
INTRODUCTION
Epidermal growth factor receptor (EGFR) is over expressed in the majority of NSCLC and genetic alterations in the tyrosine kinase domain of EGFR are associated with the sensitivity to treatment with molecular targeted agents [1]. Although tyrosine kinase inhibitor (TKI) showed encouraging clinical responses in NSCLC patients harbouring EGFR mutations [2], almost all patients developed resistance to these inhibitors after initial clinical response. About half of acquired resistance was associated with secondary T790M EGFR mutation in exon 20 [3] and 21% of such resistance was linked to c-Met amplification [4]. Beside the acquired resistance, many NSCLC patients were carrying primary resistant genetic status such as EGFR wide-type, k-ras mutation and others. Approximately 2–9% of NSCLC tumours lacked PTEN, and PTEN loss has been considered indicative of primary or acquired resistance to EGFR–TKIs [5–9]. We also noticed that increased expression of Src was reported in 50% to 80% of NSCLC. In addition, high levels of Src kinase activity have been observed in NSCLC, with the degree of kinase activity correlating with tumour size [10]. In a study of 60 cancer cell lines, the NSCLC cell lines had the highest median Src activity [11]. Src family signalling plays an important role in several signalling pathways, mainly involving in Ras/Raf/MAPK, PI3K/Akt, STAT and FAK signalling pathway. More importantly Src family kinase can interact with both EGFR and MET pathways, and may result in resistance to EGFR-TKI. Hence, each type of resistance needed to be overcome by different strategies.
Cetuximab (Erbitux®, Bristol-Myers Squibb) is a chimeric monoclonal antibody against the extracellular domain of EGFR. It has been shown to provide modest survival benefit when adding to chemotherapy for advanced NSCLC, especially in patients with high expression of EGFR. However, cetuximab is not expected to work in gefitinib or erlotinib resistant EGFR mutant cases. Dasatinib (Sprycel®, Bristol-Myers Squibb; BMS-354825), a potent, multi-targeted, oral inhibitor of Src family kinases, Bcr-Abl, c-Kit, platelet-derived growth factor receptor (PDGFR) and Eph receptors, has been shown to have anti-tumour effects in solid tumours. In preclinical studies, NSCLC cells treated with dasatinib showed decreased cell growth, substrate-dependent changes in cell morphology and changes in downstream signalling leading to a reduced capability for invasion [12, 13]. In EGFR-dependent NSCLC cell lines, treatment with dasatinib results in apoptosis. In the clinical setting, initial pharmacodynamic data have demonstrated that patients with solid tumours exposed to dasatinib showed substantially inhibitory Src activity. Furthermore, no dose-limiting toxicity was observed in a dose escalation study in patients with solid tumours [14]. However, a phase II study showed that dasatinib as monotherapy in molecularly unselected NSCLC patients did not appear promising [15]. Afatinib (Gilotrif, Boehringer Ingelheim, BIBW2992) is a novel, orally bio-available, anilinoquinazoline compound, developed and designed as an irreversible dual inhibitor covalently bound to Cys 773 of EGFR and Cys 805 of HER2 [16]. Afatinib was shown to be superior to chemotherapy, pemetrexate and cisplatin as the first line treatment for patients with advanced NSCLC with activating EGFR mutations, but it was disappointing that the drug was unable to improve OS compared with placebo, in a population of patients with prior exposure to reversible EGFR TKIs, despite the evidence of significant clinical activity such as superior response rates, time to progression as compared to placebo [17]. The response rate to afatinib alone was reported to be 7%. Osimertinib, the third generation of EGFR TKI specifically designed against EGFR activating mutation with resistant T790M mutation, was reported to have higher response rates and longer PFS than chemotherapy in these group of patients, but again majority of patients developed resistant mutation in EGFR genes and by other mechanisms [18]. All of above, the described agents alone showed limited efficacy in clinic due to development of resistance. Therefore, a new strategy to combat EGFR–TKI resistance caused by various mechanisms is needed.
