
INTRODUCTION
Dysregulated metabolism is now accepted as one of the hallmarks of cancer [1], and metabolic alterations supporting high growth rate and energy requirements are essential for the sustained tumor growth and progression. The initial observations showcasing the fact that tumor cells are highly glycolytic in nature, led to studies focusing on metabolic variations as a target for cancer therapy [2]. Glucose metabolism henceforth has been extensively studied; however, in recent years, studies have recognized that the metabolic rewiring aiding cancer growth are not just limited to glucose metabolism but also involved other metabolic fluxes including lipid and amino acids [3, 4]. The importance of lipids in cancer progression was established after several studies showing that normal cells, excluding liver and adipose, meet their requirement of lipids through uptake of free fatty acids from the diet; however, in case of cancer cells, more than 90% of their elevated lipid needs, are fulfilled by de novo lipogenesis [5-7]. Regarding prostate cancer (PCA), several studies have shown that its precursor lesions undergo exacerbated endogenous lipogenesis, irrespective of extracellular or circulating lipids levels [6-8]. The higher de novo lipogenesis in PCA cells has been linked with their increased demand for membranes, energy storage, redox balance, protection from cell death, and activation of several intracellular signaling pathways during uncontrolled cellular proliferation [6-9]. Besides, during androgen deprivation therapy, lipids (cholesterol) play an important role in the de novo synthesis of androgens by PCA cells, providing them self-sufficiency in androgen receptor (AR) signaling and hormone-refractory progression [10, 11]. This unique dependence of PCA cells on lipids for their growth and progression provides an excellent opportunity to reduce PCA burden via inhibiting lipogenesis and associated molecular regulators using non-toxic small molecules. Silibinin, isolated from the seeds of milk thistle (Silybum marianum) plant, is widely consumed as a hepatoprotective agent and has shown strong efficacy against PCA both as anti-cancer and chemopreventive agent in various cell culture and animal models, and is currently being investigated for its beneficial effects in PCA patients [12-17]. Accordingly, in the present study, employing both PCA and non-neoplastic prostate epithelial cells, for the first time we examined the detailed effect of silibinin on cellular lipid levels as well as molecular regulators of lipogenesis.
In PCA cells, glucose-derived carbons are shunted from the mitochondrial TCA cycle (as citric acid) to cytosol for lipid synthesis. Importantly, expression and activity of enzymes involved in lipid synthesis such as ATP-citrate lyase (ACLY), acetyl Co-A carboxylase (ACC), fatty acid synthase (FASN), and stearoyl-CoA desaturase 1 (SCD1) are upregulated and play an important role in PCA [6, 7, 9, 18, 19]. Furthermore, the expression of master transcriptional regulator of lipid synthesis enzymes, sterol regulatory element binding protein 1 (SREBP1), is strongly correlated with Gleason grade [7, 20]. SREBP1 overexpression alone is sufficient to increase tumorigenicity and invasiveness of PCA cells, while its inhibition decreases de novo fatty acid synthesis and causes PCA growth inhibition and apoptosis induction [6, 20]. SREBP1 is also the critical link between oncogenic signaling and tumor metabolism [7]. For example, Akt and mTORC1 promote nuclear accumulation of mature SREBP1, and in turn Akt/mTORC1 signaling is activated by SREBP1-mediated lipogenesis [21]. Similarly, a negative regulator of mTOR pathway, AMP-activated protein kinase (AMPK) is reported to phosphorylate SREBP1 and prevent its proteolytic activation [6, 8]. Our extensively published studies have shown that silibinin targets various components of oncogenic signaling in a panel of human and mouse PCA cells and animal models [22-26]; however, silibinin effect on SREBP1 expression as well as its role in the anti-cancer efficacy of silibinin have not been examined yet. Results from present study showed that silibinin effectively decreases SREBP1 expression through AMPK activation in PCA cells, and that silibinin-mediated SREBP1 inhibition is critical for its anti-cancer efficacy against PCA. Since lipid synthesis in PCA cells is controlled by androgens, and under low androgen conditions, lipogenesis regulators play an important role in androgen biosynthesis [27, 28], we also examined silibinin effect on androgen-induced lipid accumulation as well as lipogenesis regulators (SREBP1/2) expression under low androgen conditions. Our results showed that silibinin treatment strongly inhibited the synthetic androgen R1881-induced lipid accumulation as well as completely abrogated the development of androgen-independent clones via targeting SREBP1/2 expression under low androgen condition.
RESULTS
Human PCA cells exhibit lipogenic phenotype
In order to understand how PCA cells are unique in terms of their metabolic profile, we first evaluated a series of prostate/PCA cell lines for their glucose and fat uptake rates as well as endogenous lipid levels. We selected non-neoplastic benign human prostate RWPE-1, and neoplastic cells (WPE1-NA22 and WPE1-NB14) derived from RWPE-1 [29], and a panel of human PCA cell lines (androgen dependent LNCaP as well as androgen-independent DU145 and PC3 cells), and also included non-small cell lung carcinoma (NSCLC) A549 cells for comparison. As shown in Figure 1A, prostate/PCA cell lines did uptake glucose that was dependent upon their individual cell growth rate in culture; however, there was no clear trend correlating glucose consumption with aggressiveness of these cell lines, e.g. glucose consumption between non-neoplastic RWPE-1 and prostate adenocarcinoma PC3 cells was almost similar (Figure 1A). Interestingly, glucose uptake by prostate/PCA cells was much lower when compared with NSCLC A549 cells (Figure 1A), suggesting their relatively lesser dependence on glucose metabolism.
Next, we assessed the uptake of fatty acids by these cell lines and to our surprise, all prostate cell lines showed much higher fatty acid uptake compared to A549 cells (Figure 1B). Also, there was some trend in terms of higher fatty acid uptake correlating with cells’ aggressiveness, e.g. fatty acid uptake trend of WPE1-NB14 > WPE1-NA22 >RWPE-1 matches with their aggressiveness (Figure 1B). On the contrary, LNCaP cells showed higher fatty acid uptake compared with more aggressive DU145 and PC3 cells, which might be related with the functional AR signaling in LNCaP cells (Figure 1B). Together, glucose and fatty acid uptake data underline the dependence of prostate cells on fatty acid precursors compared to A549 cells, which are more glycolytic in nature.
Next, we assessed the neutral lipid content of various prostate/PCA cells by Oil red O (ORO) staining that mainly stains cellular cholesteryl esters and neutral triglycerides. In this assay, we included non-neoplastic immortalized PWR-1E cells along with other cell lines (RWPE-1, WPE1-NA22, WPE1-NB14, LNCaP, DU145 and PC3). We observed higher ORO staining in all PCA cells, with LNCaP cells showing highest lipid accumulation, while least staining was observed in non-neoplastic PWR-1E and RWPE-1 cells (Figure 1C). This data supported the notion that PCA cells have higher content of neutral lipids.
