
INTRODUCTION
The recent development of immune checkpoint inhibitors and the corresponding efficacy shown by inhibitors of the CTLA-4/B7 [1] and PD-1/PD-L1 checkpoints [2–7] in multiple tumour types has resulted in substantial investment by the pharmaceutical industry in clinical development of immunotherapeutics across tumour types and indications. Although the efficacy of inhibitors of the PD-1/PD-L1 checkpoint has been consistent across tumour types, the single agent activity of these drugs has been lacking in tumours of the central nervous system (CNS). In particular, several studies have shown less promising results in glioblastoma compared with other tumour types [8, 9]. Glioblastoma, however, poses unique challenges to the immunotherapy treatment paradigm, as traditionally the CNS has been regarded as an immune-privileged site [10]; the frequent concomitant administration of immunosuppressive medications such as corticosteroids in this patient population is an additional consideration. Although these recent trials have cast doubts over the role, if any, of immunotherapeutics in CNS malignancies, they may also serve as an opportune time to evaluate the nuances of the emerging biology surrounding the cancer-immunity cycle and the specific challenges relating to drug development in primary brain tumours.
The cancer-immunity cycle was first proposed by Chen and Mellman [11] as a paradigm for the interaction between the immune system and cancer. They argue that a series of step-wise events must occur for effective anti-tumour immunity and coined the cycle to describe these events. Cancer cells and cancer cell death initially results in the release of neoantigens, which are then presented to dendritic cells. Priming and activation subsequently occurs, leading to trafficking of T cells to tumours, and subsequent infiltration of effector cells into tumours. There is then recognition of cancer cells by effector T cells, which results in cancer cell death which reiterates the cycle. This review evaluates the challenges of developing successful immunotherapeutics for glioblastoma through the lens of the cancer-immunity cycle. We initially describe the current understanding of the immune system in the central nervous system and subsequently address unique aspects of the immune system in the brain. We then describe current clinical development of CNS immunotherapeutics and the relative lack of efficacy of immune checkpoint inhibition to date. Finally, we provide a conceptual framework through which the development of effective immunotherapeutic strategies in the CNS can be viewed, and specific considerations for clinical trial design for CNS immunotherapeutics.
The immune system and the brain – biological challenges and immune privilege
Historically, the CNS has been considered an immune-privileged site for a triumvirate of reasons [12]. Firstly, histological absence of observable lymphatics disputed lymphatic circulation in the brain, theoretically impeding functional immunity. Secondly, the blood-brain barrier (BBB) has been a major limitation since it was first described by Paul Ehrlich in the late 19th century [13]. The BBB comprises a physical barrier due to complex tight junctions between adjacent endothelial cells, which requires transcellular passage of molecules trafficking into brain tissue compared to typical paracellular trafficking in other tissue sites [14]. Practically this results in limited penetration of antibodies, immune mediators and immune cells through the BBB from the systemic circulation into the CNS [15]. The third pillar of immune privilege was the disparity between the CNS immune system compared to the rest of the body, “apparent immune absence”, supported by observations such as the paucity of dendritic cells in the brain parenchyma [16], the seminal work of Lampson demonstrating the lack of major histocompatibility complex (MHC) class I on neuronal and glial tissue, the relative paucity of MHC class II expression in resections of brain tumour patients [17] and the tight regulation of the expression of T cell co-stimulatory molecules within the brain [18]. A large body of emerging work is now challenging the traditional assumptions underlying this concept of relative CNS immune privilege with good evidence indicating that the CNS is both immune competent and actively interacts with the peripheral immune system.
Challenging lymphatic circulation as a pillar of immune privilege
Firstly, we now have clear evidence of lymphatic circulation within the brain [19]. Louveau and colleagues used sensitive imaging techniques to neatly show that the cerebrospinal fluid circulation leads to lymphatic drainage of the brain via the cervical and nasal lymphatics [20] suggesting that immune cells and tumour antigens may pass through the cerebrospinal fluid to the draining cervical lymphatics to meet with the antigen processing and presenting machinery and thereby stimulating the development of a systemic immune anti-tumour response (Figure 1). Although naive antigen-inexperienced T cells tend not to enter the healthy CNS and remained located in perivascular, subarachnoid, or meningeal spaces [21], activated CNS-specific CD4+ T cells are able to apparently chaperone naive non-CNS-specific T cells across the BBB into the CNS [22].
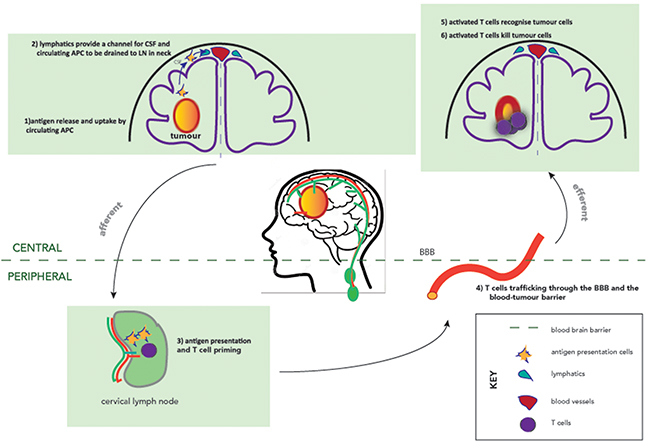
Figure 1: The afferent and efferent arms of the CNS immune system. Dashed line indicates the blood-brain-barrier. Lymphatics are shown in green, and vasculature in red. Antigen release triggers recognition of antigens by antigen presenting cells, which are channelled via CNS lymphatics to the cervical lymph nodes. Antigen presentation and T cell priming occur peripherally in the cervical lymph nodes before trafficking back to the CNS to recognise and kill tumour cells.
Challenging the BBB as a pillar of immune privilege
Importantly, it is increasingly recognised that the BBB is dynamic, with its phenotype developing from complex cell-cell interactions from adjacent astrocytes [14, 23] and with its permeability varying based upon the functional requirements of signalling systems in the brain. For instance, the fenestrated endothelial wall at the hypothalamus allows diffusion of hormones into the systemic circulation, whereas the absence of the BBB at the area postrema allows relative free perfusion of molecules from the blood into brain tissue [13]. Of particular relevance in brain tumours, the tight junctions of the BBB can be disrupted in the setting of cerebral oedema [24], pro-inflammatory cytokines, such as interferon-γ and tumor necrosis factor-α [13], anatomical disruption from direct tumour extension, as well as downregulation of tight junction proteins such as claudins 1,3 [14, 25]. These observations gel with histopathological findings in brain tumour series, which have consistently demonstrated significant quantities of infiltrating immune cells in glioblastoma specimens, both macrophages but also CD4+ and CD8+ lymphocytes [26], as well as dynamic markers of the immune response such as PD-L1 [27].
Taken together, these factors demonstrate that the BBB is a relative rather than an absolute barrier, when considering implications for the trafficking of immune cells or the delivery of cancer therapeutics.
Challenging apparent immune absence as a pillar of immune privilege
Finally, although there are definite differences in the immune system of the brain compared to other sites, this does not definitively preclude functional CNS immunity. Systemically, it is widely recognised that the critical components of the antitumour immune response are cytotoxic T cells and the adaptive immune system, and that overactivation of the innate immune system can paradoxically promote tumorigenesis [28]. Nevertheless, some degree of innate immune activation is a requisite for functional anticancer immunity. Critical components of the systemic anticancer immune response include immune recognition cells such as dendritic cells, immune effector cells such as cytotoxic CD8+ T cells and the supporting apparatus of CD4+ helper T cells. In the brain, microglia serve as the functional antigen presenting cells, having been shown by sensitive assays to avidly express MHC class II molecules, particular in the setting of inflammation, and are now thought to be able to directly present tumour antigens to T cells within the brain [29–31]. Although preclinical models of healthy mice suggested that the CNS parenchyma lacks a potent innate immune response [32], resident microglia are able to recognize “pathogen associated molecular patterns” and “danger associated molecular patterns”, which include heat shock proteins, uric acid, high-mobility group box 1 protein (HMGB1), and other structures available during tissue damage, inflammation and cell death [33]. Heat shock proteins released from tumor cells may be particularly effective chaperones for tumor-specific peptide antigens and may both activate dendritic cells and serve as antigen couriers [34, 35]. Thus, there appears to be sufficient innate immune system activation in the CNS to generate an antitumour immune response.