Drug combination may offer more benefits in the treatment of advanced NSCLC from synergistic interactions and avoidance of resistance emergence. A previous study indicated that dasatinib in combination with gefitinib achieved more inhibition of cell growth than that of either dasatinib or gefitinib alone [12]. Another study showed that combined EGFR targeting with afatinib and cetuximab achieved encouraging clinical response rate of 30% in NSCLC patients with acquired resistance (AR) to prior erlotinib or gefitinib [17]. Most recently, combination of osimertinib and gefitinib was demonstrated some efficacy in patients with C797S and T790M EGFR-mutated NSCLC, but the improvement is brief [19]; the activation of Src family kinases and focal adhesion kinase (FAK) was revealed to contribute to the resistance to afatinib, erlotinib and osimertinib in afatinib-resistant HCC827 cells, which lost the amplified, mutated EGFR genes [20]. Osimernitib combined with dasatinib was reported to overcome the resistance to first generation EGFR-TKI, gefitinib for example, in NSCLC patients with acquired T790M [21]. Up to date, there are limited reports on the efficacy of combination of EGFR-TKI and dasatinib overcoming the resistance to EGFR-TKI in NSCLC with various genetic characters.
In this study, we defined the efficacy of afatinib combined with dasatinib in gefitinib resistant NSCLC cells harbouring different genetic mutation characteristics and determined the potential biomarkers predicating synergism of the combination. Moreover, this is, to the best of our knowledge, the first report revealed the growth inhibition by combination of afatinib and dasatinib against PENT mutations in NSCLC cells, which demonstrated strong resistance to either afatinib or dasatinib alone, and we further investigated and uncovered the molecular mechanism of tumour inhibition for reversing the drug resistance.
RESULTS
Cell growth inhibition by single agent varied against 8 NSCLC cell lines
We used 5 commercial NSCLC cell lines (A549, H1975, H1650, HCC827, and H820), and 3 cell lines (As13, As87, and As125) which were established by us from NSCLC patients’ pleural effusion. Their mutation status were shown in Table 1. HCC827 carrying exon19 deletion of EGFR, is a gefitinib- sensitive cell line [4]. We used it as a positive control. Inhibition of tumour cell proliferation by single agent was tested by MTS assay (Figure 1) and IC50 of each drug in the studied cell lines was calculated (Table 1). HCC827 was sensitive to all of the four agents (gefitinib, cetuximab, afatinib and dasatinib) with the IC50 value from 0.7 nM to 50 nM. The other 7 cell lines showed resistance to both gefitinib (IC50: 4.4 ~ 25.5 μM) and cetuximab (IC50: 2.4 ~ 12 μM). H1975 carrying L858R + T790M EGFR mutation is considered as acquired resistant to gefitinib. We noted afatinib showed more inhibition against T790M mutation than gefitinib. This was mainly because afatinib irreversibly bound to Cys773 of EGFR to overcome the mechanism of resistance to gefitinib (16). H820, harbouring exon19 deletion (E746-E749) + T790M + MET amplification, also showed positive response to afatinib. Since MET amplification is able to activate HER3-dependent PI3K/Akt pathway and its occurrence is independent of T790M [4], and afatinib has proven activity of preventing phosphorylation of HER3 [16], the growth inhibition of H820 may be caused by blocking the activity of EGFR, HER2 and HER3 and their downstream signalling pathway. Afatinib was unable to reverse the resistance to gefitinib in A549, H1650, As13, As87, and As125 cell lines. In the other hand, 5 gefitinib resistant cell lines (H1975, H820, As13, As87, and As125) showed good response to dasatinib. Indicating that Src play an important role in cell proliferation of NSCLC cells. Both H1650 and A549 were resistance to all the four agents. H1650 carries exon19 deletion (delE746-A750) + deletion of exon 9 of PTEN. Loss of PTEN expression in EGFR-mutant cells correlates with decreased sensitivity to EGFR-TKI [8]. A549 harbours WT-EGFR and HER2, activating K-ras G12s point mutation. Patients with K-ras mutations have poor sensitivity to EGFR TKI [22, 23] and unfavourable prognosis [24, 25]. Overall, the efficacy of dasatinib against NSCLC cells in vitro is significantly stronger than gefitinib (p < 0.001) and cetuximab (p < 0.05), and no significant difference was found between dasatinib and afatinib.
