
INTRODUCTION
Adenoid cystic carcinoma (AdCC), although rare accounting for less than 1% of all head and neck cancers, is one of the most common carcinomas of the salivary gland [1]. Factors that often influence survival include tumor stage, node status, patient age, tumor site, large nerve perineural invasion, and surgical margins. Standard treatment for this carcinoma is surgical resection followed by post-operative radiotherapy and/or chemotherapy. While lymph node involvement is uncommon, distant metastasis to the lung is frequent [1]. AdCC is a slow-growing tumor but the long-term prognosis is poor with the 10-year survival rate being 52–65% [2]. Previous studies have revealed that MYB-NFIB and MYBL1-NFIB fusions are major alterations in this carcinoma, accounting for approximately 50% and 10% of AdCC cases, respectively [3–7]. Gene alterations involving MYB, MYBL1, or NFIB genes have been considered to be among the primary events in AdCC development. In a study employing conditional MYB-NFIB mutant transgenic mice, expression of the oncogene resulted in the development of the carcinoma in nearly 30% of the animals [8, 9]. However, oncogenetic events associated with AdCC progression have not been well recognized.
Epidermal growth factor receptor (EGFR), a trans-membrane tyrosine kinase receptor, binds with its ligands (EGF and transforming growth factor alpha), and activates downstream signaling, which stimulates mitosis, leading to cell proliferation and inhibition of apoptosis [10]. It has been well documented that mutations of genes involved in the EGFR pathway play an important role in the pathogenesis and progression of various tumors [11–14]. Recently, anti-EGFR agents, including EGFR tyrosine kinase inhibitors and anti-EGFR antibodies, have been effectively used for tumor treatment [15, 16]. In pulmonary carcinomas, EGFR mutation is associated with Asians, females, non-smokers, and the adenocarcinoma type, and it has become evident that EGFR tyrosine kinase inhibitors are highly effective for pulmonary adenocarcinomas with EGFR mutations [17, 18]. RAS proteins, encoded by three ubiquitously expressed KRAS, HRAS, and NRAS genes, are located downstream of EGFR and are frequently mutated in human cancers [19]. These proteins are GTPases that may function as a molecular switch regulating the EGFR pathways responsible for proliferation and cell survival [12]. Aberrant RAS function is probably associated with a single mutation, typically at codon 12, 13 or 61 [20], and the mutation may favor GTP binding and result in constitutive activation of RAS [20]. The prognostic significance of RAS mutations has been repeatedly reported in pulmonary carcinomas [21–24]. On the other hand, it has been firmly established that anti-EGFR antibody therapy is less effective in colonic adenocarcinomas harboring mutated RAS genes [25]. These observations suggest that detection of EGFR pathway gene mutations is critically important not only for clarifying oncogenesis and tumor progression but also for selecting the most effective molecularly targeted therapy. Unfortunately, mutations of EGFR pathway genes in salivary gland AdCC (SAdCC) have been poorly understood.
In this study, using a large cohort of SAdCC cases, we looked for mutations of the genes involved in the EGFR pathway (EGFR, RAS family, PIK3CA, BRAF, and AKT1), and correlated the results with the clinicopathological features and prognostic outcomes of the patients. These genes were chosen since the mutations have been detected in some SAdCC cases by a previous comprehensive analysis using next generation sequencing (NGS) [26–29] and have been closely associated with current molecularly targeted therapy [30]. For detection of the point mutations of the above genes, we employed the SNaPshot assay, a single-base extension multiplex assay that is highly sensitive, low-cost, and rapid [31–34].
RESULTS
Carcinoma cases
The clinicopathological characteristics of the SAdCC cases analyzed in this study (n = 70) are summarized in Table 1. The tumor cases consisted of 29 men and 41 women, with ages ranging from 27 to 82 years (median, 64). In 45 cases, the tumor had originated in the major salivary glands, and in the remaining 25, in the minor salivary glands. In 28 cases, the tumor was more than 2 cm in diameter, and in 11, there was metastasis to the regional cervical lymph nodes. Twenty-nine cases were classified as stage III/IV tumors. Surgical resection was carried out in all 70 cases and neck dissection was additionally performed in 39. On pathological evaluation, 39, 13, and 18 cases were classified as grades I, II, and III, respectively. Perineural invasion was found in 42 cases. Although all tumors were resected macroscopically with curative intent, positive surgical margins were microscopically observed in 32 cases. Post-operative radiotherapy and/or chemotherapy was carried out in 30 patients but not in the remaining 40 patients because of their poor general condition and/or personal refusal. SAdCC patients with positive surgical margins tended to receive the radiotherapy. Tumor recurrence after surgery was recorded in 28 patients (median, 22.5 months; range, 1–120). At the last follow-up (median, 60.5 months; range, 7–312), 36, 27, and 7 patients were alive with no evidence of disease, alive with disease, and had died of disease, respectively. None of the patients died of other causes.
