permeabilized and fixed, treated with a mixture of two anti-GFP mAbs, DSHB-GFP-12A6 and DSHB-GFP-AC9, and then treated with the secondary fluorescent anti-mouse IgGH+L Alexa 488 mAb. Over 80% of TPTE2-1oe and TPTE2oe-2 cells grown in tissue culture preparations exhibited bright diffuse staining (Figure 3A–3F), demonstrating that they expressed high levels of TPTE2-GFP throughout the cytoplasm. There was no staining in PTEN–/– mutant cells treated in a similar fashion (Figure 3G, 3H).
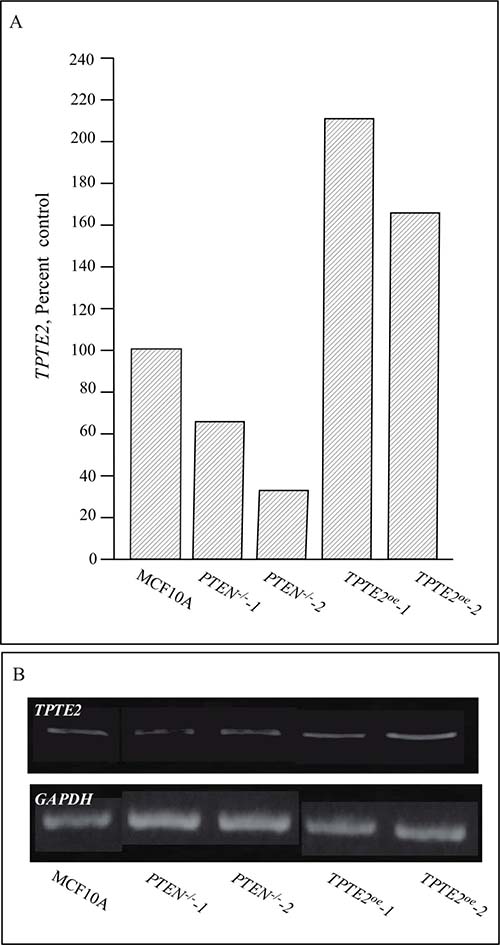
Figure 2: Expression of TPTE2 in the control strain MCF-10A, the two PTEN–/– strains and the two PTEN–/– strains in which TPTE2 is overexpressed. Expression was measured by RT-PCR. (A) Expression of TPTE2 as percent control (MCF-10A). (B) Representative gel of TPTE2 expression. GAPDH expression was assessed to verify uniform loading of gels.
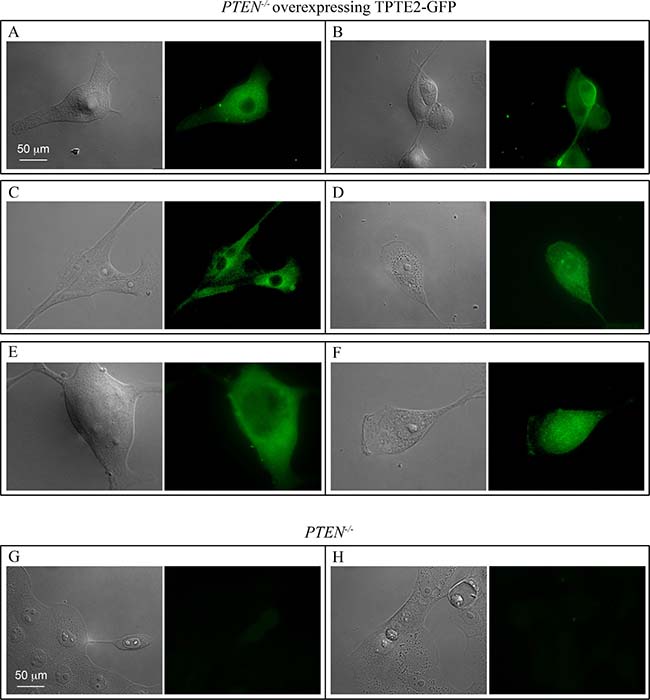
Figure 3: TPTE2oe-1 cells express the TPTE2-GFP fusion protein. (A–F) Examples of TPTE2oe cells expressing TPTE2-GFP. (G, H) Examples of PTEN–/– cells. Cells were treated with a mixture of two anti-GFP mAbs and then stained with the secondary fluorescent antibody against mouse mAb, anti H+L Alexa 488 antibody.
Wound healing
In the model system D. discoideum, it was demonstrated that individual cells of the ptenA- (human PTEN ortholog) mutant exhibited a variety of motility-associated defects, including a reduction in velocity, an increase in lateral pseudopod formation and an increase in turning, which affected the efficiency of chemotaxis and thus caused defects in multicellular morphogenesis [31–37]. Overexpressing the homolog of ptenA, lpten, in the mutant ptenA- rescued all of these phenotypic defects [30]. Identification of similarly strong motility and chemotaxis differences between individual cells of strains MCF-10A and the PTEN–/– derivative strain to test whether TPTE2 overexpression could rescue mutant defects, proved difficult because of the low velocity and weak chemotactic responses of MCF-10A and PTEN–/– cells. We therefore exploited the characteristic of collective cell migration, the basis of wound healing [49–52]. It has been suggested that collective cell migration may, in fact, more accurately reflect the motile behavior of cells during tumorigenesis and metastasis [53–55]. PTEN had previously been implicated in this behavior [56, 57]. We therefore tested for, and identified, strong behavioral differences between the parental MCF-10A line and the two mutants, PTEN–/–-1 and PTEN–/–-2 in the wound healing process. In wound healing assays, cells are either removed from a confluent monolayer by scraping a gap (“wound”) [58, 59], or by removing an insert initially positioned in a monolayer, to generate a gap (“wound”) [60]. Cells at the two edges of the opposing confluent layers in both assays then fill the gap by collective cell migration [50, 58, 59]. Leader cells move into the wound and the two opposing sheets of cells collectively move in the direction of the leader cells, filling the gap [50, 61–63]. To test wound healing, we used the latter assay [60, 64]. To assess conditions that provided the most extreme differences between parental MCF-10A cells and the two derivatives PTEN–/– strains, we grew the initial monolayer in DMEM+GFs, then monitored wound healing in the three media DMEM+GFs, DMEM+S,-other GFs and DMEM-GFs. In DMEM+GFs medium, there was an initial, small difference in the speed of wound healing between MCF-10A and the two PTEN–/– clones, observed at 10 hours (Figures 4A, 5A–5C, respectively). However, both the MCF-10A and PTEN–/– clones subsequently completed wound healing by 12.5 hours (Figures 4A, 5A–5C, respectively). Overexpressing TPTE2 in mutant clone PTEN–/–-1, in the derivatives TPTE2oe-1 and TPTE2oe-2, retarded wound healing, resulting in rates not only below that of the two PTEN–/– clones, but also below that of the parental strain MCF-10A cells (Figures 4A, 5D, 5E, respectively). Wound healing for the two PTEN–/– TPTE2oe derivatives was completed in 26 hours. These results suggested that overexpressing TPTE2-1 not only rescued the PTEN–/–-associated trait in DMEM+GFs medium, but in this case accentuated the behavioral phenotype of the parental control, MCF-10A.
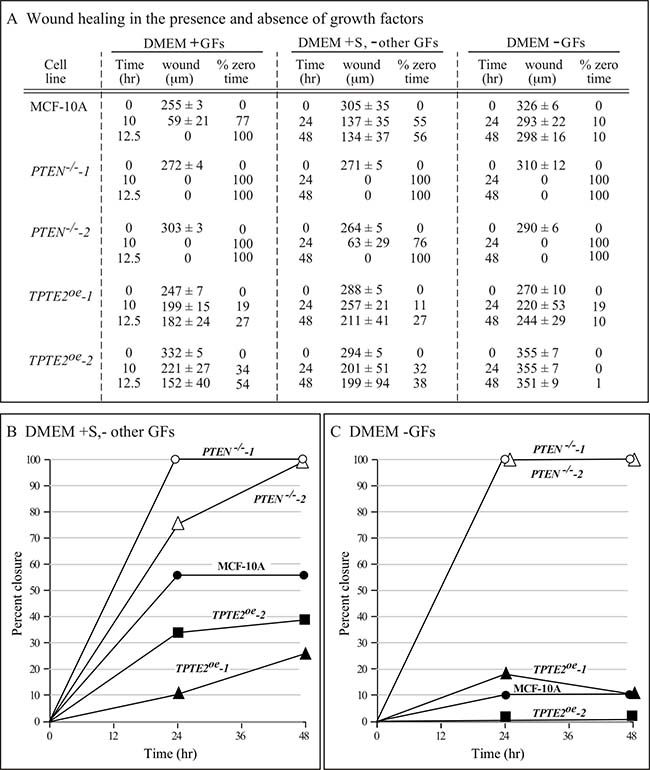
Figure 4: Overexpressing TPTE2 in PTEN–/– cells rescues mutant wound healing characteristics. Wound healing was tested in three media in which growth factors (GFs) were manipulated: DMEM+GFs medium, (DMEM+GFs), DMEM plus serum but lacking other GFs medium, (DMEM+S-other GFs) and, DMEM minus GFs medium (DMEM-GFs). (A) Wound healing data (mean ± standard deviation, N = 3) are presented as wound width in μm over time and percent of original wound width. Total time of experiment was predicated on the rate of total wound healing in the PTEN–/– mutant for each medium. (B) Data (means) graphed for wound healing as a function of time in DMEM+S, -other GFs medium. (C) Data graphed for wound healing as a function of time in DMEM-GFs medium.