Table 1: Comparison of sensitivities to 4 molecular target drugs in 8 NSCLC cell lines carrying various genetic status
IC50 Cell lines |
Gefitinib (μM) |
Dasatinib (μM) |
Afatinib (μM) |
Cetuximab (μM) |
Genetic background of cell lines |
|---|---|---|---|---|---|
A549 |
21.5 ± 1.8 |
2.2 ± 0.05 |
4.5 ± 0.9 |
5.7 ± 0.06 |
WT-EGFR and HER2, activating K-ras G12s point mutation |
H1975 |
25.5 ± 7.7 |
0.8 ± 0.1 |
0.8 ± 0.09 |
3.7 ± 0.16 |
L858R + T790M double substitution |
H1650 |
22.5 ± 1.2 |
8.8 ± 1.1 |
3.8 ± 0.4 |
5.1 ± 0.09 |
exon19 deletion (delE746-A750) + deletion of exon9 of PTEN |
HCC827 |
0.008 ± 0.004 |
0.07 ± 0.006 |
0.0007 ± 0.00007 |
0.05 ± 0.004 |
exon19 deletion |
H820 |
4.4 ± 0.9 |
0.06 ± 0.005 |
0.6 ± 0.1 |
2.4 ± 0.4 |
exon19 deletion (E746-E749) + T790M + MET amplification |
AS13 |
18.3 ± 1.5 |
0.08 ± 0.005 |
11.2 ± 0.9 |
4.2 ± 0.62 |
L858R + missense mutation (R776C) in exon20, WT-k-ras |
AS87 |
12.3 ± 5.1 |
0.6 ± 0.08 |
1.7 ± 0.2 |
6 ± 0.8 |
WT-EGFR, WT-k-ras |
AS125 |
8.8 ± 1.7 |
0.04 ± 0.02 |
5.4 ± 0.2 |
12 ± 0.4 |
WT-EGFR, WT-k-ras |
Individual IC50 values (μM) were the mean ± SD calculated from at least 3 experiments.
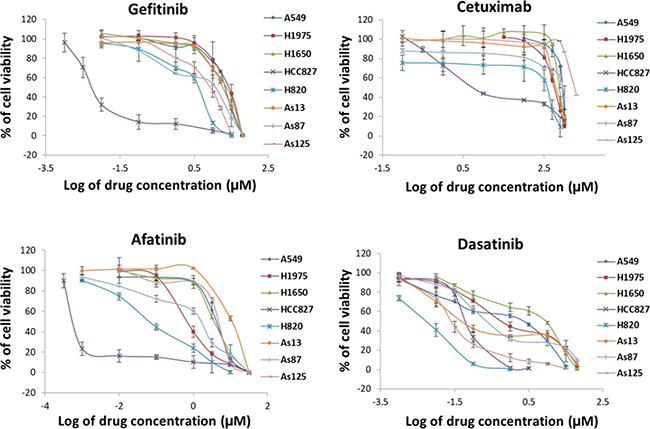
Figure 1: Comparison of growth inhibition of NSCLC cells by 4 different agents. 8 NSCLC cell lines were treated by gefitinib, cetuximab, afatinib, and dasatinib alone, respectively. Individual % of cell viability is the mean ± SD from at least three experiments.
Afatinib combined with dasatinib increased inhibition of NSCLC cell proliferation
Drug combination index (CI) was calculated for each two drug combination at the designated dosage. Combination of afatinib with dasatinib achieved significant growth inhibition against all the studied cell lines except A549 (Figure 2A). Synergistic interaction between afatinib and cetuximab was only observed in HCC827, H1975 and H820 cell lines, which were sensitive to afatinib (Figure 2B). The synergy between afatinib and dasatinib was significantly stronger than that of afatinib and cetuximab (p < 0.001).
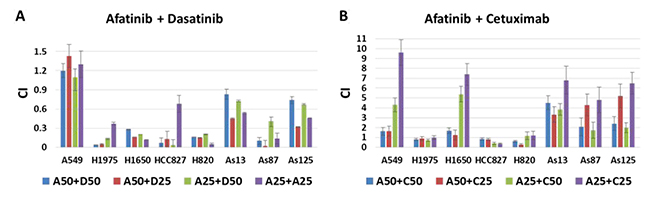
Figure 2: Combination effect of afatinib combined with either dasatinib or cetuximab in 8 NSCLC cell lines. (A) Drug interaction between afatinib and dasatinib at 4 different concentration combinations, for example, A50 + D50 indicated the combination of afatinib and dasatinib at the dosage of IC50 when treated the NSCLC cells alone. (B) Drug interaction between afatinib and cetuximab at 4 different concentration combinations, for example, A50 + C25 indicated the combination of afatinib and cetuximab at the dosage of IC50 and IC25 when treated the NSCLC cells alone, respectively. CI < 0.9, indicating the synergistic interaction between 2 drugs. Individual CI is the mean ± SD from at least 3 experiments.