It has been challenging to identify how the innate immune system activates the adaptive antitumour immune response in the brain, but preclinical models suggests that activated dendritic cells carry antigens and transit to the cervical lymph nodes where a systemic immune response is stimulated [36]. Additionally CNS-derived soluble tumour antigens may directly drain to the lymphatics where they are presented by peripheral antigen-presentation machinery [37].
A CNS-specific T-cell trafficking programme is yet to be identified, but preclinical work in auto-immune murine models suggests that activation of T cells within the cervical lymph nodes have a direct role for the neuro-inflammation seen [38]. Three potential immune entry sites into the CNS have been described, localizing to the superficial leptomeningeal vessels, parenchymal vessels and the choroid plexus [39]. In agreement with these findings, immune cell infiltrates are found in tumor tissue derived from brain cancers consisting of both macrophages and CD4+ and CD8+ lymphocytes [26, 40]. Furthermore, antibodies are able to penetrate into the CNS, albeit at lower concentrations than in the systemic circulation [41], providing evidence of the humoral component of the adaptive immune system in the brain. These factors suggest that there is a functional cellular and humoral immune response in the brain, the key components of which are demonstrated in Figure 1. Counteracting this functional adaptive immunity is the increasing recognition of a particularly immunosuppressive tumour microenvironment in the archetypal primary CNS tumour, glioblastoma.
The immunosuppressive tumour microenvironment of glioblastoma has been well documented [42] and characterized by the myriad anti-inflammatory cytokines secreted by glioma cells. Cytokines such as tumor growth factor-β (TGF-β), interleukin (IL)-6, IL-10 and prostaglandin E2 actively suppress the expression of MHC on microglia, thereby limiting antigen presentation and diminishing the cytotoxic T cell response [43, 44]. The infiltrating T cell population is over-represented by regulatory T cells (Tregs) [45], which are regulated by factors such as the enzyme indoleamine 2,3-dioxygenase (IDO) [46] and serve functionally in brain tumors to suppress the immune system [47]. This diminished response is further exacerbated by the promotion of the alternative M2 macrophage phenotype in glioblastoma [48]. There is a substantial body of literature demonstrating that phenotype switching of tumour-associated macrophages from M1 to M2 promotes tumorigenesis in diverse ways [48]. In glioblastoma, the presence of M2 macrophages has been correlated with increasing histological grade, which is thought to be driven in some part by tumoral expression of macrophage colony-stimulating factor [45, 49]. Thus, the development of clinically efficacious immunotherapeutics in the brain must both consider the unique aspects of the CNS immune system and the historical pillars of immune privilege as well as offsetting contribution of the immunosuppressive microenvironment in glioblastoma.
Current clinical developments in CNS immunotherapeutics
The use of the PD-1/PD-L1 immune checkpoint inhibitors to unleash the T cell response has been most studied immunotherapeutic strategy in glioblastoma, but has proved mostly disappointing in single agent studies presented thus far [8, 50, 51] (Table 1). Checkmate-143 was a Phase 3 study exploring nivolumab in comparison to bevacizumab in the setting of recurrent glioblastoma and demonstrated a tolerability profiles consistent with observations in other tumor types. Disappointingly however, CheckMate-143 did not meet its primary endpoint of improved overall survival, as presented by Reardon et al at World Federation of Neurooncology Societies 2017 with lower documented response rates in the nivolumab arm in spite of a hint of more durable responses in the responding patients [8, 52].
Table 1: Reported results of single agent checkpoint inhibitors trials in recurrent glioblastoma
Registration number |
Treatment |
Overall response rate* (%), (N) |
Comments |
||
|---|---|---|---|---|---|
NCT02017717 |
Nivolumab |
8% (n=153) [8] |
Longer duration of response (11.1 mo compared to 5.3 mo for bevacizumab). |
Median PFS 1.5 months. |
12-month OS 42%. |
NCT02054806 |
Pembrolizumab |
4% (n=26) [53] |
Median OS 14.4 months. |
Median PFS 2.8 months. |
|
NCT02336165 |
Durvalumab |
13.3% (n=31) [9] |
12-month OS 44.4% |
6-month OS 59.0% |
6-month PFS 20.0% |
* Overall response rate according to RANO criteria.
PFS- progression-free survival.
OS- overall survival.
Of note however, are the case reports of therapeutic successes in specific pediatric patients with biallelic mismatch repair deficiencies [54] suggesting that these antibodies do cross the BBB and penetrate into the tumour microenvironment, and are able to release a tumour specific cytotoxic T cell response. Given that these patients have hypermutated tumours with significantly high mutational load and therefore a significant immunogenic burden and thus a larger repertoire of tumor antigen-specific T cells [55, 56], the inclusion of a selected subset of glioma patients with high mutational burden into clinical trials of checkpoint inhibitors is one strategy currently being pursued (for example, in NCT02628067). As a population however, the mutational load in primary malignant brain tumours is low, approximately 10-fold lower than in melanoma and lung cancer [57, 58] with the mutational load being associated with tumour grade [59]. And although the currently available standard treatments of radiation and temozolomide are themselves mutagenic [60], and one may extrapolate that in cells that survive, the neoantigen load is likely to rise, thereby diversifying epitopes available for recognition by T cells, this has been insufficient in isolation to stimulate an adaptive immune response as demonstrated by the limited sensitivity to single agent immune checkpoint inhibition in the recurrent setting (Table 1). As such, consideration of other nodes in the CNS immunity cycle to be targeted with combinatorial strategies are urgently needed and discussed in detail in the following sections (Figure 2).
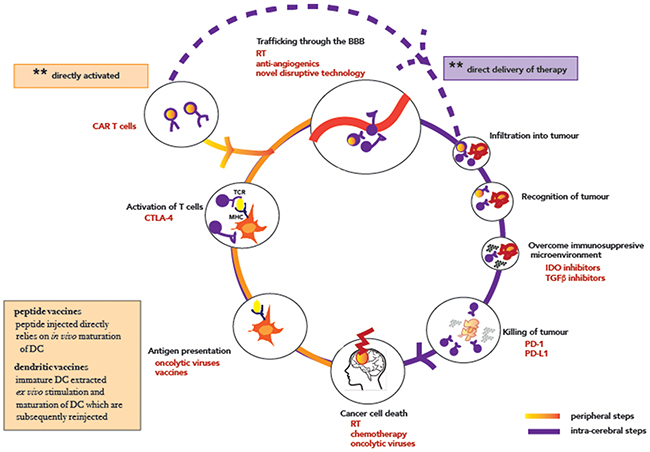
Figure 2: The cancer-immunity cycle in CNS malignancies. T lymphocytes are shown in purple, with CAR-T modified T lymphocytes highlighted with a glow. The orange half of the circle marks out steps that can be targeted systemically, while the purple indicates steps that require intra-cranial delivery/mode of action. Abbreviations: CAR Chimeric antigen receptors; RT radiotherapy; CTLA-4 cytotoxic T-lymphocyte-associated protein 4; PD-1 Programmed cell death protein 1; PD-L1 Programmed death-ligand 1; IDO Indoleamine-pyrrole 2,3-dioxygenase; TGF-β Transforming growth factor beta.