Table 1: Clinicopathological characteristics of the patients (n = 70)
Factor |
n (%) |
|
|---|---|---|
Age (years) |
Mean |
62.5 |
Median |
64 (range, 27–82) |
|
<60 |
25 (35.7) |
|
>60 |
45 (64.3) |
|
Sex |
Male |
29 (41.4) |
Female |
41 (58.6) |
|
Primary tumor site |
Parotid |
17 (24.3) |
Submandibular |
21 (30) |
|
Sublingual |
7 (10) |
|
Minor |
25 (35.7) |
|
Tumor size (cm) |
<2 |
42 (60) |
>2 |
28 (40) |
|
Nodal status |
Positive |
11 (15.7) |
Negative |
59 (84.3) |
|
Clinical stage |
I, II |
41 (58.6) |
III, IV |
29 (41.4) |
|
Neck dissection |
Performed |
39 (55.7) |
Post-operative radiation |
Performed |
30 (42.9) |
Post-operative chemotherapy |
Performed |
3 (4.3) |
Histological grade |
I |
39 (55.7) |
II |
13 (18.6) |
|
III |
18 (25.7) |
|
Perineural invasion |
Positive |
42 (60) |
Negative |
23 (32.9) |
|
Undetermined |
5 (7.1) |
|
Microscopic surgical margin |
Positive |
32 (45.7) |
Negative |
38 (54.3) |
|
Follow-up (months) |
Median |
60.5 (range, 7–312) |
Tumor recurrence |
Yes |
28 (40) |
Deceased |
Yes |
7 (10) |
Tissue FISH analysis for MYB, MYBL1, and NFIB gene translocations
We performed fluorescence in situ hybridization (FISH) analysis in 70 SAdCC cases using paraffin sections [7], and succeeded in obtaining FISH signals in 52 (74%) but failed to obtain the signals in the remaining 18 (26%) cases after repeated FISH procedures. Gene splits in MYB, MYBL1, and NFIB genes were detected in 33, 6, and 32 cases, respectively, and in 45/52 (86.5%) cases in total. In the other 7 cases, no gene splits were noted. Subsequently, we performed tissue FISH analysis for gene fusions in gene-split-positive cases. According to our previous study [7], 52 cases were divided into six gene groups; 23 MYB-NFIB (44%), 10 MYB-X (19%), 4 MYBL1-NFIB (8%), 2 MYBL1-X (4%), 6 NFIB-X (12%), and 7 (13%) cases that were completely negative (X was defined as genes whose alterations were not detected in the present FISH assay).
Mutation analysis
We looked for gene mutations in EGFR, RAS family (KRAS, HRAS, and NRAS), PIK3CA, BRAF, and AKT1, by SNaPshot assay, a highly sensitive single-base extension multiplex assay [31–34] and EGFR deletion was detected by direct sequencing. Of the 70 SAdCC cases, 16 point mutations in the EGFR pathway were detected in 13 (18.6%) cases: EGFR in one (1.4%), RAS family mutations in 10 [14.3%; KRAS in 6 (8.6%), HRAS in 4 (5.7%), and NRAS in none], PIK3CA in 5 (7.1%), BRAF in none, and AKT1 in none. All these mutations were missense mutations and none of the cases showed an EGFR deletion. The representative results of the SNaPshot assay are shown in Figure 1. While five point mutations were found to be positive by direct sequencing, all point mutations were validated by a SNaPshot analysis using both sense and anti-sense probes and with a polymerase chain reaction (PCR)-based amplification refractory mutation system (ARMS-PCR, a modified allele-specific PCR assay). Among the 13 cases positive for EGFR pathway mutations, tissue FISH analysis was successfully performed in 8 cases, all of which showed gene translocations associated with MYB, MYBL1, and NFIB genes. Details of the mutations and clinicopathological features of the 13 mutated cases are shown in Table 2.
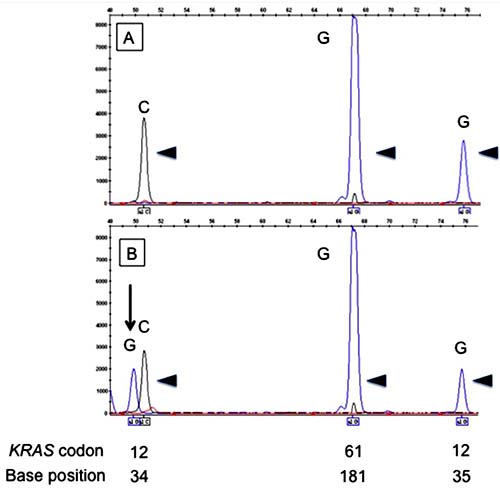
Figure 1: Electropherograms of the SNaPshot assay for KRAS codons 12 and 61. The upper panel (A) shows unmutated (arrowheads) codons 12 and 61. The lower panel (B) shows mutated codon 12 (arrow) and unmutated codon 61 (arrowheads). Positions of codons and nucleotides are indicated at the bottom of the figure.
Table 2: Salivary gland AdCC cases with gene mutations in the EGFR pathway
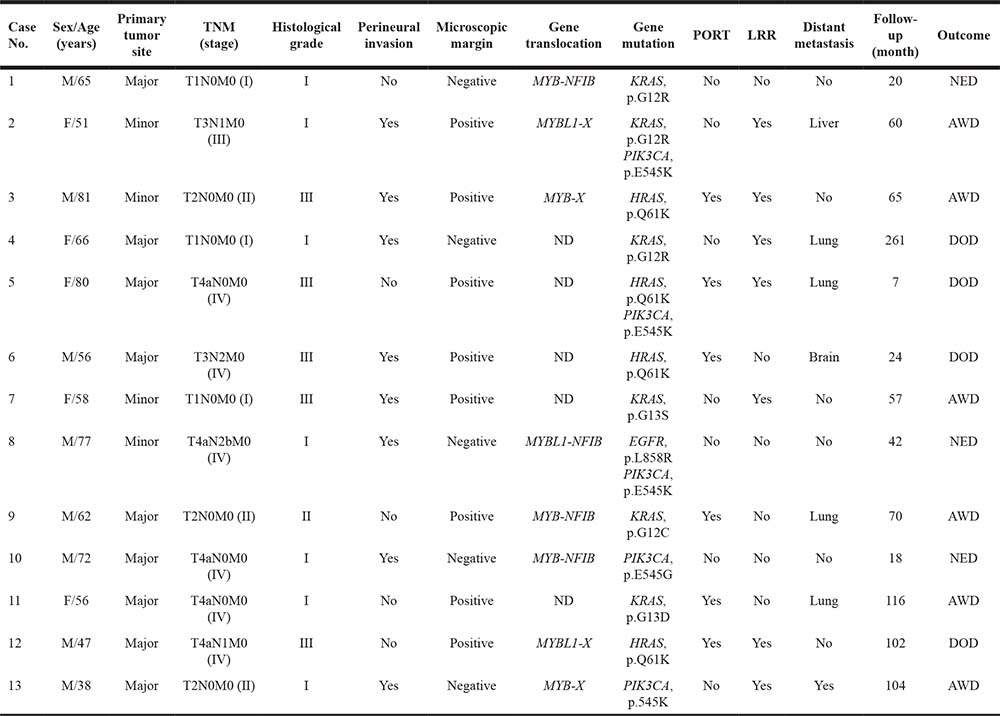
ND, not detected; PORT, post-operative radiation therapy; LRR, loco-regional recurrence; NED, no evidence of disease; AWD, alive with disease; and DOD, died of disease.
