
INTRODUCTION
In Western populations the incidence of oesophageal adenocarcinoma (OAC) in men has risen by 6-fold in the last 30 years and has become the dominant histological subtype [1, 2]. The addition of neo-adjuvant therapy prior to surgical resection has improved survival for localised disease but there are wide variations in outcomes [3, 4].
The prediction of prognosis after potentially curative surgery is currently based on the internationally accepted TNM classification system but its limited ability to stratify patients has led to the investigation of additional biomarkers. Pathological features, such as nodal involvement, lymphovascular invasion (LVI) and circumferential resection margin (CRM) status, provide additional prognostic information and in cases treated with neo-adjuvant chemotherapy a histopathological response in the tumour or lymph nodes has been shown to predict improved survival [5–7]. However, a recent analysis of the Medical Research Council Gastric Infusional Chemotherapy (MAGIC) trial demonstrated that positive lymph node status and not pathological response was the only independent predictor of survival, albeit in a predominantly gastric adenocarcinoma sample set (76%) [8]. The use of 2-[18F]fluoro-2-deoxy-D-glucose (FDG) positron emission tomography (PET) to predict benefit to chemotherapy and prognosis has also been investigated with OACs displaying persistently high levels of FDG uptake, even after the administration of chemotherapy, associated with higher rates of relapse and poor survival [9, 10]. Current prognostic strategies do not take into account the molecular features of each tumour which may explain the wide variation in survival amongst patients with similar metabolic and pathological staging. Robust prognostic biomarkers are required to enable the stratification of OAC patients following surgical resection.
Recently, comprehensive whole genome sequencing and multiple platform analyses have led to the discovery of novel molecular sub-groups in OAC and may improve prognostication [11–14]. At present the application of next-generation sequencing techniques has not extended to routine use in pathology laboratories and so there remains a role for immunohistochemical (IHC) biomarkers. A wide range of IHC prognostic markers have been investigated in OAC, including the development and validation of a three gene prognostic panel of Epidermal Growth Factor (EGFR), Tripartite Motif-containing 44 (TRIM44) and Sirtuin 2 (SIRT2), but to date none have entered routine clinical practice [15–17]. Markers may be discovered through an understanding of the molecular biology of OAC, by analysis of genomic data or from being identified as a prognostic marker in other tumour types. Once identified the studies for each marker are often limited by a small number of cases, absence of a defined scoring methodology and lack of validation in an independent dataset.
In the current study we, therefore, aimed to identify robust IHC prognostic markers which could be used to inform the management of OAC using FFPE material. Specifically, we focused on the identification of potential biomarkers from transcriptional data followed by development and validation of an IHC marker. Unlike previous studies, we examined the interaction of our biomarkers with pathological features and have developed a prognostic model which could be used in post-chemotherapy resection material to inform post-operative surveillance or treatment strategies.
RESULTS
Gene expression and gene set enrichment analysis (GSEA)
Functional analysis of gene expression data from 60 FFPE OAC biopsies (Supplementary Table 1) was performed to generate a list of candidate prognostic biomarkers. Cases were divided into pathological responders (TRG ≤2, n=7) and non-responders (TRG 3-5, n=53) and GSEA using the C2 canonical pathways gene set database identified ten pathways associated with pathological response to neo-adjuvant chemotherapy (Supplementary Table 2). The top-ranked pathway was the Hypoxia-inducible factor 1 (HIF1) pathway and a ranked gene list of HIF1 pathway genes, beginning with the most upregulated gene in the non-responders compared to the responders was generated (Supplementary Figure 1, Supplementary Table 3). The enriched genes from this pathway contained a number of genes implicated as prognostic markers in oesophageal and other cancers, such as Insulin-like Growth Factor Binding Protein 1 (IGFBP1), N-myc Downstream Regulated Gene 1 (NDRG1), Trefoil Factor 3 (TFF3) and Solute Carrier Family 2 Member 1/Glucose Transporter 1 (SLC2A1).
Candidate gene selection
To select a candidate biomarker from the HIF1 pathway an MA plot of gene expression levels corresponding to the HIF1 pathway was performed and a False Discovery Rate (FDR) of <0.1 applied (Supplementary Figure 2). We further assessed the list of candidates according to the published literature for genes with a biological role in oesophageal cancer, potential as a prognostic marker and for availability of a validated antibody. Four candidates were selected (SLC2A1, IGFBP1, NDRG1, TFF3) of which SLC2A1 was the only gene to have FDR <0.1 and expression of SLC2A1 was strongly correlated with both IGFBP1 and NDRG1 (Spearman’s correlation co-efficient; FDR <0.1) indicating its expression was representative of these HIF1 pathway candidates. SLC2A1 encodes the membrane protein Glucose Transporter 1 (GLUT1) and high expression of GLUT1 has been associated with poor survival in oesophageal squamous cell carcinoma and progression of Barrett’s oesophagus to adenocarcinoma but GLUT1 has not been studied as a prognostic marker in OAC [18–23]. The primary mechanism for the accumulation of FDG in cancer cells during PET scans is through GLUT1-mediated transport and so the persistently high levels of FDG uptake associated with a poor prognosis in OAC may be mediated by high GLUT1 expression [24, 25]. Considering the prognostic ability of GLUT1 in a range of tumour types, its role in PET response and the availability of a robust and validated antibody we selected GLUT1 for further investigation as a prognostic marker in OAC.