The CNS cancer-immunity cycle- a framework for immunotherapeutic strategies in CNS tumour
Cancer cell death- DNA damaging agents and immunogenic cell death
Initiating the cancer immunity cycle is cell death and immunogenic cell death refers to activation of the immune system by apoptotic cells or pre-apoptotic cells resulting in tumor cell death [61]. DNA damaging agents including radiation and temozolomide can cause immunogenic cell death and the release of danger signals including “damage-associated molecular patterns” that stimulate the recruitment of APCs where they process and present tumour neoantigens, thereby priming an adaptive immune response [62]. It is worth noting that to date there does not appear to be any evidence that immunogenic cell death is affected by mutational load [62]. In preclinical murine glioma models, combined PD-1 blockade and stereotactic radiosurgery (SRS) have been shown to improve antitumor immunity and produce long-term survivors [63, 64] and this concept is now in early clinical testing in patients with malignant brain tumours. The focus on augmenting immunogenic cell death in glioblastoma to negate the limited single agent efficacy of PD-1 inhibition is translating into ongoing early phase clinical trials. Sahebjam et al recently presented preliminary findings from one such phase I study evaluating the concomitant use of hypofractionated SRS, pembrolizumab, and bevacizumab for recurrent, high-grade gliomas noting that all patients tolerated the regimen, and and an impressive durable response rate (response for ≥ 6 months) of 53% was noted [65, 66]. Numerous other combination trials of immunotherapy in combination with DNA damaging agents for CNS malignancies are ongoing including with temozolomide (e.g. NCT02311920), radiotherapy (NCT02617589, NCT02336165) and the combination of temozolomide and radiation (NCT02667587).
Antigen presentation- oncolytic virotherapy and vaccine strategies
Cell death can kickstart the cancer-immunity cycle in the brain by activating the adaptive immune system via antigen presentation. There are several complementary therapeutic strategies that are focussing upon this component of the cancer-immunity cycle in the brain. Oncolytic virotherapy makes use of non-pathogenic viruses to selectively invade or specifically express proteins in brain tumor cells that can directly kill cancer cells or otherwise stimulate an immune response, therefore marrying the concepts of immunogenic cell death with antigen presentation. The oncolytic polio virus utilizes the aberrant expression of the poliovirus receptor, CD155, in solid tumours to mediate viral cell entry [67]. In humans, infection of tumor macrophages and dendritic cells is sublethal and eventually leads to induction of MHC class II expression and the stimulation of a tumor antigen-specific T cell response [67, 68]. A Phase I clinical trial of a poliovirus chimera, PVSRIPO for recurrent glioblastoma showed that this approach was safe, with initial promising results, with 10 out of the initial 13 patients treated still alive at the end of the trial [69]. To overcome the attenuated immune responses within the brain, groups are attempting to engineer virotherapy with inducible inflammatory cytokines, for example the Ad-RTS-hIL-12, an inducible adenoviral vector that expresses IL12 in the presence of an orally-administered activator ligand, veledimex. This early phase trial showed evidence of systemic increases of IL-12, IFNγ as well as increased number of CD8+T-cells in circulation, with an impressive 100% 6-month survival for the 13 patients thus far [70]. The challenge here is that virotherapy for brain tumors relies heavily on viral migration to the tumor site and has mostly been explored by intratumoural injection which is not always achievable. Efforts are therefore underway to explore the feasibility of systemic intravenous delivery approaches to overcome this (e.g. REOGLIO ISRCTN70044565).
Apart from tumour cell lysis mediated by oncolytic viruses, there are complementary methods of targeting antigen presentation in the brain. The identification of a growing number of potentially unique immunoreactive tumor-associated antigens expressed by human gliomas make cancer vaccines including peptide, dendritic cell, tumor cell, and neoantigen vaccines a very exciting strategy. Moreover, this approach can be utilized peripherally, bypassing the logistical challenges of delivering therapeutics directly intracranially. Peptide vaccines induce a T-cell response at the tumor site by releasing peptides specific to tumor-associated antigens. These are commonly coupled with carrier proteins and adjuvants, are taken up by APCs and presented on the cell surface by MHC molecules. APCs navigate the lymphatic system to prime T-cells, which then recognize the tumor cell from its antigen [71]. Glioblastoma represents an attractive therapeutic target for peptide vaccination as the unique epidermal growth factor receptor (EGFR) variant, EGFRvIII,is expressed in approximately 30% of patients with glioblastoma [72]. The most advanced therapeutic candidate peptide vaccine is rindopepimut, which targets a neoepitope created by a 13 amino-acid sequence unique to EGFRvIII, chemically conjugated to KLH which serves as an immune adjuvant [73]. Although initially heralded as a major breakthrough on the back of positive early phase studies [74], recent published large phase three studies have failed to show a survival benefit and argue against rindopepimut’s efficacy [75], and this may be largely due to the heterogenous nature of glioblastoma. To address this issue of heterogeneously-expressed tumor-associated antigens, multi-peptide vaccine strategies such as the IMA950 vaccine which contains 11 human leukocyte antigen (HLA)-restricted tumor-associated peptides are being explored with some initial hints of benefit, particularly in a sub-group of patients with marked injection site reactions [76]. Other candidate peptide vaccines are also showing initial promise in early phase clinical trials [77, 78] and the results of larger studies are eagerly waited.
The alternative vaccine strategy is of dendritic cell vaccination. Instead of injecting a peptide that is presented to an APC, autologous dendritic cells sourced from peripheral blood monocytes are primed with tumour lysate from the patients’ own tumour in the presence of growth factors such as interleukin-4 and granulocyte macrophage colony stimulating factor, [79]. Immature dendritic cells can uptake and process tumour-associated antigens, and mature ex vivo, thus becoming capable of proper antigen presentation for T-cell recognition in a MHC-restrictive manner [80]. These pools of dendritic cells are subsequently autologously transplanted into patients. Studies performed in glioblastoma patients have typically involved injection intradermally [79, 81] in proximity to the draining cervical lymph nodes, or occasionally in patients with Ommaya reservoirs, directly into the cerebrospinal fluid [81]. In these studies, although unarguable clinical benefit could not be observed, there was clear evidence of increases in tumour-lysate specific T cells in the periphery [81] and tumour lysate specific memory T cells and cytotoxic T cells intratumorally [79]. One example is the ICT-107 autologous dendritic cell vaccine pulsed with six tumor-associated antigens for which ten-year follow-up data is available for the initial Phase I vaccine trial. 19% of 16 patients remained disease free for 8 years with a median overall survival of 38.4 months [82]. These durable responses have fueled combination studies with checkpoint inhibitors which are ongoing (for example NCT02529072).
T cell activation
Antigen presentation is followed by T cell activation in the cancer immunity cycle, which represents another potential target of immunotherapeutic strategies in the brain. The inhibitory cell surface protein CTLA-4 primarily regulates the amplitude of the early stages of T cell activation [83] and is expressed solely by T-cells localized primarily within secondary lymphoid tissues. It binds preferentially to CD80/CD86 on the surface of APCs, thus preventing their binding to the T-cell co-stimulatory receptor CD28, leading to decreased T-cell activation and proliferation in the context of antigen-presenting MHC class [84–87]. CTLA-4 also contributes to immune modulation by enhancing the suppressor functions of Tregs [88].