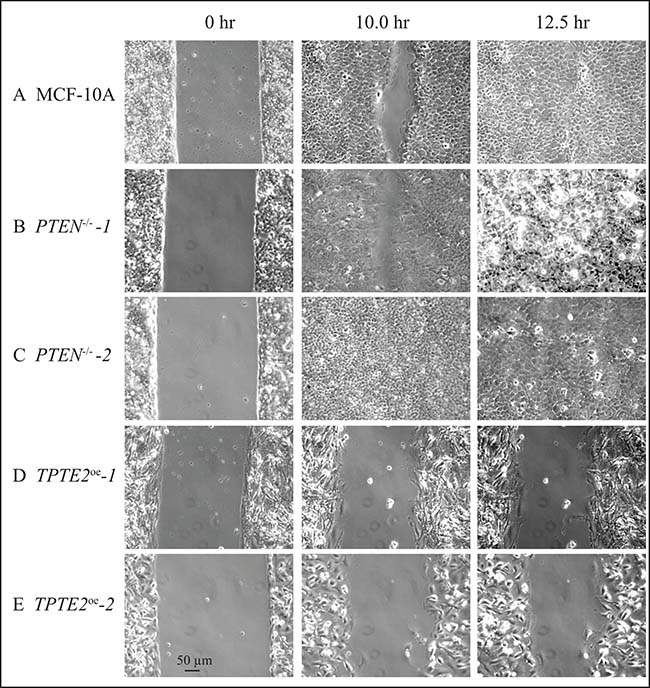
Figure 5: Representative images in DMEM+GFs medium reveal that the PTEN-/- mutant undergoes wound healing faster than parental MCF-10A cells and that overexpressing TPTE2 in PTEN-/- cells actually retards wound healing, accentuating the parental (PTEN+/+) phenotype. (A) MCF-10A, (B) PTEN-/--1 (C) PTEN-/--2, (D) TPTE2oe-1, (E) TPTE2oe-2.
When the GFs other than serum were removed, in the medium DMEM + S,-other GFs, the rate of wound healing by MCF-10A cells was dramatically reduced (Figures 4A, 4B; 6A). Parental MCF-10A cells underwent only 56% closure after 48 hours (Figures 4A, 4B, 6A). In contrast, the two PTEN–/– strains underwent either near or complete closure in 24 hours (Figures 4A, 4B, 6B, 6C, respectively). Overexpressing TPTE2, in TPTE2oe-1 and TPTE2oe-2, reversed rapid closure of the gap and again accentuated the characteristic of slow closure exhibited by the parental line MCF-10A (Figures 4A, 4B, 6A, 6D, 6E, respectively). Whereas closure by MCF-10A was 56% complete after 48 hours, that of the two PTEN–/–TPTE2OE lines was 27% and 38%, respectively (Figures 4A, 4B, 6A, 6D, 6E, respectively).
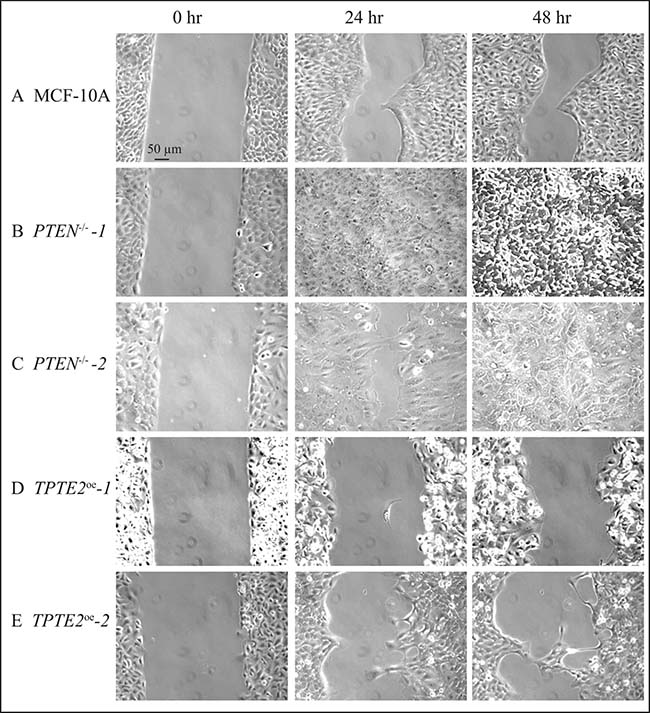
Figure 6: Representative images in DMEM+S, -other GFs medium reveal that overexpressing TPTE2 in PTEN-/- cells slows the wound healing process, thus reestablishing and actually accentuating the slower rate of the parental MCF-10A cell line. (A) MCF-10A, (B) PTEN-/--1 (C) PTEN-/--2, (D) TPTE2oe-1, (E) TPTE2oe-2.
When all GFs, including serum, were omitted, in DMEM-GFs medium, MCF-10A cells underwent only 10% closure by 48 hours (Figures 4A, 7A), whereas cells of the two PTEN–/– mutants underwent 100% closure by 24 hours (Figures 4A, 4C, 7B and 7C, respectively). Overexpressing TPTE2 in the two overexpression strains reinstated the MCF-10A phenotype (Figures 4A, 4C, 7A, 7D, 7E, respectively). The percent closure was 10 and 1%, respectively in the two TPTE2 overexpression lines, after 48 hours (Figures 4A, 4C, 7D and 7E, respectively). Together, the results demonstrate that the slightly lower rate of wound healing by MCF-10A cells in the presence of GFs, and the extremely slow rate in the absence of GFs were lost in the PTEN–/– mutants, which undergo rapid wound healing in the presence of GFs, but reestablished in PTEN–/– mutants in which TPTE2 was overexpressed.
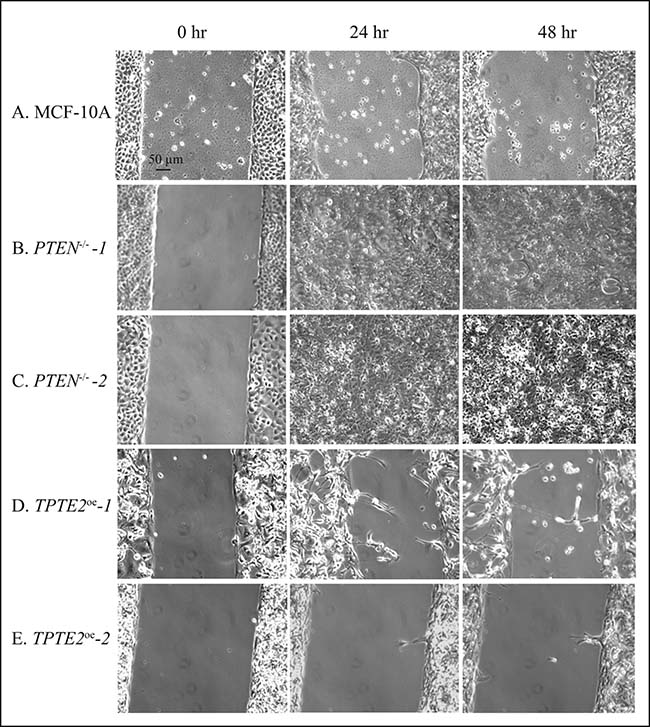
Figure 7: Representative images in DMEM-GFs medium reveal that overexpressing TPTE2 in PTEN-/- cells reverses completely GF-independent wound healing exhibited by PTEN-/- cells, resulting in the absence of wound healing, the phenotype of parental MCF-10A cells. (A) MCF-10A, (B) PTEN-/- -1 (C) PTEN-/- -2, (D) TPTE2oe -1, (E) TPTE2oe -2
Rate of cell division in the presence of GFs
It was previously demonstrated that loss of PTEN, in PTEN–/– mutant cells, conferred growth factor-independent proliferation [10]. We observed, both in tissue culture and in suspension cultures in DMEM+GFs medium, that it took longer for MCF-10A cells than PTEN–/– cells to reach confluence in a tissue culture dish in DMEM+GFs medium. To test whether this difference was related to a shorter cell cycle and if so, whether overexpression of TPTE2 reinstated the slower rate of parental MCF-10A cells, we measured the time between cleavage furrows for 10 dividing cells in preconfluent preparations in DMEM+GFs medium on the plastic surface of tissue culture dishes. Examples of cleavage and the times of sequential cleavage furrows are presented for representative MCF-10A, PTEN–/– and TPTE2oe-1 cells in Figure 8A–8C, respectively. No differences were evident in the actual cellular mechanics of cell division. However, the average interval times between cleavage furrows differed. The mean ± standard deviation of interval times for 10 MCF-10A cells and 10 PTEN–/–-1 cells were 17.3 ± 2.9 (N = 10) and 11.6 ± 0.7 (N = 10) hours, respectively, a decrease of 33%. The difference was significant (p value 3 × 10–5). The interval time for 10 PTEN–/–TPTE2OE -1 cells was 21.0 ± 4.3 hours, which was not significantly different from that of MCF-10A cells (p value of 5 × 10–2), but significantly different from that of the PTEN–/–-1 cells (p value 9 × 10–6). These results indicate that at the cellular level in the presence of GFs, the division rate of individual PTEN–/– cells was higher than that of MCF-10A cells and that overexpressing TPTE2 in a PTEN–/– background returned the rate of division to that of parental MCF-10A cells.
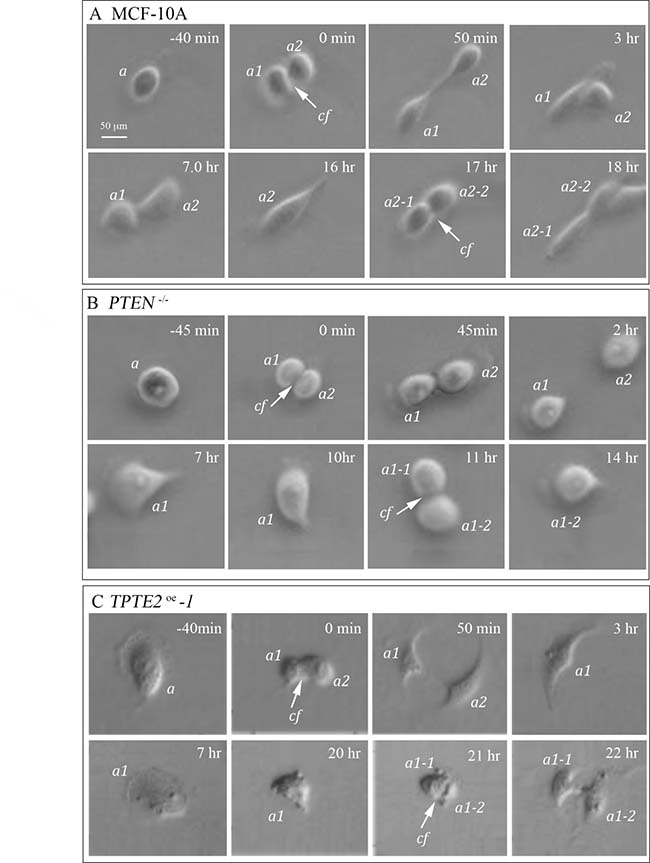
Figure 8: Overexpressing TPTE2 in PTEN–/– cells slows the accelerated rate of cell division to that of parental MCF-10A cells. The time between division furrows was assessed microscopically for 10 cells of MCF-10A, PTEN–/– and TPTE2oe, and found to be 17.3 ± 2.9, 11.6 ± 0.7 and 21.0 ± 4.3 hours, respectively. (A) Representative division of MCF-10A cells. (B) Representative division of a PTEN–/– cell. (C) Representative division of PTEN–/– cells in which TPTE2 is overexpressed (TPTE2oe-1). a, parent cell; a1 and a2, daughter cells from first division; a1-1 and a1-2, and a2-1 and a2-2, daughter cells of second division; cf arrow, cleavage furrow.