Identification of potential biomarkers predicating the synergism between afatinib and dasatinib
In order to identify potential biomarkers predicating the synergetic effects between afatinib and dasatinib, we measured the expression level of total (T) proteins and phosphorylated (P) proteins in the signalling pathways which may be affected by afatinib or dasatinib (Figure 3A–3D). Strong synergism between afatinib and dasatinib was correlated with high expression level of T-MAPK (p < 0.05) (Figure 3E) in 6 gefitinib-resistant cell lines which positively responded to the combination of afatinib and dasatinib. We also found that baseline expression level of T-Src significantly correlated with T-Stat3 (p < 0.001) (Figure 3F). These findings might imply the synergistic interaction between afatinib and dasatinib on the signaling pathways affected by Src, Stat3 and MAPK.
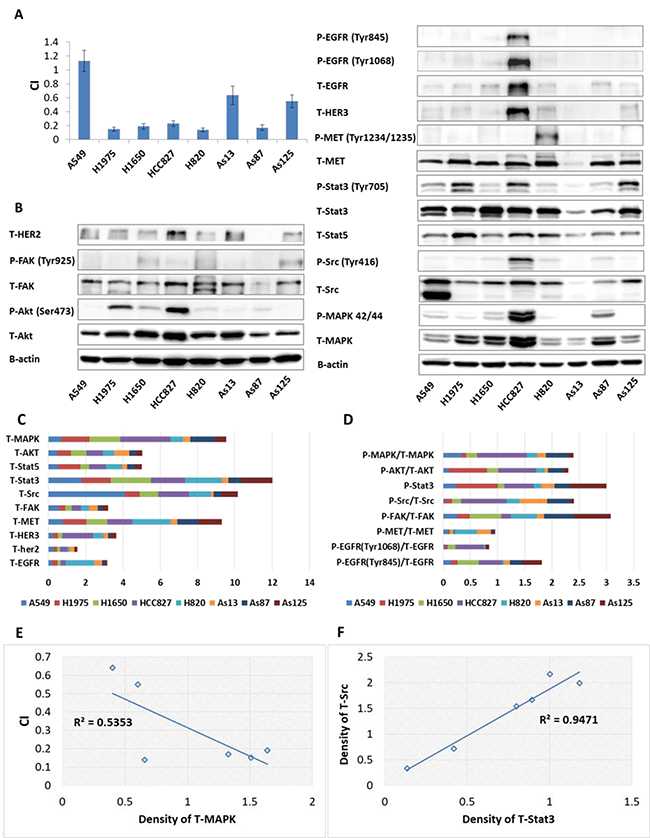
Figure 3: Baseline proteins expressions as well as combination index (CI) in NSCLC cells. (A) CI indicated the interaction between afatinib and dasatinib in 8 NSCLC cell lines. (B) Baseline expression of receptor tyrosine kinases and downstream signaling molecules determined by western blot, β-actin was used as the loading control. (C) The expression ratio of the studied proteins to β-actin quantified by ImageJ software. (D) The expression ratio of phosphorylated protein to total protein quantified by ImageJ software. (E) Significant correlation between the synergistic interaction of afatinib plus dasatinib and baseline expression of MAPK (p < 0.05). The Pearson correlation coefficient (r) was equal to 0.733. (F) Significant correlation between baseline expression level of Src and Stat3 (p < 0.001). The Pearson correlation coefficient (r) was equal to 0.972. The p-value correlation probability from r was calculated by student’s t test. Results represented the mean ± SD from at least three experiments.