The combination of anti-CTLA-4 plus anti-PD-1 has demonstrated encouraging activity in preclinical murine models of orthotopic transplanted gliomas [45, 63, 64, 89, 90], however this has failed to translate to substantial clinical benefit (8). In the Phase I CheckMate-143 study, 90% of patients who received combination therapy had grade 3 or 4 treatment-related adverse events, and 50% of patients in that arm had to discontinue treatment early due to intolerability leading to the exclusion of this combination in the subsequent phase II/III study [52]. In patients with an overall poor prognosis, this limited efficacy combined with significant toxicity is unacceptable and as such, needs tweaking to deliver tangible clinical benefits to patients. One approach to minimize the risk of increased systemic toxicity from these combination is to use intra-tumoral delivery of anti-CTLA-4 following the resection of the recurrent glioblastoma which is currently ongoing (NCT03233152).
Lymphocyte-trafficking into the CNS: BBB
Following T cell activation, the CNS cancer immunity cycle needs to consider trafficking into the CNS and crossing the BBB. The therapeutic strategy most advanced in glioblastoma that may theoretically affect the BBB is anti-angiogenic therapy. Although, initially uptake of anti-angiogenics was met by optimism due to unprecedented response rates [91], subsequent large randomised trials have failed to demonstrate evidence of benefit [92, 93] and a large meta-analysis has shown no overall survival benefit for these agents [94]. Nevertheless, emerging data support a strong rationale for combining therapies targeting vessel normalization with immunotherapies [95]. In particular, abnormal tumour vasculature promotes the production of cytokines which preferentially recruit immunosuppressive lymphoid populations [95] and polarize tumor associated macrophages to the immunosuppressive M2 phenotype [48]. As such, combinations of anti-angiogenics together with checkpoint inhibitors are actively being pursued in early phase clinical trials (NCT02336165, NCT02337491). It is however, worth noting that glioblastoma is a highly invasive tumour, and that anti-angiogenic agents may paradoxically promote invasiveness [96, 97] thus impeding the efficacy of this combination.
Other ingenious out-of-the-box solutions are being explored to overcome the impediment of the BBB in drug delivery. Armed with the knowledge that some of the activity of radiotherapy in brain tumours is due to disruption of tight junctions and therefore vessel permeability [98], the hypotheses that low dose radiotherapy could increase drug delivery to the CNS was recently tested [99]. Preliminary results in a cohort of resected brain metastases patients has demonstrated substantially (~20x) higher tissue afatinib concentrations compared to plasma, thereby validating this hypothesis. Other viable strategies to disrupt the BBB undergoing clinical evaluation include the combination of microbubble injections with pulsed ultrasound, which has been shown to functionally disrupt the BBB on serial contrast-enhanced MRI [100]. These trials provide proof-of-principle that augmentation of drug delivery into the CNS could be achieved and is likely to be used in combination strategies soon.
Infiltration and recognition of tumour- adoptive cell therapy
Once lymphocytes have been trafficked to the tumour, the effector components of the immune system must infiltrate into the tumour and recognise the tumour to propagate the CNS cancer immunity cycle. One strategy targeting this component of the cycle is adoptive cell therapy. Instead of relying on the afferent of the neuro-immune system, adoptive cell therapy aims to engineer and directly activate T cells which are then able to home back to the tumour (Figure 2). This technology, first developed by Gross et al [101] utilizes a chimeric construct consisting of a single-chain variable fragment of a high affinity antibody recognizing a tumour antigen fused to one/multiple co-stimulatory domains that directly activate T cells (CAR-T) in a non-MHC restricted manner [101] They have exhibited striking activity in hematological malignancies and the first CAR-T therapy recently being approved by the FDA for use in relapsed B cell precursor acute lymphoblastic leukemia [102]. Efforts in solid tumours are ongoing (see Table 2), but suffer from lack of well described cell surface targets which are solely expressed on tumour cells and absent from normal tissue [103]. In some ways, glioblastoma is relatively fortunate compared to other solid malignancies, with the well described truncating EGFRvIII variant [72] exhibiting characteristics of an opportune target – high frequency aberration in target disease and absence in normal tissue. Consequently, CAR-T cells targeting this variant are undergoing clinical development [104]. The first-in-human Phase I study of CAR-T EGFRvIII cells demonstrated the safety of this approach, without evidence of off-target toxicity or cytokine release syndrome with one patient having stable disease at 18 months [105].
Other antigens being targeted in current clinical trials include Eph-A2 and IL13Rα2 (see Table 2). Preliminary results from a Phase I trial of a first-generation CAR-T cells targeting the glioblastoma tumor antigen IL13Rα2 reported safe intracranial delivery of the CAR-T cells with one particular patient exhibiting a 79% regression of recurrent tumour mass [106]. Building on this, a 2nd generation CAR-T incorporating a 4-1BB (CD137) costimulatory domain and a mutated IgG4-Fc linker to reduce off-target Fc-receptor interaction is in testing with a dramatic transient clinical response in a patient with recurrent multifocal glioblastoma [107]. Two important lessons can be drawn from this study – firstly, the challenge of T cell homing as this patient did not respond to the initial intercavitary delivery of CAR-T cells, but responded dramatically when this was switched to an intra-ventricular mode of delivery. And secondly, despite the incredible radiological response, the patient relapsed with tumours that had significantly decreased IL13Rα2 expression suggesting that antigenic heterogeneity may be a significant hurdle to the success of this approach. Technical advances in cellular engineering may help overcome some of these challenges, for example a recent preclinical study has shown that trivalent CAR-T cells targeting commonly expressed glioma antigens including HER-2, IL13Rα2 and Eph-A2 can overcome tumour heterogeneity and target nearly all tumour cells in patient-derived xenograft models compared to bispecific or single-epitope targeting CARs [108].
Table 2: Ongoing trials of CART cells in glioblastoma
NCT number |
Tumour type |
Target |
Mode of delivery |
|---|---|---|---|
NCT02331693 |
Advanced Glioma |
EGFR |
Systemic infusion (IV) |
NCT02209376 |
Glioblastoma Multiforme |
EGFRvIII |
Systemic infusion (IV) |
NCT02844062 |
Glioblastoma Multiforme |
EGFRvIII |
Systemic infusion (IV) |
NCT01454596 |
Glioblastoma Multiforme |
EGFRvIII |
Systemic infusion with aldesleukin (IL-2) (IV) |
NCT02937844 |
Glioblastoma Multiforme |
PD-L1 |
Systemic infusion (three-day split) (IV) |
NCT02575261 |
Glioma |
EphA2 |
Systemic infusion (IV) |
NCT02664363 |
Glioblastoma Multiforme |
EGFRvIII |
Systemic infusion (IV) Companion imaging study. |
NCT01082926 |
Brain tumours |
IL13Rα2 |
Intratumoral |
NCT02208362 |
High grade glioma |
IL13Rα2 |
Intratumoral |
NCT02442297 |
Glioblastoma Multiforme |
Her2 |
Intratumoral |
NCT01109095 |
Glioblastoma Multiforme |
Her2 (CMV specific T cells) |
Systemic infusion (IV) |
Overcoming the suppressive immune microenvironment
Finally, for ongoing cell death to perpetuate the CNS cancer immunity cycle, the immunosuppressive microenvironment must be overcome. The challenge of the immunosuppressive microenvironment has been particularly highlighted by the early phase CAR-T trials. O’Rourke and colleagues found evidence of trafficking of CAR-T-EGFRvIII cells to regions of active glioblastoma, with antigen decrease in five of these seven patients who proceeded to surgery, but in all cases in situ evaluation of the tumour microenvironment demonstrated increased and robust expression of inhibitory molecular and infiltration by regulatory T cells after CAR-T-EGFRvIII infusion, compared to pre-infusion specimens [105]. As such, novel strategies targeting the immune microenvironment are urgently required. Components of the immunosuppressive microenvironment include Tregs, monocytes as well as signaling molecules, all of which could theoretically be targeted to enhance anti-cancer immunity in glioblastoma.