GLUT1 immunohistochemistry
Expression of GLUT1 was observed in the cytoplasm and at the cell membrane of OAC cells and heterogeneous staining was noted. (Supplementary Figure 3) [24]. GLUT1 expression was assessed in a discovery set consisting of resection specimens from 141 oesophageal and gastro-oesophageal junction adenocarcinoma patients (Table 1) and no GLUT1 staining was observed in 27 cases (19.1%) with weak, moderate and strong staining observed in 49 (34.8%), 37 (26.2%) and 29 (20.6%) cases respectively (Figure 1). Close agreement was observed between three independent pathologists (Concordance correlation coefficient= 0.88; 95% CI 0.83-0.91). No significant association was found between GLUT1 and expression of the clinically validated markers Her2, p53 or MET (Supplementary Table 4).
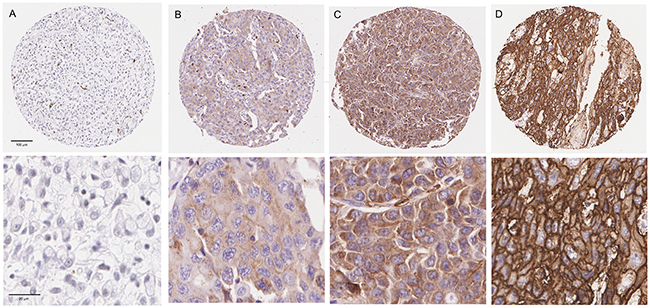
Figure 1: Tissue Microarray GLUT1 staining. Representative 10X and 40X views of tumours showing negativity for GLUT1 (A) and weak (B), moderate (C) and strong (D) staining for GLUT1.
Table 1: Comparison of discovery and validation tissue microarrays
Discovery TMA NICC |
Validation TMA OCCAMS |
p-value |
|
|---|---|---|---|
n = 141 (%) |
n = 262 (%) |
||
Age |
|||
<60 |
42 (30) |
80 (31) |
0.034 |
60-69 |
67 (47) |
92 (35) |
|
≥ 70 |
32 (24) |
86 (33) |
|
Unknown |
0 |
4 (2) |
|
Median |
63 |
66 |
0.058† |
Range |
28-83 |
33-88 |
|
Sex |
|||
Male |
110 (78) |
213 (81) |
0.431 |
Female |
31 (22) |
49 (19) |
|
Unknown |
0 |
6 (2) |
|
Tumour Site |
|||
Oesophagus |
22 (17) |
262 (100) |
NA |
GOJ, Siewert 1 |
72 (52) |
||
GOJ, Siewert 2 |
35 (23) |
||
GOJ, Siewert 3 |
12 (8) |
||
Depth of Invasion (T stage) |
|||
pT0/1 |
15 (11) |
21 (8) |
0.578 |
pT2 |
27 (19) |
56 (21) |
|
pT3 |
94 (67) |
180 (69) |
|
pT4 |
5 (4) |
5 (2) |
|
Lymph node Involvement (N stage) |
|||
N0 |
51 (36) |
72 (27) |
0.008 |
N1 |
29 (21) |
92 (35) |
|
N2/3 |
61 (43) |
96 (37) |
|
Unknown |
0 |
2 (1) |
|
Differentiation |
|||
Well |
6 (4) |
21 (8) |
0.189 |
Moderate |
53 (38) |
79 (30) |
|
Poor |
81 (57) |
151 (58) |
|
Unknown |
1 (1) |
11 (4) |
|
Lymphovascular Invasion |
|||
Negative |
47 (33) |
98 (37) |
0.004 |
Positive |
93 (66) |
101 (39) |
|
Unknown |
1 (1) |
63 (24) |
|
Circumferential Margin Involvement |
|||
Negative |
77 (55) |
113 (43) |
0.073 |
Positive |
63 (45) |
61 (23) |
|
Unknown |
1 (1) |
88 (34) |
|
Neo-Adjuvant chemotherapy |
|||
Yes |
141 (100) |
127 (48) |
<0.0001 |
No |
0 |
135 (52) |
|
†Mann-Whitney U Test
NICC- Northern Ireland Cancer Centre
OCCAMS- Oesophageal Cancer Clinical and Molecular Stratification Consortium.
To increase the clinical utility of GLUT1 as a biomarker, and to enable a more reliable scoring methodology, we defined GLUT1 positivity as any cancer cells staining for GLUT1, regardless of the intensity or percentage of cells. With the exception of tumour differentiation, there were no significant differences in any major clinicopathological factors between GLUT1 negative and positive patients (Supplementary Table 5).