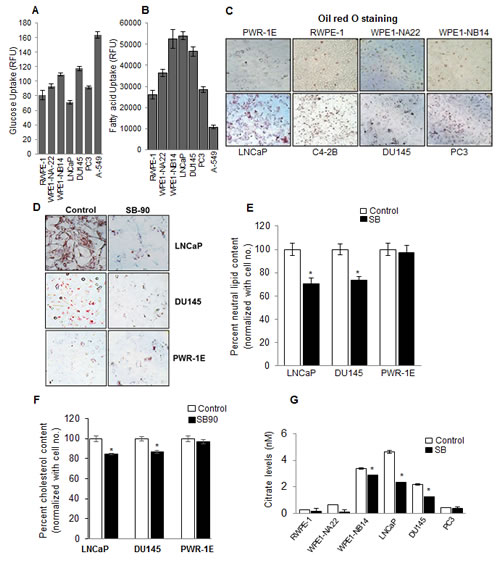
Figure 1: PCA cells exhibit a lipogenic phenotype, and silibinin inhibits neutral lipids, free cholesterol and citrate levels selectively in human PCA cells. In each case, equal number of cells were plated and analyzed for (A) glucose uptake, (B) fatty acid uptake, and (C) neutral lipids by ORO staining following procedures detailed in the ‘Materials and Methods’. (D-E) Photomicrographs (at 200x) and quantification of ORO staining in human PCA cells (LNCaP and DU145) and normal prostate epithelial PWR-1E cells following silibinin (90 µM) treatment for 48 h. Quantification data shown for ORO staining was normalized with respective cell number for each group. (F) Percent cholesterol content measured by Filipin III staining following silibinin (90 µM) treatment for 48 h in LNCaP, DU145 and PWR-1E, cells. Quantification data shown for cholesterol content was normalized with respective cell number for each group. (G) Cells were treated with DMSO or silibinin (90 µM) for 48 h; cell lysates were prepared and analyzed for intracellular citrate levels. *, p≤0.001. Abbreviations: SB: Silibinin, RFU: Relative fluorescence units.
Silibinin specifically inhibits neutral lipid, cholesterol and citrate levels in PCA cells
Since we observed that PCA cells possess higher turnover rates of lipid and cholesterol discussed above, we next chose to assess whether silibinin treatment could affect the lipid content in these cells. As shown in Figure 1D and 1E, silibinin treatment significantly reduced the neutral lipid level in PCA LNCaP and DU145 cells but not in non-neoplastic PWR-1E cells. Since ORO dye stains for cholesteryl esters, we next assessed the effect of silibinin on unesterified cholesterol by Filipin III staining. Similar to the results obtained with neutral lipids, we observed a significant decrease in the unesterified cholesterol following silibinin treatment in both LNCaP and DU145 cells but not in PWR-1E cells (Figure 1F).
Normal prostate cells are also known to produce and secrete high amount of citrate into the seminal fluid; however, PCA cells overexpress ACLY, an enzyme that converts citrate to acetyl CoA, which is the substrate for fatty acid biosynthesis. ACLY-catalyzed reaction also generates energy (NADPH) required for fatty acid synthesis. Therefore, next, we measured the level of intracellular citrate in prostate/PCA cells. We observed that the cancer cells especially WPE1-NB14, LNCaP and DU145 have higher level of intracellular citrate (Figure 1G), corresponding to higher lipid accumulation in these cells (Figure 1C), and importantly, silibinin treatment significantly reduced the citrate levels in WPE1-NB14, LNCaP and DU145 cells (Figure 1G).
Silibinin decreases nuclear SREBP1 level via inhibiting its proteolytic activation, and thus down-regulates SREBPs target genes involved in fatty acid and cholesterol metabolism in PCA cells
SREBPs regulate the transcriptional activation of several genes involved in fatty acid biosynthesis and cholesterol metabolism; therefore, we next analyzed the effect of silibinin on SREBP1 and 2 expression levels. Silibinin treatment did not significantly alter the total levels of the SREBP1/2 in both LNCaP and DU145 cells (data not shown); however, there was a profound decrease in the nuclear SREBP1 following silibinin treatment in both LNCaP and DU145 cells but not in PWR-1E cells (Figure 2A). Similarly, a decrease in SREBP2 protein level was evident only in the nuclear fractions of LNCaP and DU145 cells but not in PWR-1E cells following silibinin treatment (Figure 2A). The observed decrease in nuclear SREBP1 levels by silibinin could be through its effect on SREBP1 transcription and/or through post-translational modifications. To address that, we first examined silibinin effect on SREBP1 mRNA level after 24 and 48 h of its treatment, but found no effect on SREBP1 mRNA level (Figure 2B).
The mammalian SREBP1 is synthesized as 125 kDa precursor with an N-terminal transcription factor domain. In its inactive form, the precursor SREBP1 is present in a complex with SREBP cleavage-activating protein (SCAP), which in turn is bound with another protein, INSIG, and this complex remains bound to the ER membrane [30]. During starvation or sterol depleted condition, there is a decrease in the SCAP-INSIG interaction and the SCAP-SREBP complex translocates to the Golgi apparatus where two sequential proteolytic cleavage marks the activation of SREBPs [31]. The mature soluble SREBP1 (68 kDa) rapidly translocate to the nucleus and activates the target genes by binding to the sterol response element (SRE) within their promoter regions [32]. Since we did not observe any change in SREBP1 mRNA levels with silibinin treatment, but there was a decrease in nuclear SREBP1 levels, we next examined the SREBP1 precursor level after treatment with silibinin and found that while the precursor SREBP1 level was increased, the mature SREBP1 level was decreased, further validating that silibinin inhibits the proteolytic cleavage of SREBP1 (Figure 2C). Next, to confirm inhibitory effect of silibinin on nuclear SREBP1, we employed confocal microscopy, and as shown in Figure 2D (images and bar diagram), SREBP1 expression was mainly nuclear in untreated PCA cells and silibinin treatment reduced the nuclear SREBP1 level while significantly increasing the cytoplasmic SREBP1 level.
Next, we examined the effect of silibinin on the protein expression of several genes whose expression is controlled transcriptionally by SREBPs. As shown in Figure 2E, silibinin treatment strongly decreased the FASN level in both LNCaP and DU145 cells. ACC is another important enzyme involved in maintaining fatty acid homeostasis by catalyzing the carboxylation of acetyl-CoA to form malonyl-CoA. This step precedes the fatty acid synthesis steps catalyzed by FASN, therefore, ACC is considered an important target in PCA [33]. ACC is inactivated by a phosphorylation at Serine-79 site by AMPK or at Serine-1200 site by protein kinase A [34]. We found that there was a significant up-regulation of ACC phosphorylation at Serine-79 site especially in LNCaP cells by silibinin without any significant change in total ACC level (Figure 2E), suggesting an inactivation of ACC following silibinin treatment. Another key regulator of de novo fatty acid synthesis is ACLY [35]. We found that silibinin inhibited the phosphorylation of ACLY at Serine-455 site (Figure 2E), thereby stopping the utilization of citrate for synthesis of acetyl-CoA. 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR) catalyzes the rate limiting step in cholesterol biosynthesis. It is suggested to be an important player in PCA progression, as it is shown to contribute to intra-tumoral androgen synthesis [10]. We found that silibinin strongly decreased the HMGCR level in both LNCaP and DU145 cells (Figure 2E). Another metabolic gene α-methylacyl-CoA racemase (AMACR) is reported to be overexpressed in PCA [36]. It is a peroxisomal and mitochondrial enzyme capable of racemizing the α-carbon of various α-methylacyl-CoA derivatives. We found that silibinin also strongly down-regulated the expression of AMACR especially in LNCaP cells.
