
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common cancers worldwide [1, 2]. Chronic infection with hepatitis B virus (HBV) is one of the major risk factors for HCC and plays an important part in the prevalence of HCC [3–5].
Cytokines can mediate a variety of biological behavior of cells and have a role in the development of HBV-related HCC. It was reported that HBV can upregulate the expression of cytokines and cause liver damage directly through the cytokines signaling pathway. For instance, we previously reported that HBx induced IL-32 expression through NF-κB activation [6]. HBV surface antigen (HBsAg) could efficiently induce IL-23 secretion in a mannose receptor (MR)-dependent manner [7]. HBx induced IL-23 secretion through the activation of the ERK/NF-κB pathway [8]. The baseline level of IL-23 in serum and hepatic tissue was found to be higher in patients with CHB than in controls [8, 9]. IL-23 belongs to the IL-12 cytokine family, which shares the p40 subunit of IL-12 but is distinguished from the latter by its cytokine subunit, p19 [10]. Consistent with the structural and biological similarities of IL-12 and IL-23, IL-23 receptor is composed of the IL-12Rβ1 subunit and the novel IL-23R subunit which is different from IL-12 receptor [10, 11]. IL-23 and its receptor mainly express in monocytes, DCs, T cells, natural killer cells and myeloid cells [11, 12].
IL-23 was reported to play conflictive roles in carcinogenesis. For instance, exogenously overexpressed IL-23 exerts potent antitumor and anti-metastatic effects to inhibit cancer progression [13–16]. However, Langowski et al. reported that mice deficient in IL-23p19 were resistant to tumor induction and treated with anti-IL-23p19 show decreased tumor growth and increased tumor rejection [17]. And IL-23 promoted growth and proliferation of human squamous carcinoma cells of the oral cavity [18]. Through its receptor expressed on cancer cells, IL-23 participated in the progress of colorectal cancer [19] and regulated the proliferation of lung cancer in a concentration-dependent manner [20].
Given the important role of HBV in the prevalence of HCC and the upregulation of IL-23 induced by HBV, it merits to investigate if IL-23 could affect the biological behavior of hepatoma cells and, if so, the underlying mechanisms. In this article, we found that IL-23 did enhance the malignant properties of hepatoma cell lines HepG2 and Huh-7. This enhancement promoted hepatoma cells progressing into invasive cell by the attenuation of HNF4α, which is essential for liver development and hepatocyte function [21]. These findings identified potential targets of interventional strategies for treating hepatitis B patients through manipulation of the IL-23.
RESULTS
IL-23 expression is elevated in HBV-integrated HepG2.215 cells
Previous studies have revealed the correlation between elevated expression of IL-23 and HBV infection [7–9]. To explore the role of IL-23 in progression of HBV-related HCC, elevated IL-23 expression was confirmed in hepatoma cell lines HepG2 and HBV-integrated HepG2.215 cells. As shown in Figure 1A, the mRNA levels of inflammatory cytokines (such as TNF, IL-23, HMGB1, IL-1β) in HepG2.215 cells were higher than those in HepG2 cells. Among them, IL-23 increased more evidently. We then evaluated the expression of IL-23 receptor (IL-23R) on these cells lines. RT-PCR results showed that the mRNA of IL-23R could be detected in HepG2, HepG2.215 and Huh-7 cells. The mRNA level of IL-23R showed no statistically difference between HepG2 and HepG2.215 (Figure 1B) but was decreased in Huh-7 (P < 0.05). Flow cytometry results (Figure 1C) manifested that IL-23R expression levels on HepG2 and HepG2.215 cells were parallel to that on A549 cells which were reported to show strong positive expression of the IL-23R [20]. Immunofluorescence staining confirmed positive expression of IL-23R on these 3 hepatoma cell lines (Figure 1D). Expression of the IL-23R in liver cancer cells inferred that hepatoma cells might be the potential targets of IL-23.
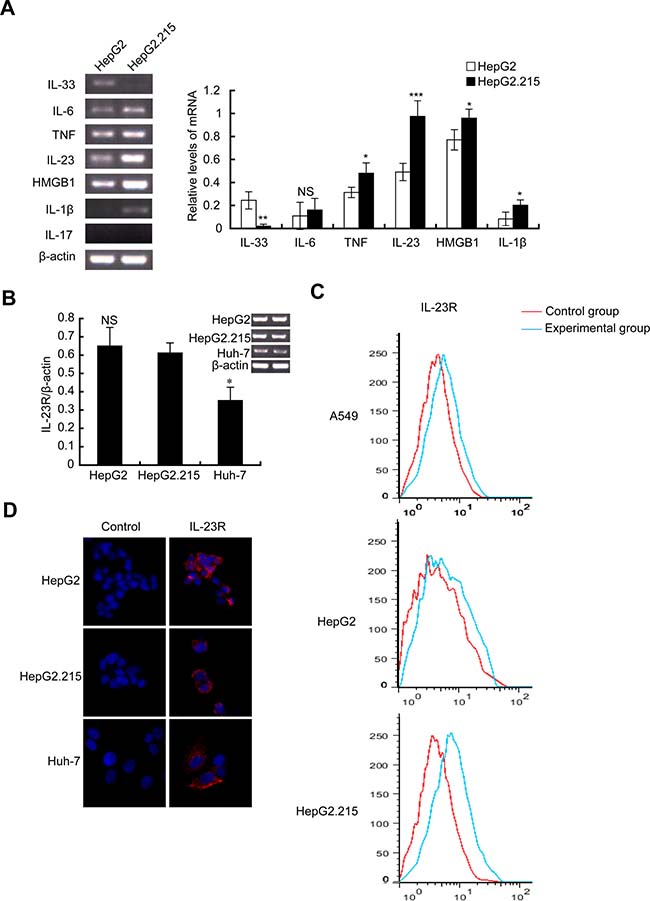
Figure 1: IL-23 expression was elevated in HBV-integrated HepG2.215 cells. (A) RT-PCR and statistical analysis showed the expression of inflammatory cytokines (IL-1β, IL-6, TNF, IL-23, HMGB1, IL-17 and IL-33) in the hepatoma cell lines HepG2 and HepG2.215. Photograph is one represent of three independent experiments. IL-23R expression was detected by RT-PCR (B), flow cytometer (C) and immunofluorescence (D) in HepG2, Huh-7 and HepG2.215. Hochest33342 was used to stain nuclei. Magnification, ×400. *P < 0.05, **P < 0.01, ***P < 0.001, NS, non-significant difference.