Afatinib combined with dasatinib inhibits the activity of EGFR, HER2, Src and downstream signaling in H1650 cells
In order to study the mechanism underlying synergetic tumor inhibition by combination of afatinib and dasatinib, H1650 cells were treated by afatinib, dasatinib and their combination at the designated doses. The targeted proteins were analyzed by western blotting and the ratio of P-protein to T-protein was calculated by ImageJ software (Figure 4A–4C). Phosphorylation of EGFR at Tyr845 (P-EGFR845) was completely inhibited by afatinib alone at the dosage of 0.1 μM (p < 0.01), slightly decreased by dasatinib (1 μM) alone, and the complete inhibition was observed by the combinations (p < 0.05). The baseline expression level of both P-EGFR (Tyr1068) and P-HER2 (Tyr1221/1222) was very weak. Their phosphorylation was completely abolished by afatinib alone and the combinations (p < 0.01), even though it was slightly increased by dasatinib alone. Src activity (P-Src) was inhibited by dasatinib at the dosage of 1 μM (p < 0.05) but not afatinib. The combination of afatinib (1 μM) and dasatinib (1 μM) demonstrate the inhibition of P- Src (p < 0.05). However, there was no further inhibition was observed by the combination comparing with the treatment by dasatinib alone. There was a dose-dependent inhibition of phosphorylation of FAK at tyr925 by dasatinib, and the inhibition level at both of the dosage 0.1 μM and 1 μM were significant (p < 0.01), enhanced inhibition by the combination was clearly demonstrated (p < 0.01).The activity of Akt (P-Akt) was reduced by afatinib alone (p < 0.01), and further deduction was observed by the combinations (p < 0.05). Afatinib also showed a strong inhibition on activating of MAPK. 0.1 μM of afatinib significantly inhibited P-MAPK (Tyr42/44) (p < 0.01), which wasn’t affected by dasatinib. Furthermore, 0.1 μM of afatinib combined with 0.1 μM of dasatinib enhanced afatinib’s inhibition effect on phosphorylation of MAPK (p < 0.05). Afatinib showed 15% of inhibition on P-Stat3 (Try705) at the dosage of 0.1 μM (p < 0.05), and no significant inhibition was shown at the dosage of 1 μM; dasatinib demonstrated more reduction of the activity of Stat3 at both 0.1 μM (23%) and 1 μM (48%), respectively (p < 0.01) when compared with the control; the combination of afatinib and dasatinib at the lower concentration (afatinib: 0.1 μM, dasatinib:0.1 μM) induced more inhibition of the activity of Stat3 than either afatinib or dasatinib alone (p < 0.01). The increasing of cleaved PARP was observed by each individual drug treatment (p < 0.05), and further increasing was induced by the combinations (p < 0.01). The activated HER3, MET, PTEN, and Stat5 were not detected in H1650 cells.
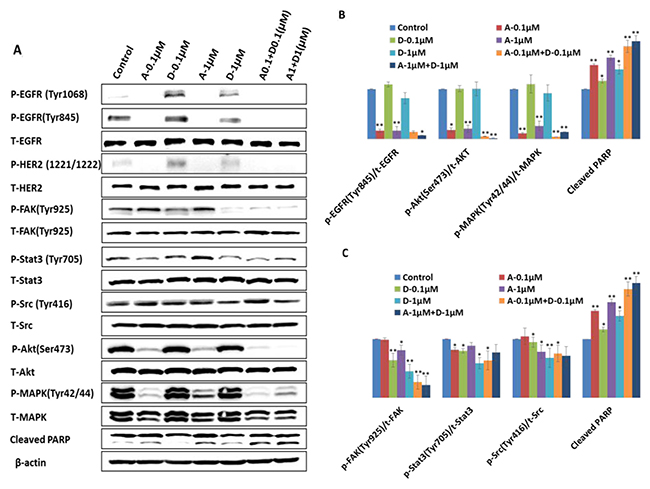
Figure 4: Different effect of afatinib, dasatinib and their combination on the targeted proteins and downstream molecules in H1650 cells. (A) Western blot analysis showed different expression level of the targeted proteins and downstream molecules upon the designated treatments. For example, A-0.1 μM indicates the treatment of afatinib at the dosage of 0.1 μM, and A0.1 + D0.1 (μM) means the combination of 0.1μM of afatinib and 0.1 μM of dasatinib. (B, C) Quantification of the ratio of phosphorylated protein to total protein by ImageJ software. The results were mean ± SD obtained from at least three experiments. P-value was calculated by student’s t test (**p < 0.01; *p < 0.05).
Afatinib in combination with dasatinib enhanced apoptosis of H1650 cells
H1650 cells were treated as described in methods. Early stage of cell apoptosis was induced (Figure 5), they were 2.61% (control), 7.11% (0.1 μM of afatinib), 4.6% (0.1 μM of dasatinib) and 13.44% (afatinib + dasatinib). Afatinib combined with dasatinib significantly enhanced apoptosis of H1650 cells (p < 0.05).