Clinicopathological correlation and prognostic impact of gene mutations
As shown in Table 3, no correlation was found between EGFR pathway mutations and clinicopathological factors. RAS mutations were significantly associated with a microscopically positive margin (p = 0.036). Noting the case number of each gene group and taking into consideration that MYBL1 is a member of the MYB family, we analyzed the clinicopathological impact of MYB-NFIB (n = 23), MYB-split-positive (n = 33), and MYB/MYBL1-split-positive (n = 39) mutations (Table 4). The MYB-NFIB fusion was found to have no association with the clinicopathological factors examined and the MYB-split-positive group was associated with a histological grade III status (p = 0.018). The MYB/MYBL1-split-positive group was associated with a histological grade III status (p = 0.023), a positive perineural invasion (p = 0.037), and a microscopically positive margin (p = 0.008). None of these gene groups was associated with an EGFR pathway mutation or RAS mutation.
Table 3: Association between EGFR pathway mutations and clinicopathological factors
Factor |
EGFR pathway mutations |
p |
RAS mutations |
p |
|||
|---|---|---|---|---|---|---|---|
Positive |
Negative |
Positive |
Negative |
||||
Age |
>60y |
7 |
38 |
0.523 |
5 |
40 |
0.477 |
<60y |
6 |
19 |
5 |
20 |
|||
Sex |
Male |
8 |
21 |
0.127 |
5 |
24 |
0.731 |
Female |
5 |
36 |
5 |
36 |
|||
Tumor site |
Major |
9 |
36 |
0.759 |
7 |
38 |
1.00 |
Minor |
4 |
21 |
3 |
22 |
|||
Tumor size |
I/II |
6 |
36 |
0.349 |
5 |
37 |
0.507 |
III/IV |
7 |
21 |
5 |
23 |
|||
Nodal metastasis |
Positive |
4 |
7 |
0.198 |
3 |
8 |
0.186 |
Negative |
9 |
50 |
7 |
52 |
|||
Clinical stage |
I/II |
6 |
35 |
0.361 |
5 |
36 |
0.731 |
III/IV |
7 |
22 |
5 |
24 |
|||
PORT |
Performed |
6 |
24 |
1.00 |
6 |
24 |
0.308 |
Not received |
7 |
33 |
4 |
36 |
|||
Histological grade |
I/II |
8 |
44 |
0.296 |
5 |
47 |
0.111 |
III |
5 |
13 |
5 |
13 |
|||
Perineural invasion |
Positive |
8 |
34 |
1.00 |
5 |
37 |
0.710 |
Negative |
4 |
19 |
4 |
19 |
|||
Microscopic margin |
Positive |
8 |
24 |
0.232 |
8 |
24 |
0.036 |
Negative |
5 |
33 |
2 |
36 |
|||
PORT, post-operative radiation therapy.
Table 4: Association of gene alteration group with clinicopathological factors (52 cases in total)
Gene group (p) |
|||
|---|---|---|---|
MYB-NFIB |
MYB-split-positive |
MYB/MYBL1-split-positive |
|
Factor |
(n = 23) |
(n = 33) |
(n = 39) |
Age >60 years |
0.77 |
0.546 |
0.334 |
Sex |
1.00 |
0.771 |
0.743 |
Tumor site |
0.77 |
0.382 |
0.177 |
Tumor size |
0.778 |
0.766 |
0.099 |
Nodal metastasis |
0.307 |
0.729 |
0.051 |
Clinical stage |
0.574 |
0.558 |
0.057 |
PORT |
0.397 |
0.371 |
0.328 |
Histological grade |
0.524 |
0.018 (grade III) |
0.023 (grade III) |
Perineural invasion |
0.398 |
0.070 |
0.037 (positive) |
Microscopic margin |
0.400 |
0.149 |
0.008 (positive) |
EGFR pathway mutations |
1.00 |
1.00 |
0.177 |
RAS mutations |
1.00 |
1.00 |
0.314 |
PORT, post-operative radiation therapy; DFS, disease-free survival; and OS, overall survival.
The disease-free survival (DFS) rates of the SAdCC patients at 5 years and 10 years were 57.9% and 31.4%, respectively, and overall survival (OS) rates at these points were 94.7% and 72.7%, respectively. Results of the prognostic analysis are shown in Table 5. With respect to DFS, T3/4 tumors (p = 0.006), clinical stage III/IV (p = 0.006) tumors, histological grade III (p = 0.009) tumors, tumors with a microscopically positive margin (p = 0.013), and those with EGFR pathway mutations (p = 0.011, Figure 2A) and RAS mutations (p = 0.010, Figure 2B) were all significantly associated with a shorter patient survival. For OS, histological grade III (p < 0.0001), microscopically positive margins (p = 0.023), EGFR pathway mutations (p = 0.049, Figure 2C), and RAS mutations (p = 0.024, Figure 2D) were selected as risk factors with statistical significance. An MYB-NFIB, MYB-split-positive, and MYB/MYBL1-split-positive status had no prognostic impact on either DFS or OS (Table 5).
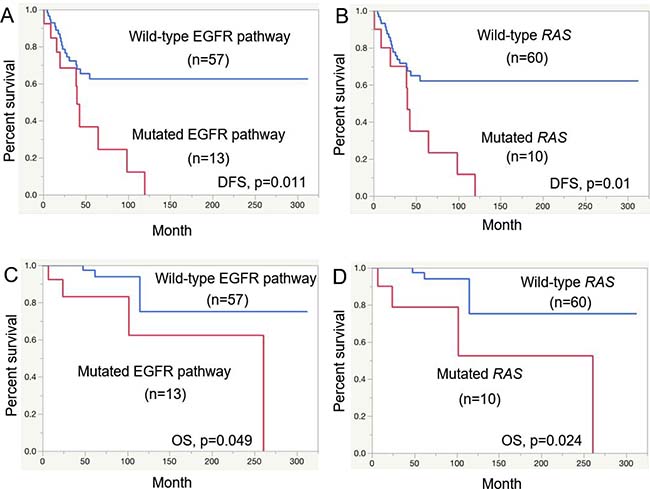
Figure 2: Prognostic analysis for salivary gland AdCC patients. Disease-free survival (DFS) for EGFR pathway mutations (A) and RAS mutations (B) and overall survival (OS) for EGFR pathway mutations (C) and RAS mutations (D).