hrIL-23 enhances growth of hepatoma cells
To address whether IL-23 could affect the progression of hepatoma cells in vitro, the proliferation of cells pre-treated with human recombinant IL-23 (hrIL-23) was assessed by CCK-8 assay and colony-forming assay. As showed in Figure 2A, hrIL-23 promoted the proliferation of HepG2 cells. This promotion peaked at 20 ng/ml. Clonogenic assay also showed that when HepG2 cells were exposed to hrIL-23 for 5–7 days, more and bigger colonies formed (Figure 2B). Furthermore, hrIL-23 promoted progression of the cell cycle at the G1/S transition (Figure 2C). The same results were observed in Huh-7 cell line (Figure 2A-2C). In addition, hrIL-23 increased the expression of Ki-67 at dose-dependent manner (data not shown). Our results confirmed that hrIL-23 promoted proliferation and clone formation of hepatoma cells. The optimal concentration was 20 ng/ml.
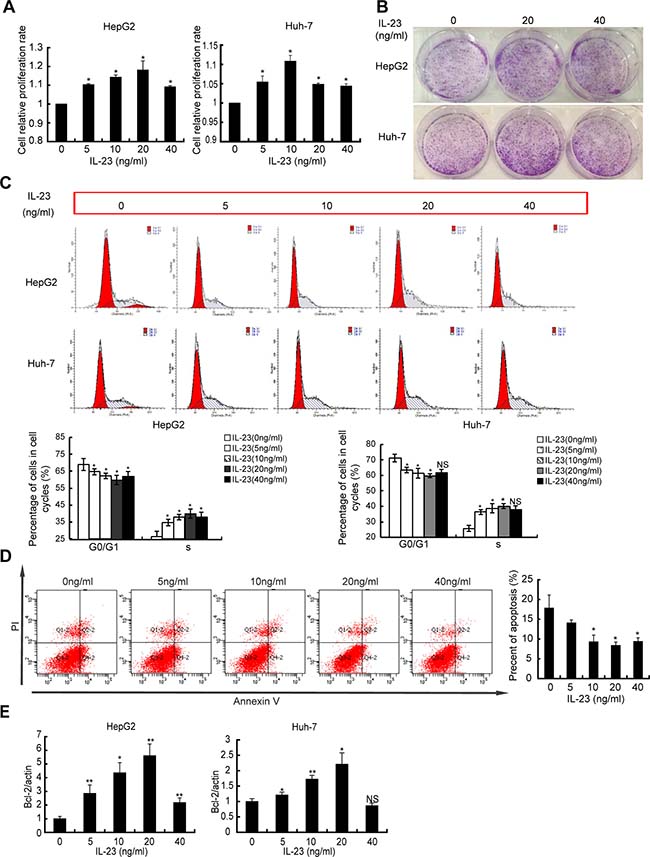
Figure 2: hrIL-23 regulates growth of hepatoma cells. Cells were treated with hrIL-23 (0 ng/ml, 5 ng/ml, 10 ng/ml, 20 ng/ml, 40 ng/ml) for 48 h. (A) CCK8 proliferation assay was performed. Data were expressed as fold change in proliferation rate in hrIL-23-treated cells versus untreated ones (arbitrary value = 1). Each value represents the mean ± SD for triplicate samples. (B) Cells were seeded at 500 cells/cm2 and incubated for 5–7 days to form colonies. A representative experiment was shown. The experiment was performed two times yielding similar results. (C) Flow cytometric analysis of DNA content in hepatoma cells in the presence of hrIL-23. The percentages of cells in each cycle phases were shown. (D, E) Cells were cultured in serum-free medium containing different concentration of hrIL-23 for 48 h. Then cells were stained with Annexin V and PI to detect the percentage of apoptotic cells by FCM (D) and Bcl-2 level was detected by realtime PCR (E). Data were expressed as fold change in Bcl-2 expression in hrIL-23-treated versus untreated cells (arbitrary value = 1). Each value represents the mean ± SD for triplicate samples. *P < 0.05, **P < 0.01, NS, non-significant difference vs negative control (Student t test).
As to the effect of hrIL-23 on apoptosis of hepatoma cells, we observed that apoptotic HepG2 cells declined ~20% from baseline by 5 ng/ml hrIL-23 treatment and continued to decline to 8.4% ± 0.9% by 20 ng/ml hrIL-23, with slight recovery detected by 40 ng/ml hrIL-23 (Figure 2D). Furthermore, mRNA level of anti-apoptosis related gene Bcl-2 was observed to elevate in hrIL-23 treated hepatoma cells (Figure 2E). No obvious changes were observed in the expression of p53 and Survivin in these cells (data not shown).
hrIL-23 induces motility and invasivity of hepatoma cells
In order to study the biological effects of IL-23 on cellular properties associated with the malignant phenotypes, scratch wound assays were carried out. It was ascertained that 20 ng/ml hrIL-23 pretreated hepatoma cells acquired good motility, but 40 ng/ml hrIL-23 treated cells did not (Figure 3A). The enhancement of hrIL-23 on cell motility and invasivity could be observed by a transwell assay in the presence of matrigel, which mimicks the extracellular matrix microenvironment (Figure 3B).