Growth, adhesion and viability in the absence of GFs
It was previously demonstrated by measuring MTT reduction in cell preparations in the wells of tissue culture plates, that the viability of MCF-10A cells was dependent upon GFs, but the viability of mutant PTEN–/– cells was independent [10]. We first analyzed whether overexpression of TPTE2 reversed this mutant-associated characteristic, using the MTT reduction assay. In the assay MCF-10A, PTEN–/– and PTEN–/–TPTE2OE cells were plated in the wells of a 96 well tissue culture dish containing DMEM-GFs medium. Cultures were assayed at zero, one, three and five days for MTT reduction. MCF-10A cells lost approximately 90% of MTT reduction activity after the first day (Figure 9A). In PTEN–/– cultures, MTT reduction activity increased continuously over the five days of incubation, almost doubling by five days (Figure 9A). In PTEN–/–TPTE2OE cultures, MTT reductase activity was reduced by approximately 75% in one day, and by 100% in five days (Figure 9A), suggesting a loss in metabolic activity at a rate higher than in MCF-10A cultures (Figure 9A). Low magnification images of cells at the substratum of undisturbed preparations suggested that cells of all three cell lines adhered to the dish bottom for five days in DMEM-GFs, but that the density was higher in PTEN–/– cultures (Figure 9B). In a longer analysis of MCF-10A cell cultures in DMEM-GFs medium, imaged at high magnification on the substratum, we found that spread cells were still present on the substratum, but three days after changing the medium with fresh DMEM-GF medium, cell density on the substratum decreased (Figure 9C). Low magnification images of the medium revealed that cells had released into the medium and had rounded-up (Figure 9D). Similar results were obtained for PTEN–/–TPTE2OE cells, but few released in PTEN–/– cultures. These results together demonstrate that MCF-10A cells lose viability and release from the substratum when deprived of growth factors, but PTEN–/– derivatives are growth factors-independent, retain viability, continue to grow and remain attached to the substratum. Overexpression of TPTE2 in PTEN–/– cells reverse GF-independence, returning the phenotype to that of MCF-10A cells and actually speeding up the loss of metabolic activity.
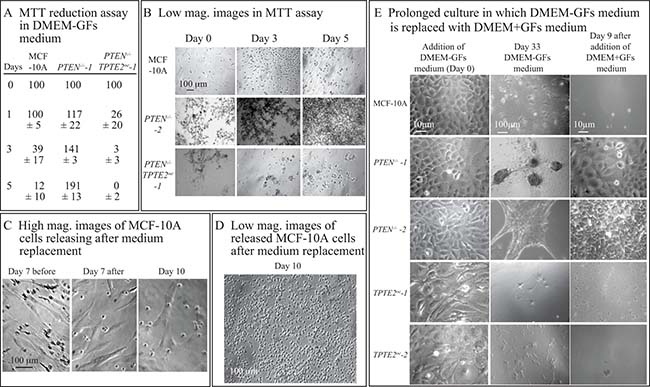
Figure 9: Overexpressing TPTE2 in PTEN–/– cells reverses PTEN–/–-associated increases in viability, adhesion and growth on a 2D substrate in the absence of GFs. (A) MTT reduction activity of undisturbed cell preparations over five days in DMEM-GFs medium. Measurements were made on total cells in the preparation. (B) Low magnification images of MTT preparations in panel A. (C) High magnification images of MCF-10A preparations on a 2D substrate, in which DMEM-GFs medium is replaced with fresh DMEM-GFs medium at seven days and incubated three additional days. Before and after medium change is noted. (D) Low magnification image of the supernatant of an MCF-10A cell preparation on a 2D substrate in which DMEM-GFs was replaced with fresh DMEM-GFs medium at seven days and incubated three additional days. (E) Images of cell preparations grown to confluency on a 2D substrate in DMEM+GFs medium (0 days), then cultured for 33 days in DMEM-GFs medium. During the 33 days, the DMEM-GFs medium was replaced every seven days with fresh DMEM-GFs. At 33 days the medium was replaced with DMEM+GFs and incubated nine additional days.
In a final experiment to demonstrate that long term viability acquired by PTEN–/– cells is reversed by overexpressing TPTE2, cells were grown to confluency in DMEM+GFs medium, and the medium then substituted with DMEM-GFs medium and cultured for 33 additional days. The DMEM-GFs medium was replaced every seven days. At the end of this period, there were very few cells on the substratum of MCF-10A cultures, whereas PTEN–/– cultures contained cell that had formed aggregates (Figure 9E). Reintroduction of GFs after 33 days resulted in essentially no growth in the MCF-10A cultures, which were visually devoid of cells, but caused the cells in the PTEN–/– aggregates to spread and grow as polylayers (Figure 9E). Overexpression of TPTE2 and PTEN–/– cells reestablished the scenario observed for MCF-10A cells (Figure 9E).
Viability in a 3D Matrigel matrix
Transformed cells have been demonstrated to acquire anchorage-independent growth in soft agar [65]. Previous experiments in soft agar suggested that deletion of PTEN in MCF-10A cells did not confer this characteristic [10]. Here, however, using a 3D model [66, 67] of transparent Matrigel, which is composed primarily of laminin, collagen and heparin sulfate proteoglycans [68], in DMEM-GF medium, we found that PTEN–/– cells acquired anchorage independent growth and viability. When seeded in 3D Matrigel and incubated for 50 days, the great majority of MCF-10A cells died off (Figure 10A). The dramatic decrease in cells was verified by the negligible levels of staining with C12-resazurin (Figure 10A). C12-resazurin identifies metabolically active cells [69]. In marked contrast the two PTEN–/– strains formed large aggregates after 50 days, which stained with C12 resazurin (Figure 10B, 10C). Overexpression of TPTE2 reinstated the characteristics of cell death (Figure 10D, 10E). Similar results were obtained for 10 regions of duplicate preparations for each tested strain.
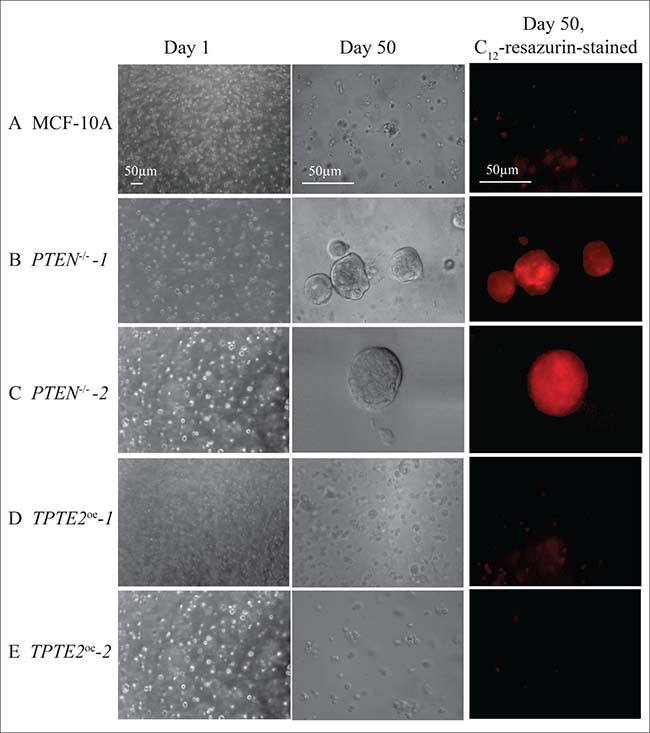
Figure 10: Overexpression of TPTE2 in the PTEN–/– mutant reverses the mutant-acquired capacity of cells to form aggregates and survive for extended periods in the absence of anchorage in a 3D Matrigel model, in DMEM-GFs medium. Cells were seeded in transparent 3D Matrigel in DMEM-GFs medium and incubated for 50 days, then analyzed by light microscopy and by staining with C12-resazurin, which identifies metabolically active cells.
Apoptosis marker
Previous studies [10] and those described here demonstrated that deletion of PTEN results in the loss of the dependency of growth and viability on GFs and anchorage to a substratum, characteristics associated with apoptosis [38, 39]. Overexpressing TPTE2 in PTEN–/– cells reestablished the parental charcteristics. We therefore tested whether MCF-10A cells and PTEN–/– cells overexpressing TPTE2 expressed apoptosis-associated annexin V binding sites and whether these sites were absent in PTEN–/– cells. This was indeed the case. The majority of MCF-10A cells grown to subconfluence in DMEM+GFs stained with fluorescently conjugated annexin V (Figure 11A). PTEN–/– cells did not bind annexin (Figure 11B). When TPTE2 was overexpressed in PTEN–/–, the majority again stained with annexin V (Figure 11C). Over 100 cells were analyzed for each cell line.
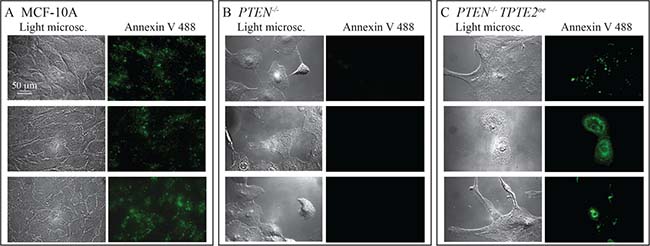
Figure 11: Overexpression of TPTE2 in the PTEN–/– mutant reverses the mutant-acquired loss of annexin V binding, the latter a characteristic associated with apoptosis. Three pairs of representative images, light microscopic and fluorescent, are provided for annexin V-stained preparations. (A) MCF-10A cells. (B) PTEN–/– cells. PTEN–/– TPTE2oe-1 cells.