Table 5: Prognostic analysis (70 cases in total)
Factor |
p |
||
|---|---|---|---|
DFS |
OS |
||
Age |
>60y |
0.066 |
0.976 |
Sex |
Male |
0.183 |
0.646 |
Tumor site |
Major |
0.510 |
0.482 |
Tumor size |
T3/4 |
0.006 |
0.086 |
Nodal metastasis |
Positive |
0.267 |
0.057 |
Clinical stage |
III/IV |
0.006 |
0.089 |
Histological grade |
III |
0.009 |
<0.0001 |
Perineural invasion |
Positive |
0.146 |
0.869 |
Microscopic margin |
Positive |
0.013 |
0.023 |
Mutations in EGFR pathway |
Positive |
0.011 |
0.049 |
RAS mutations |
Positive |
0.010 |
0.024 |
MYB-NFIB (n = 52) |
Positive |
0.646 |
0.609 |
MYB-split (n = 52) |
Positive |
0.562 |
0.499 |
MYB/MYBL1-split (n = 52) |
Positive |
0.220 |
0.170 |
DFS, disease-free survival; OS, overall survival
DISCUSSION
In examining our 70 SAdCC cases, we searched for mutations in genes involved in the EGFR pathway [EGFR, RAS family (KRAS, HRAS, and NRAS), PIK3CA, BRAF, and AKT1]. All AdCC cases included in this study were retrieved from salivary gland cases since surgical procedures [35], TNM classifications [36], and prognoses [37, 38] are different between SAdCC cases of the salivary gland and those of other head and neck sites. Of the 70 SAdCC cases, gene mutations were detected in 18.6% (RAS in 14.3%, PIK3CA in 7.1%, and EGFR in 1.4%) but not in BRAF or AKT1. All these mutations were missense mutations and none of the cases showed an EGFR deletion. Concurrent mutations were found in 4.3%. In the prognostic analysis, EGFR pathway mutations were selected as a risk factor for DFS (p = 0.011) and OS (p = 0.049) as were RAS mutations, (p = 0.010) and OS (p = 0.024), respectively. In the 70 AdCC cases, the fusion status was successfully determined in 52 using custom-made FISH probes. An MYB-NFIB and MYB-split-positive status had no or only a weak clinicopathological impact whereas an MYB/MYBL1-split-positive status was associated with a higher tumor grade and a local aggressiveness of the tumor but failed to have a prognostic impact.
One of the important findings of this study was that RAS mutations were frequently detected (14.3%) in SAdCC cases. Anti-EGFR antibodies, including cetuximab and panitumumab, are promising molecularly targeted drugs for head and neck carcinomas including AdCC. Hitre et al. recently reported that in a phase II trial with cetuximab combined with conventional chemotherapy and/or radiotherapy, an objective response was obtained in >40% of patients with locally advanced or metastatic head and neck AdCCs [39]. Locati et al. reported treating 23 patients with recurrent and/or metastatic salivary gland AdCCs with cetuximab alone and 12 of these patients remained in a stable disease state for more than six months [40]. Jacob et al. reported treating 18 patients with recurrent and/or metastatic salivary gland AdCCs with orally administered gefitinib and seven of these patients showed prolonged disease stabilization for more than nine months [41]. Unfortunately, the RAS mutation status was not examined in these trials. It is well known that colorectal adenocarcinomas with RAS mutations often respond poorly to anti-EGFR antibody therapy. In head and neck squamous cell carcinomas, RAS mutations may confer resistance to therapies using EGFR tyrosine kinase inhibitors or anti-EGFR antibodies in experimental and clinical studies [42]. It should be determined whether RAS mutations have a similar value in predicting efficacy in SAdCC patients. On the other hand, RAS genes may be important therapeutic targets. For some carcinomas, cancer vaccines that stimulate immunity against mutant RAS proteins and antisense therapies that block the translation of mutant RAS genes can be applied in treatment [43]. Evidence using cancer cell lines has indicated that KRAS antisense oligodeoxynucleotide inhibits KRAS expression, tumor growth, and tumor invasiveness [44, 45], and that the oligodeoxynucleotide can also suppress peritoneal dissemination of cancer cells in vivo [45, 46].
Another important finding of this study was that the RAS mutations were identified as risk factors for both DFS and OS. To the best of our knowledge, this finding has not been reported for SAdCC. The prognostic significance of RAS mutations varies according to the cancer type; and while it has not been established in colonic adenocarcinoma [47], it has been repeatedly reported in pulmonary carcinomas [21–24]. The salivary glands are developmentally close to the lungs where many salivary gland type tumors including AdCC frequently occur [48]. In addition, RAS mutation rates may be similar in pulmonary adenocarcinoma and SAdCC [21–24]. We speculate that the significance of RAS mutations in SAdCC may be similar to that in pulmonary carcinomas. Although the biological significance of RAS mutations in SAdCC oncogenesis remains to be elucidated, according to lung carcinoma oncogenesis [49, 50], RAS mutations may be a relatively late genetic event. Gene fusions involving MYB, MYBL1, and NFIB, which were found in nearly 90% of our SAdCC cases, are currently considered to be the primary oncogenetic event in SAdCC [8, 9], and RAS mutations may confer progressive or invasive features on the tumor cells, resulting in a worse prognosis for SAdCC patients.