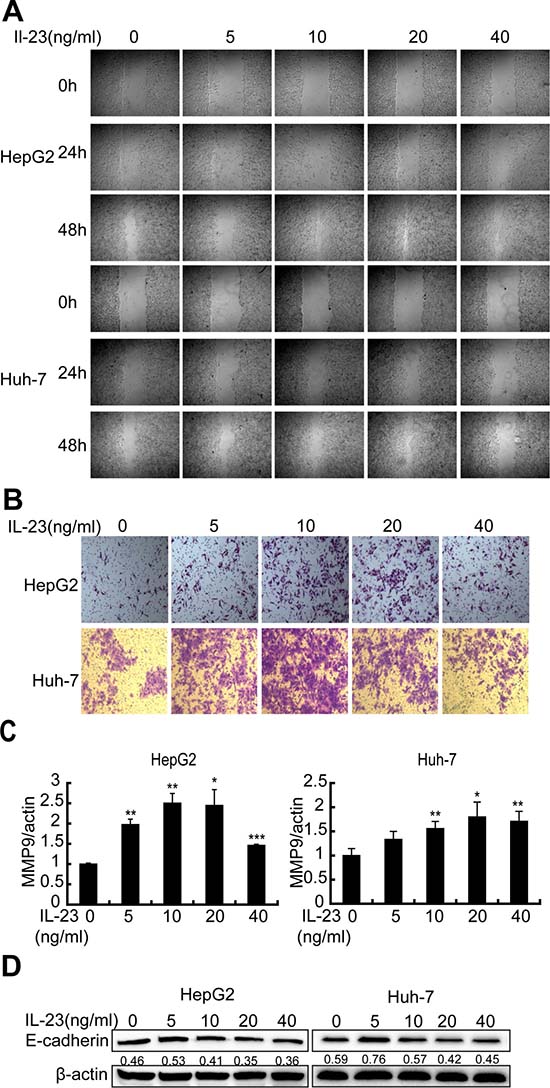
Figure 3: hrIL-23 induces motility and invasivity of hepatoma cells. (A) Wound healing assays. Cells were plated in 12-well plates and a scratch was made 24 h later. And then cells were cultured in 2% FBS medium containing hrIL-23 for 24 h and 48 h to check wound closure with an inverted microscope. (B) Cell motility assays. 1 × 105 HepG2 or Huh-7 cells pretreated with increasing doses of hrIL-23 (0–40 ng/ml) for 24 h were seeded in the upper chamber in serum free medium and 2.5% FBS medium was added to the lower chamber. 24 h later, migrated cells were fixed with 4% paraformaldehyde and then stained with 0.1% Crystal Violet. (C) Realtime PCR analysis for MMP9 expression in hrIL-23 treated hepatoma cells. Data were expressed as fold changes in MMP9 expression in hrIL-23-treated versus untreated cells (arbitrary value = 1). Each value represents the mean ± SD for triplicate samples. *P < 0.05, **P < 0.01, ***P < 0.001 vs negative control (Student t test). (D) Western blot showed that the expression of E-cadherin protein in hrIL-23 treated hepatoma cells. The band densitometry readings were normalized to β-actin loading control.
Transcriptional and translational changes observed coherently in the two lines demonstrated that hrIL-23-induced tumor promoting effects were associated with an EMT gene expression profile: upregulation of invasivity marker matrix metalloproteinase 9 (MMP9) mRNA (Figure 3C) and downregulation of liver-specific and epithelial marker E-cadherin protein (Figure 3D) in HepG2 and Huh-7 cells.
hrIL-23 increases the proportion of stem/progenitor cells in hepatoma cells
CD133 [22–24] is a well-known marker for the identification of cancer stem/progenitor cells in HCC. CD133 expression confers malignant potential which may contribute to the survival of HCC cells [25]. To study the effect of IL-23 on CD133 expression, CD133 level was detected by real-time PCR and flow cytometry on hrIL-23 pretreated HepG2 and Huh-7 cells. As showed in Figure 4A, significant upregulation in CD133 mRNA level was detected in both cells. Similarly, flow cytometry confirmed the increment in the mean fluorescence index (MFI) and the proportions of CD133+ cells in both cells. These enhancements achieved maximum when cells were pretreated by hrIL-23 at 20 ng/ml (Figure 4B and 4C).
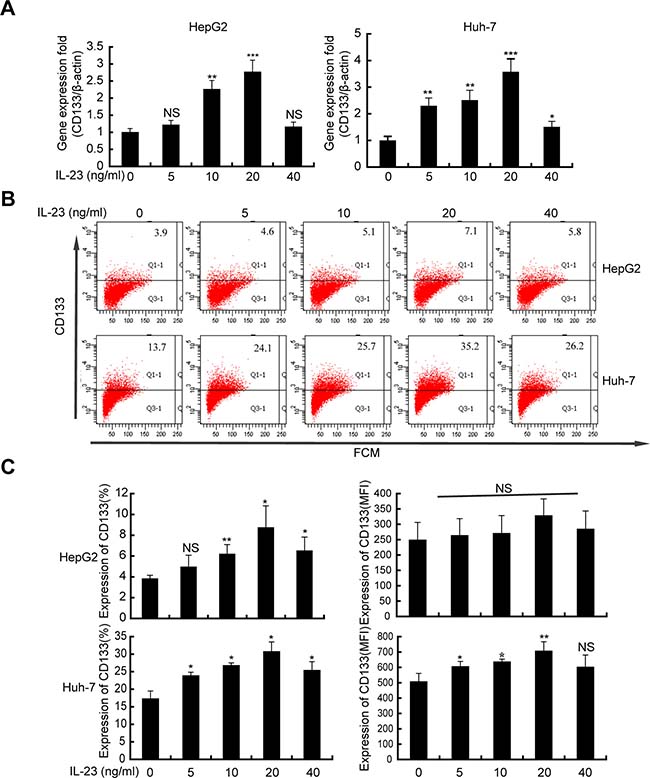
Figure 4: hrIL-23 promotes the “stemness” gene CD133 expression in hepatoma cells. Cells were treated with hrIL-23 at different concentrations as indicated for 24 h or 48 h. CD133 expression was analyzed by realtime PCR (A) and flow cytometer (B). Bar graphs (C) are the percentage and MFI of CD133 expression. Each column and bar represents the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, NS, non-significant difference vs negative control.
Attenuation of HNF4α by HBV-related IL-23
Hepatocyte nuclear factor 4 alpha (HNF4α) is known as liver-enriched transcription factor [21, 26]. Downregulation of HNF4α is closely related to progress of HCC [27]. Our data showed that the expressions of HNF4α and its target gene G6Pase were severely suppressed in HepG2.215 cells which expressed high concentrations of Hbs and Hbe proteins (Figure 5A). After transiently transfected with plasmids encoding for the full length of HBV genome, HepG2 and Huh-7 cells reduced their HNF4α expression in protein levels (Figure 5B) albeit no changes in their mRNA levels (Figure 5C). When cultured in medium mixed with different proportions of HepG2.215 culture supernatant, HepG2 cells also decreased their HNF4α levels and this decrease was abolished in the presence of anti-IL-23p19 antibody (Figure 5D). These data suggested that HBV infected cells could down-regulate their HNF4α expression, which may be mediated by the secretion of IL-23. To confirm the impact of IL-23 on its expression, HNF4α was detected in hrIL-23-treated hepatoma cells. Western blot showed that HNF4α level gradually decreased as hrIL-23 concentration increased in a certain range (0–20 ng/ml, Figure 5E), as well as the levels of E-cadherin, PTEN but not GRP78. Restoration of HNF4α expression (at 40 ng/ml hrIL-23) could block the reduction of E-cadherin and PTEN (Figure 5F). Contrary to decrease in HNF4α protein level, MMP9 level increased as hrIL-23 concentration increased in a certain range. However, the levels of N-cadherin and Bcl-2 were not altered (Figure 5F).