DISCUSSION
PTEN is a phosphoinositide 3 (PIP3) phosphatase that regulates the level of PIP3 by counteracting the phosphorylation of PIP2 to PIP3 by the kinase PI3K [70]. PIP3 is a cytoplasmic signal, which functions primarily by activating the AKT pathway, which regulates membrane trafficking, membrane-cytoskeletal interactions and a number of other basic cellular processes [71–73], some of which are involved in tumorigenesis [74–76]. By maintaining a low level of PIP3, PTEN suppresses motility [6, 8, 30, 36, 77, 78], cell multiplication [4, 10, 12, 15], adhesion [10], aggregation [30, 36], and resistance to apoptosis [4, 10, 18]. Mutations in PTEN are found in a variety of cancers, and is considered a secondary mutation involved in metastasis and tumorigenesis [3, 5, 7, 79–81]. PTEN localizes to the plasma membrane, endoplasmic reticulum, and mitochondrial membrane [82–85]. It attaches to the plasma membrane through interactions with the CBR3 loop associated with the PTEN-C2 binding domain [85–87]. Adhesion appears to be mediated by electrostatic interactions [88]. Reversing the effects of a PTEN mutation, therefore, could suppress tumorigenesis, either inhibiting or retarding it.
One direct approach to reversing the loss of PTEN function would be to replace the mutated gene with a normal copy. In the application of this general approach to ovarian cancer involving a mutated P53 gene, the normal P53 gene, regulated by a strong constitutive promoter in the adenovirus vector SCH58500, was used to infect mutated cells in order to reestablish P53 function. Using this general technique in clinical trials, there were indications of increased patient survival [89–91], but the general approach has not matured to a level that leads to reliable inhibition or elimination of tumors [92–95]. Recently, we considered an alternative approach. To reestablish normal function in a PTEN mutant, we hypothesized that upregulating a functional homolog of PTEN, presumably by identifying a soluble signal or monoclonal antibody that accomplishes this, might reestablish PTEN function [30]. If this approach has any validity, it would first have to be demonstrated that a homolog of the mutated gene, when up-regulated or overexpressed, reversed the alterations in phenotype caused by the loss of PTEN function in a mutated cell. Our initial attempt to explore this hypothesis was performed in the model system D. discoideum [29]. D. discoideum contains ptenA, an ortholog of human PTEN, and lpten, a homolog of ptenA [31, 37]. We found that overexpressing lpten rescued all of the behavioral defects associated with deletion of ptenA, in the mutant ptenA- [30]. The defects reversed by overexpressing lpten in the ptenA- mutant included a decrease in persistence of motility and increased turning, an increase in lateral pseudopod formation, decreased chemotactic efficiency, and an inability to undergo normal multicellular morphogenesis [30].
Given these results in D. discoideum, we have here applied the strategy to test whether overexpressing a homolog of PTEN in a human PTEN–/– human cell line, generated by targeted mutagenesis [10], would rescue the behavioral changes caused by the mutation. In past studies, it was demonstrated that the mutant PTEN–/– cell lines differed from the parent cell line by exhibiting growth factor-independent growth, increased adherence to tissue culture plastic, and increased resistance to apoptosis in the absence of growth factors [10]. It was also demonstrated that PTEN–/– cells abnormally remodeled the cortical actin cytoskeleton to form microtentacles that adhered to a coated tissue culture dish substrate [9], a change akin to the increase in the frequency of lateral pseudopod formation by D. discoideum ptenA- cells. To test our hypothesis in the human PTEN–/– cell line PTEN–/–-1, a homolog of PTEN, TPTE2 was overexpressed in PTEN–/–-1, generating two individual TPTE2 overexpressor cell lines, TPTE2oe-1 and TPTE2oe-2. We hypothesized that because TPTE2 harbored the conserved catalytic domain, CDC14, of PTEN, it had the potential to function in a similar fashion to mediate the dephosphorylation of PIP3 to PIP2, and because it contained the conserved membrane binding domain C2, it had the potential to bind to PTEN binding sites. We further hypothesized that overexpression of TPTE2 would overwhelm the TPTE2 binding sites, and excess TPTE2 would then bind to unoccupied PTEN binding sites in the PTEN–/– mutant, even if the avidity was lower. To this end, we first identified phenotypic characteristics of the PTEN–/– mutant that differed markedly from the parental strain MCF-10A. The PTEN–/– characteristics we selected included 1) accelerated wound healing in the presence of GFs; 2) independence of wound healing on GFs; 3) a decrease in the rate of cytokinesis in the presence of GFs; 4) adhesion and viability in the absence of GFs; 5) anchorage-independent growth in the absence of GFs; and 7) loss of annexin V binding sites, a characteristic associated with apoptosis (Table 1). We demonstrate that overexpression of TPTE2 in PTEN–/– cells, in the cell lines TPTE2oe-1 and TPTE2oe-2, reversed all of the changes associated with the PTEN–/– mutation and for at least one case actually accentuated the normal characteristic expressed in parental MCF-10A cells (Table 1).
Table 1: The mutant-acquired PTEN–/– characteristics reversed by overexpression TPTE2
Characteristic |
Relative levelb |
||
|---|---|---|---|
MCF-10A |
PTEN–/– |
PTEN–/– TPTE2oe |
|
Wound healing, +GFsa |
+++ |
++++ |
++ |
Wound healing, +S, –other GFsa |
++ |
++++ |
+ |
Wound healing, –GFsa Division rate, +GFsa |
– ++ |
++++ ++++ |
– ++ |
Viability, on 2D substrate, –GFsa, undisturbed |
– |
++++ |
– |
Adhesion on 2D substrate and long term viability, –GFsa |
– |
++++ |
– |
Viability in 3D matrix, –GFsa |
– |
++++ |
– |
Expression of annexin V surface binding site, +GFsa |
+++ |
– |
++++ |
a +GFs: plus growth factors. +S, –other GFs: plus serum, minus growth factors.
b “Relative level” represents a comparison to the maximum measured amongst the three genotypes. Maximum is ++++, descending (+++, ++, +) to zero (–).
Mechanistic model
Based upon the conserved domains in the PTEN homologs TPTE2, a mechanistic model of how TPTE2 overexpression compensates for the loss of PTEN function, is developed in Figure 12. In parental cells, PTEN (black arrows) occupies membrane binding sites (blue cups) through interactions mediated by the PTEN-C2 domain (Figure 12A). At these sites, PTEN catalyzes the dephosphorylation of PIP3 to PIP2, which keeps in check the concentration of PIP3, thus suppressing PIP3-activated pathways, most notably the AKT pathway [4, 10, 96, 97], resulting in the “normal” (control) phenotype (Figure 12A). TPTE2 (red arrows), which we have shown here is expressed in parental MCF-10A cells, binds to TPTE2 membrane receptors (yellow cups) in the endoplasmic reticulum [27] (Figure 12A). The selective binding of TPTE2 to these receptors is presumably facilitated in part by the four transmembrane domains (TMs) not present in PTEN (Figure 1), and, possibly, by the PTEN-C2 domain as well. In the PTEN–/– mutant, PIP3 levels increase due to the absence of PTEN dephosphorylation, a result of the absence of PTEN binding to PTEN sites (Figure 12B). Increased PIP3 lends to activation of the AKT pathway that results in the mutant phenotype (Figure 12B). We have shown that TPTE2 is expressed in PTEN–/– cells, but at levels approximately half that of parental cells. We presume that in PTEN–/– cells, TPTE2 would still target its normal binding sites and, in so doing, not replace PTEN at PTEN binding sites (Figure 12B). We assume that the avidity of TPTE2 to its own binding sites would be higher than its avidity to PTEN binding sites. However, when TPTE2 is overexpressed in a PTEN–/– background, TPTE2 saturates TPTE2 binding sites, and excess TPTE2 then binds to the unoccupied PTEN binding sites (Figure 12C). It is assumed that at the PTEN binding sites, TPTE2 substitutes functionally for PTEN, resulting in a reduction in the level of PIP3, thus reinstating the control cell phenotype (Figure 12C). There are of course alternative explanations for the results obtained. For instance, the 50% reduction in TPTE2 expression caused by deletion of PTEN may be basic to the PTEN–/– phenotype (i.e., the decrease in TPTE2 function results in the PTEN–/– phenotype), and overexpression of TPTE2 rescues the phenotype simply by restoring normal TPTE2 function, not by substituting for PTEN. This alternative explanation seems less plausible since it suggests that a 50% reduction in TPTE2 expression results in the PTEN–/– phenotype. Unfortunately, there have been no reports of a TPTE2–/– null mutant generated by targeted mutation. Our attempts to delete TPTE2 by targeted mutation have so far failed, possibly either as a result of the high level of redundancy, which includes two TPTE genes, TPTE1 and TPTE2, one TPTE1 pseudogene, and seven TPTE2 pseudogenes [23–28] (www,ncbi.nim.nih.gov/gene; /www.uniprot.orgQ6xP23.www.ensemble.org), or the fact that the TPTE2 genes map to C-heterochromatin regions [98], which are less accessible to integration and, therefore, less amenable to targeted mutagenesis [99, 100].
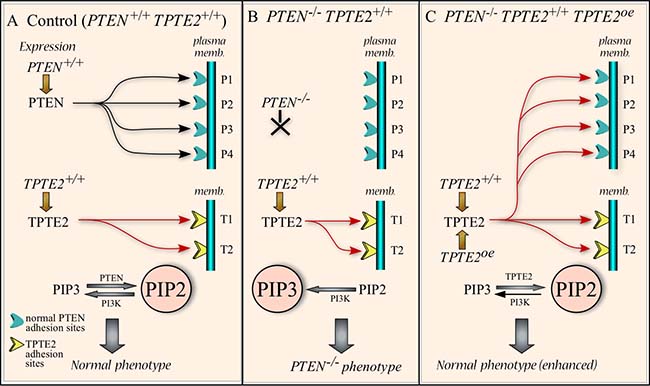
Figure 12: Working model for the rescue of the PTEN–/– mutant phenotype by overexpressing TPTE2. (A) Model of PTEN and TPTE2 binding in control cells (PTEN+/+TPTE2+/+). PIP2 dominates because of PTEN dephosphorylation of PIP3. In this model of control cells, all PTEN binding sites are filled by PTEN and all TPTE2 sites by TPTE2. (B) Model of binding in PTEN–/– cells (PTEN–/–TPTE2+/+). PTEN binding sites are not occupied and PIP3 dominates because of the absence of PTEN. PTEN receptors are unoccupied whereas TPTE2 receptors are occupied by TPTE2. (C) Model of binding in the TPTE2 overexpressor (PTEN–/–TPTE2+/+TPTE2oe). Overexpressed TPTE2 leads to TPTE2 binding to both PTEN binding sites as well as TPTE2 binding sites. PIP2 dominates, resulting in a phenotype equivalent to that of control cells. P1, P2, P3, P4: PTEN binding sites. T1, T2: TPTE2 binding sites. PI3K, PI3 kinase.