Given the prominence of the M2 macrophage phenotype in glioblastoma [49], strategies aiming to switch macrophage polarization are being explored. Preclinical models implicate the macrophage colony stimulating factor 1 receptor (CSF-1R) in macrophage/monocyte polarization to the pro-tumorigenic M2 phenotype and antagonists to this are in clinical testing (NCT02526017). Other signaling molecules including the phosphoinositide 3-kinase (PI3K) signaling pathway also have a role in directly polarization of macrophages to the M2 phenotype [109] and despite limited single agent activity of multiple PI3K pathway inhibitors in glioblastoma [110], these may have value combinatorially.
TGFβ, secreted by tumour cells in an autocrine loop is a potent immunosuppressive cytokine and inhibits the efficacy of immune effector cells [111]. A bispecific antibody targeting PD-L1 and TGFβR2 has shown preclinical evidence of enhancing antibody-dependent cellular cytotoxicity mediated by both PD-L1 and TGFβR2 preclinically [111] and is now in clinical trials including a glioma cohort of patients [112] (NCT02517398). Other ongoing trials include combinations with the TGFβR1 inhibitor galunersertib (NCT02423343).
The immunoregulatory enzyme IDO has been heavily associated with immune tolerance [113] and has been specifically associated with controlling the functional status of Tregs in response to inflammatory stimuli [46]. Inhibitors of this enzyme are amongst the most advanced novel immunotherapeutics in clinical development with multiple clinical trials ongoing in numerous tumour types including in glioblastoma (NCT02052648). Although single agent activity of IDO inhibitors have not been promising in solid tumours [114], recent reports of significantly higher response rates in combination with PD-1 inhibitors have prompted excitement [115] and this strategy may have utility in combinations for glioblastoma.
Table 3: Current strategies targeting the cancer-immunity cycle in glioblastoma
Cancer immunity cycle component |
Possible therapeutic strategy |
Examples of current trials |
|---|---|---|
Cell death |
• Combination with DNA damaging agents • Combination with stereotactic radiosurgery |
• NCT02311920, NCT02617589, NCT02336165 NCT02667587 • NCT02313272 |
Antigen presentation |
• Oncolytic viruses • Vaccines |
• ISRCTN70044565 • NCT02529072 |
T cell activation |
• Intratumoural CTLA-4 combination |
• NCT03233152 |
Lymphocyte trafficking |
• Combination with antiangiogenic agents |
• NCT02336165, NCT02337491 |
Infiltration and recognition of tumour |
• CART cells/ adoptive cell therapy |
• NCT02209376 |
Overcoming the suppressive immune microenvironment |
• Macrophage polarization • Bispecific antibodies • Immunoregulatory inhibitors |
• NCT02526017 • NCT02517398 • NCT02052648 |
Considerations for CNS drug development
In this review, we have presented a framework for understanding the CNS-cancer immunity cycle to effectively develop immunotherapeutics for CNS tumours. Table 3 summarises the components of the cancer immunity cycle and current strategies targeting these components. Rational strategies backed by strong preclinical data for combinations must be developed to optimize efficacy. Specific challenges unique to brain tumours must be considered. One of the major hurdles in developing preclinical insights is the lack of biologically relevant models for hypothesis testing. Moreover, although in other solid tumours sequential tumour biopsies are increasingly used to compress clinical development timelines and improve pharmacodynamic studies [116], given the relative importance of brain tissue and associated difficulty with tissue sampling, this strategy is simply not feasible in CNS tumours. Nevertheless, there are ways to combat this specific issue. Having optional research biopsy components in patients who are undergoing re-resections for clinical reasons can bypass this problem. Pharmacokinetic information can also be established with cerebrospinal fluid samples, which has previously added useful information to pharmacokinetic profiles [117].
Other specific challenges unique to the CNS include the BBB, which is an impediment to effective CNS penetration of numerous drugs. There are many approaches that that may mitigate this problem. Firstly, several trials are currently being performed on small molecule inhibitors, with the compound being delivered immediately in the pre-operative period prior to re-resection, thus allowing for a more substantial study of pharmacodynamic endpoints. Secondly, given a substantial component of the cancer-immunity cycle occurs peripherally, there is no reason why therapeutics targeting the periphery cannot have central activity.
There are also some unique clinical considerations in glioblastoma patients that can impede effective drug delivery and drug development. Many patients with brain tumours have uncontrolled seizures requiring numerous anti-epileptic medications. These represent a challenge in early phase clinical trials, as typically the use of such drugs is prohibited due to the uncertain pharmacokinetic profiles that they result in, particularly in the development of drugs predicted to be metabolized by the hepatic cytochrome p450 system. However, it is important to note that second and third generation anti-epileptic medications are typically not enzyme inducing and therefore limit the risks of adverse drug-drug interactions and eligibility for participation in early phase clinical trials.
Additionally, there is specific concern regarding the use of immunotherapeutics. A major impediment to effective in vivo activity in patients with primary brain tumours is the oft-needed baseline use of corticosteroids to control intra-cerebral edema. It is well known that corticosteroids diminish immune activity and therefore their presence at baseline could impair the robustness of any anti-tumour immune response. In this respect, combination strategies with drugs such as bevacizumab which may have a steroid sparing effect [118] may augment anti-tumour immunity. Moreover, if a response was nevertheless to occur, there remains concern that tumour flare may present with mass effect like symptoms, which can be quite significant in a patient population already suffering from cerebral edema, or auto-immune neurotoxicity. Caution must continue, though it is reassuring that most reported studies of checkpoint inhibitors in glioblastoma to date have not shown an adverse event profile substantially dissimilar to other solid tumours which mitigates the latter point [8, 119].
Finally, although the various immune combination strategies described in this review hold promise due to their underlying biological rationale, implementation of any of these strategies needs to take into account the cost of these technologies with a keen focus on the ultimate value delivered to be patients [120].
CONCLUSION
In conclusion, despite the disappointing results of single agent immunotherapeutics to date, there remain reasons to be not only be optimistic, but excited. Understanding the CNS cancer immunity cycle provides a suitable framework upon which the various approaches and challenges to CNS drug development can be expounded and will be the foundation for the development of rational combination strategies to improve patient outcomes in this disease.
CONFLICTS OF INTEREST
The authors declared that there has no conflicts of interest.
REFERENCES
1. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 2010:711–23.
2. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N Engl J Med. 2015; 372:2018–28.
3. Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, Vaishampayan UN, Drabkin HA, George S, Logan TF, Margolin KA, Plimack ER, Lambert AM, et al. Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J Clin Oncol. 2014; 33:1430–7.
4. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, Savage KJ, Hernberg MM, Lebbé C, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015; 372:320–30.
5. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013; 369:122–33.
6. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, Barlesi F, Kohlhäufl M, Arrieta O, et al. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N Engl J Med. 2015; 373:1627–39.
7. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, Castellano D, Choueiri TK, Gurney H, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015; 373:1803–13.
8. Reardon D, Omuro A, Brandes A, Rieger J, Wick A, Sepulveda J, Phuphanich S, de Souza P, Ahluwalia M, Vlahovic LG, Sampson J. OS10. 3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: checkmate 143. Neuro Oncol. 2017; 19:iii21–III.
9. Reardon DA, Kaley TJ, Dietrich J, Clarke JL, Dunn GP, Lim M, Cloughesy TF, Gan HK, Park AJ, Schwarzenberger P, Ricciardi T, Macri MJ, Ryan A, et al. Phase 2 study to evaluate safety and efficacy of medi4736 (durvalumab [DUR]) in glioblastoma (GBM) patients: an update. Am Soc Clin Oncol. 2017.
10. Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006; 213:48–65.
11. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013; 39:1–10.