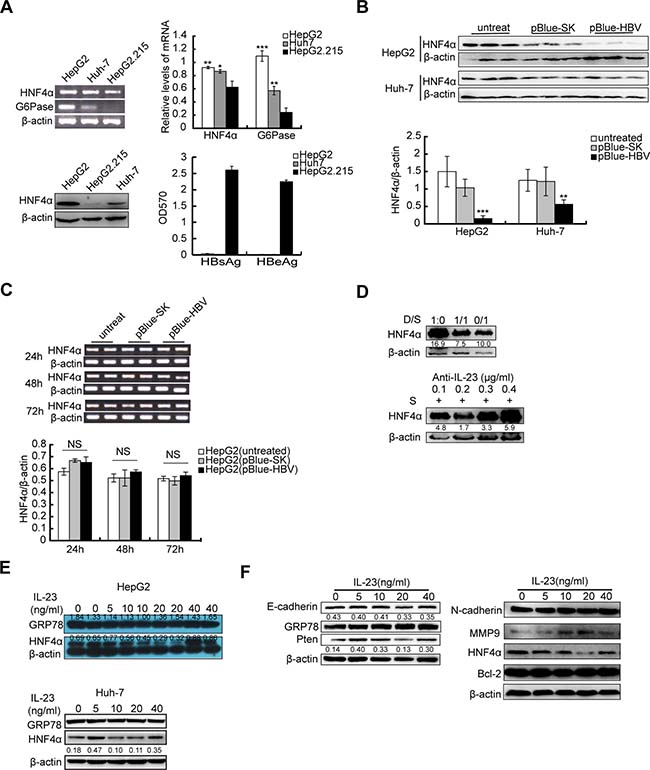
Figure 5: Downregulation of HNF4α by HBV-related IL-23. (A) RT-PCR and Western blot show the expression of HNF4α and/or G6Pase in hepatoma cells. *P < 0.05, **P < 0.01, ***P < 0.001, NS vs HepG2.215. ELISA shows the levels of HBsAg and HBeAg in culture supernatant of hepatoma cells. (B) 72 h after transfection with pBlue-HBV, protein level of HNF4α in hepatoma cells was detected with western blot and counted with bar graph. **P < 0.01, ***P < 0.001, vs mock plasmids. (C) RT-PCR and photograph showed the mRNA expression of HNF4α in HepG2 transfected with pBlue-HBV for 24 h, 48 h or 72 h. (D) Western blot was used to detect the expression of HNF4α in HepG2 cells cultured in DMEM supplemented with different proportions of HepG2.215 supernatant (D/S) or cultured in HepG2.215 supernatant supplemented with different concentration of anti-hIL-23p19 antibody for 24 h. (E) Expressions of HNF4α in hrIL-23 treated HepG2 and Huh-7 cells were detected by western blot. (F) Expressions of E-cadherin, PTEN, N-cadherin, MMP9, Bcl-2 and HNF4α in hrIL-23 treated HepG2 cells. The WB band densitometry readings were normalized to β-actin loading control. Each column and bar represents mean ± SD.
DISCUSSION
HBV-related chronic hepatitis is one of major risk factors for HCC [3–5]. Tumor development is accompanied with production of a great number of cytokines, chemokines and growth factors, favoring increased cellular proliferation. Among these, IL-23 plays an important role in chronic inflammation and is the hub of tumor-associated inflammation and tumor immune escape [17, 28]. mDCs and macrophages are the main sources of IL-23. Xia et al. [8] reported that the hepatocytes were one of the main cell source that secreted IL-23 in CHB patients. In this paper, our data confirmed that IL-23 was produced by hepatoma cells and its expression was upregulated in HBV-infected cells. This increment was in accordance with previous reports that IL-23 can be induced by HBx and upregulated in the serum and liver tissues of CHB patients [7, 8].
There is a debate about roles of IL-23 in carcinogenesis. Some studies have shown that IL-23 inhibits tumor growth [14–16], and some suggest that IL-23 promotes carcinogenesis [17–20]. Our findings suggested that hrIL-23 enhanced the proliferation of HepG2 and Huh-7 cells in a dose-dependent manner. Furthermore, IL-23 could affect multiple aspects of their malignant phenotypes, such as promoting colony formation, inhibiting apoptosis through enforced expression of BCl-2, inducing motility and invasivity through upregulation of MMP9 and downregulation of E-cadherin, increasing the proportion of stem/progenitor cells. Based on these findings, IL-23 was suggested to participate in the progression of HCC. That could partly explain why chronic infection with HBV is one of the major risk factors for HCC.
As demonstrated in this study, IL-23 promoted carcinogenesis of hepatoma cells. However, this promotion peaked at 20 ng/ml and slightly decreased at 40 ng/ml. Li et al. [20] reported that low concentrations of IL-23 bound to the higher affinity IL-23R resulting in promotion of human lung cancer cells proliferation, whereas a high concentration of IL-23 bound to both IL-23R and IL-12Rβ1 resulting in inhibition of their proliferation. Whether the different affinities of the two receptors affect the action of IL-23 on hepatoma cells is worth of our further investigation.