The RAS gene mutation rate (14.3%) in our SAdCC cases was somewhat higher than those reported in previous studies (0–9.1%), as summarized in Supplementary Table 1 [26–30, 51, 52]. This difference may be explained partly in terms of the heterogeneity of the carcinomas and the detection methods employed. For detection of point mutations, we used the SNaPshot assay [31–34]. This is a highly sensitive single-base extension multiplex assay that requires less than 5% mutant alleles to identify mutations [32]. Allele-specific PCR assays (including ARMS-PCR that we used for the mutation validation) may have a sensitivity similar to that of SNaPshot while conventional direct sequencing requires more than 20% mutant alleles [53]. NGS offers simultaneous sequencing of thousands to millions of short nucleic acid sequences in a multi-parallel fashion [54, 55]. In NGS, however, the detection sensitivity is largely dependent on the depth-of-coverage and the default calling parameters for an automatic analysis because NSG has the inherent weak points of short-length amplicons and a low reliability of the sequencing data of each read. In two landscape studies of AdCC [26, 27] where the depths-of-coverage were low, RAS mutations were infrequent (0% and 1.7%) while in two target sequence studies of AdCC [28, 29], RAS mutations were frequent (6.1% and 9.1%), similar to our study (14.3%) (Supplementary Table 1). We rigorously confirmed the mutations by (1) testing all samples in at least two separate experiments, (2) validating the point mutation results with the ARMS-PCR assay, and (3) carrying out a SNaPshot assay using both sense and anti-sense probes (Supplementary Figure 1).
While gene mutation rates of EGFR, BRAF, and AKT1 were low in our study, the PIK3CA mutation was detected in 5/70 (7.1%) SAdCC cases. These rates were similar to those of previous AdCC studies (Supplementary Table 1) [26–30, 51, 52]. Owing to the small number of mutated cases, we were unable to clarify the clinicopathological significance of the PIK3CA mutation in SAdCC. The prognostic impact of this mutation has not been established in other carcinomas, but constitutive activation of PI3K by PIK3CA mutation has been associated with resistance to trastuzumab therapy targeting HER2 [56]. However, since HER2 overexpression and HER2 mutation are rare in SAdCC [57], the presence of a PIK3CA mutation may not be very important as an efficacy-predicting biomarker. Similarly, mutations in EGFR, BRAF, and AKT1 were found to be very rare in our SAdCC cases, suggesting their limited role in the diagnosis and treatment of these cases.
As we recently described [7], we divided our SAdCC cases (n = 52) into six gene groups. Since the case numbers in some gene groups were small, it was difficult to draw a definite clinicopathological contour of each group. We focused our attention on the MYB-NFIB, MYB-split-positive, and MYB/MYBL1-split-positive groups. While the former two groups had no or only a weak clinicopathological impact, the latter group was associated with a higher histological grade, perineural tumor invasion, and a positive microscopic margin (Table 4). These findings were in accord with our previous finding that MYB/MYBL1-split-positive tumors may be locally aggressive [7]. This is difficult to explain but it is intriguing to speculate that MYBL1 alterations may be associated with adverse features as Brayer et al. previously suggested [5]. Further studies are needed to clarify whether MYBL1 gene alterations are associated with aggressive behavior in SAdCC.
On performing FISH analysis of our 70 SAdCC cases using paraffin sections, we successfully obtained FISH signals in 52 (74%) but failed to obtain these signals in the remaining 18 (26%). This failure may be partly owing to a suboptimal DNA quality of the samples and the lengths of the FISH probes used. Our FISH probes were custom-made and the lengths (range, 180 kb to 192 kb) were shorter than those of commercially available FISH probes (usually longer than 400 kb). We randomly selected four FISH-negative cases and performed a preliminary FISH assay using commercially available ETV6-split probes (486 kb and 632 kb in length, Zytovision, Bremerhaven, Germany) and successfully obtained FISH signals in three cases (data not shown).
In summary, we showed that RAS mutations were frequent (14.3%) in our SAdCC cases and that the mutations were associated with a shorter DFS and OS in these patients. These findings may be useful in developing novel therapeutic strategies against this lethal tumor. Clarification is warranted as to whether the RAS mutation status would be useful in predicting the efficacy of treatments targeting EGFR pathway molecules in SAdCC patients.
MATERIALS AND METHODS
Case selection
From the pathology files of the authors’ institutions, we retrieved 70 cases previously diagnosed as SAdCC. Cases involving the sinonasal cavity, lung or other sites were not included in this study. All cases were carefully reviewed by two independent pathologists (TM and HI) according to WHO classification criteria for salivary gland tumors [1]. None of the cases showed distant metastasis at the initial treatment. The following clinicopathological factors were analyzed: age, sex, primary tumor site, tumor size, metastasis to regional lymph nodes, clinical stage, histological grade, perineural invasion, microscopic surgical margin, post-operative treatment (radiotherapy and/or chemotherapy), and follow-up. Tumors were histopathologically classified as grade I, II, or III; grade I tumors mainly showed a tubular and cribriform pattern without solid tumor components, grade II tumors were defined as cribriform with solid components of <30%, and grade III tumors, as those showing solid components of >30% [58]. This study was approved by the institutional review board of Nagoya City University and conducted in accordance with the Declaration of Helsinki.
Tissue FISH analysis for MYB, MYBL1, and NFIB gene translocations
Using paraffin tumor sections, we performed interphase FISH analysis for the gene splits in MYB, MYBL1, and NFIB, as well as gene fusions between these genes when the gene splits were present. FISH procedures have been described elsewhere [7, 59] and the FISH probes that we used are shown in Supplementary Table 2. The frequencies of gene abnormalities were determined by counting >100 tumor cells. SAdCC cases known to possess gene alterations and normal parotid glands were used as positive and negative controls, respectively. Cut-off values were evaluated in 100 non-overlapping cell nuclei of 10 normal salivary gland tissues. The samples were considered positive if >10% (mean + 3SD, rounded-up) of examined nuclei showed abnormal signals.