12. Streilein JW. Immune privilege as the result of local tissue barriers and immunosuppressive microenvironments. Curr Opin Immunol. 1993; 5:428–32.
13. Pachter JS, de Vries HE, Fabry Z. The blood-brain barrier and its role in immune privilege in the central nervous system. J Neuropathol Exp Neurol. 2003; 62:593–604.
14. Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006; 7:41.
15. Muldoon LL, Alvarez JI, Begley DJ, Boado RJ, del Zoppo GJ, Doolittle ND, Engelhardt B, Hallenbeck JM, Lonser RR, Ohlfest JR, Prat A, Scarpa M, Smeyne RJ, et al. Immunologic privilege in the central nervous system and the blood–brain barrier. J Cereb Blood Flow Metab. 2012; 33:13–21.
16. D’Agostino PM, Gottfried-Blackmore A, Anandasabapathy N, Bulloch K. Brain dendritic cells: biology and pathology. Acta Neuropathol. 2012; 124:599–614.
17. Lampson L, Hickey W. Monoclonal antibody analysis of MHC expression in human brain biopsies: tissue ranging from "histologically normal" to that showing different levels of glial tumor involvement. J Immunol Res. 1986; 136:4054–62.
18. Ebner F, Brandt C, Thiele P, Richter D, Schliesser U, Siffrin V, Schueler J, Stubbe T, Ellinghaus A, Meisel C, Sawitzki B, Nitsch R. Microglial activation milieu controls regulatory t cell responses. J Immunol. 2013; 191:5594–602.
19. Engelhardt B, Carare RO, Bechmann I, Flugel A, Laman JD, Weller RO. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. 2016; 132:317–38.
20. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, Harris TH, Kipnis J. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015; 523:337–41.
21. Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Disproportionate recruitment of cd8+ t cells into the central nervous system by professional antigen-presenting cells. Am J Pathol. 1999; 154:481–94.
22. Krakowski ML, Owens T. Naive T lymphocytes traffic to inflamed central nervous system, but require antigen recognition for activation. Eur J Immunol. 2000; 30:1002–9.
23. Dehouck MP, Meresse S, Delorme P, Fruchart JC, Cecchelli R. An easier, reproducible, and mass-production method to study the blood-brain barrier in vitro. J Neurochem. 1990; 54:1798–801.
24. Huber JD, Egleton RD, Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood–brain barrier. Trends Neurosci. 2001; 24:719–25.
25. Schneider SW, Ludwig T, Tatenhorst L, Braune S, Oberleithner H, Senner V, Paulus W. Glioblastoma cells release factors that disrupt blood-brain barrier features. Acta Neuropathol. 2004; 107:272–6.
26. Rossi M, Hughes J, Esiri M, Coakham H, Brownell D. Immunohistological study of mononuclear cell infiltrate in malignant gliomas. Acta Neuropathol. 1987; 74:269–77.
27. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wöhrer A, Dieckmann K, Filipits M, Brandstetter A, Weller M, Kurscheid S, Hegi ME, Zielinski CC, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2014; 17:1064–75.
28. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006; 6:24.
29. Aloisi F. Immune function of microglia. Glia. 2001; 36:165–79.
30. Hayes G, Woodroofe M, Cuzner M. Microglia are the major cell type expressing MHC class II in human white matter. J Neurol Sci. 1987; 80:25–37.
31. Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Rev. 1995; 20:269–87.
32. Andersson P, Perry V, Gordon S. The acute inflammatory response to lipopolysaccharide in cns parenchyma differs from that in other body tissues. Neuroscience. 1992; 48:169–86.
33. Esen N, Kielian T. Central role for MyD88 in the responses of microglia to pathogen-associated molecular patterns. J Immunol. 2006; 176:6802–11.
34. Serot JM, Foliguet B, Béné MC, Faure GC. Ultrastructural and immunohistological evidence for dendritic-like cells within human choroid plexus epithelium. Neuroreport. 1997; 8:1995–8.
35. Hussain SF, Heimberger AB. Immunotherapy for human glioma: innovative approaches and recent results. Expert Rev Anticancer Ther. 2005; 5:777–90.
36. Karman J, Ling C, Sandor M, Fabry Z. Initiation of immune responses in brain is promoted by local dendritic cells. J Immunol. 2004; 173:2353–61.
37. de Vos AF, van Meurs M, Brok HP, Boven LA, Hintzen RQ, van der Valk P, Ravid R, Rensing S, Boon L, 't Hart BA, Laman JD. Transfer of central nervous system autoantigens and presentation in secondary lymphoid organs. J Immunol. 2002; 169:5415–23.
38. van Zwam M, Huizinga R, Heijmans N, van Meurs M, Wierenga-Wolf AF, Melief MJ, Hintzen RQ, 't Hart BA, Amor S, Boven LA, Laman JD, et al. Surgical excision of CNS-draining lymph nodes reduces relapse severity in chronic-relapsing experimental autoimmune encephalomyelitis. J Pathol. 2009; 217:543–51.
39. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. 2017; 18:123–31.
40. Yang I, Tihan T, Han SJ, Wrensch MR, Wiencke J, Sughrue ME, Parsa AT. CD8+ T-cell infiltrate in newly diagnosed glioblastoma is associated with long-term survival. J Clin Neurosci. 2010; 17:1381–5.
41. Stemmler HJ, Schmitt M, Willems A, Bernhard H, Harbeck N, Heinemann V. Ratio of trastuzumab levels in serum and cerebrospinal fluid is altered in HER2-positive breast cancer patients with brain metastases and impairment of blood–brain barrier. Anticancer Drugs. 2007; 18:23–8.
42. Gustafson MP, Lin Y, New KC, Bulur PA, O’Neill BP, Gastineau DA, Dietz AB. Systemic immune suppression in glioblastoma: the interplay between CD14+ HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncology. 2010; 12:631–44.
43. Heimberger AB, Sampson JH. Immunotherapy coming of age: what will it take to make it standard of care for glioblastoma? Neuro Oncology. 2010; 13:3–13.
44. Hao C, Parney IF, Roa WH, Turner J, Petruk KC, Ramsay DA. Cytokine and cytokine receptor mRNA expression in human glioblastomas: evidence of Th1, Th2 and Th3 cytokine dysregulation. Acta Neuropathol. 2002; 103:171–8.
45. Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, Herndon JE 2nd, Bigner DD, Dranoff G, Sampson JH. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006; 66:3294–302.
46. Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, Mellor A. IDO activates regulatory t cells and blocks their conversion into Th17-like t cells. J Immunol. 2009; 183:2475–83.
47. Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, Tobias AL, Han Y, Lesniak MS. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012; 18:6110–21.
48. Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van den Bossche J, Mack M, Pipeleers D, In't Veld P, De Baetselier P, Van Ginderachter JA. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C (high) monocytes. Cancer Res. 2010; 70:5728–39.
49. Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol. 2008; 216:15–24.
50. Reardon D, Kaley T, Dietrich J, Lim M, Dunn G, Gan H, Cloughesy T, Clarke J, Park A, Macri M, Ryan A, Ricciardi T, Reddy V, Venhaus R. Atim-04. phase 2 study to evaluate the clinical efficacy and safety of MEDI4736 (durvalumab [dur]) in patients with glioblastoma (GBM): results for cohort b (dur monotherapy), bevacizumab (BEV) naïve patients with recurrent GBM. Neuro Oncol. 2016; 18:vi18–vi.
51. Reardon DA, De Groot JF, Colman H, Jordan JT, Daras M, Clarke JL, Nghiemphu PL, Gaffey SC, Peters KB. Safety of pembrolizumab in combination with bevacizumab in recurrent glioblastoma (rGBM). J Clin Oncol. 2016.