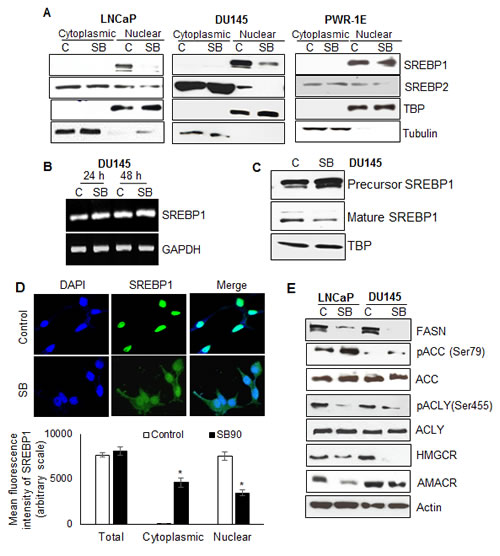
Figure 2: Silibinin decreases SREBP1/2 nuclear localization specifically in PCA cells, and modulates expression/phosphorylation of key molecules involved in lipogenesis. (A) LNCaP, DU145 and PWR-1E cells were treated with silibinin (90 μM) for 24 h, and nuclear and cytoplasmic fractions were prepared and analyzed for SREBP1 and SREBP2 expression by Western blotting. Membranes were stripped and reprobed for TBP and tubulin as nuclear and cytoplasmic loading controls, respectively. (B) RT-PCR analysis showing mRNA expression of SREBP1 after treatment with silibinin (90 μM) for 24 and 48 h. GAPDH was used as loading control. (C) Western blot for precursor SREBP1 (125 kDa) and mature SREBP1 (68 kDa) in DU145 cells after treatment with silibinin (90 μM) for 24 h. TBP was used as loading control. (D) Confocal images (upper panel) and quantitative fluorescence intensity (lower panel) for SREBP1 (green- AlexaFluor 488) in LNCaP cells after 24 h of treatment with silibinin (90 μM); DAPI (in blue) stains nuclei. (E) LNCaP and DU145 cells were treated with silibinin (90 μM) for 48 h, and total cell lysates were prepared and analyzed for the total and/or phosphorylated FASN, ACC, ACLY, HMGCR and AMACR by Western blotting. Actin was used as loading control. *, p≤0.001. Abbreviations: SB: Silibinin, TBP: TATA binding protein.
Silibinin regulates SREBP1 expression via activating AMPK
To examine potential mechanisms of silibinin-mediated decrease in SREBP1 expression, we focused on signaling mechanisms that regulate SREBP1 expression at post-translational level. One of the key signaling molecules, known to regulate SREBP1, is AMPK, which is an energy-sensing molecule with serine/threonine kinase activity and is activated in the cells in response to metabolic stress (with a higher AMP/ATP ratio) [37]. Once activated via phosphorylation at Threonine-172 site, AMPK inhibits bioenergetic pathways, especially lipogenesis by either direct phosphorylation of ACC or indirectly (via targeting SREBP1) to reduce the transcriptional activity of lipogenic genes such as FASN and ACLY [38]. Since we found down-regulation of SREBP1 and its target genes by silibinin, we inferred that this effect could be mediated by up-regulation of AMPK. Indeed, treatment with silibinin resulted in a robust up-regulation of AMPK phosphorylation in LNCaP and DU145 cells at 12 h that was completely inhibited in the presence of AMPK inhibitor, compound C (Figure 3A), prior to a decrease in SREBP1 expression by ~16 h following silibinin treatment (data not shown). Activated AMPK is known to inhibit SREBP1 activation and nuclear translocation by phosphorylating it at Serine-372 site [39], and thus we also employed confocal microscopy to assess whether SREBP1 was phosphorylated by AMPK at Serine-372 site. A prolific increase in the phosphorylated levels of SREBP1 was observed in both LNCaP and DU145 cells in response to silibinin treatment (Figure 3B). Consistent with these findings, next we examined whether inhibiting silibinin-caused AMPK activation would reverse silibinin effect on lipid accumulation. Indeed AMPK inhibitor compound C alone treatment resulted in a significant increase in lipid accumulation in both LNCaP and DU145 cells as measured by ORO staining, and when combined with silibinin, it reversed the inhibitory effect of silibinin on lipid accumulation in these PCA cell lines (Figure 3C, left and right panels).
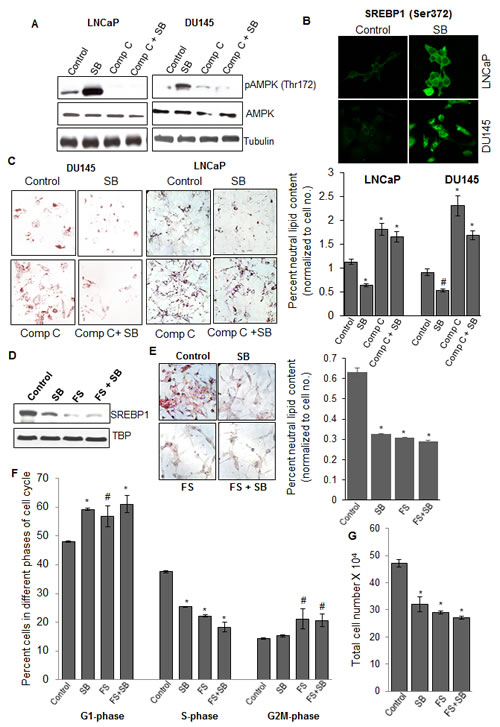
Figure 3: Silibinin decreases SREBP1 expression via activating AMPK, and does not show additional efficacy against PCA cells in the presence of SREBP1 inhibitor fatostatin. (A) LNCaP and DU145 cells were treated with silibinin (90 μM) with or without compound C (10 μM), and at the end of 12 h, total cell lysates were prepared and analyzed for phosphorylated and total AMPK levels by Western blotting. Membranes were stripped and probed with tubulin antibody to assess protein loading. (B) Confocal microscopy images (at 1000x) showing SREBP1 phosphorylation (Serine-372) following treatment with silibinin (90 μM) for 24 h in LNCaP and DU145 cells. (C) Photomicrographs (at 200x) (left panel) and quantification (right panel) of ORO staining in LNCaP and DU145 cells after 48 h of treatment with silibinin and/or compound C. Quantification data shown for ORO staining was normalized with respective cell number for each group. (D) LNCaP cells were treated with silibinin (90 μM) and/or fatostatin (20 μM), and 24 h later, nuclear lysates were prepared and analyzed for SREBP1 expression by Western blotting. Membranes were stripped and probed with TBP antibody to assess protein loading. LNCaP cells were treated with silibinin (90 μM) and/or fatostatin (20 μM) for 48 h and analyzed for: (E) ORO staining (photomicrographs at 200x, left panel; quantification, right panel), (F) percent of cell in different phases of cell cycle, and (G) total cell number. *, p≤0.001; #, p≤0.01. Abbreviations: Comp C: Compound C, SB: Silibinin, TBP: TATA binding protein, FS: Fatostatin.
Silibinin does not show additional efficacy against PCA cells in the presence of SREBP inhibitor fatostatin
Fatostatin (FS) is a synthetic inhibitor of SREBPs and has been shown to block the activation of SREBPs, thereby decreasing the transcription activity of downstream effectors [40]. Since we observed that silibinin mediates a similar action on SREBPs, we did an analogue experiment to further confirm that silibinin inhibits lipogenesis in PCA cells solely via SREBPs inhibition. Fatostatin treatment alone or in combination with silibinin resulted in a decrease in nuclear SREBP1, with combination showing a better decrease compared with either fatostatin or silibinin alone (Figure 3D). However, the decrease in neutral lipid level observed with combination was comparable to either agent alone, where fatostatin alone reduced the lipid content by 48%, silibinin by 51% and there combination by 54%, suggesting that there was no significant additional effect in combination (Figure 3E). Since inhibiting lipid synthesis would essentially deprive the cells of cellular biomolecules for membrane synthesis and other processes important for cell division, we propounded that silibinin-mediated reduction in lipid level would be associated with its (earlier reported) PCA cell growth inhibitory effects [12, 22, 23]. Accordingly, we next examined the effect of silibinin and fatostatin on cell cycle progression and cell proliferation. Fatostatin and silibinin alone or in combination resulted in comparable G1 phase arrest; 56%, 59% and 61% cells in G1 phase compared to 48% in control, respectively (Figure 3F). Similarly, individual treatment of fatostatin or silibinin resulted in 38% and 32% decrease in total cell number and their combination caused 42% decrease in total cell number (Figure 3G). Together, these results validated that both fatostatin and silibinin act by targeting the same molecule, i.e. SREBP1.