The relationship between GLUT1 expression, four known pathological factors (LVI, CRM, differentiation and nodal status) and prognosis were examined further by univariate (Supplementary Table 6) and multivariate analysis (Table 2). In univariate analysis GLUT1 positivity was a significant predictor of reduced relapse-free survival (RFS) (HR 2.07, 95% CI 1.09-3.93; p=0.026) with a median RFS of 21.2 (95% CI 15.9-29.2) and 63.7 (95%CI 28.1-NA) months for GLUT1 positive and negative patients respectively (Supplementary Figure 4A). Overall survival (OS) was also significantly reduced for GLUT1 positive patients (HR 1.85, 95% CI 1.01-3.39; p=0.047) with a median OS of 31.6 (95%CI 23.4-38.4) months in GLUT1 positive patients and 43.9 (95%CI 32.7-NA) months in GLUT1 negative cases (Supplementary Figure 4B). Following multivariate analysis GLUT1 positivity remained an independent prognostic factor alongside CRM and nodal status in this cohort uniformly treated with neo-adjuvant chemotherapy and surgical resection (Table 2). Variables were selected using the elastic net penalty method (Supplementary Table 7) and the concordance index to assess the predictive fit of each model (Supplementary Table 8). The relative contribution of each variable was also assessed by the log likelihood ratio, with the greatest effect observed for nodal status (Supplementary Table 9).
Table 2: Multivariate analysis of clinicopathological factors, GLUT1 expression, relapse-free and overall survival in the discovery cohort
Relapse-free Survival |
Overall Survival |
|||||
|---|---|---|---|---|---|---|
Hazard Ratio |
95% CI |
p-value |
Hazard Ratio |
95% CI |
p -value |
|
CRM |
||||||
Negative |
1 |
1 |
||||
Positive |
2.059 |
1.167-3.631 |
0.01 |
1.897 |
1.067-3.373 |
0.03 |
N Stage |
||||||
0 |
1 |
1 |
||||
1 |
1.542 |
0.749-3.176 |
0.24 |
2.189 |
0.999-4.796 |
0.05 |
2/3 |
3.962 |
2.017-7.782 |
<0.001 |
5.694 |
2.742-11.823 |
<0.001 |
Differentiation |
||||||
Poor |
1 |
1 |
||||
Moderate |
1.19 |
0.717-1.967 |
0.502 |
1.05 |
0.194-3.703 |
0.827 |
Well |
0.862 |
0.198-3.763 |
0.844 |
0.849 |
0.624-1.764 |
0.855 |
GLUT1 |
||||||
Low |
1 |
1 |
||||
High |
2.352 |
1.191-4.647 |
0.014 |
2.059 |
1.08-3.922 |
0.028 |
Probability nomograms for RFS and OS (Supplementary Figure 5 and Table 2) combining GLUT1, CRM and nodal status were derived from the respective multivariate Cox regression models. Prognostic indices for OS and RFS were developed from the respective nomograms, based on combinations of GLUT1, CRM and nodal status. Each prognostic index was used to separate patients into three groups - good, intermediate and poor prognosis (Table 3). The poor prognosis group was associated with an RFS and OS of 13 and 16.6 months respectively following surgical resection (Figure 2).
Table 3: Prognostic model incorporating N stage, CRM and GLUT1 in the discovery cohort
Prognostic Group |
Variable Combination |
Median RFS (95% CI months) |
Median OS (95% CI months) |
|
|---|---|---|---|---|
N Stage |
GLUT1/CRM Status |
|||
Group 1 |
N Stage 0 |
GLUT1 Negative AND CRM Negative |
Not reached |
Not reached |
N Stage 1 |
||||
Group 2 |
N Stage 2/3 |
GLUT1 Negative AND CRM Negative |
39 (21.2-NA) |
39.2 (34.5-NA) |
N Stage 0 |
GLUT1 Positive AND CRM Positive |
|||
N Stage 1 |
GLUT1 Positive OR CRM Positive |
|||
Group 3 |
N Stage 1 |
GLUT1 Positive AND CRM Positive |
13 (9.9-15.9) |
16.6 (11.7-20.9) |
N Stage 2/3 |
GLUT1 Positive AND/OR CRM Positive |
|||
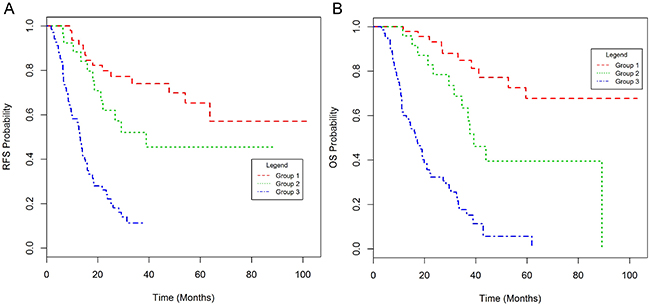
Figure 2: Kaplan-Meier plots of relapse-free (A) and overall survival (B) for the prognostic score in the discovery set.