52. Reardon DA, Sampson JH, Sahebjam S, Lim M, Baehring JM, Vlahovic G, Cloughesy TF, Strauss LC, Latek RR, Paliwal P. Safety and activity of nivolumab (nivo) monotherapy and nivo in combination with ipilimumab (ipi) in recurrent glioblastoma (GBM): updated results from checkmate-143. Am Soc Clin Oncol; 2016.
53. Reardon DA, Kim TM, Frenel JS, Santoro A, Lopez J, Subramaniam DS, Siu LL, Rodon J, Tamura K, Saraf S, Morosky A, Stein K, Soria JC. ATIM-35. Results of the phase IB keynote-028 multi-cohort trial of pembrolizumab monotherapy in patients with recurrent PD-L1-positive glioblastoma multiforme (GBM). Neuro Oncology. 2016; 18:vi25–vi6.
54. Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V, Zhukova N, Mason G, Farah R, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016; 34:2206–11.
55. Shlien A, Campbell BB, de Borja R, Alexandrov LB, Merico D, Wedge D, Van Loo P, Tarpey PS, Coupland P, Behjati S, Pollett A, Lipman T, Heidari A, et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat Genet. 2015; 47:257–62.
56. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science. 2015; 348:124–8.
57. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–21.
58. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, Asthana S, Jalbert LE, Nelson SJ, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014; 343:189–93.
59. Draaisma K, Wijnenga MM, Weenink B, Gao Y, Smid M, Robe P, van den Bent MJ, French PM. PI3 kinase mutations and mutational load as poor prognostic markers in diffuse glioma patients. Acta Neuropathol Commun. 2015; 3:88.
60. Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, Huse JT, de Groot J, Li S, Overwijk WW, Spetzler D, Heimberger AB. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol. 2017; 19:1047–57.
61. Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, Kroemer G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2007; 15:3.
62. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017; 17:97–111.
63. Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, Liu A, Sankey EW, Tam A, Xu H, Mathios D, Jackson CM, Harris-Bookman S, et al. Combination therapy with anti-PD-1, anti-tim-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017; 23:124–36.
64. Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E, Pradilla G, Ford E, Wong J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013; 86:343–9.
65. Sahebjam S, Johnstone PA, Forsyth P, Arrington J, Jaglal M, Tran ND, Vrionis FD, Etame AB, Wicklund M, Gatewood AL, Macaulay R, Chinnaiyan P, Yu M. Atim-15. A Phase I Trial of Hypofractionated Stereotactic Irradiation (HFSRT) with Pembrolizumab and Bevacizumab in patients with recurrent high grade gliomas. Neuro Oncol. 2016; 18:vi21–vi.
66. Sahebjam S, Forsyth P, Arrington J, Jaglal M, Tran ND, Etame AB, Wicklund M, Drury-Sibiga A, Long W, Gatewood BE, Macaulay R, Chinnaiyan P, Yu M. Atim-18. A Phase I Trial of Hypofractionated Stereotactic Irradiation (HFSRT) with Pembrolizumab and Bevacizumab in patients with recurrent high grade glioma (NCT02313272). Neuro Oncol. 2017; 19:vi30–vi.
67. Brown MC, Holl E, Boczkowski D, Walton R, Bigner DD, Gromeier M, Nair SN. Oncolytic poliovirus directs tumor antigen presentation and T cell activation in vitro. J ImmunoTher Cancer. 2015; 3:P332.
68. Brown MC, Holl EK, Boczkowski D, Dobrikova E, Mosaheb M, Chandramohan V, Bigner DD, Gromeier M, Nair SK. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen–specific CTLs. Sci Transl Med. 2017; 9:eaan4220.
69. Desjardins A, Sampson JH, Peters KB, Vlahovic G, Randazzo D, Threatt S, Herndon JE, Boulton S, Lally-Goss D, McSherry F, Lipp ES, Friedman AH, Bigner DD. Patient survival on the dose escalation phase of the oncolytic polio/rhinovirus recombinant (PVSRIPO) against WHO grade IV malignant glioma (MG) clinical trial compared to historical controls. Am Soc Clin Oncol. 2016.
70. Lebel FM, Barrett JA, Chiocca EA, Yu J, Lukas RV, Nagpal S, Kumthekar P, Krishnan S, Cooper JN. Effect of controlled intratumoral viral delivery of Ad-RTS-hIL-12+ oral veledimex in subjects with recurrent or progressive glioma. Am Soc Clin Oncol. 2016.
71. Babu R, Adamson DC. Rindopepimut: an evidence-based review of its therapeutic potential in the treatment of EGFRviii-positive glioblastoma. Core Evid. 2012; 7:93–103.
72. Gan HK, Kaye AH, Luwor RB. The EGFRviii variant in glioblastoma multiforme. J Clin Neurosci. 2009; 16:748–54.
73. Heimberger AB, Crotty LE, Archer GE, Hess KR, Wikstrand CJ, Friedman AH, Friedman HS, Bigner DD, Sampson JH. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin Cancer Res. 2003; 9:4247–54.
74. Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, Mrugala MM, Jensen R, Baehring JM, Sloan A, Archer GE, Bigner DD, Cruickshank S, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the act III study. Neuro Oncol. 2015; 17:854–61.
75. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, Ashby L, Mechtler L, Goldlust SA, Iwamoto F, Drappatz J, O'Rourke DM, Wong M, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017; 18:1373–85.
76. Migliorini D, Dutoit V. Atim-21. ima950 peptide-based vaccine adjuvanted with poly-iclc in combination with standard therapy in newly diagnosed hla-a2 glioblastoma patients: preliminary results. Neuro Oncol. 2016; 18:vi22–vi.
77. Terasaki M, Shibui S, Narita Y, Fujimaki T, Aoki T, Kajiwara K, Sawamura Y, Kurisu K, Mineta T, Yamada A, Itoh K. Phase I trial of a personalized peptide vaccine for patients positive for human leukocyte antigen–a24 with recurrent or progressive glioblastoma multiforme. J Clin Oncol. 2010; 29:337–44.
78. Sampson JH, Mitchell DA. Vaccination strategies for neuro-oncology. Neuro Oncol. 2015; 17:vii15–vii25.
79. Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic t-cells in patients with malignant glioma. Cancer Res. 2004; 64:4973–9.
80. Wheeler CJ, Black KL, Liu G, Mazer M, Zhang XX, Pepkowitz S, Goldfinger D, Ng H, Irvin D, Yu JS. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008; 68:5955–64.
81. Yamanaka R, Homma J, Yajima N, Tsuchiya N, Sano M, Kobayashi T, Yoshida S, Abe T, Narita M, Takahashi M, Tanaka R. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res. 2005; 11:4160–7.
82. Phuphanich S, Wheeler C, Rudnick J, Hu J, Mazer M, Sanchez C, Nuno M, Chu R, Black K, Yu J. ATIM-25. ten-year follow up with long term remission in patients with newly diagnosed glioblastoma (GBM) treated with ICT-107 vaccine (phase I). Neuro Oncol. 2016; 18-:vi23–vi.
83. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, Golstein P. A new member of the immunoglobulin superfamily—CTLA-4. Nature. 1987; 328:267–70.
84. Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human b7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994; 1:793–801.
85. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over foxp3+ regulatory t cell function. Science. 2008; 322:271–5.
86. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti–CTLA-4 antibodies. J Exp Med. 2009; 206:1717–25.
87. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009; 229:12–26.
88. Tai X, Van Laethem F, Pobezinsky L, Guinter T, Sharrow SO, Adams A, Granger L, Kruhlak M, Lindsten T, Thompson CB, Feigenbaum L, Singer A. Basis of CTLA-4 function in regulatory and conventional CD4+ T cells. Blood. 2012; 119:5155–63.