MATERIALS AND METHODS
Growth and maintenance of cell lines
Two independent PTEN–/– mutants were previously generated by targeted mutagenesis, clone 1 (PTEN–/–-1) and clone 2 (PTEN–/–-2). Both contained targeted disruption of exon 2 of both PTEN alleles [10]. The parental strain of the mutants, MCF-10A, a non-tumorigenic human cell line derived from normal breast epithelium [101], the two derivative PTEN–/– strains and the two TPTE2-overexpressing lines generated here from PTEN–/–-1 (TPTE2OE -1, and TPTE2OE -2), were cultured in DMEM/F12 (DMEM) medium (Life Technologies, Carlsbad, CA, USA) supplemented with horse serum (S), human recombinant EGF (EGF), insulin (I), hydrocortisone (HC) and cholera toxin (CT), all obtained from Sigma Aldrich (St. Louis, MO, USA), and a penicillin- streptomycin mixture, obtained from Thermo-Fischer (Grand Island, NY) [10]. This fully supplemented medium will be referred to as “DMEM+ GFs medium”, “GFs” referring to the full complement of growth factors. Medium containing serum, but lacking the other GFs (otherGFs: E, I, HC, CT) will be referred to as “DMEM+S,-otherGFs medium”, and medium lacking all growth factors will be referred to as “DMEM-GFs medium”. In the case of “DMEM+S,-otherGFs medium”, growth factors were charcoal-stripped (Valley Biomedical, Winchester, VA) from serum prior to its addition to DMEM medium.
Generating PTEN–/–TPTE2OE strains
RNA of the parental strain MCF-10A was isolated as previously described in detail [30, 102], using Trizol (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol. A 3ʹ RACE system with reverse transcriptase (Life Technologies, Carlsbad, CA, USA) was used to generate cDNA using 4 μg of total RNA and the primer AUP. The three identified TPTE2 variant cDNAs (TPTE2 variant 1 accession number KX610659, TPTE2 variant 2 accession number KX752070 and TPTE2 variant 3 accession number NM_199254) were amplified from the cDNA template using primers specific for the 5ʹ and 3ʹ ends of the TPTE2 transcript. Primers were designed using information provided by the EMBL (http://www.ensembl.org) for the TPTE2-1 variant. cDNA was amplified using the primers TPTE2-1 FW (5ʹ -TCCACCCACCCATGAATTATCAGGGAGTGAACCCAGAGGCACGTATGA ATGAAAGTCC-3ʹ) and TPTE2-1 RT (5ʹ -ATTTTCTCGCCAAAAAGTATCTCCACAGCAAATTCTGG-3ʹ). PCR was performed using a Long-Range Polymerase (Roche, Indianapolis, IA, USA) /Taq-Polymerase (NEB) mixture, and a regime that included 5 min denaturation at 95° C and 40 cycles, each consisting of 20 seconds at 92° C, 30 seconds at 47° C and 4 min at 70° C. This was followed by a final elongation step of 10 min at 72° C. Fragments were gel extracted (Qiagen, Ventura, CA, USA) and cloned into the plasmid pCR4.0 (LifeScience, Carlsbad, CA) for sequencing. Sequencing was performed at the Roy J. Carver Center of Genomics in the Department of Biology of the University of Iowa (http://biology.uiowa.edu/ccg). The TPTE2 variant TPTE2-1 was used for insertion into the transforming vector. The 5ʹ-3ʹprimers were designed to begin at nucleotide 1383 in order to obtain inframe fusion with GFP in the plasmid pcDNA3.1/CT-GFP TOPO (Lifescience, Carlsbad, CA). The stop codon was omitted to ensure that the TPTE2 and GFP sequences were inframe and produced as a fusion protein. PTEN–/– cells were grown in OptiMEM medium (Life Science, Carlsbad, CA) in 24 well tissue culture plates overnight. Fugene (Promega, Madison, WI) was employed to transfect cells with 1 μg/ml of plasmid containing the TPTE2-1-GFP fusion, according to the manufacturer’s instructions. PTEN–/– cells were subjected to a G418 (Sigma-Aldrich, St. Louis, MO, USA) kill curve. The concentration of G418 used for selection was 150 μg/ml. Two TPTE2OE clones were selected for further analysis using G418, TPTE2OE -1 and TPTE2OE -2. Note that both produced TPTE2-GFP transcripts.
RT-PCR
RNA was prepared as described in the previous section, using 2-step LongRange RT-PCR (Qiagen, Ventura, CA, USA). For reverse transcription, an Oligo-dT primer provided by the manufacturer was employed. Amplification was performed using the Roche Expand Long Template Kit (Roche, Indianapolis, IN). Primers used to amplify a 1.135bp TPTE2-GFP fusion fragment were ATG120TPTE2 5ʹ-ATGGACACATTTAGTTCGACT TCTACG-3ʹstarting at position 415 of the TPTE2-1 cDNA and GFP Reverse(CT-GFP) 5ʹ-GGTAAGCTTTCCGTAT GTAGC-3ʹending at position 131 of the GFP cDNA. To analyze expression levels by RT-PCR, RNA was isolated as described above. Reverse Transcription was performed using Quantitec RT-PCR (Qiagen, Ventura, CA, USA). One μg of total RNA was denatured at 65° C for 5 min, incubated in gDNA wipeout buffer for 2 min at 42° C and immediately placed on ice. Reverse transcription was performed for 30 min at 42° C with an Oligo-dT primer. The reaction was terminated by incubation at 95° C for 3 min. Fifty ng of cDNA was used for amplification of a 200 bp TPTE2 fragment. The primers were RTFw 5ʹ-TGGTTTGTGCCCTCCTTATTGCC-3ʹand RTRv 5ʹ-
TCACATCATCATACAGAGGTGGACCG -3ʹ. GAPDH was used as a loading control, using the primers GAPDH Fw 5ʹ-CATCTTCTTTTGCGTCGCC-3ʹand GAPDHrv 5ʹ-CTACACTGAGCACCAGGTGGTCTC-3ʹ, to amplify an 891 base pair fragment. PCR was performed as described above, with minor changes. Primer elongation was performed for 1 min through 30 cycles for a GAPDH control fragment and through 40 cycles for the TPTE2 fragment. Gel-Electrophoresis was performed using a 3% agarose gel in TBE buffer. Images were taken with UV light under subsaturating conditions. Gel band image analysis was performed using Adobe Photoshop. To measure TPTE2 expression, integrated pixel density of the band area was determined. The integrated density is the sum of the value of the pixels in a band, divided by the area of the band. TPTE2 expression was normalized to GAPDH expression determined in the same manner.
Immunostaining TPTE2-GFP
To immunostain the fusion protein in overexpression strains, cells were washed 3 times in TBS pH7.6/0.05% Tween 20 to remove growth media. Cells were fixed in 3.8% formaldehyde (Sigma Aldrich, St.Louis, MO) for 12 min. Fixed cells were washed 3 times in tris-buffered saline (TBS) pH7.6 containing 1% BSA (ACROS, Geel, Belgium), 0.05% Tween 20, to remove growth media. Fixed cells were washed 3 times in the TBS-Tween 20 buffer. Preparations were incubated for 2 hours in TBS pH7.6/1% BSA, 0.05% Tween 20 and 0.1% Triton X-100. Cells were then washed 3 times in the TBS-Tween 20 buffer. One ml of a 1:1 mixture of the anti-GFP monoclonal antibody (mAb) DSHB-GFP-12A6 and the anti-GFP mAb DSHB-GFP-4C9 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA 52245, USA [103], containing 10 μg of mAbs in TBS (pH7.6) plus 1% BSA, was added and cells incubated overnight. Excess primary antibody was removed by washing the cells 3 times with TBS (pH7.6) plus 0.05% Tween 20. The affinity-purified fluorescent anti-IgG H+L Alexa 488 antibody (Jackson ImmunoResearch, Westgrove, PA, USA) was used as a secondary antibody. It was diluted 1:300 in TBS (pH 7.6) plus 0.05% Tween 20, added and the preparation incubated for 2 hours. Cells were then washed 3 times in TBS (pH7.6) plus 0.05% Tween 20, and analyzed using a Nikon TE2000 inverted epifluorescent microsope (Nikon Instruments, Melville, NY, USA). Images were taken with a with an EOS Rebel T3i/EOS 600D camera (Canon, Lake Success, NY, USA).
Wound healing assay
A wound healing assay was employed that consisted of a two well culture dish in which the wells were separated by an insert, which, when removed, formed a gap (“wound”) (IBIDI, Madison, WI, USA). The assay monitored collective directional migration into the gap between the opposing confluent layers of cells [60]. Experiments were performed in an incubator at 37° C in 5% CO2. The assay was performed according to the manufacturer’s directions. In brief, 80 μl aliquots, containing 4 × 104 cells were inoculated into each of the two wells and grown for 24 hours to confluency in DMEM+GFs medium. The insert was then removed, leaving a 200 μm gap between the opposing confluent layers of cells. Two ml of fresh medium (DMEM+GFs, DMEM+S,-otherGFs or DMEM-GFs) were then added. Migration into the wound was monitored microscopically through an Olympus CK2 inverted microscope equipped with a digital XCD-V50 camera (Sony, San Diego, CA, USA). Images were obtained at noted time intervals.
Assessment of the rate of cell division
Cells at a concentration of 104 per ml were inoculated into DMEM+GFs medium in a tissue culture dish and incubated in 5% CO2 at 37° C. Individual cells, which distributed independently on the dish bottom, were monitored through an Olympus CK2 inverted microscope equipped with a XCD-V50 digital camera (Sony, San Diego, CA, USA). Images were obtained every 45 seconds. The time of cell division was taken as the period between the formation of cleavage furrows. Ten dividing cells of each line were analyzed. Images were obtained as previously described.