Validation cohort
The validation set consisted of 262 OAC patients of whom 127 (69.8%) were treated with neo-adjuvant chemotherapy followed by surgery and 135 (74.2%) by surgery alone (Table 1, Supplementary Table 10). Patients in the validation set were older, had a significantly higher nodal status and a lower proportion of cases were LVI positive. GLUT1 expression did not show a significant correlation with any pathological factor but higher GLUT1 positivity was observed in chemotherapy-naïve patients (Supplementary Table 11). GLUT1 positive patients had a significantly worse prognosis (HR 1.85, 95% CI 1.11-3.08, p = 0.018) with a median OS of 18 (95% CI 17-24) and 39 (95% CI 33-53) months in GLUT1 positive and negative patients respectively (Supplementary Figure 6). GLUT1 was confirmed as a statistically significant prognostic indicator of worse OS in both the univariate and multivariate analysis for the neo-adjuvant chemotherapy and surgery treated patients but did not retain significance in the multivariate analysis for the chemotherapy-naïve patients (Supplementary Table 12). However, the prognostic model developed in the NICC cohort successfully stratified patients into three prognostic groups regardless of whether the patient received neo-adjuvant chemotherapy or not (p<0.001) (Figure 3, Supplementary Table 13).
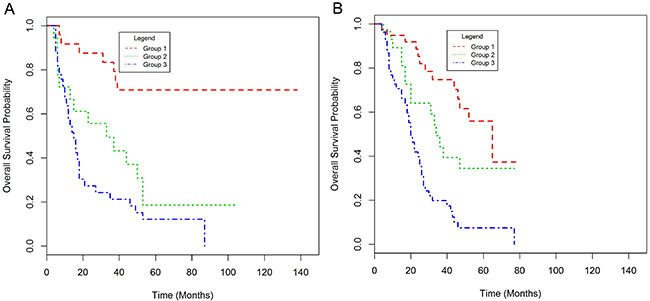
Figure 3: Kaplan-Meier plots of overall survival for the prognostic score in patients treated with neo-adjuvant chemotherapy and surgery (A) and those treated with surgery alone (B) in the validation set.
These results confirm the ability of our prognostic score incorporating GLUT1, CRM and nodal status to stratify patients following oesophagectomy and, in particular, to identify a poor prognosis group of patients.
DISCUSSION
In this study we have performed biomarker selection using gene expression data and identified GLUT1 as an immunohistochemical marker. We have developed a prognostic model incorporating GLUT1 expression, CRM and nodal status which stratifies patients into three prognostic groups when applied to OAC resection material in two independent cohorts.
It has long been recognized that the upregulation of glycolysis, known as the Warburg effect, is a characteristic feature of cancer cells [26]. Intra-tumoural hypoxia leads to increased glycolytic enzyme activity in many cancer types and in response cancer cells upregulate GLUT1, under the control of HIF-1α [26, 27]. The presence of a high degree of intra-tumoural hypoxia has been correlated with invasion and metastases of malignant tumours and may promote refractoriness to anti-cancer therapies [28–30]. The heterogeneous staining of GLUT1 observed in our study is in keeping with evidence suggesting that GLUT1 is expressed primarily in hypoxic areas of the tumour [31].
Strengths of our study include the definition of GLUT1 positivity as any tumour cells staining for GLUT1, increasing the clinical applicability of the assay. The development of a prognostic model increased the ability to stratify patients compared to GLUT1 status alone and validation in an independent sample set was a further strength. Due to the greater age of the samples, a proportion of patients in the validation set did not receive neo-adjuvant chemotherapy and this may account for the differing levels of GLUT1 positivity in the discovery (80.9%) and validation (69.5%) sets as chemo-sensitive tumours which demonstrated a pathological response will not be represented on the discovery TMA. The finding of a significantly lower proportion of N stage 0 patients in the validation cohort may also reflect insufficient pre-operative staging prior to the introduction of PET scans and the reduced administration of neo-adjuvant chemotherapy. In keeping with its discovery in a neo-adjuvant chemotherapy treated cohort, GLUT1 expression predicted survival in the validation set in the context of neo-adjuvant chemotherapy and surgery but not with surgery alone. However, the prognostic model successfully stratified patients in both settings indicating its robustness.
Limitations of our study included the use of pre-chemotherapy biopsy material for the discovery of candidate biomarkers by gene expression analysis followed by the investigation of their protein expression in post-chemotherapy resection material. It was not possible to examine the expression of GLUT1 in the biopsies due to the limited amount of tumour tissue available and the assessment of markers of poor prognosis in post-chemotherapy tissue is consistent with current data indicating the persistence of oncogenic drivers which contribute to prognosis throughout neo-adjuvant chemotherapy [32]. Therefore, we surmised that a dominant marker in a poor prognosis subgroup would be consistently detected in biopsy material and residual tumour tissue which did not respond to chemotherapy.