89. Agarwalla P, Barnard Z, Fecci P, Dranoff G, Curry WT Jr. Sequential immunotherapy by vaccination with GM-CSF expressing glioma cells and CTLA-4 blockade effectively treats established murine intracranial tumors. J Immunother. 2012; 35:385.
90. Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, Jones KL, Conway AS, Liao X, Zhou J, Wen PY, Van Den Abbeele AD, Hodi FS, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2016; 4:124–35.
91. Khasraw M, Ameratunga M, Grommes C. Bevacizumab for the treatment of high-grade glioma: an update after phase III trials. Expert Opin Biol Ther. 2014; 14:729–40.
92. Chinot O, de La Motte Rouge T, Moore N, Zeaiter A, Das A, Phillips H, Modrusan Z, Cloughesy T. AvAglio: phase 3 trial of bevacizumab plus temozolomide and radiotherapy in newly diagnosed glioblastoma multiforme. Adv Ther. 2011; 28:334–40.
93. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, Jeraj R, Brown PD, Jaeckle KA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014; 370:699–708.
94. Khasraw M, Ameratunga MS, Grant R, Wheeler H, Pavlakis N. Antiangiogenic therapy for high-grade glioma. Cochrane Libr. 2014.
95. Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013; 73:2943–8.
96. Keunen O, Johansson M, Oudin A, Sanzey M, Rahim SA, Fack F, Thorsen F, Taxt T, Bartos M, Jirik R, Miletic H, Wang J, Stieber D, et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc Natl Acad Sci U S A. 2011; 108:3749–54.
97. Talasila KM, Røsland GV, Hagland HR, Eskilsson E, Flønes IH, Fritah S, Azuaje F, Atai N, Harter PN, Mittelbronn M, Andersen M, Joseph JV, Hossain JA, et al. The angiogenic switch leads to a metabolic shift in human glioblastoma. Neuro Oncol. 2017; 19:383–93.
98. Rubin P, Gash D, Hansen J, Nelson D, Williams J. Disruption of the blood-brain barrier as the primary effect of CNS irradiation. Radiother Oncol. 1994; 31:51–60.
99. Baird RD, Garcia-Corbacho J, Linossi C, Kumar SS, Smith D, Williams M, Qian W, Machin A, Ahmad S, Matys T, Jena R, Pacey S, Caldas C, et al. Cambridge brain mets trial 1 (CamBMT1): a proof of principle study of afatinib penetration into cerebral metastases (mets) for patients (pts) undergoing neurosurgical resection, combined with low-dose, targeted radiotherapy (RT)—Phase 1b results. Am Soc Clin Oncol; 2017.
100. Carpentier A, Canney M, Vignot A, Reina V, Beccaria K, Horodyckid C, Karachi C, Leclercq D, Lafon C, Chapelon JY, Capelle L, Cornu P, Sanson M, et al. Clinical trial of blood-brain barrier disruption by pulsed ultrasound. Sci Transl Med. 2016; 8:343re2.
101. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989; 86:10024–8.
102. Yang XH. A new model T on the horizon? Cell. 2017; 171:1–3.
103. Maus MV, Fraietta JA, Levine BL, Kalos M, Zhao Y, June CH. Adoptive immunotherapy for cancer or viruses. Annu Rev Immunol. 2014; 32:189–225.
104. Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, Nace AK, Dentchev T, Thekkat P, Loew A, Boesteanu AC, Cogdill AP, Chen T, et al. Rational development and characterization of humanized anti–EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Trans Med. 2015; 7:275ra22–ra22.
105. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJ, Martinez-Lage M, Brem S, Maloney E, Shen A, Isaacs R, Mohan S, Plesa G, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Trans Med. 2017; 9:eaaa0984.
106. Brown C, Alizadeh D, Starr R, Weng L, Wagner J, Naranjo A, Blanchard S, Kilpatrick J, Simpson J, Ressel JA, Jensen M, Portnow J, D'Apuzzo M, et al. Atim-13. Phase I study of chimeric antigen receptor-engineered T cells targeting iL13rα2 for the treatment of glioblastoma. Neuro Oncol. 2016; 18:vi20–vi.
107. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, Kurien A, Priceman SJ, Wang X, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016; 375:2561–9.
108. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, Wu MF, Orange JS, Sumazin P, Man TK, Joseph SK, Hegde M, Ahmed N, et al. Trivalent CAR T-cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2017:nox182–nox.
109. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011; 11:750.
110. Zhao HF, Wang J, Shao W, Wu CP, Chen ZP, To ST, Li WP. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: current preclinical and clinical development. Mol Cancer. 2017; 16:100.
111. Jochems C, Tritsch SR, Pellom ST, Su Z, Soon-Shiong P, Wong HC, Gulley JL, Schlom J. Analyses of functions of an anti-PD-L1/TGFβr2 bispecific fusion protein (M7824). Oncotarget. 2017; 8:75217-75231. https://doi.org/10.18632/oncotarget.20680.
112. Gulley JL, Heery CR, Schlom J, Madan RA, Cao L, Lamping E, Marte JL, Cordes LM, Christensen O, Helwig C, Strauss J. Preliminary results from a phase 1 trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting pd-l1 and tgf-β, in advanced solid tumors. J Clin Oncol. 2017; 35:3006.
113. Munn DH, Mellor AL. IDO and tolerance to tumors. Trends Mol Med. 2004; 10:15–8.
114. Beatty GL, O’Dwyer PJ, Clark J, Shi JG, Bowman KJ, Scherle PA, Newton RC, Schaub R, Maleski J, Leopold L, Gajewski TF. First-in-human phase 1 study of the oral inhibitor of indoleamine 2, 3-dioxygenase-1 epacadostat (INCB024360) in patients with advanced solid malignancies. Clin Cancer Res. 2017; 23:3269–76.
115. Zakharia Y, McWilliams R, Shaheen M, Grossman K, Drabick J, Milhem M, Rixie O, Khleif S, Lott R, Kennedy E, Munn D, Vahanian N, Link C. Abstract CT117: interim analysis of the phase 2 clinical trial of the IDO pathway inhibitor indoximod in combination with pembrolizumab for patients with advanced melanoma. AACR; 2017.
116. Dowlati A, Haaga J, Remick SC, Spiro TP, Gerson SL, Liu L, Berger SJ, Berger NA, Willson JK. Sequential tumor biopsies in early phase clinical trials of anticancer agents for pharmacodynamic evaluation. Clin Cancer Res. 2001; 7:2971–6.
117. Reardon DA, Fink KL, Mikkelsen T, Cloughesy TF, O’Neill A, Plotkin S, Glantz M, Ravin P, Raizer JJ, Rich KM, Schiff D, Shapiro WR, Burdette-Radoux S, et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol. 2008; 26:5610–7.
118. Vredenburgh JJ, Cloughesy T, Samant M, Prados M, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, Das A, Friedman HS. Corticosteroid use in patients with glioblastoma at first or second relapse treated with bevacizumab in the BRAIN study. Oncologist. 2010; 15:1329–34.
119. Reardon DA, Kaley TJ, Dietrich J, Clarke JL, Dunn GP, Lim M, Cloughesy TF, Gan HK, Park AJ, Schwarzenberger P, Ricciardi T, Macri MJ, Ryan A, et al. Phase 2 study to evaluate safety and efficacy of MEDI4736 (durvalumab [DUR]) in glioblastoma (GBM) patients: an update. J Clin Oncol. 2017; 35:2042.
120. Schnipper LE, Davidson NE, Wollins DS, Tyne C, Blayney DW, Blum D, Dicker AP, Ganz PA, Hoverman JR, Langdon R, Lyman GH, Meropol NJ, Mulvey T, et al. American society of clinical oncology statement: a conceptual framework to assess the value of cancer treatment options. J Clin Oncol. 2015; 33:2563–77.