HNF4α, as a master transcriptional regulator of hepatocyte differentiation, is a target gene of cytokines. It is essential for liver development and hepatocyte function [21], and has links to cancer [27, 29]. Substantial evidences had revealed that the expression of HNF-4α was decreased in human HCC compared to the adjacent noncancerous tissues [27, 30, 31]. Loss of HNF-4α facilitates HCC progression [27], whereas introduction of HNF-4α obviously prevented the development of HCC [30, 32]. Engagement of IL-23R by IL-23 results in phosphorylation of tyrosine residues to induce downstream effector molecules for STAT, MAPK, PI3K and NF-kB signaling [11, 33–35]. STAT3 is a key molecule of IL-23 signaling pathway [36, 37] and its activation inhibits expression of HNF4α [38]. Our results manifested that HepG2 and Huh-7 cells showed significantly higher expression of HNF4α than HepG2.215 cells, and introduction of HBV dramatically down-regulated their HNF4α expression. These findings were consistent with previous reports that overexpression of HBx in HepG2 cells inhibited HNF-4α expression, and HNF-4α levels were inversely related with viral proteins both in HBV-infected HepG2.215 cells and HBV positive HCC liver tissues [39, 40]. However, other studies showed that HNF4α plays important roles in transcription and replication of HBV by binding and upregulating the HBV promoter activity [41, 42]. Our results confirmed that HepG2.215 did express lower level of HNF-4α and high amount of HBsAg and HBeAg. It is suggested that the malignant properties of HepG2.215 maybe correlated with loss of HNF4α. Hence, it is worthy of our further investigation to explain how HBV, IL-23 and HNF4α talk with each other to affect HCC progression.
In summary, our results suggested that HBV infection could attenuate the expression of HNF4α via secretion of IL-23. Hence, it is extrapolated that 1) HBV drives infected hepatocytes to produce more inflammatory cytokines, especially IL-23, then 2) IL-23 engages in IL-23R to turn down the expression of HNF4α, and finally 3) inhibition of HNF4α induces progression of hepatocellular carcinoma by increasing ratio of stem/progenitor cells, promoting the proliferation and colony formation of hepatoma cells in vitro, and preventing apoptosis and cell cycle arrest of hepatoma cells. Although conflicting reports have assigned HNF4α tumor-promoting role in liver cancer [43], our data support the tumor-suppressing role of HNF4α in line with other reports [30, 32, 38, 44]. Whether IL-23 affects the expression of HNF4α through STAT3 pathway to regulate differentiation of cells needs further study.
Our research still could not answer how HNF4α participates in tumor progression. Hatziapostolou M et al. suggested that a HNF4α-miRNA inflammatory feedback loop circuit was essential [38]. F Garibaldi et al. [45] inferred that HNF4α directly repressed Snail gene to stabilize the hepatocytic phenotype. Walesky et al. suggested that some proliferation-related genes came into play [44]. Downregulation of HNF4α plays a critical role in promoting EMT and affecting apoptosis and cell cycle progression [46]. In this article, we identified that anti-apoptosis related gene Bcl-2, and tumor suppressor protein PTEN participated in this IL-23 and HNF4α mediated tumor progression. The expression change of HNF4α might be positively correlated with E-cadherin, and negatively with MMP9 in IL-23 mediated tumor progression. However, significant work remains to identify the underlying mechanisms involved in tumor progression of HBV related IL-23.
In conclusion, we demonstrated that HBV drove infected hepatoma cells to produce more IL-23. At low concentration, hrIL-23 enhanced malignant properties of hepatoma cell lines HepG2 and Huh-7, which might promote their progression into invasive carcinoma. And this effect could be mediated through attenuation of HNF4α. The findings identified potential targets of interventional strategies for treating hepatitis B patients through manipulation of the IL-23 and HNF4α.
MATERIALS AND METHODS
Cell culture and plasmids
Human hepatoma cell lines HepG2, Huh-7, HepG2.215 were obtained from the China Center for Type Culture Collection (Wuhan, China). They were maintained in DMEM medium (Gibco, BRL Co. Ltd, USA) containing 10% fetal bovine serum (Gibco), 100 units/ml penicillin and 100 mg/ml streptomycin (Sigma, St Louis, MO). Cells were cultured at 37° C in a humidified atmosphere of 5% CO2.
For analyzing HNF4α expression affected by HBV-related IL-23, HepG2.215 cells were cultured with serum-free medium for 48 h. Then supernatant was collected and mixed with DMEM in different proportions (D/S). After that, HepG2 cells were cultured with D/S or supernatant (S) with IL-23 neutralization antibody (anti-IL-23p19, R&D, Minnesota, USA) for 24 h.
Plasmid pBlue-HBV encoding the full length of HBV was preserved by our lab.
RNA extraction, reverse transcription, RT-PCR and Real-time PCR
Total RNA was extracted with TRIzol (Invitrogen, California, USA) and reverse transcribed with MMLV reverse-transcriptase (Cwbiotech, Wuhan, China) according to the manufacturer’s protocol. cDNA was amplified as described previously [47, 48]. Equal amounts of cDNA were submitted to real-time PCR in the presence of the SYBR Green qPCR mix (ToYoBo, Osaka, Japan) or RT-PCR in the presence of BioRad Miniopticon (Cwbiotech). The specific primers used were as follows: HNF4α, G6Pase, IL-33, IL-6, TNF, IL-23p19, IL-1β, IL-17, HMGB1, β-actin1, IL-23R, MMP9, CD133, Bcl-2, β-actin2 (Table 1).