MTT reduction assay
The MTT reduction assay was performed according to procedures previously described [104, 105]. In brief, 3 × 103 cells in 150 μl of DMEM-GFs medium from cultures grown to near confluency in DMEM+GFs, were plated into wells of a 96 well tissue culture plate (Midwest Scientific, Valley Park, MO, USA). After 0, 1, 3 and 5 days 20 μl of 5 mg/ml MTT, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltrtrazolium bromide (Life Technologies, Carlsbad, CA, USA), were added. The plates were then incubated subsequently for 3.5 hour at 37° C in the absence of light. Microscopic images, were then taken using an Olympus CK2 inverted microscope equipped with a digital XCD-V50 camera (Sony, San Diego, CA, USA), and then150μl of a MTT solvent containing 8mM HCl, 0.2% Nonidet P-40 (NP40, Amresco, Solon, OH) in isopropanol was immediately added to each culture. The 96 well plates were then shaken for 15 min at 100 rpm protected from light at room temperature. The absorbance was determined using a Spectra max Plus 384 multiwell plate reader (Molecular Devices, Sunnyvale, CA, USA) at 590 nm. All cultures were compared to day 0. Experiments were performed in triplicate.
Viability in the absence of growth factors on a 2D substrate
Cells were first grown in DMEM+GFs medium to confluency in the wells of a 24 well tissue culture dish. In a first approach, the DMEM+GFs medium was gently replaced with DMEM-GFs medium (zero hours), cultured undisturbed for seven days, the DMEM-GFs medium gently replaced at seven days with fresh DMEM-GFs medium and cultured three additional days. Microscopic images were taken at high magnification at seven days before and after fresh medium replacement, and at 10 days. Microscopic images were also taken of supernatant at low magnification at 10 days. In a second, long term approach, DMEM+GFs medium was replaced with DMEM-GFs, and the DMEM-GFs medium gently replaced at 9, 18 and 24 days. At 33 days, the DMEM-GFs medium was replaced with DMEM+GFs medium and incubated for nine additional days. The cell preparations were monitored microscopically with time and images obtained as described for the wound healing assay.
Cell cultures a 3D Matrigel model
Cells grown in DMEM+GFs medium were randomly dispersal in a 3D Matrigel model in DMEM-GFs, as described previously in detail [66, 67, 106–108]. In brief, a 30 mm glass insert at the bottom of a customized culture dish was coated with Matrigel previously hydrated in DMEM-GFs for 20 min at 5° C. This preparation was then incubated at 37° C for one hour to cause gelation. Then 5 × 106 cells, grown in DMEM+GFs medium, were resuspended in 250 μl of DMEM-GFs medium, added to 500 μl of (5%) hydrated GF-reduced Matrigel (Corning, Life Science, Corning, NJ) at 5° C and the cell-Matrigel mixture distributed atop the basal cushion of Matrigel and incubated at 37° C in 5% CO2 for 30 min, which caused Matrigel gelation. After gelation, cells were dispersed randomly throughout the 3D gel. At 50 days C12-Resazurin (Life Technologies, CA) was added directly to the dishes at a final concentration of 500 nM and incubated for 1 hour. Metabolically active cells reduce nonfluorescent C12-Resazurin to fluorescent C12-resorufin. Preparations were imaged with a Nikon TE2000 inverted fluorescence microscope (Nikon Instruments, Melville, NY, USA) equipped with an EOS Rebel T3i/EOS 600D camera (Canon, Lake Success, NY, USA).
Annexin V binding
Cells were seeded on a 30mm glass botton dish (Cellvis, Mountain View, CA, USA) and grown for 2 days in DMEM+GFs. Cell death was assessed using Annexin V CF 488A conjugate (Biotium, Fremont, CA, USA) on unfixed cells according to the manufacturer’s instructions. In brief, media was first removed and supernatant examined and substrate microscopically imaged. Cells on the substratum were then washed two times in 1X binding buffer (Biotium, Fremont, CA, USA). Cells were incubated in staining solution containing binding buffer and 1.25 μg/ml Annexin V conjugate for 30 min at room temperature. Stained cells were washed two times with binding buffer. Cells were imaged using a Nikon TE2000 inverted epifluorescent microsope (Nikon Instruments, Melville, NY, USA). Images were taken with an EOS Rebel T3i/EOS 600D camera (Canon, Lake Success, NY, USA) within 1 hour, as recommended by the manufacturer
CONCLUSIONS
We have explored the hypothesis that loss of function resulting from a mutation or an epigenetic effect, of a gene that suppresses metastasis and/or tumorigenesis, may be reversed by up-regulating or overexpressing a functional homolog of that gene in the mutated cell. We previously demonstrated that this could be accomplished in a model system for a mutant of a PTEN ortholog [30], and here we have demonstrated it in a human epithelial cell line for a targeted PTEN mutant. These results support the possibility that this strategy may have the potential to be translated into a therapy for tumorigenesis. The next step in this strategy is to screen for a compound or antibody that, through interaction with a receptor, up-regulates TPTE2 in PTEN–/– cells. This general approach may also be applicable to other cancer-associated genes with known homologs.
ACKNOWLEDGMENTS
Research in the Soll laboratory was supported by the Developmental Studies Hybridoma Bank, a self-funded National Resource created by the NIH. Research in the Vitolo lab was supported by NIH grant K01-CA166567.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Chetram MA, Odero-Marah V, Hinton CV. Loss of PTEN permits CXCR4-mediated tumorigenesis through ERK1/2 in prostate cancer cells. Mol Cancer Res. 2011; 9:90–102. https://doi.org/10.1158/1541-7786.MCR-10-0235.
2. Christensen M, Najy AJ, Snyder M, Movilla LS, Kim HR. A critical role of the PTEN/PDGF signaling network for the regulation of radiosensitivity in adenocarcinoma of the prostate. Int J Radiat Oncol Biol Phys. 2014; 88:151–8. https://doi.org/10.1016/j.ijrobp.2013.10.019.
3. Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997; 57:2124–9.
4. Li DM, Sun H. PTEN/MMAC1/TEP1 suppresses the tumorigenicity and induces G1 cell cycle arrest in human glioblastoma cells. Proc Natl Acad Sci U S A. 1998; 95:15406–11.
5. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997; 275:1943–7.
6. Schneider E, Keppler R, Prawitt D, Steinwender C, Roos FC, Thuroff JW, Lausch E, Brenner W. Migration of renal tumor cells depends on dephosphorylation of Shc by PTEN. Int J Oncol. 2011; 38:823–31. https://doi.org/10.3892/ijo.2010.893.
7. Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997; 15:356–62. https://doi.org/10.1038/ng0497-356.
8. Takei Y, Saga Y, Mizukami H, Takayama T, Ohwada M, Ozawa K, Suzuki M. Overexpression of PTEN in ovarian cancer cells suppresses i.p. dissemination and extends survival in mice. Mol Cancer Ther. 2008; 7:704–11. https://doi.org/10.1158/1535-7163.MCT-06-0724.
9. Vitolo MI, Boggs AE, Whipple RA, Yoon JR, Thompson K, Matrone MA, Cho EH, Balzer EM, Martin SS. Loss of PTEN induces microtentacles through PI3K-independent activation of cofilin. Oncogene. 2013; 32:2200–10. https://doi.org/10.1038/onc.2012.234.
10. Vitolo MI, Weiss MB, Szmacinski M, Tahir K, Waldman T, Park BH, Martin SS, Weber DJ, Bachman KE. Deletion of PTEN promotes tumorigenic signaling, resistance to anoikis, and altered response to chemotherapeutic agents in human mammary epithelial cells. Cancer Res. 2009; 69:8275–83. https://doi.org/10.1158/0008-5472.CAN-09-1067.
11. Worby CA, Dixon JE. Pten. Annu Rev Biochem. 2014; 83:641–69. https://doi.org/10.1146/annurev-biochem-082411-113907.
12. Zhang Y, Zhang J, Xu K, Xiao Z, Sun J, Xu J, Wang J, Tang Q. PTEN/PI3K/mTOR/B7-H1 signaling pathway regulates cell progression and immuno-resistance in pancreatic cancer. Hepatogastroenterology. 2013; 60:1766–72.
13. Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, Salmena L, Sampieri K, Haveman WJ, Brogi E, Richardson AL, Zhang J, Pandolfi PP. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010; 42:454–8. https://doi.org/10.1038/ng.556.
14. Cheney IW, Johnson DE, Vaillancourt MT, Avanzini J, Morimoto A, Demers GW, Wills KN, Shabram PW, Bolen JB, Tavtigian SV, Bookstein R. Suppression of tumorigenicity of glioblastoma cells by adenovirus-mediated MMAC1/PTEN gene transfer. Cancer Res. 1998; 58:2331–4.
15. Cheney IW, Neuteboom ST, Vaillancourt MT, Ramachandra M, Bookstein R. Adenovirus-mediated gene transfer of MMAC1/PTEN to glioblastoma cells inhibits S phase entry by the recruitment of p27Kip1 into cyclin E/CDK2 complexes. Cancer Res. 1999; 59:2318–23.
16. Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001; 27:222–4. https://doi.org/10.1038/84879.
17. Freeman D, Lesche R, Kertesz N, Wang S, Li G, Gao J, Groszer M, Martinez-Diaz H, Rozengurt N, Thomas G, Liu X, Wu H. Genetic background controls tumor development in PTEN-deficient mice. Cancer Res. 2006; 66:6492–6. https://doi.org/10.1158/0008-5472.CAN-05-4143.
18. Lu Y, Lin YZ, LaPushin R, Cuevas B, Fang X, Yu SX, Davies MA, Khan H, Furui T, Mao M, Zinner R, Hung MC, Steck P, et al. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene. 1999; 18:7034–45. https://doi.org/10.1038/sj.onc.1203183.
19. Tian XX, Pang JC, To SS, Ng HK. Restoration of wild-type PTEN expression leads to apoptosis, induces differentiation, and reduces telomerase activity in human glioma cells. J Neuropathol Exp Neurol. 1999; 58:472–9.
20. Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van Dyke T, Cordon-Cardo C, Pandolfi PP. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003; 1:E59. https://doi.org/10.1371/journal.pbio.0000059.
21. Lambricht L, Lopes A, Kos S, Sersa G, Preat V, Vandermeulen G. Clinical potential of electroporation for gene therapy and DNA vaccine delivery. Expert Opin Drug Deliv. 2016; 13:295–310. https://doi.org/10.1517/17425247.2016.1121990.
22. Teo PY, Cheng W, Hedrick JL, Yang YY. Co-delivery of drugs and plasmid DNA for cancer therapy. Adv Drug Deliv Rev. 2016; 98:41–63. https://doi.org/10.1016/j.addr.2015.10.014.
23. Chen H, Rossier C, Morris MA, Scott HS, Gos A, Bairoch A, Antonarakis SE. A testis-specific gene, TPTE, encodes a putative transmembrane tyrosine phosphatase and maps to the pericentromeric region of human chromosomes 21 and 13, and to chromosomes 15, 22, and Y. Hum Genet. 1999; 105:399–409.
24. Mishra RR, Chaudhary JK, Rath PC. Cell cycle arrest and apoptosis by expression of a novel TPIP (TPIP-C2) cDNA encoding a C2-domain in HEK-293 cells. Mol Biol Rep. 2012; 39:7389–402. https://doi.org/10.1007/s11033-012-1571-6.
25. Pulido R, Baker SJ, Barata JT, Carracedo A, Cid VJ, Chin-Sang ID, Dave V, den Hertog J, Devreotes P, Eickholt BJ, Eng C, Furnari FB, Georgescu MM, et al. A unified nomenclature and amino acid numbering for human PTEN. Sci Signal. 2014; 7:pe15. https://doi.org/10.1126/scisignal.2005560.
26. Tapparel C, Reymond A, Girardet C, Guillou L, Lyle R, Lamon C, Hutter P, Antonarakis SE. The TPTE gene family: cellular expression, subcellular localization and alternative splicing. Gene. 2003; 323:189–99.
27. Walker SM, Downes CP, Leslie NR. TPIP: a novel phosphoinositide 3-phosphatase. Biochem J. 2001; 360:277–83.
28. Dunham A, Matthews LH, Burton J, Ashurst JL, Howe KL, Ashcroft KJ, Beare DM, Burford DC, Hunt SE, Griffiths-Jones S, Jones MC, Keenan SJ, Oliver K, et al. The DNA sequence and analysis of human chromosome 13. Nature. 2004; 428:522–8. https://doi.org/10.1038/nature02379.
29. Williams RS, Boeckeler K, Graf R, Muller-Taubenberger A, Li Z, Isberg RR, Wessels D, Soll DR, Alexander H, Alexander S. Towards a molecular understanding of human diseases using Dictyostelium discoideum. Trends Mol Med. 2006; 12:415–24. https://doi.org/10.1016/j.molmed.2006.07.003.
30. Lusche DF, Wessels D, Richardson NA, Russell KB, Hanson BM, Soll BA, Lin BH, Soll DR. PTEN Redundancy: Overexpressing lpten, a Homolog of Dictyostelium discoideum ptenA, the Ortholog of Human PTEN, Rescues All Behavioral Defects of the Mutant ptenA. PLoS One. 2014; 9:e108495. https://doi.org/10.1371/journal.pone.0108495.
31. Funamoto S, Meili R, Lee S, Parry L, Firtel RA. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell. 2002; 109:611–23.
32. Hoeller O, Kay RR. Chemotaxis in the absence of PIP3 gradients. Curr Biol. 2007; 17:813–7. https://doi.org/10.1016/j.cub.2007.04.004.
33. King JS, Teo R, Ryves J, Reddy JV, Peters O, Orabi B, Hoeller O, Williams RS, Harwood AJ. The mood stabiliser lithium suppresses PIP3 signalling in Dictyostelium and human cells. Dis Model Mech. 2009; 2:306–12. https://doi.org/10.1242/dmm.001271.
34. Kortholt A, King JS, Keizer-Gunnink I, Harwood AJ, Van Haastert PJ. Phospholipase C regulation of phosphatidylinositol 3,4,5-trisphosphate-mediated chemotaxis. Mol Biol Cell. 2007; 18:4772–9. https://doi.org/10.1091/mbc.E07-05-0407.
35. Pramanik MK, Iijima M, Iwadate Y, Yumura S. PTEN is a mechanosensing signal transducer for myosin II localization in Dictyostelium cells. Genes Cells. 2009; 14:821–34. https://doi.org/10.1111/j.1365-2443.2009.01312.x.
36. Wessels D, Lusche DF, Kuhl S, Heid P, Soll DR. PTEN plays a role in the suppression of lateral pseudopod formation during Dictyostelium motility and chemotaxis. J Cell Sci. 2007; 120:2517–31. https://doi.org/10.1242/jcs.010876.
37. Iijima M, Devreotes P. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 2002; 109:599–610.
38. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972; 26:239–57.
39. Rello S, Stockert JC, Moreno V, Gamez A, Pacheco M, Juarranz A, Canete M, Villanueva A. Morphological criteria to distinguish cell death induced by apoptotic and necrotic treatments. Apoptosis. 2005; 10:201–8. https://doi.org/10.1007/s10495-005-6075-6.
40. Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992; 148:2207–16.
41. Verhoven B, Schlegel RA, Williamson P. Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. J Exp Med. 1995; 182:1597–601.
42. Futterer K, Machesky LM. "Wunder" F-BAR domains: going from pits to vesicles. Cell. 2007; 129:655–7. https://doi.org/10.1016/j.cell.2007.05.006.
43. Hopkins BD, Fine B, Steinbach N, Dendy M, Rapp Z, Shaw J, Pappas K, Yu JS, Hodakoski C, Mense S, Klein J, Pegno S, Sulis ML, et al. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science. 2013; 341:399–402. https://doi.org/10.1126/science.1234907.
44. Liang H, He S, Yang J, Jia X, Wang P, Chen X, Zhang Z, Zou X, McNutt MA, Shen WH, Yin Y. PTENalpha, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab. 2014; 19:836–48. https://doi.org/10.1016/j.cmet.2014.03.023.
45. Wu Y, Dowbenko D, Pisabarro MT, Dillard-Telm L, Koeppen H, Lasky LA. PTEN 2, a Golgi-associated testis-specific homologue of the PTEN tumor suppressor lipid phosphatase. J Biol Chem. 2001; 276:21745–53. https://doi.org/10.1074/jbc.M101480200.
46. Halaszovich CR, Leitner MG, Mavrantoni A, Le A, Frezza L, Feuer A, Schreiber DN, Villalba-Galea CA, Oliver D. A human phospholipid phosphatase activated by a transmembrane control module. J Lipid Res. 2012; 53:2266–74. https://doi.org/10.1194/jlr.M026021.
47. Duffy EE, Rutenberg-Schoenberg M, Stark CD, Kitchen RR, Gerstein MB, Simon MD. Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry. Mol Cell. 2015; 59:858–66. https://doi.org/10.1016/j.molcel.2015.07.023.
48. Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, Asplund A, Sjostedt E, Lundberg E, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014; 13:397–406. https://doi.org/10.1074/mcp.M113.035600.
49. Ashby WJ, Zijlstra A. Established and novel methods of interrogating two-dimensional cell migration. Integr Biol (Camb). 2012; 4:1338–50. https://doi.org/10.1039/c2ib20154b.
50. Poujade M, Grasland-Mongrain E, Hertzog A, Jouanneau J, Chavrier P, Ladoux B, Buguin A, Silberzan P. Collective migration of an epithelial monolayer in response to a model wound. Proc Natl Acad Sci U S A. 2007; 104:15988–93. https://doi.org/10.1073/pnas.0705062104.
51. Rodriguez LG, Wu X, Guan JL. Wound-healing assay. Methods Mol Biol. 2005; 294:23–9.
52. Trepat X, Fredberg JJ. Plithotaxis and emergent dynamics in collective cellular migration. Trends Cell Biol. 2011; 21:638–46. https://doi.org/10.1016/j.tcb.2011.06.006.
53. Dauer DJ, Ferraro B, Song L, Yu B, Mora L, Buettner R, Enkemann S, Jove R, Haura EB. Stat3 regulates genes common to both wound healing and cancer. Oncogene. 2005; 24:3397–408. https://doi.org/10.1038/sj.onc.1208469.
54. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986; 315:1650–9. https://doi.org/10.1056/NEJM198612253152606.
55. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57–70.
56. Bloomekatz J, Grego-Bessa J, Migeotte I, Anderson KV. Pten regulates collective cell migration during specification of the anterior-posterior axis of the mouse embryo. Dev Biol. 2012; 364:192–201. https://doi.org/10.1016/j.ydbio.2012.02.005.
57. Mayor R, Etienne-Manneville S. The front and rear of collective cell migration. Nat Rev Mol Cell Biol. 2016; 17:97–109. https://doi.org/10.1038/nrm.2015.14.
58. Holmes SJ. The Cultivation of Tissues from the Frog. Science. 1914; 39:107–8. https://doi.org/10.1126/science.39.994.107.
59. Vaughan RB, Trinkaus JP. Movements of epithelial cell sheets in vitro. J Cell Sci. 1966; 1:407–13.
60. Ebeling S, Naumann K, Pollok S, Wardecki T, Vidal-Y-Sy S, Nascimento JM, Boerries M, Schmidt G, Brandner JM, Merfort I. From a traditional medicinal plant to a rational drug: understanding the clinically proven wound healing efficacy of birch bark extract. PLoS One. 2014; 9:e86147. https://doi.org/10.1371/journal.pone.0086147.
61. Gov NS. Collective cell migration patterns: follow the leader. Proc Natl Acad Sci U S A. 2007; 104:15970–1. https://doi.org/10.1073/pnas.0708037104.
62. Omelchenko T, Vasiliev JM, Gelfand IM, Feder HH, Bonder EM. Rho-dependent formation of epithelial "leader" cells during wound healing. Proc Natl Acad Sci U S A. 2003; 100:10788–93. https://doi.org/10.1073/pnas.1834401100.
63. Stoker M, Gherardi E, Perryman M, Gray J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. 1987; 327:239–42. https://doi.org/10.1038/327239a0.
64. Todaro GJ, Lazar GK, Green H. The initiation of cell division in a contact-inhibited mammalian cell line. J Cell Physiol. 1965; 66:325–33.