Our work establishes a role for GLUT1 in more accurate prognostication for patients and clinicians. When combined with nodal status and CRM into a prognostic model GLUT1 contributes to the separation of patients into three prognostic groups. The poorest prognostic group has a median OS of 16.6 (95% CI 11.7-20.9) and 20 (95% CI 18-26) months in the discovery and validation sets respectively, calling into question the validity of treating these patients with the high morbidity approach of neo-adjuvant chemotherapy and surgery. It is tempting to suggest that patients identified as having a higher risk of recurrence should be selected for more intensive follow-up or receive alternative post-operative treatment. Whilst intensive surveillance regimes have been shown to detect cases of local recurrence following radical chemo-radiotherapy suitable for salvage oesophagectomy the role of such follow-up strategies following surgical resection for OAC remains controversial [33]. Considering its role in tumour progression and resistance to therapy, GLUT1, and intra-tumoural hypoxia in general, may also represent a potential adjuvant therapeutic target [34].
In conclusion we have identified GLUT1 expression as a potential biomarker for poor prognosis in resected OAC. We have established a promising assay incorporating IHC and pathological factors which is informative for patient care following chemotherapy and surgical resection.
PATIENTS AND METHODS
This study was performed and reported in line with the REMARK recommendations (Supplementary Figure 7 and Supplementary Table 14) [35, 36].
Gene expression profiling from FFPE tissue
Transcriptional profiling of sixty pre-treatment endoscopic OAC FFPE biopsies from patients treated with neo-adjuvant chemotherapy followed by surgical resection at the Northern Ireland Cancer Centre (NICC) from 2004-2010 was performed. (Supplementary Table 1). A Mandard score of ≤2 indicated a pathological response in the corresponding resection specimen [37]. Total RNA was extracted using the Ambion Recoverall Kit (Thermo Fisher Scientific, Waltham, MA) and amplified using the NuGEN WT-Ovation FFPE System (NuGEN, San Carlos, CA). The amplified product was hybridized to the Almac Xcel™ Array (Almac, Craigavon, United Kingdom) and analysed using the Affymetrix 7G scanner (Affymetrix, Santa Clara, CA). Expression data is available at ArrayExpress (Accession Number E-MTAB-4666).
Discovery and validation tissue microarrays
The discovery set of 152 FFPE OAC resection specimens (matched gene expression data available for 58 cases) were collected from 2004-2012 at the NICC. All patients received neo-adjuvant chemotherapy prior to surgical resection and had a median follow up time of 48.8 months. Pathological staging was defined according to International Union Against Cancer (UICC) TNM staging, 7th edition. Eight specimens had no tumour tissue identifiable and 3 died within 3 months of surgery of causes unrelated to their cancer, resulting in 141 cases entering the final analysis (Table 1).
Validation was performed using a TMA generated from 262 OAC samples from patients who underwent potentially curative surgery at one of six Oesophageal Cancer Clinical and Molecular Stratification (OCCAMS) study group centres (Table 1). The TMAs were constructed as previously described [16, 38].
Immunohistochemistry
A 3-μm thick section was deparaffinised and endogenous peroxidase activity was quenched with 0.3% hydrogen peroxide prior to staining using the Ventana Discovery XT® automated immunostainer (Ventana Medical Systems Inc, Tuscon, AZ). Antibodies to GLUT1 (Ventana), c-MET (CONFIRM anti-Total c-MET (SP44), Ventana) and Her2 (anti-HER2/neu (4B5), Ventana) were used according to the manufacturer’s instructions. Staining for p53 was performed as previously described [39]. Sections were incubated with GLUT1 antibody at 37ºC for 8 minutes prior to use of the Omnimap® anti-rabbit HRP conjugate detection kit (Ventana). Lung adenocarcinoma tissue was used as a positive control and test tissue with no primary antibody was used as a negative control.
GLUT1 staining was scored by three independent observers (RT, SMcQ & JJ) who were blinded to the clinical data. In cases of discordance a consensus score was reached after discussion. GLUT1 immunoreactivity was considered positive when strong homogeneous staining was observed at the cancer cell membrane or cytoplasm. Scoring was based on intensity (0 = no staining, 1 = weak, 2 = moderate and 3 = strong staining observed) with the highest intensity from the three cores used for analysis.
Statistical analysis
Microarray data analysis was performed using Partek Genomics Suite software, version 6.6 (Partek Inc., St Louis, MO). Data was normalized using the Robust Microarray Averaging (RMA) method. Statistical analysis was performed using R (‘RMS’, ‘HMISC’, ‘COXNET’, ‘Survival’, ‘epiR’ and ‘powerSurvEpi’ packages). Gene Set Enrichment Analysis (GSEA) using the Molecular Signature Database (MSigDB v5.0) was performed as previously described [40]. Associations between GLUT1 status and the clinicopathological characteristics were calculated using chi-squared tests, with p-value adjustment for multiple comparisons using the Bonferroni correction method. Relapse-free and overall survival were calculated from the date of surgery to the date of clinical or pathological recurrence or death from any cause, respectively.