SREBP1 overexpression abrogates anti-PCA effects of silibinin
To test directly whether SREBP1 plays an essential role in the down-regulation of lipid accumulation and inhibition of cell growth in PCA cells by silibinin, we transduced DU145 cells with a lentiviral expression vector constitutively driving the expression of SREBP1. After lentiviral infection and selection, positive clones were checked for SREBP1 expression by immunoblot analysis to confirm the transduction and SREBP1 overexpression in SREBP1-DU145 cells compared to vector control-DU145 (VC-DU145) cells (Figure 4A). Next, effect of silibinin treatment was examined on neutral lipid content in both VC-DU145 and SREBP1-DU145 cells. We observed a 22% increase in the lipid content in SREBP1-DU145 cells compared with VC-DU145 cells, but importantly, the induced SREBP1 over-expression abrogated silibinin-mediated inhibitory effect on the lipid content (Figure 4B). The lipid content was reduced by 32% in VC-DU145 by silibinin, but only by 10% in SREBP1-DU145 cells (Figure 4B).
As mentioned above, since silibinin also induces a cytostatic effect in PCA cells by inducing a G1 arrest, we next questioned whether over-expression of SREBP1 would abrogate this effect of silibinin. We found that silibinin treatment of VC-DU145 cells resulted in a G1 arrest (59% cells in G1 phase compared to 51% in DMSO vehicle control, p≤0.001), but this effect of silibinin was compromised in SREBP1-DU145 cells showing no change in the cell cycle distribution (Figure 4C). Next, we compared the effect of silibinin treatment on cell growth in VC-DU145 and SREBP1-DU145 cells. Photomicrographs captured at the end of silibinin treatment clearly showed that it has a strong effect on the morphology and growth of VC-DU145 cells but not SREBP1-DU145 cells (Figure 4D, upper panel). Further quantitative analysis confirmed that whereas silibinin causes a strong and dose-dependent decrease in the cell growth of VC-DU145 cells, there was almost no growth inhibitor effect of silibinin in SREBP1-DU145 cells (Figure 4D, lower panel). Together, these results clearly suggested that silibinin-caused PCA inhibition is mainly via inhibiting SREBP1 and lipid accumulation in PCA cells.
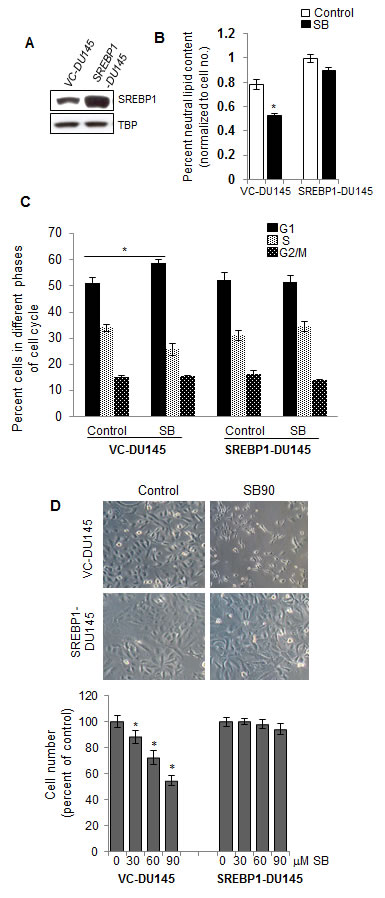
Figure 4: SREBP1 overexpression abrogates silibinin efficacy against PCA cells. (A) DU145 cells were transfected with pLX304-SREBP1 plasmid or empty vector using lentiviral transfection system and stable clones were identified through blasticidin-based selection. Western blotting was performed to show nuclear SREBP1 expression in vector control (VC-DU145) and SREBP1 overexpressing DU145 cells (SREBP1-DU145).TBP was used as loading control. (B) Quantification (normalized with respective cell number) of neutral lipid content in VC-DU145 and SREBP1-DU145 cells following treatment with silibinin (90 μM) for 48 h. (C) Cell cycle analysis by saponin-PI method after treatment with silibinin (60 μM) for 48 h in VC-DU145 and SREBP1-DU145 cells. (D) Representative pictures and total cell number quantification after treatment with various doses of silibinin (30-90 μM) for 48 h in VC-DU145 and SREBP1-DU145 cells. *, p≤0.001. Abbreviations: SB: Silibinin, TBP: TATA binding protein.
Silibinin inhibits androgen-induced lipid accumulation as well as androgen independent growth of LNCaP cells via targeting SREBPs
Androgens such as testosterone and dihydrotestosterone (DHT) are known to induce neutral lipid accumulation in LNCaP cells, while androgen antagonist Casodex inhibits lipogenesis [41]. This suggests that lipid accumulation and synthesis in PCA cells is influenced by androgens. In light of the fact that silibinin inhibits AR signaling in PCA cells [42, 43], we also sought to understand whether the inhibitory effect of silibinin on lipid content was mediated via its inhibitory effect on androgen-AR signaling. To study this, we stimulated LNCaP cells with 10 nM R1881 under charcoal stripped serum (cFBS) condition. We observed a robust accumulation of lipid in LNCaP cells in response to R1881 treatment, but silibinin addition along with R1881 completely inhibited the R1881-induced neutral lipid accumulation in these cells (Figure 5A). In another experiment, we cultured these cells in media containing R1881 for 96 h and treated them with silibinin after 24 h (i.e. for last 72 h) or after 72 h (i.e. only for last 24 h); we nonetheless found inhibition in lipid accumulation (Figure 5A, lower panel), advocating that silibinin could inhibit the androgen-induced lipid accumulation in PCA cells.
It is now believed that androgen-independent growth of PCA cells also depends upon lipogenesis, as lipids (such as cholesterol) act as precursors for the synthesis of androgens [44]. Accordingly, next we also examined whether silibinin could inhibit the hormone-refractory growth of PCA cells through its inhibitory effect on key lipogenesis regulator SREBP1/2. LNCaP cells were grown to 70% confluency and then shifted to androgen free 10% cFBS containing media and maintained in this media till androgen-independent clones emerged. Androgen withdrawal led to inhibition in cell growth, but significant increase in cell death started only after 3-4 weeks of cFBS treatment. Throughout the study, cells were washed and fresh media was added every 3rd day. In the control group, after 18 weeks of androgen withdrawal, we found development and re-growth of androgen-independent clones; however, in the flasks with 10 µM silibinin treatment, there was no re-growth or appearance of colonies even after 23rd week (Figure 5B), which suggests that silibinin inhibited the attainment of androgen-independent phenotype. RNA analysis of LNCaP cells collected at different time-points following androgen withdrawal, showed that SREBP1 and SREBP2 expression did not decrease much and/or remained higher even under androgen deprived conditions, but silibinin treatment reduced their expression at all the time-points (Figure 5C). This finding further supported our hypothesis that silibinin treatment specifically inhibited SREBP1 and SREBP2, which are important for the androgen-independent growth of these LNCaP PCA cells.
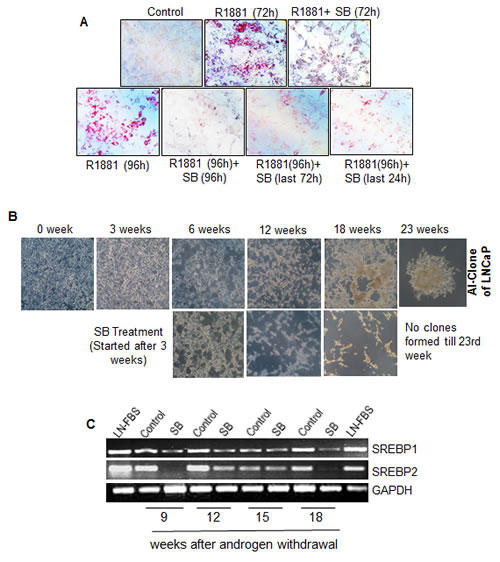
Figure 5: Silibinin inhibits R1881-induced lipid accumulation and androgen independent growth of LNCaP cells via decreasing the SREBP1/2 expression. (A) LNCaP cells (2X105 per plate) were seeded, and 24 h later, cells were starved for 24 h in 10% cFBS media and then treated daily with R1881 (10 nM) with or without silibinin (90 µM) for 72 or 96 h (upper panel) or induced with R1881 for 96 h and silibinin (90 µM) was added after 24 h or after 72 h (lower panel). At the end of each treatment, cells were fixed and stained with ORO. Representative photomicrographs of ORO staining are shown (at 200x). (B) LNCaP cells were grown to 70% confluency in T-75 flasks under 10% FBS condition, and then transferred to low androgen media (10% cFBS) and maintained till androgen independent clones developed. One set of flask was also treated with 10 µM silibinin, starting from the 3rd week of androgen deprivation. Representative pictures at different time intervals of the experiments are presented. (C) Employing the samples from the experiment shown in panel B, RNA was isolated at different time intervals and RT-PCR was performed to analyze the expression pattern of SREBP1 and SREBP2. GAPDH was used as a loading control. AI-clones of LNCaP cells are labeled as ‘control’ in the gels shown. Abbreviations: SB: Silibinin, GAPDH: Glyceraldehyde 3-phosphate dehydrogenase, AI: Androgen independent, LN-FBS: LNCaP cells grown under 10%FBS condition.