SNaPshot multiplex assay for point mutations
Formalin-fixed, paraffin-embedded tumor samples were cut at 4 μm, and tissue sections were deparaffinized and lightly stained with methyl green. Tumor tissues only were scraped under a dissecting microscope using a serial hematoxylin and eosin-stained section as a guide. DNA was extracted by incubating tumor tissues at 56° C overnight in digestion buffer containing proteinase K. PCR for the β-globin gene (300 bp) was performed to test the DNA quality. Primers were designed to amplify DNA fragments containing codons 746–750 and 858 for EGFR; codons 12, 13, and 61 for KRAS and HRAS; codons 12 and 61 for NRAS; codons 542, 545, and 1047 for PIK3CA; codon 600 for BRAF; and codon 17 for AKT1 (Supplementary Table 3). The PCR products were treated with exonuclease I (Exo-I, Takara Bio, Kusatsu, Japan) and shrimp alkaline phosphatase (SAP, Takara Bio) to remove unincorporated primers and deoxynucleotide triphosphates. A single-base extension multiplex assay was performed using a SNaPshot Multiplex Kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. The final reaction mix (10 μl) contained 3 μl of the treated PCR product, 5 μl of SNaPshot ready reaction premix containing fluorescent dideoxynucleotides (A = ddR6G, green; C = ddTAMRA, black; G = ddR110, blue; and T = ddROX, red) and probe primers. The reaction was performed for 25 cycles under stringent conditions (96° C for 10 sec, 50° C for 5 sec, and 60° C for 30 sec). An aliquot of the SNaPshot extension reaction mixture (10 μl) was then treated with SAP, followed by enzyme inactivation for 15 min at 75° C. The probe primer sequences are listed in Supplementary Table 4. The fluorescence and size of the extended products were determined by capillary electrophoresis on an ABI PRISM 3130 genetic analyzer (Applied Biosystems). Data thus obtained were analyzed using GeneMapper v4.0 (Applied Biosystems) with specific detection parameters. The point mutations detected with the SNaPshot assay were validated using (1) a SNaPshot assay using both sense and antisense probes and (2) direct sequencing or ARMS-PCR. Primers for the ARMS-PCR assay (Supplementary Table 5) were specifically designed for each of the detected point mutations. All samples were tested in at least two separate experiments.
Deletion analysis for the EGFR gene
Tumor DNA was extracted as described above. The PCR reaction was carried out using primers (Supplementary Table 3) for amplifying DNA fragments containing exon 19 of the EGFR gene. The PCR products were analyzed by DNA-chip-based electrophoresis (Agilent Bioanalyzer 2100, Agilent Technologies, Santa Clara, CA). Cases suspected of a gene deletion (E746-A750del) were subjected to direct sequencing to confirm the deletions.
Statistical analysis
Clinicopathological features were compared using Fisher’s exact test or the Mann–Whitney U-test. Survival curves were calculated using the Kaplan-Meier method. To identify variables significantly associated with patient survival, a log-rank test was performed. A value of p < 0.05 in each test was regarded as statistically significant. All statistical analyses were two-tailed and performed with the statistical software JMP (ver. 10, SAS, Cary, NC, USA).
Author contributions
Conception and design: Saida K, Murase T, and Inagaki H; acquisition of data: Saida K, Fujii K, Takino H, Masaki A, Kusafuka K, and Yatabe Y; analysis and interpretation of data: Saida K, Iida Y, Onitsuka T, Hanai N, Hasegawa Y; Shinomiya H, Nibu K, Kawakita D, Ijichi K, Tada Y, Shimozato K, and Inagaki H; writing, review, and revision of the manuscript: Saida K, Murase T, and Inagaki H.
CONFLICTS OF INTEREST
None to declare
FUNDING
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan awarded to Murase T (17K08746), Fujii K (17K15647), and Inagaki H (15K08351).
REFERENCES
1. Stenman G, Licitra L, Said-Al-Naief N, van Zante A, Yarbrough WG. Adenoid cystic carcinoma. In: El-Naggar AK, Chan J, Grandis JR, Takata T, Slootweg PJ. editors. WHO Classification of Head and Neck Tumours. Lyon: IARC Press. 2017; 164–5.
2. Coca-Pelaz A, Rodrigo JP, Bradley PJ, Vander Poorten V, Triantafyllou A, Hunt JL, Strojan P, Rinaldo A, Haigentz M Jr, Takes RP, Mondin V, Teymoortash A, Thompson LD, et al. Adenoid cystic carcinoma of the head and neck--An update. Oral Oncol. 2015; 51:652–661.
3. Persson M, Andrén Y, Mark J, Horlings HM, Persson F, Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci USA. 2009; 106:18740–18744.
4. Rettig EM, Tan M, Ling S, Yonescu R, Bishop JA, Fakhry C, Ha PK. MYB rearrangement and clinicopathological characteristics in head and neck adenoid cystic carcinoma. Laryngoscope. 2015; 125:292–299.
5. Brayer KJ, Frerich CA, Kang H, Ness SA. Recurrent fusions in MYB and MYBL1 define a common, transcription factor-driven oncogenic pathway in salivary gland adenoid cystic carcinoma. Cancer Discov. 2016; 6:176–187.
6. Mitani Y, Liu B, Rao PH, Borra VJ, Zafereo M, Weber RS, Kies M, Lozano G, Futreal PA, Caulin C, El-Naggar AK. Novel MYBL1 gene rearrangements with recurrent MYBL1-NFIB fusions in salivary adenoid cystic carcinomas lacking t(6;9) translocations. Clin Cancer Res. 2016; 22:725–733.
7. Fujii K, Murase T, Beppu S, Saida K, Takino H, Masaki A, Ijichi K, Kusafuka K, Iida Y, Onitsuka T, Yatabe Y, Hanai N, Hasegawa Y, Inagaki H. MYB, MYBL1, MYBL2 and NFIB gene alterations and MYC overexpression in salivary gland adenoid cystic carcinoma. Histopathology. 2017; 71:823–34.
8. Mikse OR, Tchaicha JH, Akbay EA, Chen L, Bronson RT, Hammerman PS, Wong KK. The impact of the MYB-NFIB fusion proto-oncogene in vivo. Oncotarget. 2016; 7:31681–88. https://doi.org/10.18632/oncotarget.9426.