Table 1: Sequence of specific primers used for RT-PCR and real-time PCR
Gene |
Forward |
Reverse |
Bp |
|---|---|---|---|
HNF4α |
5′-GCCTACCTCAAAGCCATCAT-3′ |
5′-GACCCTCCCAGCAGCATCTC-3′ |
275 |
G6Pase |
5′-GCCACATCCACAGCATCTATAA-3′ |
5′-CCAGAGTCCACAGGAGGTCTAC-3′ |
129 |
IL-33 |
5′-TTCCCTCTGTATAACTGC-3′ |
5′-AGTGTTTGAGCCTATCGT-3′ |
283 |
IL-6 |
5′-TGAAAGCAGCAAAGAGGC-3′ |
5′-TGAAAGCAGCAAAGAGGC-3′ |
259 |
TNF |
5′-CGAGTCTGGGCAGGTCTA-3′ |
5′-GAAGTGGTGGTCTTGTTGC-3′ |
199 |
IL-23p19 |
5′-GCTTCAAAATCCTTCGCAG-3′ |
5′-GATCTGAGTGCCATCCTTGAG-3′ |
119 |
IL-1β |
5′-ACAGTGGCAATGAGGATG-3′ |
5′-TGTAGTGGTGGTCGGAGA-3′ |
243 |
IL-17 |
5′-CAACCGATCCACCTCACC-3′ |
5′-AGCCCACGGACACCAGTA-3′ |
470 |
IL-23R |
5′-ATCGTGAATGAGGAGTTGCC-3′ |
5′-TGTGCCTGTATGTGTGACCA-3′ |
470 |
β-actin1 |
5′-CTGGGGCGCCCCAGGCACCA-3′ |
5′-CTCCTTAATGTCACGCACGATTTC-3′ |
540 |
HMGB1 |
5′-TATGGCAAAAGCGGACAAGG-3′ |
5′-CTTCGCAACATCACCAATGGA-3′ |
196 |
MMP9 |
5′-CGCTACCACCTCGAACTTTG-3′ |
5′-GCCATTCACGTCGTCCTTAT-3′ |
196 |
CD133 |
5′-ACATGAAAAGACCTGGGGG-3′ |
5′-GATCTGGTGTCCCAGCATG-3′ |
200 |
Bcl-2 |
5′-CTGGTGGGAGCTTGCATCAC-3′ |
5′-ACAGCCTGCAGCTTTGTTTC-3′ |
150 |
β-actin2 |
5′-GCAAAGACCTGTACGCCAAC-3′ |
5′-GTACTTGCGCTCAGGAGGAG-3′ |
120 |
Western blot
Cells were lysed in RIPA buffer containing 1mM protease inhibitors PMSF (Beyotime, Shanghai China). Equal amounts of protein were separated and then blotted with antibodies as follows: antibodies against HNF4α (C19, 1:500), E-cadherin (1:500) and β-actin (1:500) from Santa Cruz Biotechnology (Santa Cruz, USA), anti-GRP78 (1:1000) from Abcam (Cambridge, UK), antibodies against N-cadherin (1:1000), MMP9 (1:1000) and Bcl-2 (1:1000) from Cell Signaling Technology (Beverly, MA, USA) and HRP-conjugated species-specific secondary antibodies (1:2000 dilution, ProteinTech Group, Wuhan, China). After enhanced chemiluminescence reaction (Tiangen, Beijing, China), relative amounts of protein were quantified using Image Gel-Pro analyzer software (Media Cybernetics, Florida, USA).
Cell proliferation assay
Cell proliferation was assessed using the Cell Counting Kit-8 (CCK8, Zoman Biotechnology, Beijing, China). Briefly, 5 × 103 cells were cultured in DMEM containing less than 1% FBS for 12 h for synchronization. 24 h after supplement of hrIL-23 (Peprotech, New Jersey, USA), CCK-8 solution was added. 2.5 h later, cell proliferation was determined by microplate reader at a wavelength of 450 nm.
Colony Formation assay
Huh-7 or HepG2 cells (5 × 103) were cultured in DMEM medium supplemented with hrIL-23 and 2% FBS for 5–7 days. Cells were fed fresh medium every 2–3 days. Colonies were fixed with 4% paraformaldehyde and visualized via crystal violet staining.
Cell cycle analysis
Synchronized cells were exposed to hrIL-23 for 24 h. After fixation, cells were treated with RNAase (10 μg/ml) and then stained with propidium iodide (PI, 10 μg/ml, BD, New York, USA). Cell cycle was analyzed by flow cytometer (LSR II, BD Biosciences) using software Flowjo.
Apoptosis assay
Cells (1.5 × 105) were incubated in serum-free culture supplemented with hrIL-23 for 48 h. Then cells were stained with Annexin V and PI and analyzed by flow cytometer. Plots from the gated cells showed the populations corresponding to viable (Annexin V–PI–), early and late apoptotic (Annexin V+PI–, Annexin V+PI+) cells.
Immunofluorescence assay
For detection of IL-23R, cells were cultured with 1:100 diluted goat anti-IL-23R (Abcam) at 4° C and then with 1:100 diluted rabbit anti-goat PE-conjugated secondary antibody (ProteinTech Group, Wuhan, China) at 4° C in the dark. After that, half cells were analyzed by flow cytometer and half cells were further stained with hochest33342 (5 μg/ml, Beyotime) for 10min followed by observation under the fluorescence microscope (LSM 710 and ConfoCor 3, Zeiss, Germany).
For detection of CD133, saponin permeabilized cells were incubated with PE labeled anti-CD133 (Miltenyi Biotec, Germany) and then were analyzed by FACS Calibur.
Cell culture wound closure assay
For the wound healing assays, cells were plated in 12-well plates and a scratch was made 24 h later. A low serum (2% FBS) culture medium was added to the plates to inhibit cell proliferation. hrIL-23 was supplemented at different concentrations. Wound closure was checked under an inverted microscope 24 h and 48 h later.
Transwell cell migration and invasion assays
24-well plates (8 μm pore; Corning Inc, NY, USA) were coated with matrigel (100 μg/cm2; BD, NJ, USA). hrIL-23-pretreated cells (1 × 105) were then plated in the upper chamber in serum free medium. 2.5% FBS medium was added to the lower chamber. 24 h later, migrated cells were fixed with 4% paraformaldehyde and then stained with 0.1% Crystal Violet.
Enzyme-linked immunosorbent assay (ELISA)
HBsAg and HBeAg in the culture medium were measured by ELISA kits (Kehua Bioengineering, Shanghai, China), respectively. In Briefly, HepG2, Huh-7 and HepG2.2.15 cells were seeded in 24-well plates at 70–80% confluence. The culture media were collected, removed precipitate after centrifugation for 20 minutes at 2500 rpm and measured levels of HBsAg and HBeAg according to the manufacturer’s instructions.
Statistical analysis
Values were expressed as means ± SD. Student’s t-test was applied to all analyses except for percentage data which were analyzed by Mann-Whitney U test using SPSS 17.0 statistical software (SPSS Inc., USA). P < 0.05 was considered significant.
Abbreviations
HCC: Hepatocellular carcinoma; HBV:Hepatitis B virus; HCV: Hepatitis C virus; HBsAg: HBV surface antigen; HNF4α: Hepatocyte nuclear factor 4 alpha; CCK-8: Cell Counting Kit-8; PI: Propidium iodide; MMP9: Matrix metalloproteinase 9; MFI: Mean fluorescence index; ELISA: Enzyme-Linked Immunosorbent Assay.