65. Shin SI, Freedman VH, Risser R, Pollack R. Tumorigenicity of virus-transformed cells in nude mice is correlated specifically with anchorage independent growth in vitro. Proc Natl Acad Sci U S A. 1975; 72:4435–9.
66. Kuhl S, Voss E, Scherer A, Lusche DF, Wessels D, Soll DR. 4D Tumorigenesis model for quantitating coalescence, quantitating directed cell motility and chemotaxis, identifying unique cell behaviors and testing anti-cancer drugs. In: Hereld D, Jin D, editors. Chemotaxis: Methods and Protocols. Springer; 2016.
67. Wessels DJ, Lusche DF, Kuhl S, Scherer A, Voss E, Soll DR. Quantitative Motion Analysis in Two and Three Dimensions. Methods Mol Biol. 2016; 1365:265–92. https://doi.org/10.1007/978-1-4939-3124-8_14.
68. Hughes CS, Postovit LM, Lajoie GA. Matrigel: a complex protein mixture required for optimal growth of cell culture. Proteomics. 2010; 10:1886–90. https://doi.org/10.1002/pmic.200900758.
69. Kumar M, Khan IA, Verma V, Qazi GN. Microplate nitrate reductase assay versus Alamar Blue assay for MIC determination of Mycobacterium tuberculosis. Int J Tuberc Lung Dis. 2005; 9:939–41.
70. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998; 273:13375–8.
71. Bassi C, Mak TW. Regulation of the Phosphatidylinositide 3-Kinase Pathway by the Lipid Phosphatase PTEN. Clin Chem. 2016; 62:884–5. https://doi.org/10.1373/clinchem.2015.253237.
72. Czech MP. PIP2 and PIP3: complex roles at the cell surface. Cell. 2000; 100:603–6.
73. Rodriguez-Escudero I, Roelants FM, Thorner J, Nombela C, Molina M, Cid VJ. Reconstitution of the mammalian PI3K/PTEN/Akt pathway in yeast. Biochem J. 2005; 390:613–23. https://doi.org/10.1042/BJ20050574.
74. Cao J, Zhu S, Zhou W, Li J, Liu C, Xuan H, Yan J, Zheng L, Zhou L, Yu J, Chen G, Huang Y, Yu Z, et al. PLZF mediates the PTEN/AKT/FOXO3a signaling in suppression of prostate tumorigenesis. PLoS One. 2013; 8:e77922. https://doi.org/10.1371/journal.pone.0077922.
75. Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006; 6:184–92. https://doi.org/10.1038/nrc1819.
76. David O, Jett J, LeBeau H, Dy G, Hughes J, Friedman M, Brody AR. Phospho-Akt overexpression in non-small cell lung cancer confers significant stage-independent survival disadvantage. Clin Cancer Res. 2004; 10:6865–71. https://doi.org/10.1158/1078-0432.CCR-04-0174.
77. Liliental J, Moon SY, Lesche R, Mamillapalli R, Li D, Zheng Y, Sun H, Wu H. Genetic deletion of the Pten tumor suppressor gene promotes cell motility by activation of Rac1 and Cdc42 GTPases. Curr Biol. 2000; 10:401–4.
78. Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998; 280:1614–7.
79. Bonneau D, Longy M. Mutations of the human PTEN gene. Hum Mutat. 2000; 16:109–22. https://doi.org/10.1002/1098-1004(200008)16:2<109::AID-HUMU3>3.0.CO;2-0.
80. Ciuffreda L, Falcone I, Incani UC, Del Curatolo A, Conciatori F, Matteoni S, Vari S, Vaccaro V, Cognetti F, Milella M. PTEN expression and function in adult cancer stem cells and prospects for therapeutic targeting. Adv Biol Regul. 2014; 56:66–80. https://doi.org/10.1016/j.jbior.2014.07.002.
81. Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S, Cognetti F, Ciuffreda L. PTEN: Multiple Functions in Human Malignant Tumors. Front Oncol. 2015; 5:24. https://doi.org/10.3389/fonc.2015.00024.
82. Bononi A, Pinton P. Study of PTEN subcellular localization. Methods. 2015; 77–78: 92–103. https://doi.org/10.1016/j.ymeth.2014.10.002.
83. Planchon SM, Waite KA, Eng C. The nuclear affairs of PTEN. J Cell Sci. 2008; 121:249–53. https://doi.org/10.1242/jcs.022459.
84. Bassi C, Stambolic V. PTEN, here, there, everywhere. Cell Death Differ. 2013; 20:1595–6. https://doi.org/10.1038/cdd.2013.127.
85. Gil A, Lopez JI, Pulido R. Assessing PTEN Subcellular Localization. Methods Mol Biol. 2016; 1388:169–86. https://doi.org/10.1007/978-1-4939-3299-3_12.
86. Nguyen HN, Afkari Y, Senoo H, Sesaki H, Devreotes PN, Iijima M. Mechanism of human PTEN localization revealed by heterologous expression in Dictyostelium. Oncogene. 2014; 33:5688–96. https://doi.org/10.1038/onc.2013.507.
87. Yasui M, Matsuoka S, Ueda M. PTEN hopping on the cell membrane is regulated via a positively-charged C2 domain. PLoS Comput Biol. 2014; 10:e1003817. https://doi.org/10.1371/journal.pcbi.1003817.
88. Das S, Dixon JE, Cho W. Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci U S A. 2003; 100:7491–6. https://doi.org/10.1073/pnas.0932835100.
89. Buller RE, Shahin MS, Horowitz JA, Runnebaum IB, Mahavni V, Petrauskas S, Kreienberg R, Karlan B, Slamon D, Pegram M. Long term follow-up of patients with recurrent ovarian cancer after Ad p53 gene replacement with SCH 58500. Cancer Gene Ther. 2002; 9:567–72. https://doi.org/10.1038/sj.cgt.7700473.
90. Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014; 13:217–36. https://doi.org/10.1038/nrd4236.
91. Yamaguchi M, Osuka S, Weitzmann MN, El-Rayes BF, Shoji M, Murata T. Prolonged survival in pancreatic cancer patients with increased regucalcin gene expression: Overexpression of regucalcin suppresses the proliferation in human pancreatic cancer MIA PaCa-2 cells in vitro. Int J Oncol. 2016; 48:1955–64. https://doi.org/10.3892/ijo.2016.3409.
92. Birkeland AC, Ludwig ML, Spector ME, Brenner JC. The potential for tumor suppressor gene therapy in head and neck cancer. Discov Med. 2016; 21:41–7.
93. Cross D, Burmester JK. Gene therapy for cancer treatment: past, present and future. Clin Med Res. 2006; 4:218–27.
94. Guo XE, Ngo B, Modrek AS, Lee WH. Targeting tumor suppressor networks for cancer therapeutics. Curr Drug Targets. 2014; 15:2–16.
95. Husain SR, Han J, Au P, Shannon K, Puri RK. Gene therapy for cancer: regulatory considerations for approval. Cancer Gene Ther. 2015; 22:554–63. https://doi.org/10.1038/cgt.2015.58.
96. Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998; 95:29–39.
97. Persad S, Attwell S, Gray V, Delcommenne M, Troussard A, Sanghera J, Dedhar S. Inhibition of integrin-linked kinase (ILK) suppresses activation of protein kinase B/Akt and induces cell cycle arrest and apoptosis of PTEN-mutant prostate cancer cells. Proc Natl Acad Sci U S A. 2000; 97:3207–12. https://doi.org/10.1073/pnas.060579697.
98. Lana E, Mégarbané A, Tourrière H, Sarda P, Lefranc G, Claustres M, De Sario A. DNA replication is altered in Immunodeficiency Centromeric instability Facial anomalies (ICF) cells carrying DNMT3B mutations. European Journal of Human Genetics. 2012; 20:1044–50. https://doi.org/10.1038/ejhg.2012.41.
99. Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002; 110:521–9.
100. Srikantha T, Morrow B, Schroppel K, Soll DR. The frequency of integrative transformation at phase-specific genes of Candida albicans correlates with their transcriptional state. Mol Gen Genet. 1995; 246:342–52.
101. Soule HD, Maloney TM, Wolman SR, Peterson WD Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990; 50:6075–86.
102. Lusche DF, Wessels D, Scherer A, Daniels K, Kuhl S, Soll DR. The IplA Ca2+ channel of Dictyostelium discoideum is necessary for chemotaxis mediated through Ca2+, but not through cAMP, and has a fundamental role in natural aggregation. J Cell Sci. 2012; 125:1770–83. https://doi.org/10.1242/jcs.098301.
103. Sanchez P, Daniels KJ, Park YN, Soll DR. Generating a battery of monoclonal antibodies against native green fluorescent protein for immunostaining, FACS, IP, and ChIP using a unique adjuvant. Monoclon Antib Immunodiagn Immunother. 2014; 33:80–8. https://doi.org/10.1089/mab.2013.0089.
104. Slater TF, Sawyer B, Straeuli U. Studies on Succinate-Tetrazolium Reductase Systems. Iii. Points of Coupling of Four Different Tetrazolium Salts. Biochim Biophys Acta. 1963; 77:383–93.
105. Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988; 48:589–601.
106. Ambrose J, Livitz M, Wessels D, Kuhl S, Lusche DF, Scherer A, Voss E, Soll DR. Mediated coalescence: a possible mechanism for tumor cellular heterogeneity. Am J Cancer Res. 2015; 5:3485–504.
107. Scherer A, Kuhl S, Wessels D, Lusche DF, Hanson B, Ambrose J, Voss E, Fletcher E, Goldman C, Soll DR. A Computer-Assisted 3D Model for Analyzing the Aggregation of Tumorigenic Cells Reveals Specialized Behaviors and Unique Cell Types that Facilitate Aggregate Coalescence. PLoS One. 2015; 10:e0118628. https://doi.org/10.1371/journal.pone.0118628.
108. Wessels D, Lusche DF, Voss E, Kuhl S, Buchele EC, Klemme MR, Russell KB, Ambrose J, Soll BA, Bossler A, Milhem M, Goldman C, Soll DR. Melanoma cells undergo aggressive coalescence in a 3D Matrigel model that is repressed by anti-CD44. PLoS One. 2017; 12:e0173400. https://doi.org/10.1371/journal.pone.0173400.