In the discovery cohort, univariate and multivariate analyses were performed using Cox proportional hazards regression, with hazard ratios and p-values reported. Proportional hazards assumptions were tested using Schoenfeld residuals. In multivariate Cox proportional hazards analysis variable selection was performed using a lasso penalty approach with cross-validation [41]. The concordance index (c-index) was used to assess the predictive fit of each model. Ranging from 0 to 1, a c-index of 0.5 suggests a model that is no better than random, while a c-index between 0.6 and 0.7 is common in survival models. Observed c-indices for each multivariate model combination were validated using bootstrap resampling validation (n = 150). Nomograms were constructed from the Cox proportional hazards models, from which prognostic indices were derived. Survival curves were estimated using the Kaplan-Meier method and compared using the log-rank test. In the validation cohort, using the variables identified in the discovery cohort, a multivariate Cox proportional hazards regression analysis (stratified by site) was performed and a prognostic index (OS) calculated and survival compared as before. Significance was set at p = 0.05, unless otherwise stated.
Author contributions
Conception and Design: DMcM, RT, JKB
Provision of study materials or patients: CJP, CAJO, RCF, CC, JJ, SMcQ, KA, MST, RT
Collection and Assembly of Data: LC, NMcC, LS, NBR, VJS, CC, DMcM, JJ, SMcQ, SC, KA, PLS, RT
Data analysis and Interpretation: JKB, LC, GL, NMcC, LS, NBR, VJS, CC, DMcA, PH, RT
Manuscript writing: All authors
Final approval of manuscript: All authors.
ACKNOWLEDGMENTS
This work was supported by the Gastrointestinal Cancer Research Charitable Fund administered by the Belfast Health and Social Care Trust, the Cancer Research UK Experimental Cancer Medicine Centre Initiative, Invest Northern Ireland and Almac Diagnostics. Further support was received from the Cambridge University Hospitals Human Research Tissue Bank, which is funded by the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre. R.C.F. has programmatic funding from the Medical Research Council and infrastructure support from the NIHR Biomedical Research Centre and the Cambridge Experimental Medicine Centre.
Tissue samples used in this research were received from the Northern Ireland Biobank (NIB) which is funded by HSC Research and Development Division of the Public Health Agency in Northern Ireland and Cancer Research UK through the Belfast Cancer Research UK Centre and the Northern Ireland Experimental Cancer Medicine Centre; additional support was received from the Friends of the Cancer Centre. The Northern Ireland Molecular Pathology Laboratory has received funding from Cancer Research UK, the Friends of the Cancer Centre and the Sean Crummey Foundation.
The Oesophageal Cancer Clinical and Molecular Stratification (OCCAMS) Study Group is a multicentre UK collaboration. In addition to the listed authors, OCCAMS members involved in the validation dataset were P. M. Safranek, N. Carroll, and S. Dwerryhouse (Addenbrookes Hospital, Cambridge, UK); S. J. Darnton and R. S. Steyn (Birmingham Heartlands, Birmingham, UK); J. Going and M. McKernan (Glasgow Royal Infirmary, Glasgow, UK); R. Stuart (Ross Hall Hospital, Glasgow, UK); M. Moorghen, J. Blazeby, and C. P. Barham (Bristol Royal Infirmary, Bristol, UK); and C. Rajaguru, N. Imrit, and N. Maynard (Oxford Radcliffe Hospitals NHS Trust, Oxford, UK).
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
FUNDING
Belfast Health and Social Care Trust Gastrointestinal Cancer Research Fund
Medical Research Council
Cancer Research UK
HSC Research and Development Division of the Public Health Agency in Northern Ireland
National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre
Invest Northern Ireland.
REFERENCES
1. Cancer Research UK. Oesophageal cancer statistics. [cited 2017 Aug 1]. Available 2017 August 1, from http://www.cancerresearchuk.org/health-professional/oesophageal-cancer-statistics.
2. Brown LM, Devesa SS, Chow WH. Incidence of adenocarcinoma of the esophagus among white americans by sex, stage, and age. J Natl Cancer Inst. 2008; 100:1184–7. https://doi.org/10.1093/jnci/djn211.
3. Cunningham D, Allum WH, Stenning SP, Thompson JN, Van de Velde CJ, Nicolson M, Scarffe JH, Lofts FJ, Falk SJ, Iveson TJ, Smith DB, Langley RE, Verma M, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med. 2006; 355:11–20. https://doi.org/10.1056/NEJMoa055531.
4. van Hagen P, Hulshof MC, van Lanschot JJ, Steyerberg EW, van Berge Henegouwen MI, Wijnhoven BP, Richel DJ, Nieuwenhuijzen GA, Hospers GA, Bonenkamp JJ, Cuesta MA, Blaisse RJ, Busch OR, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Engl J Med. 2012; 366:2074–84. https://doi.org/10.1056/NEJMoa1112088.
5. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, Vastrik I, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007; 446:153–8. https://doi.org/10.1038/nature05610.
6. Noble F, Nolan L, Bateman AC, Byrne JP, Kelly JJ, Bailey IS, Sharland DM, Rees CN, Iveson TJ, Underwood TJ, Bateman AR. Refining pathological evaluation of neoadjuvant therapy for adenocarcinoma of the esophagus. World J Gastroenterol. 2013; 19:9282–93. https://doi.org/10.3748/wjg.v19.i48.9282.
7. Reid T, Chan D, Roberts S. Prognostic significance of circumferential resection margin involvement following oesophagectomy for cancer and the predictive role of endoluminal ultrasonography. Br J Cancer. 2012; 107:1925–31. https://doi.org/10.1038/bjc.2012.511.