Author contributions
QJ and YLS performed most of the experiments, summarized the results and wrote most of the manuscript. PL and GXS conceived the idea for the experiments, coordinated the study and edited the manuscript. CR, ZLG, SMM and MPF conducted the rest of the experiments. HFZ and NJ contributed to experimental data analysis and edited the manuscript. PL and GXS provided critical review of the manuscript. All authors approved the final version of the manuscript.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the Natural Science Foundation of China (81373152); Program for Changjiang Scholars and Innovative Research Team in University (IRT_14R20); the Fundamental Research Funds for the Central Universities of China (2015TS124) and Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College, HUST.
CONFLICTS OF INTEREST
The authors do not have any potential conflicts of interest to disclose.
REFERENCES
1. Yang JD, Roberts LR. Hepatocellular carcinoma: A global view. Nat Rev Gastroenterol Hepatol. 2010; 7:448–458.
2. Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, Gores G. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016; 2:16018.
3. El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012; 142:1264–1273.
4. Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellularcarcinoma. J Hepatol. 2016; 64:S84–S101.
5. World Health Organization. Hepatitis B fact sheet. Revised July. World HealthOrganization: Geneva, Switzerland, 2015, Available http://www.who.int/mediacentre/factsheets/fs204/en/. Accessed: 10 April 2016.
6. Pan X, Cao H, Lu J, Shu X, Xiong X, Hong X, Xu Q, Zhu H, Li G, Shen G. Interleukin-32 expression induced by hepatitis B virus protein X is mediated through activation of NF-kappa B. Mol Immunol. 2011; 48:1573–1577.
7. Wang Q, Zhou J, Zhang B, Tian Z, Tang J, Zheng Y, Huang Z, Tian Y, Jia Z, Tang Y, van Velkinburgh JC, Mao Q, Bian X, et al. Hepatitis B virus induces IL-23 production in antigen presenting cells and causes liver damage via the IL-23/IL-17 axis. PLoS Pathog. 2013; 9:e1003410.
8. Xia L, Tian D, Huang W, Zhu H, Wang J, Zhang Y, Hu H, Nie Y, Fan D, Wu K. Upregulation of IL-23 expression in patients with chronic hepatitis B is mediated by the HBx/ERK/NF-kappaB pathway. J Immunol. 2012; 188:753–764.
9. Yu C, Gong X, Yang Q, Lian J, Xu K, Ruan B, Li L. The serum IL-23 level predicts the response to pegylated interferon therapy in patients with chronic hepatitis B. Liver Int. 2015; 35:1549–1556.
10. Oppmann B, Lesley R. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000; 13:715–725.
11. Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, To W, Wagner J, O’Farrell AM, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12R beta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002; 168:5699–5708.
12. Duvallet E, Semerano L, Assier E, Falgarone G, Boissier MC. Interleukin-23: a key cytokine in inflammatory diseases. Ann Med. 2011; 43:503–511.
13. Cocco C, Canale S, Frasson C, Di Carlo E, Ognio E, Ribatti D, Prigione I, Basso G, Airoldi I. Interleukin-23 acts as antitumor agent on childhood B-acute lymphoblastic leukemia cells. Blood. 2010; 116:3887–3898.
14. Lo CH, Chang CM, Tang SW, Pan WY, Fang CC, Chen Y, Wu PY, Chen KY, Ma HI, Xiao X, Tao MH. Differential antitumor effect of interleukin-12 family cytokines on orthotopic hepatocellular carcinoma. J Gene Med. 2010; 12:423–434.
15. Lo CH, Lee SC, Wu PY, Pan WY, Su J, Cheng CW, Roffler SR, Chiang BL, Lee CN, Wu CW, Tao MH. Antitumor and antimetastatic activity of IL-23. J Immunol. 2003; 171:600–607.
16. Ringkowski S, Thomas PS, Herbert C. Interleukin-12 family cytokines and sarcoidosis. Front Pharmacol. 2014; 5:233.
17. Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, Basham B, McClanahan T, Kastelein RA, Oft M. IL-23 promotes tumour incidence and growth. Nature. 2006; 442:461–465.
18. Fukuda M, Ehara M, Suzuki S, Ohmori Y, Sakashita H. IL-23 promotes growth and proliferation in human squamous cell carcinoma of the oral cavity. Int J Oncol. 2010; 36:1355–1365.
19. Lan F, Zhang L, Wu J, Zhang J, Zhang S, Li K, Qi Y, Lin P. IL-23/IL-23R: potential mediator of intestinal tumor progression from adenomatous polyps to colorectal carcinoma. Int J Colorectal Dis. 2011; 26:1511–1518.
20. Li J, Zhang L, Zhang J, Wei Y, Li K, Huang L, Zhang S, Gao B, Wang X, Lin P. Interleukin 23 regulates proliferation of lung cancer cells in a concentration-dependent way in association with the interleukin-23 receptor. Carcinogenesis. 2013; 34:658–666.
21. Parviz F, Matullo C, Garrison WD, Savatski L, Adamson JW, Ning G, Kaestner KH, Rossi JM, Zaret KS, Duncan SA. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet. 2003; 34:292–296.
22. Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, Zheng BJ, Guan XY. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007; 132:2542–2556.
23. Suetsugu A, Nagaki M, Aoki H, Motohashi T, Kunisada T, Moriwaki H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem Biophys Res Commun. 2006; 351:820–824.
24. Yin S, Li J, Hu C, Chen X, Yao M, Yan M, Jiang G, Ge C, Xie H, Wan D, Yang S, Zheng S, Gu J. CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int J Cancer. 2007; 120:1444–1450.
25. Kohga K, Tatsumi T, Takehara T, Tsunematsu H, Shimizu S, Yamamoto M, Sasakawa A, Miyagi T, Hayashi N. Expression of CD133 confers malignant potential by regulating metalloproteinases in human hepatocellular carcinoma. J Hepatol. 2010; 52:872–879.
26. Battle MA, Konopka G, Parviz F, Gaggl AL, Yang C, Sladek FM, Duncan SA. Hepatocyte nuclear factor 4alpha orchestrates expression of cell adhesion proteins during the epithelial transformation of the developing liver. Proc Natl Acad Sci U S A. 2006; 103:8419–8424.
27. Lazarevich NL, Cheremnova OA, Varga EV, Ovchinnikov DA, Kudrjavtseva EI, Morozova OV, Fleishman DI, Engelhardt NV, Duncan SA. Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology. 2004; 39:1038–1047.