9. Wysocki PT, Izumchenko E, Meir J, Ha PK, Sidransky D, Brait M. Adenoid cystic carcinoma: emerging role of translocations and gene fusions. Oncotarget. 2016; 7:66239–54. https://doi.org/10.18632/oncotarget.11288.
10. Yewale C, Baradia D, Vhora I, Patil S, Misra A. Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials. 2013; 34:8690–8707.
11. Carcereny E, Morán T, Capdevila L, Cros S, Vilà L, de Los Llanos Gil M, Remón J, Rosell R. The epidermal growth factor receptor (EGRF) in lung cancer. Transl Respir Med. 2015; 3:1. https://doi.org/10.1186/s40247-015-0013-z.
12. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012; 72:2457–2467.
13. Samuels Y, Waldman T. Oncogenic mutations of PIK3CA in human cancers. Curr Top Microbiol Immunol. 2010; 347:21–41.
14. Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer cell. 2014; 25:282–303.
15. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005; 353:123–132.
16. Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, Tebbutt NC, van Hazel G, Wierzbicki R, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007; 357:2040–2048.
17. Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004; 304:1497–1500.
18. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004; 350:2129–2139.
19. Fernández-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer. 2011; 2:344–358. https://doi.org/10.1177/1947601911411084.
20. Quinlan MP, Settleman J. Isoform-specific ras functions in development and cancer. Future Oncol. 2009; 5:105–116.
21. Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, Ince WL, Jänne PA, Januario T, Johnson DH, Klein P, Miller VA, Ostland MA, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone in combination with erlotinib. J Clin Oncol. 2005; 23:5900–5909.
22. Martin P, Leighl NB, Tsao MS, Shepherd FA. KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J Thorac Oncol. 2013; 8:530–542.
23. Kadota K, Sima CS, Arcila ME, Hedvat C, Kris MG, Jones DR, Adusumilli PS, Travis WD. KRAS mutation is a significant prognostic factor in early-stage lung adenocarcinoma. Am J Surg Pathol. 2016; 40:1579–1590.
24. Hames ML, Chen H, Iams W, Aston J, Lovly CM, Horn L. Correlation between KRAS mutation status and response to chemotherapy in patients with advanced non-small cell lung c cancer. Lung Cancer. 2016; 92:29–34.
25. Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008; 359:1757–1765.
26. Stephens PJ, Davies HR, Mitani Y, Van Loo P, Shlien A, Tarpey PS, Papaemmanuil E, Cheverton A, Bignell GR, Butler AP, Gamble J, Gamble S, Hardy C, et al. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest. 2013; 123:2965–2968.
27. Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, Ramaswami D, Walsh LA, Eng S, Huse JT, Zhang J, Dolgalev I, Huberman K, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013; 45:791–798.
28. Grünewald I, Vollbrecht C, Meinrath J, Meyer MF, Heukamp LC, Drebber U, Quaas A, Beutner D, Hüttenbrink KB, Wardelmann E, Hartmann W, Büttner R, Odenthal M, et al. Targeted next generation sequencing of parotid gland cancer uncovers genetic heterogeneity. Oncotarget. 2015; 6:18224–37. https://doi.org/10.18632/oncotarget.4015.
29. Kato S, Elkin SK, Schwaederle M, Tomson BN, Helsten T, Carter JL, Kurzrock R. Genomic landscape of salivary gland tumors. Oncotarget. 2015; 6:25631–45. https://doi.org/10.18632/oncotarget.4554.
30. Cros J, Sbidian E, Hans S, Roussel H, Scotte F, Tartour E, Brasnu D, Laurent-Puig P, Bruneval P, Blons H, Badoual C. Expression and mutational status of treatment-relevant targets and key oncogenes in 123 malignant salivary gland tumours. Ann Oncol. 2013; 24:2624–2629.
31. Zinsky R, Bölükbas S, Bartsch H, Schirren J, Fisseler-Eckhoff A. Analysis of KRAS mutations of exon 2 codons 12 and 13 by SNaPshot analysis in comparison to common DNA sequencing. Gastroenterol Res Pract. 2010; 2010:789363. https://doi.org/10.1155/2010/789363.
32. Magnin S, Viel E, Baraquin A, Valmary-Degano S, Kantelip B, Pretet JL, Mougin C, Bigand M, Girardo B, Borg C, Ferrand C. A multiplex SNaPshot assay as a rapid method for detecting KRAS and BRAF mutations in advanced colorectal cancers. J Mol Diagn. 2011; 13:485–492.
33. Lurkin I, Stoehr R, Hurst CD, van Tilborg AA, Knowles MA, Hartmann A, Zwarthoff EC. Two multiplex assays that simultaneously identify 22 possible mutation sites in the KRAS, BRAF, NRAS and PIK3CA genes. PLoS One. 2010; 5:e8002. https://doi.org/10.1371/journal.pone.0008802.
34. Chang YS, Yeh KT, Hsu NC, Lin SH, Chang TJ, Chang JG. Detection of N-, H-, and KRAS codons 12, 13, and 61 mutations with universal RAS primer multiplex PCR and N-, H-, and KRAS-specific primer extension. Clin Biochem. 2010; 43:296–301.
35. Trivedi N. Atlas of Head and Neck Cancer Surgery: The Compartment Surgery for Resection in 3-D. Berlin, Germany: Springer. 2014.
36. Brierley JD, Gospodarowicz MK, Wittekind C, editors. TNM Classification of Malignant Tumours. 8th ed. Hoboken (NJ): Wiley-Blackwell. 2016.
37. Ellington CL, Goodman M, Kono SA, Grist W, Wadsworth T, Chen AY, Owonikoko T, Ramalingam S, Shin DM, Khuri FR, Beitler JJ, Saba NF. Adenoid cystic carcinoma of the head and neck: Incidence and survival trends based on 1973–2007 Surveillance, Epidemiology, and End Results data. Cancer. 2012; 118:4444–4451.