DISCUSSION
PCA is the most common non-cutaneous cancer in US men and according to the American Cancer Society reports, 233,000 new cases and 29,480 deaths from PCA are estimated in the United States in 2014 [45]. Although curative measures exist for PCA management, significant toxicity is associated with them and thousands of patients succumb to this deadly malignancy every year. Clearly, additional approaches and strategies are needed to better understand and target PCA. One such unique opportunity is to target aberrant lipogenesis in PCA, because a plethora of studies in recent years have shown that this metabolic feature is essential for PCA proliferation, survival, bioenergetics, chemoresistance, etc. [6-9, 19, 20, 46, 47]. However, studies focusing on understanding and targeting aberrant lipid metabolism in PCA cells by non-toxic natural agents are lacking, although a series of such agents have been studied extensively for their PCA efficacy [22, 48]. Results from the present study, for the first time, clearly suggest that natural flavonoid silibinin targets cellular lipid accumulation and, thereby, inhibits PCA cell proliferation.
Prostate tissue is unique in terms of endogenous citrate secretion and turnover. The human prostate gland accumulates and secrets extraordinarily high levels of citrate and zinc in the prostatic fluid (citrate concentration ranges from 40 to 150 mM), which is not found in any other tissues in the body [49]. However, PCA cells, instead of secreting citrate, divert it towards lipogenesis. In PCA cells, citric acid, produced in mitochondrion, is transported to the cytoplasm via citrate-pyruvate shuttle, where it is cleaved by ACLY to generate energy and acetyl CoA, a precursor for lipid biosynthesis. In the present study, we observed higher intracellular citrate level in PCA cells (WPE1-NB14, LNCaP and DU145 cells) compared to non-neoplastic RWPE-1 and less aggressive WPE1-NA22 cells. This suggests that PCA cells utilize citrate for de novo lipogenesis, and silibinin treatment reduced the intracellular citrate level via down-regulating ACLY expression. The decrease in intracellular citrate level by silibinin was also well reflected in the inhibition of neutral lipid droplets in PCA cells. Importantly, PCA cells consistently showed a higher lipid level compared with non-neoplastic prostate cells, which is in line with earlier published report showing an aberrant accumulation of esterified cholesterol in lipid droplets of high-grade PCA and metastases [50]. Importantly, the inhibitory effect of silibinin on neutral lipid and cholesterol level was quite specific to PCA cells and no significant change was observed in neutral lipid and cholesterol level in non-neoplastic PWR-1E cells following silibinin treatment. Moreover, a decrease in lipid level by silibinin correlated with its PCA specific growth inhibitory effects reported in this study as well as earlier [51]. This could be explained by the fact that most of the normal cells do not require de novo lipogenesis, and therefore, normal cells are less sensitive to inhibition of this pathway by silibinin.
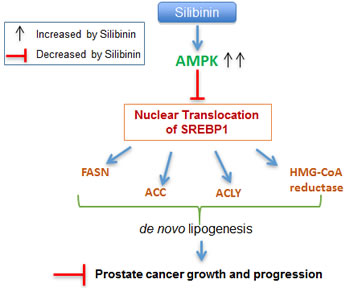
Figure 6: Schematic representation of silibinin-mediated action against lipogenesis and cell growth inhibition in PCA. Silibinin treatment activated AMPK, enhanced SREBP1 phosphorylation, decreased SREBP1 nuclear translocation and expression of SREBP1 regulated genes, thereby inhibited lipid accumulation and cell proliferation, and induced cell cycle arrest in PCA cells.
Lipogenesis is mainly controlled by SREBPs, and aberrant activation of SREBPs as well as their target genes has been reported in several cancer types, including PCA [52]. Huang et al have reported that SREBP1 is highly expressed in the nuclei of prostate tumor cells with higher Gleason grades, which correlates with poor prognosis and androgen-independent PCA progression [20]. Results from the present study showed that silibinin decreases the nuclear SREBP1/2 expression specifically in PCA cells. Earlier studies have clearly established that mTORC1 promotes nuclear accumulation of mature SREBP1 [21], while a negative regulator of mTOR pathway, AMPK, physically interacts and phosphorylates SREBP1 (at Ser-372 site) and suppresses its proteolytic cleavage and nuclear translocation [6, 8]. You et al reported that AMPK inhibition leads to activation of SREBP1-mediated lipogenesis [53]. Similarly, AMPK inhibition has been suggested as a causal event in ethanol-induced fatty liver [53]. AMPK activation restricts anabolic pathways including lipogenesis and represses cancer cells growth and induces apoptosis [54-58]. Therefore, we conjectured that silibinin might inhibit SREBP1-mediated lipogenesis through AMPK activation. Indeed, our results showed that silibinin strongly up-regulates AMPK activation in PCA cells, followed by robust inhibitory phosphorylation of SREBP1 at Serine-372 site. AMPK inhibition by compound C also resulted in up-regulation of lipid biosynthesis. This is in line with earlier report by Shi et al that inhibiting AMPK in hepatocytes resulted in lipid droplet accumulation [59]. Importantly, AMPK-mediated SREBP1 inactivation by silibinin seems to be central to its anti-PCA effects (Figure 6), as AMPK inhibitor reversed the inhibitory effect of silibinin on lipid accumulation, and also silibinin did not exert any additional efficacy against PCA cells in the presence of SREBP1 inhibitor fatostatin. Furthermore, growth inhibitory effects of silibinin were completely reversed in PCA cells overexpressing SREBP1. Together, these results suggest that SREBP1 is a novel silibinin target and plays a central role in silibinin efficacy against PCA cells (Figure 6).
De novo androgen synthesis and sustained activation of AR signaling are the central events during PCA re-growth under low androgen conditions during hormonal ablation therapy [60-65]. There are several studies suggesting that intra-tumoral androgens could be derived from de novo synthesized cholesterol as well as circulating cholesterol from diet [10, 44, 66]. Ettinger et al reported that in LNCaP xenograft model, expression levels of both SREBP1a and SREBP1c were significantly enhanced in mice 2-3 weeks following castration compared to their basal levels [28]. Similar results were also observed in clinical PCA specimens showing an up-regulation of SREBPs and their downstream effector genes during progression to androgen independence [28]. Results from the present study suggested that silibinin could prevent PCA hormone-refractory progression via targeting SREBP1/2 expression. Further studies are however needed in future to confirm silibinin effect on de novo synthesis of androgens from cholesterol and resultant activation of AR signaling in PCA cells especially under in vivo conditions. In conclusion, our results clearly show that silibinin decreases nuclear level of SREBP1 and lipid accumulation in PCA cells as a novel mechanism of its action against PCA growth and progression. Together, these mechanistic findings suggest that non-toxic silibinin could be useful in PCA prevention and treatment via targeting aberrant lipogenesis.