28. Teng MW, Andrews DM, McLaughlin N, von Scheidt B, Ngiow SF, Moller A, Hill GR, Iwakura Y, Oft M, Smyth MJ. IL-23 suppresses innate immune response independently of IL-17A during carcinogenesis and metastasis. Proc Natl Acad Sci U S A. 2010; 107:8328–8333.
29. Darsigny M, Babeu JP, Seidman EG, Gendron FP, Levy E, Carrier J, Perreault N, Boudreau F. Hepatocyte nuclear factor-4alpha promotes gut neoplasia in mice and protects against the production of reactive oxygen species. Cancer Res. 2010; 70:9423–9433.
30. Yin C, Lin Y, Zhang X, Chen YX, Zeng X, Yue HY, Hou JL, Deng X, Zhang JP, Han ZG, Xie WF. Differentiation therapy of hepatocellular carcinoma in mice with recombinant adenovirus carrying hepatocyte nuclear factor-4alpha gene. Hepatology. 2008; 48:1528–1539.
31. Lazarevich NL, Shavochkina DA, Fleishman DI, Kustova IF, Morozova OV, Chuchuev ES, Patyutko YI. Deregulation of hepatocyte nuclear factor 4 (HNF4)as a marker of epithelial tumors progression. Exp Oncol. 2010; 32:167–171.
32. Ning BF, Ding J, Yin C, Zhong W, Wu K, Zeng X, Yang W, Chen YX, Zhang JP, Zhang X, Wang HY, Xie WF. Hepatocyte nuclear factor 4 alpha suppresses the development of hepatocellular carcinoma. Cancer Res. 2010; 70:7640–7651.
33. Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol. 2008; 8:458–466.
34. Floss DM, Mrotzek S, Klöcker T, Schröder J, Grötzinger J, Rose-John S, Scheller J. Identification of canonical tyrosine-dependent and non-canonical tyrosine-independent STAT3 activation sites in the intracellular domain of the interleukin 23 receptor. J Biol Chem. 2013; 288:19386–19400.
35. Cho ML, Kang JW, Moon YM, Nam HJ, Jhun JY, Heo SB, Jin HT, Min SY, Ju JH, Park KS, Cho YG, Yoon CH, Park SH, et al. STAT3 and NF-kappaB signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. J Immunol. 2006; 176:5652–5661.
36. Gee K, Guzzo C, Che Mat NF, Ma W, Kumar A. The IL-12 family of cytokines in infection, inflammation and autoimmune disorders. Inflamm Allergy Drug Targets. 2009; 8:40–52.
37. O’Shea JJ, Lahesmaa R, Vahedi G, Laurence A, Kanno Y. Genomic views of STAT function in CD4+ T helper cell differentiation. Nat Rev Immunol. 2011; 11:239–250.
38. Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA, Ogata H, Karin M, Struhl K, Hadzopoulou-Cladaras M, Iliopoulos D. An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell. 2011; 147:1233–1247.
39. Liu H, Lou G, Li C, Wang X, Cederbaum AI, Gan L, Xie B. HBx inhibits CYP2E1 gene expression via downregulating HNF4α in human hepatoma cells. PLoS One. 2014; 9:e107913.
40. Wu Q, Liu HO, Liu YD, Liu WS, Pan D, Zhang WJ, Yang L, Fu Q, Xu JJ, Gu JX. Decreased expression of hepatocyte nuclear factor 4α (Hnf4α)/microRNA-122 (miR-122) axis in hepatitis B virus-associated hepatocellular carcinoma enhances potential oncogenic GALNT10 protein activity. J Biol Chem. 2015; 290:1170–1185.
41. Long Y, Chen E, Liu C, Huang F, Zhou T, He F, Liu L, Liu F, Tang H. The correlation of hepatocyte nuclear factor 4 alpha and 3 beta with hepatitis B virus replication in the liver of chronic hepatitis B patients. J Viral Hepat. 2009; 16:537–546.
42. Chen EQ, Sun H, Feng P, Gong DY, Liu C, Bai L, Yang WB, Lei XZ, Chen LY, Huang FJ, Tang H. Study of the expression levels of Hepatocyte nuclear factor 4 alpha and 3 beta in patients with different outcome of HBV infection. Virol J. 2012; 9:23.
43. Xu L, Hui L, Wang S, Gong J, Jin Y, Wang Y, Ji Y, Wu X, Han Z, Hu G. Expression profiling suggested a regulatory role of liver-enriched transcription factors in human hepatocellular carcinoma. Cancer Res. 2001; 61:3176–3181.
44. Walesky C, Gunewardena S, Terwilliger EF, Edwards G, Borude P, Apte U. Hepatocyte-specific deletion of hepatocyte nuclear factor-4alpha in adult mice results in increased hepatocyte proliferation. Am J Physiol Gastrointest Liver Physiol. 2013; 304:26–37.
45. Garibaldi F, Cicchini C, Conigliaro A, Santangelo L, Cozzolino AM, Grassi G, Marchetti A, Tripodi M, Amicone L. An epistatic mini-circuitry between the transcription factors Snail and HNF4alpha controls liver stem cell and hepatocyte features exhorting opposite regulation on stemness-inhibiting microRNAs. Cell Death Differ. 2012; 19:937–946.
46. Yao HS, Wang J, Zhang XP, Wang LZ, Wang Y, Li XX, Jin KZ, Hu ZQ, Wang WJ. Hepatocyte nuclear factor 4α suppresses the aggravation of colon carcinoma. Mol Carcinog. 2016; 55:458–472.
47. Zhou Y, Wang S, Ma JW, Lei Z, Zhu HF, Lei P, Yang ZS, Zhang B, Yao XX, Shi C, Sun LF, Wu XW, Ning Q, et al. Hepatitis B virus protein X-induced expression of the CXC chemokine IP-10 is mediated through activation of NF-kappaB and increases migration of leukocytes. J Biol Chem. 2010; 285:12159–12168.
48. Wang Q, Shu R, He H, Wang L, Ma Y, Zhu H, Wang Z, Wang S, Shen G, Lei P. Co-silencing of Birc5 (survivin) and Hspa5 (Grp78) induces apoptosis in hepatoma cells more efficiently than single gene interference. Int J Oncol. 2012; 41:652–660.