38. Thompson LD, Penner C, Ho NJ, Foss RD, Miettinen M, Wieneke JA, Moskaluk CA, Stelow EB. Sinonasal tract and nasopharyngeal adenoid cystic carcinoma: a clinicopathologic and immunophenotypic study of 86 cases. Head Neck Pathol. 2014; 8:88–109.
39. Hitre E, Budai B, Takácsi-Nagy Z, Rubovszky G, Tóth E, Remenár É, Polgár C, Láng I. Cetuximab and platinum-based chemoradio- or chemotherapy of patients with epidermal growth factor receptor expressing adenoid cystic carcinoma: a phase II trial. Br J Cancer. 2013; 109:1117–1122.
40. Locati LD, Bossi P, Perrone F, Potepan P, Crippa F, Mariani L, Casieri P, Orsenigo M, Losa M, Bergamini C, Liberatoscioli C, Quattrone P, Calderone RG, et al. Cetuximab in recurrent and/or metastatic salivary gland carcinomas: A phase II study. Oral Oncol. 2009; 45:574–578.
41. Jakob JA, Kies MS, Glisson BS, Kupferman ME, Liu DD, Lee JJ, El-Naggar AK, Gonzalez-Angulo AM, Blumenschein GR Jr. Phase II study of gefitinib in patients with advanced salivary gland cancers. Head Neck. 2015; 37:644–49.
42. Boeckx C, Baay M, Wouters A, Specenier P, Vermorken JB, Peeters M, Lardon F. Anti-epidermal growth factor receptor therapy in head and neck squamous cell carcinoma: focus on potential molecular mechanisms of drug resistance. Oncologist. 2013; 18:850–864.
43. Liu SX, Xia ZS, Zhong YQ. Gene therapy in pancreatic cancer. World J Gastroenterol. 2014; 20:13343–13368.
44. Wang YX, Gao L, Ji ZZ. Inhibitary effects of antisense oligonucleotide specific to K-ras point mutation on the target gene expression in human pancreatic carcinoma cells. Chin Med J (Engl). 2007; 120:1448–1450.
45. Morioka CY, Machado MC, Saito S, Nakada Y, Matheus AS, Jukemura J, Bacchella T, Takahara T, Watanabe A. Suppression of invasion of a hamster pancreatic cancer cell line by antisense oligonucleotides mutation-matched to K-ras gene. In Vivo. 2005; 19:535–538.
46. Miura Y, Ohnami S, Yoshida K, Ohashi M, Nakano M, Ohnami S, Fukuhara M, Yanagi K, Matsushita A, Uchida E, Asaka M, Yoshida T, Aoki K. Intraperitoneal injection of adenovirus expressing antisense K-ras RNA suppresses peritoneal dissemination of hamster syngeneic pancreatic cancer without systemic toxicity. Cancer Lett. 2005; 218:53–62.
47. Schirripa M, Lenz HJ. Biomarker in colorectal cancer. Cancer J. 2016; 22:156–164.
48. Molina JR, Aubry MC, Lewis JE, Wampfler JA, Williams BA, Midthun DE, Yang P, Cassivi SD. Primary salivary gland-type lung cancer: spectrum of clinical presentation, histopathologic and prognostic factors. Cancer. 2007; 110:2253–2259.
49. Nickell-Brady C, Hahn FF, Finch GL, Belinsky SA. Analysis of K-ras, p53 and c-raf-1 mutations in beryllium-induced rat lung tumors. Carcinogenesis. 1994; 15:257–262.
50. Sugio K, Kishimoto Y, Virmani AK, Hung JY, Gazdar AF. K-ras mutations are a relatively late event in the pathogenesis of lung carcinomas. Cancer Res. 1994; 54:5811–5815.
51. Dahse R, Driemel O, Schwarz S, Kromeyer-Hauschild K, Berndt A, Kosmehl H. KRAS status and epidermal growth factor receptor expression as determinants for anti-EGFR therapies in salivary gland carcinomas. Oral Oncol. 2009; 45:826–829.
52. Shalmon B, Drendel M, Wolf M, Hirshberg A, Cohen Y. Mutational analysis of PI3K/AKT and RAS/RAF pathway activation in malignant salivary gland tumours with a new mutation of PIK3CA. Int J Oral Maxillofac Surg. 2016; 45:721–725.
53. Pao W, Ladanyi M. Epidermal growth factor receptor mutation testing in lung cancer: searching for the ideal method. Clin Cancer Res. 2007; 13:4954–4955.
54. Thomas RK, Nickerson E, Simons JF, Jänne PA, Tengs T, Yuza Y, Garraway LA, LaFramboise T, Lee JC, Shah K, O’Neill K, Sasaki H, Lindeman N, et al. Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat Med. 2006; 12:852–855.
55. Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010; 11:31–46.
56. Loibl S, Majewski I, Guarneri V, Nekljudova V, Holmes E, Bria E, Denkert C, Schem C, Sotiriou C, Loi S, Untch M, Conte P, Bernards R, et al. PIK3CA mutations are associated with reduced pathological complete response rates in primary HER2-positive breast cancer: pooled analysis of 967 patients from five prospective trials investigating lapatinib and trastuzumab. Ann Oncol. 2016; 27:1519–1525.
57. Clauditz TS, Reiff M, Gravert L, Gnoss A, Tsourlakis MC, Münscher A, Sauter G, Bokemeyer C, Knecht R, Wilczak W. Human epidermal growth factor receptor 2 (HER2) in salivary gland carcinomas. Pathology. 2011; 43:459–464.
58. Szanto PA, Luna MA, Tortoledo ME, White RA. Histologic grading of adenoid cystic carcinoma of the salivary glands. Cancer. 1984; 54:1062–1069.
59. Ito Y, Ishibashi K, Masaki A, Fujii K, Fujiyoshi Y, Hattori H, Kawakita D, Matsumoto M, Miyabe S, Shimozato K, Nagao T, Inagaki H. Mammary analogue secretory carcinoma of salivary glands: a clinicopathologic and molecular study including 2 cases harboring ETV6-X fusion. Am J Surg Pathol. 2015; 39:602–610.