8. Smyth EC, Fassan M, Cunningham D, Allum WH, Okines AF, Lampis A, Hahne JC, Rugge M, Peckitt C, Nankivell M, Langley R, Ghidini M, Braconi C, et al. Effect of pathologic tumor response and nodal status on survival in the medical research council adjuvant gastric infusional chemotherapy trial. J Clin Oncol. 2016; 34:2721–7. https://doi.org/10.1200/JCO.2015.65.7692.
9. Weber W, Ott K, Becker K. Prediction of response to preoperative chemotherapy in adenocarcinomas of the esophagogastric junction by metabolic imaging. J Clin Oncol. 2001; 19:3058–65.
10. Wieder HA, Ott K, Lordick F, Becker K, Stahl A, Herrmann K, Fink U, Siewert JR, Schwaiger M, Weber WA. Prediction of tumor response by FDG-PET: comparison of the accuracy of single and sequential studies in patients with adenocarcinomas of the esophagogastric junction. Eur J Nucl Med Mol Imaging. 2007; 34:1925–32. https://doi.org/10.1007/s00259-007-0521-3.
11. Weaver JM, Ross-Innes CS, Shannon N, Lynch AG, Forshew T, Barbera M, Murtaza M, Ong CJ, Lao-Sirieix P, Dunning MJ, Smith L, Smith ML, Anderson CL, et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat Genet. 2014; 46:837–43. https://doi.org/10.1038/ng.3013.
12. Ross-Innes CS, Becq J, Warren A, Cheetham RK, Northen H, O’Donovan M, Malhotra S, di Pietro M, Ivakhno S, He M, Weaver JM, Lynch AG, Kingsbury Z, et al. Whole-genome sequencing provides new insights into the clonal architecture of Barrett’s esophagus and esophageal adenocarcinoma. Nat Genet. 2015; 47:1–11. https://doi.org/10.1038/ng.3357.
13. Stachler MD, Taylor-Weiner A, Peng S, McKenna A, Agoston AT, Odze RD, Davison JM, Nason KS, Loda M, Leshchiner I, Stewart C, Stojanov P, Seepo S, et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat Genet. 2015; 47:1–12. https://doi.org/10.1038/ng.3343.
14. Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C, Bandla S, Imamura Y, Schumacher SE, Shefler E, McKenna A, Carter SL, Cibulskis K, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013; 45:478–86. https://doi.org/10.1038/ng.2591.
15. Matthews LH, Noble F, Tod J, Jaynes E, Harris S, Primrose JN, Ottensmeier C, Thomas GJ, Underwood TJ. Systematic review and meta-analysis of immunohistochemical prognostic biomarkers in resected oesophageal adenocarcinoma. Br J Cancer. 2015; 113:1–12. https://doi.org/10.1038/bjc.2015.179.
16. Peters C, Rees J, Hardwick R. A 4-gene signature predicts survival of patients with resected adenocarcinoma of the esophagus, junction, and gastric cardia. Gastroenterology. 2010; 139:1995–2004.e15. https://doi.org/10.1053/j.gastro.2010.05.080.
17. Ong C, Shapiro J. Three-gene immunohistochemical panel adds to clinical staging algorithms to predict prognosis for patients with esophageal adenocarcinoma. J Clin Oncol. 2013; 31:1576–82. https://doi.org/10.1200/JCO.2012.45.9636.
18. Tohma T, Okazumi S, Makino H. Overexpression of glucose transporter 1 in esophageal squamous cell carcinomas: a marker for poor prognosis. Dis Esophagus. 2005; 18:185–9.
19. Sawayama H, Ishimoto T. High expression of glucose transporter 1 on primary lesions of esophageal squamous cell carcinoma is associated with hematogenous recurrence. Ann Surg Oncol. 2013. https://doi.org/10.1245/s10434-013-3371-1.
20. Griffiths E, Pritchard S. Increasing expression of hypoxia-inducible proteins in the barrett’s metaplasia–dysplasia–adenocarcinoma sequence. Br J Cancer. 2007; 96:1377–83. https://doi.org/10.1038/sj.bjc.6603744.
21. Elimova E, Wang X, Etchebehere E, Shiozaki H, Shimodaira Y, Wadhwa R, Planjery V, Charalampakis N, Blum MA, Hofstetter W, Lee JH, Weston BR, Bhutani MS, et al. 18-Fluorodeoxy-glucose positron emission computed tomography as predictive of response after chemoradiation in oesophageal cancer patients. Eur J Cancer. 2015. https://doi.org/10.1016/j.ejca.2015.07.044.
22. Wang J, Ye C, Chen C, Xiong H, Xie B, Zhou J, Chen Y, Zheng S, Wang L. Glucose transporter GLUT1 expression and clinical outcome in solid tumors: a systematic review and meta-analysis. Oncotarget. 2017; 8:16875–86. https://doi.org/10.18632/oncotarget.15171.