MATERIALS AND METHODS
Cell lines and reagents
All the cell lines were from from ATCC (Manassas, VA). LNCaP, DU145, PC3 and A549 cells were cultured in RPMI1640 media with 10% FBS (fetal bovine serum). PWR-1E, RWPE-1, WPE-NA22 and WPE-NB14 were maintained in keratinocyte growth medium with 5 ng/mL human recombinant epidermal growth factor and 0.05 mg/mL bovine pituitary extract; and as per experimental requirements, these cells were also cultured with 10% FBS or serum free keratinocyte growth medium. Media, serum and other cell culture materials were from Invitrogen-Life Technologies (Carlsbad, CA). Silibinin and fatostatin were from Sigma (St Louis, MO) and dissolved in DMSO as stock solution. The final concentration of DMSO in the culture medium during different treatments did not exceed 0.1% (v/v). Antibodies for SREBP1, HMGCR, and TATA-binding protein (TBP) were obtained from Abcam (Cambridge, MA).Total and/or phosphorylated antibodies for SREBP1, SREBP2, FASN, ACC, ACYL, AMPK, and AMACR were from Cell Signaling Technologies (Danvers, MA). Tubulin antibody was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Compound C was from Calbiochem (La Jolla, CA). R1881 was from Perkin Elmer (Waltham, MA). ECL detection system and anti-mouse HRP-conjugated secondary antibody were from GE Healthcare (Buckinghamshire, UK).
Glucose uptake assay
Glucose uptake was measured using the glucose assay-cell based assay kit from Caymen chemicals (Ann Arbor, MI). Cells were seeded at the density of 10,000 cells per well in a 96-well plate and maintained at 37ºC in CO2 incubator for 24 h. Next day, the media was removed and replaced with glucose free RPMI. After the end of 6 h, media containing 2-NBDG (150 µg/ml) was added to each well and incubated for 30 minutes in dark. Thereafter, media was removed and processed as per manufacture description. The fluorescence was measured using a plate reader at λex=485 nm and λem=535 nm.
Fatty acid uptake assay
Fatty acid uptake was measured using QBT™ fatty acid uptake assay from Molecular devices (Sunnyvale, CA) following manufacturer’s protocol. Briefly, cells were seeded at 50,000 cells/well in a 96-well plate. After 24 h of cells seeding, complete media was replaced with serum free RPMI media for 6 h. The cells were treated with 100 µl of 1x loading buffer and transferred to a fluorescent plate reader for kinetic reading every 60 seconds at λex=485nm and λem=515nm, cutoff 495 nm.
Citrate measurement
Citrate content in the cells was measured using the citrate colorimetric/fluorometric assay kit from Biovision (Milpitas, CA). Cells were treated with 0.1% DMSO or silibinin (90 µM) for 48 h. Thereafter, cells were homogenized with 100 µl of citrate assay buffer and centrifuged at 16,000 rpm for 10 minutes to remove cell debris. Cell lysates containing equal amount of proteins were used for the deproteinization step which was done with 10 kDa molecular weight cut off spin columns (BioVision, Cat # 1997-25). The samples were then processed as per the manufacturer’s protocol, and final concentration of citrate was obtained after extrapolation from the citrate standard curve.
Oil Red O (ORO) staining
Cells were seeded at 50,000 cells/ well in a 6 well plate. At the end of desired treatment, cells were fixed in 10% buffered formalin for 15 minutes at room temperature, washed twice with distilled water and then with 60% isopropanol for 5 minutes. After isopropanol wash, the plate was completely dried and stained with ORO stain (0.3% ORO in 100% isopropanol, diluted with distilled water in the ratio of 3:2) for 30 minutes. After staining, cells were washed with distilled water to get a clear background. Pictures were captured at 200x magnification under a microscope and lipid content quantitation was carried out by dye elution using 100% isopropanol and absorbance was measured by spectrophotometer at 500 nm.
Cholesterol estimation
Cells were seeded at the density of 10,000 cells per well in a 96 well plate. After 24 h of cell seeding, complete media was replaced with serum free media with or without silibinin treatment for 48 h. Cholesterol content in cells was detected using cholesterol cell based detection assay kit from Caymen chemicals (Ann Arbor, MI) following manufacturer’s protocol.
Cell growth and cell cycle distribution assays
Cells were plated at a density of 50,000 cells per well in 6-well plate. At the end of desired treatments, total cell number was determined using a hemocytometer. Cell cycle distribution (saponin/PI staining) was analyzed by FACS using University of Colorado Cancer Center Flow Cytometry Core Facility following published method [12].
Confocal imaging
Cells were grown on cover slips and treated with DMSO or silibinin (90 µM). At the end, cells were fixed in 4% buffered formalin for 30 minutes at room temperature followed by gentle PBS wash three times, and permeabilized with PBST (PBS + 0.3% Triton X-100) for 2 h with gentle shaking; and then blocked for 1 h in CAS block buffer (Invitrogen, 1:1 in PBS). Next, cells were incubated with respective primary antibodies, SREBP1 (1: 500) and pSREBP1 (1: 200) after diluting in PBST (with 1% BSA) overnight with gentle shaking. Thereafter, cells were washed with PBS with 0.1% Triton X-100, and finally cells were incubated with rabbit Alexa-Fluor 488 secondary antibody for 1 h. The coverslips were washed with PBS containing 0.1% Triton X-100 and mounted on slides with ProLong® Gold Antifade Mountant from Invitrogen-Life Technologies (Carlsbad, CA). Images were captured at 1000x magnification on a Nikon inverted confocal microscope using 488/405 nm laser wavelengths to detect SREBP1 or pSREBP1 (green) and DAPI (blue) emissions, respectively. Average fluorescence intensity was quantified using Image J software. In both control and treatment groups, the intensity plot was generated from 20 cells captured from different areas. The corrected cell fluorescence (CF) was obtained after normalizing to the background fluorescence using the formula: CF= Integrated Density - (Area of selected cell x Mean fluorescence of background readings).
R1881 stimulation and androgen-deprivation growth studies
LNCaP cells (2X105 per plate) were seeded, and 24 h later, cells were starved for 24 h in 10% cFBS media, and then treated daily with R1881 (10 nM in ethanol) with or without silibinin (90 µM) for 3 or 4 days or induced with R1881 for 4 days and silibinin (90 µM) added only for last 72 or 24 h. At the end of each treatment, cells were fixed and stained with ORO. For androgen deprivation studies, LNCaP cells were grown to 70% confluency and then shifted to androgen free 10% cFBS containing media, and maintained in this media till androgen independent clones emerged. Cells were washed and fresh media was added after every 3rd day. In the silibinin treatment group, 10 µM silibinin was added every time to the fresh media. Cells were collected and RNA was isolated from cells at different time-points following androgen withdrawal.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from desired cells using TRIzol® Reagent (Invitrogen-Life Technologies, Carlsbad, CA). cDNA was produced from 1 μg of total RNA by using High capacity cDNA reverse transcription assay kit from Life Technologies. SREBP1 (Forward primer: TCAGCGAGGCGGCTTTGGAGCAG; Reverse primer: TCAGCGAGGCGGCTTTGGAGCAG) and SREBP2 (Forward primer: AACGGTCATTCACCCAGGTC; Reverse primer: GGCTGAAGAATAGGAGTTGCC) mRNA levels were quantified by RT-PCR following standard procedures. GAPDH was used as the loading control.