23. Yu M, Yongzhi H, Chen S, Luo X, Lin Y, Zhou Y, Jin H, Hou B, Deng Y, Tu L, Jian Z. The prognostic value of GLUT1 in cancers: a systematic review and meta-analysis. Oncotarget. 2017; 8:43356–67. https://doi.org/10.18632/oncotarget.17445.
24. Szablewski L. Expression of glucose transporters in cancers. Biochem Biophys Acta. 2012; 1835:164–9. https://doi.org/10.1016/j.bbcan.2012.12.004.
25. Flier JS, Mueckler MM, Usher P, Lodish HF. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science. 1987; 235:1492–5. https://doi.org/10.1126/science.3103217.
26. Cairns R, Harris I, Mak T. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011; 11:85–95. https://doi.org/10.1038/nrc2981.
27. Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1: interaction between H-ras and hypoxia. J Biol Chem. 2001; 276:9519–25. https://doi.org/10.1074/jbc.M010144200.
28. Lu X, Kang Y. Hypoxia and hypoxia-inducible factors: master regulators of metastasis. Clin Cancer Res. 2010; 16:5928–35. https://doi.org/10.1158/1078-0432.CCR-10-1360.Hypoxia.
29. Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. An integrative genomics approach identifies Hypoxia inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009; 37:4587–602. https://doi.org/10.1093/nar/gkp425.
30. Vleugel MM, Greijer AE, Shvarts A, van der Groep P, van Berkel M, Aarbodem Y, van Tinteren H, Harris AL, van Diest PJ, van der Wall E. Differential prognostic impact of hypoxia induced and diffuse HIF-1alpha expression in invasive breast cancer. J Clin Pathol. 2005; 58:172–7. https://doi.org/10.1136/jcp.2004.019885.
31. Airley R, Loncaster J, Davidson S, Bromley M, Roberts S, Patterson A, Hunter R, Stratford I, West C. Glucose transporter Glut-1 expression correlates with tumor hypoxia and predicts metastasis-free survival in advanced carcinoma of the cervix. Clin Cancer Res. 2001; 7:928–34.
32. Murugaesu N, Wilson GA, Birkbak NJ, Watkins TB, McGranahan N, Kumar S, Abbassi-Ghadi N, Salm M, Mitter R, Horswell S, Rowan A, Phillimore B, Biggs J, et al. Tracking the genomic evolution of esophageal adenocarcinoma through neoadjuvant chemotherapy. Cancer Discov. 2015; 5:821–31. https://doi.org/10.1158/2159-8290.CD-15-0412.
33. Sudo K, Xiao L, Wadhwa R, Shiozaki H, Elimova E, Taketa T, Blum MA, Lee JH, Bhutani MS, Weston B, Ross WA, Komaki R, Rice DC, et al. Importance of surveillance and success of salvage strategies after definitive chemoradiation in patients with esophageal cancer. J Clin Oncol. 2014; 32:3400–5. https://doi.org/10.1200/JCO.2014.56.7156.
34. Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N. Crystal structure of the human glucose transporter GLUT1. Nature. 2014; 510:121–5. https://doi.org/10.1038/nature13306.
35. Pepe M, Etzioni R, Feng Z. Phases of biomarker development for early detection of cancer. J Natl Cancer Inst. 2001; 93:1054–61.
36. McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. REporting recommendations for tumor MARKer prognostic studies (REMARK). Br J Cancer. 2006; 93:387–91. https://doi.org/10.1007/s10549-006-9242-8.
37. Mandard A, Dalibard F, Mandard J, Marnay J, Henry-Amar M, Petiot J, Roussel A, Jacob J, Segol P, Samama G. Pathologic assessment of tumor regression after preoperative chemoradiotherapy of esophageal carcinoma. Clinicopathologic correlations. Cancer. 1994; 73:2680–6.
38. Ilyas M, Grabsch H, Ellis IO, Womack C, Brown R, Berney D, Fennell D, Salto-Tellez M, Jenkins M, Landberg G, Byers R, Treanor D, Harrison D, et al. Guidelines and considerations for conducting experiments using tissue microarrays. Histopathology. 2013; 62:827–39. https://doi.org/10.1111/his.12118.
39. Boyle DP, Mcart DG, Irwin G, Wilhelm-Benartzi CS, Lioe TF, Sebastian E, Mcquaid S, Hamilton PW, James JA, Mullan PB, Catherwood MA, Harkin DP, Salto-Tellez M. The prognostic significance of the aberrant extremes of p53 immunophenotypes in breast cancer. Histopathology. 2014; 65:340–52. https://doi.org/10.1111/his.12398.
40. Stevenson L, Allen WL, Turkington R, Jithesh PV, Proutski I, Stewart G, Lenz HJ, Van Schaeybroeck S, Longley DB, Johnston PG. Identification of galanin and its receptor galr1 as novel determinants of resistance to chemotherapy and potential biomarkers in colorectal cancer. Clin Cancer Res. 2012; 18:5412–26. https://doi.org/10.1158/1078-0432.CCR-12-1780.
41. Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010; 33:1–22. https://doi.org/10.18637/jss.v033.i01.