Western blotting
Cells were treated with DMSO or silibinin and whole cell or nuclear/cytoplasmic extracts were prepared and immunoblotting was performed following published methods [67]. As needed, 60 µg of protein lysate per sample was denatured in 2x sample buffer and subjected to sodium dodecylsulfate–polyacrylamide gel electrophoresis (SDS-PAGE) on 8-16% Tris–glycine gel (as required based upon the protein molecular weight). The separated proteins were transferred on to nitrocellulose membrane followed by blocking with 5% non-fat milk powder (w/v) in Tris-buffered saline (10 mM Tris–HCl, pH 7.5, 100 mM NaCl, 0.1% Tween 20) for 1 h at room temperature. Membranes were probed for the protein levels of desired molecules using specific primary antibodies followed by the appropriate peroxidase-conjugated secondary antibody and visualized by ECL detection system. As required, membranes were also stripped and re-probed again for protein of interest or with appropriate loading control antibody. The autoradiograms/bands were scanned with Adobe Photoshop 6.0(Adobe Systems, San Jose, CA). In each case, blots were subjected to multiple exposures on the film to make sure that the band density is in the linear range.
Generation of DU145-vector control and SREBP1 overexpressing stable cell lines
DU145 cells were transfected with lentiviral particles containing pLX304-SREBP1 (Clone ID: ccsbBroad304_06995) or vector control pLX304 (Addgene, Cambridge, MA). Stable transduced cells were selected using blasticidin (10 µg/ml) in complete media for 3-4 weeks. The individual resistant clones were picked and grown separately and maintained in the same selection medium.
Statistical analysis
All statistical analyses were carried out with Sigma Stat software version 2.03 (Jandel scientific, San Rafael, CA). Mean and SEM were used to describe the quantitative data. One-way ANOVA followed by Tukey’s test was used for multiple comparisons and statistically significant difference was considered at p≤0.05.
Conflict of Interest
No potential conflicts of interest were identified by any of the authors.
Grant support
This work was supported by NCI RO1 CA102514. DKN was supported by Fulbright Fellowship and a Senior Research Fellowship from CSIR, New Delhi, India.
Abbreviations
ACC: Acetyl Co-A carboxylase; ACLY: ATP-citrate lyase; AI: Androgen independent; AMACR: α-methylacyl-CoA racemase; AMPK: AMP-activated protein kinase; ANOVA: Analysis of variance; AR: Androgen receptor; DMSO: Dimethyl sulfoxide; ECL: Enhanced chemiluminescence; ER: Endoplasmic reticulum; FASN: Fatty acid synthase; FS: Fatostatin; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; HMGCR: 3-hydroxy-3-methyl-glutaryl-CoA reductase; mTORC1: Mammalian target of rapamycin complex 1; NADPH: Nicotinamide adenine dinucleotide phosphate; ORO: Oil red O; PCA: Prostate cancer; PI: Propidium iodide; RT-PCR: Reverse transcription-polymerase chain reaction; SB: Silibinin; SCAP: SREBP cleavage-activating protein; SCD1: Stearoyl-CoA desaturase 1; SDS-PAGE: Sodium dodecylsulfate–polyacrylamide gel electrophoresis, SEM: Standard error of the mean; SRE: Sterol response element; SREBP: Sterol regulatory element binding protein; TBP: TATA-binding protein.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144: 646-674.
2. Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nature reviews Drug discovery. 2013; 12: 829-846.
3. Zhang F, Du G. Dysregulated lipid metabolism in cancer. World journal of biological chemistry. 2012; 3: 167-174.
4. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell metabolism. 2008; 7: 11-20.
5. Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer research. 1953; 13: 27-29.
6. Zadra G, Photopoulos C, Loda M. The fat side of prostate cancer. Biochimica et biophysica acta. 2013; 1831: 1518-1532.
7. Suburu J, Chen YQ. Lipids and prostate cancer. Prostaglandins & other lipid mediators. 2012; 98: 1-10.
8. Flavin R, Zadra G, Loda M. Metabolic alterations and targeted therapies in prostate cancer. The Journal of pathology. 2011; 223: 283-294.
9. Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D, Daniels VW, Machiels J, Vanderhoydonc F, Smans K, Waelkens E, Verhoeven G, Swinnen JV. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer research. 2010; 70: 8117-8126.
10. Leon CG, Locke JA, Adomat HH, Etinger SL, Twiddy AL, Neumann RD, Nelson CC, Guns ES, Wasan KM. Alterations in cholesterol regulation contribute to the production of intratumoral androgens during progression to castration-resistant prostate cancer in a mouse xenograft model. The Prostate. 2010; 70: 390-400.
11. Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, Marck B, Matsumoto AM, Simon NI, Wang H, Chen S, Balk SP. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer research. 2011; 71: 6503-6513.
12. Deep G, Singh RP, Agarwal C, Kroll DJ, Agarwal R. Silymarin and silibinin cause G1 and G2-M cell cycle arrest via distinct circuitries in human prostate cancer PC3 cells: a comparison of flavanone silibinin with flavanolignan mixture silymarin. Oncogene. 2006; 25: 1053-1069.
13. Singh RP, Raina K, Deep G, Chan D, Agarwal R. Silibinin suppresses growth of human prostate carcinoma PC-3 orthotopic xenograft via activation of extracellular signal-regulated kinase 1/2 and inhibition of signal transducers and activators of transcription signaling. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009; 15: 613-621.
14. Raina K, Rajamanickam S, Singh RP, Deep G, Chittezhath M, Agarwal R. Stage-specific inhibitory effects and associated mechanisms of silibinin on tumor progression and metastasis in transgenic adenocarcinoma of the mouse prostate model. Cancer research. 2008; 68: 6822-6830.
15. Singh RP, Raina K, Sharma G, Agarwal R. Silibinin inhibits established prostate tumor growth, progression, invasion, and metastasis and suppresses tumor angiogenesis and epithelial-mesenchymal transition in transgenic adenocarcinoma of the mouse prostate model mice. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008; 14: 7773-7780.
16. Flaig TW, Gustafson DL, Su LJ, Zirrolli JA, Crighton F, Harrison GS, Pierson AS, Agarwal R, Glode LM. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Invest New Drugs. 2007; 25: 139-146.
17. Flaig TW, Glode M, Gustafson D, van Bokhoven A, Tao Y, Wilson S, Su LJ, Li Y, Harrison G, Agarwal R, Crawford ED, Lucia MS, Pollak M. A study of high-dose oral silybin-phytosome followed by prostatectomy in patients with localized prostate cancer. The Prostate. 2010; 70: 848-855.
18. Fritz V, Benfodda Z, Rodier G, Henriquet C, Iborra F, Avances C, Allory Y, de la Taille A, Culine S, Blancou H, Cristol JP, Michel F, Sardet C, Fajas L. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Molecular cancer therapeutics. 2010; 9: 1740-1754.
19. Tamura K, Makino A, Hullin-Matsuda F, Kobayashi T, Furihata M, Chung S, Ashida S, Miki T, Fujioka T, Shuin T, Nakamura Y, Nakagawa H. Novel lipogenic enzyme ELOVL7 is involved in prostate cancer growth through saturated long-chain fatty acid metabolism. Cancer research. 2009; 69: 8133-8140.
20. Huang WC, Li X, Liu J, Lin J, Chung LW. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Molecular cancer research : MCR. 2012; 10: 133-142.
21. Lewis CA, Griffiths B, Santos CR, Pende M, Schulze A. Regulation of the SREBP transcription factors by mTORC1. Biochemical Society transactions. 2011; 39: 495-499.
22. Ting H, Deep G, Agarwal C, Agarwal R. The strategies to control prostate cancer by chemoprevention approaches. Mutation research. 2014; 760: 1-15.
23. Deep G, Agarwal R. Antimetastatic efficacy of silibinin: molecular mechanisms and therapeutic potential against cancer. Cancer metastasis reviews. 2010; 29: 447-463.
24. Ting H, Deep G, Agarwal R. Molecular mechanisms of silibinin-mediated cancer chemoprevention with major emphasis on prostate cancer. The AAPS journal. 2013; 15: 707-716.
25. Singh RP, Agarwal R. A cancer chemopreventive agent silibinin, targets mitogenic and survival signaling in prostate cancer. Mutation research. 2004; 555: 21-32.
26. Deep G, Agarwal R. Targeting tumor microenvironment with silibinin: promise and potential for a translational cancer chemopreventive strategy. Current cancer drug targets. 2013; 13: 486-499.
27. Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, Ettinger SL, Gleave ME, Nelson CC. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer research. 2008; 68: 6407-6415.
28. Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, Nelson CC. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer research. 2004; 64: 2212-2221.
29. Webber MM, Quader ST, Kleinman HK, Bello-DeOcampo D, Storto PD, Bice G, DeMendonca-Calaca W, Williams DE. Human cell lines as an in vitro/in vivo model for prostate carcinogenesis and progression. The Prostate. 2001; 47: 1-13.
30. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997; 89: 331-340.
31. Sun LP, Li L, Goldstein JL, Brown MS. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. The Journal of biological chemistry. 2005; 280: 26483-26490.
32. Amemiya-Kudo M, Shimano H, Hasty AH, Yahagi N, Yoshikawa T, Matsuzaka T, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Sato R, Kimura S, Ishibashi S, et al. Transcriptional activities of nuclear SREBP-1a, -1c, and -2 to different target promoters of lipogenic and cholesterogenic genes. Journal of lipid research. 2002; 43: 1220-1235.
33. Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, Verhoeven G, Swinnen JV. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer research. 2007; 67: 8180-8187.
34. Ha J, Daniel S, Broyles SS, Kim KH. Critical phosphorylation sites for acetyl-CoA carboxylase activity. The Journal of biological chemistry. 1994; 269: 22162-22168.
35. Zaidi N, Swinnen JV, Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer research. 2012; 72: 3709-3714.
36. Carnell AJ, Kirk R, Smith M, McKenna S, Lian LY, Gibson R. Inhibition of Human alpha-Methylacyl CoA Racemase (AMACR): a Target for Prostate Cancer. ChemMedChem. 2013.
37. Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes & development. 2011; 25: 1895-1908.
38. Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. The Journal of biological chemistry. 1996; 271: 27879-27887.
39. Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, Gao B, Wierzbicki M, Verbeuren TJ, Shaw RJ, Cohen RA, Zang M. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell metabolism. 2011; 13: 376-388.
40. Kamisuki S, Mao Q, Abu-Elheiga L, Gu Z, Kugimiya A, Kwon Y, Shinohara T, Kawazoe Y, Sato S, Asakura K, Choo HY, Sakai J, Wakil SJ, Uesugi M. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chemistry & biology. 2009; 16: 882-892.
41. Swinnen JV, Van Veldhoven PP, Esquenet M, Heyns W, Verhoeven G. Androgens markedly stimulate the accumulation of neutral lipids in the human prostatic adenocarcinoma cell line LNCaP. Endocrinology. 1996; 137: 4468-4474.
42. Zi X, Agarwal R. Silibinin decreases prostate-specific antigen with cell growth inhibition via G1 arrest, leading to differentiation of prostate carcinoma cells: implications for prostate cancer intervention. Proceedings of the National Academy of Sciences of the United States of America. 1999; 96: 7490-7495.
43. Zhu W, Zhang JS, Young CY. Silymarin inhibits function of the androgen receptor by reducing nuclear localization of the receptor in the human prostate cancer cell line LNCaP. Carcinogenesis. 2001; 22: 1399-1403.
44. Twiddy AL, Leon CG, Wasan KM. Cholesterol as a potential target for castration-resistant prostate cancer. Pharmaceutical research. 2011; 28: 423-437.
45. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: a cancer journal for clinicians. 2014; 64: 9-29.
46. Fritz V, Benfodda Z, Henriquet C, Hure S, Cristol JP, Michel F, Carbonneau MA, Casas F, Fajas L. Metabolic intervention on lipid synthesis converging pathways abrogates prostate cancer growth. Oncogene. 2013; 32: 5101-5110.
47. Liu Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate cancer and prostatic diseases. 2006; 9: 230-234.
48. Ozten-Kandas N, Bosland MC. Chemoprevention of prostate cancer: Natural compounds, antiandrogens, and antioxidants - In vivo evidence. Journal of carcinogenesis. 2011; 10: 27.
49. Costello LC, Franklin RB. The intermediary metabolism of the prostate: a key to understanding the pathogenesis and progression of prostate malignancy. Oncology. 2000; 59: 269-282.
50. Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, Cheng L, Masterson TA, Liu X, Ratliff TL, Cheng JX. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell metabolism. 2014; 19: 393-406.
51. Roy S, Kaur M, Agarwal C, Tecklenburg M, Sclafani RA, Agarwal R. p21 and p27 induction by silibinin is essential for its cell cycle arrest effect in prostate carcinoma cells. Molecular cancer therapeutics. 2007; 6: 2696-2707.
52. Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Current opinion in clinical nutrition and metabolic care. 2006; 9: 358-365.
53. You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004; 127: 1798-1808.
54. Rattan R, Giri S, Singh AK, Singh I. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. The Journal of biological chemistry. 2005; 280: 39582-39593.
55. Lee SK, Lee J, Lee SI, Bae WJ, Lee YM, Park JS, Park SJ, Min SK, Kim EC. N(1)-guanyl-1,7,-diamineoheptane, an inhibitor of deoxyhypusine synthase, suppresses differentiation and induces apoptosis via mitochondrial and AMPK pathways in immortalized and malignant human oral keratinocytes. Journal of oral pathology & medicine : official publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology. 2009; 38: 792-800.
56. Chen MB, Shen WX, Yang Y, Wu XY, Gu JH, Lu PH. Activation of AMP-activated protein kinase is involved in vincristine-induced cell apoptosis in B16 melanoma cell. Journal of cellular physiology. 2011; 226: 1915-1925.
57. Xiang X, Saha AK, Wen R, Ruderman NB, Luo Z. AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochemical and biophysical research communications. 2004; 321: 161-167.
58. Huang CH, Tsai SJ, Wang YJ, Pan MH, Kao JY, Way TD. EGCG inhibits protein synthesis, lipogenesis, and cell cycle progression through activation of AMPK in p53 positive and negative human hepatoma cells. Mol Nutr Food Res. 2009; 53: 1156-1165.
59. Shi T, Fan GQ, Xiao SD. SIRT3 reduces lipid accumulation via AMPK activation in human hepatic cells. Journal of digestive diseases. 2010; 11: 55-62.
60. Attard G, Richards J, de Bono JS. New strategies in metastatic prostate cancer: targeting the androgen receptor signaling pathway. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011; 17: 1649-1657.
61. Mohler JL, Titus MA, Bai S, Kennerley BJ, Lih FB, Tomer KB, Wilson EM. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer research. 2011; 71: 1486-1496.
62. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer research. 2008; 68: 4447-4454.
63. Cai C, Wang H, Xu Y, Chen S, Balk SP. Reactivation of androgen receptor-regulated TMPRSS2:ERG gene expression in castration-resistant prostate cancer. Cancer research. 2009; 69: 6027-6032.
64. Page ST, Lin DW, Mostaghel EA, Hess DL, True LD, Amory JK, Nelson PS, Matsumoto AM, Bremner WJ. Persistent intraprostatic androgen concentrations after medical castration in healthy men. J Clin Endocrinol Metab. 2006; 91: 3850-3856.
65. Mohler JL, Gregory CW, Ford OH, 3rd, Kim D, Weaver CM, Petrusz P, Wilson EM, French FS. The androgen axis in recurrent prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004; 10: 440-448.
66. Krycer JR, Brown AJ. Cholesterol accumulation in prostate cancer: a classic observation from a modern perspective. Biochimica et biophysica acta. 2013; 1835: 219-229.
67. Kavitha CV, Agarwal C, Agarwal R, Deep G. Asiatic Acid inhibits pro-angiogenic effects of VEGF and human gliomas in endothelial cell culture models. PloS one. 2011; 6: e22745.