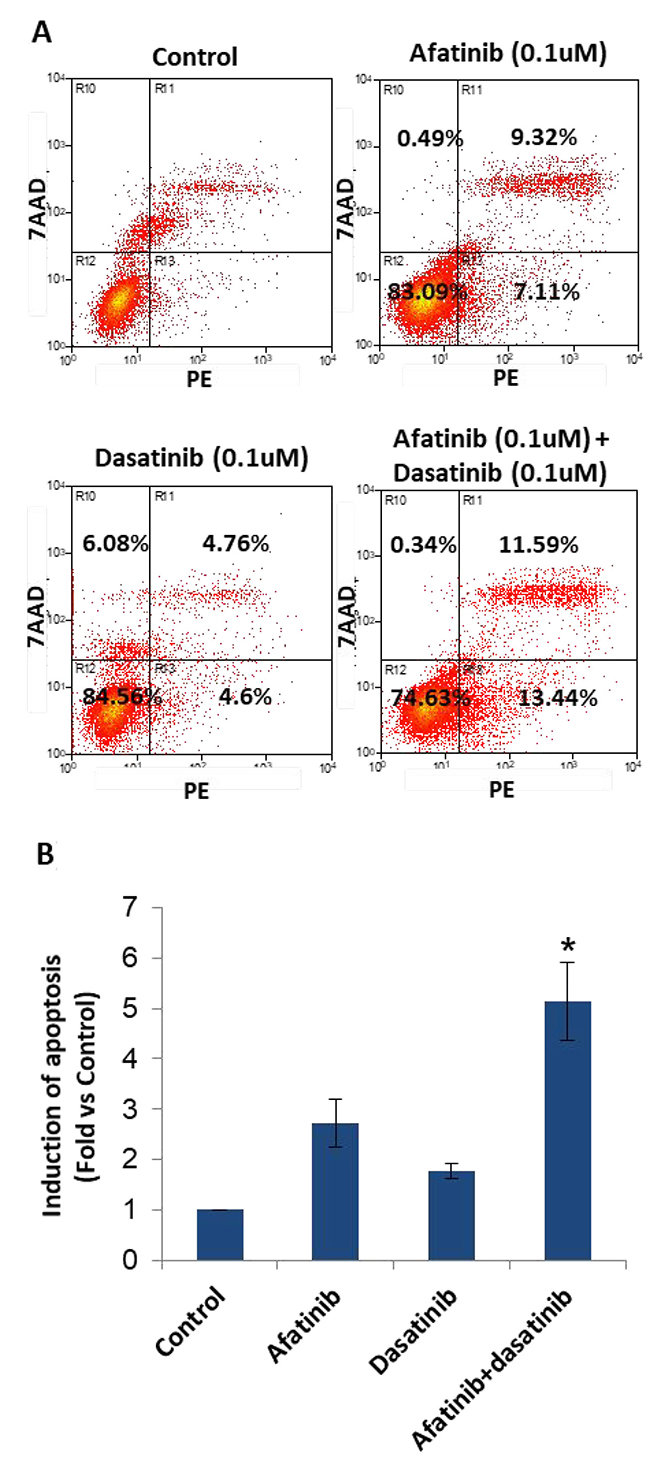
Figure 5: Induction of apoptosis by the treatment of afatinib, dasatinib, and their combination in H1650 cells. (A) Flow cytometry analysis showed the induction of apoptosis upon the designated treatment. (B) Statistic chart showed the significant induction of apoptosis by the combination of afatinib and dasatinib comparing with afatinib or dasatinib alone. The results were mean ± SD obtained from at least three experiments. P-value was calculated by student’s t test (*p < 0.05).
Effects of combined afatinib and dasatinib on cell adhesion, migration and invasion of H1650
In general, both afatinib and dasatinib showed dose dependent inhibition of cell adhesion to collagen I. Particularly, 0.1μM of dasatinib significantly reduced cell adhesion by 74%, 44% and 10% after 0.5 h (p < 0.01), 1 h (p < 0.01) and 2 h (p < 0.05) treatments, respectively. In contrast, 0.1 μM of afatinib increased H1650 cells adhere to collagen I after 0.5 h, 1 h and 2 h (p < 0.05), the combination didn’t show more effect than dasatinib alone (75%, 32%, 9% respectively), but it reversed the negative effect of afatinib (Figure 6A). With increasing the dosage to 1 μM, dasatinib further inhibited cell adhesion by 90%, 91% and 51% respectively at above time points, afatinib didn’t show any significant effect (12%, 2%, 0%) on cell adhesion; more inhibition by the combination of afatinib and dasatinib (66%) was observed on cell adhesion after 2 h (p < 0.05), but no significant difference was found after 0.5 h and 1 h (Figure 6B).
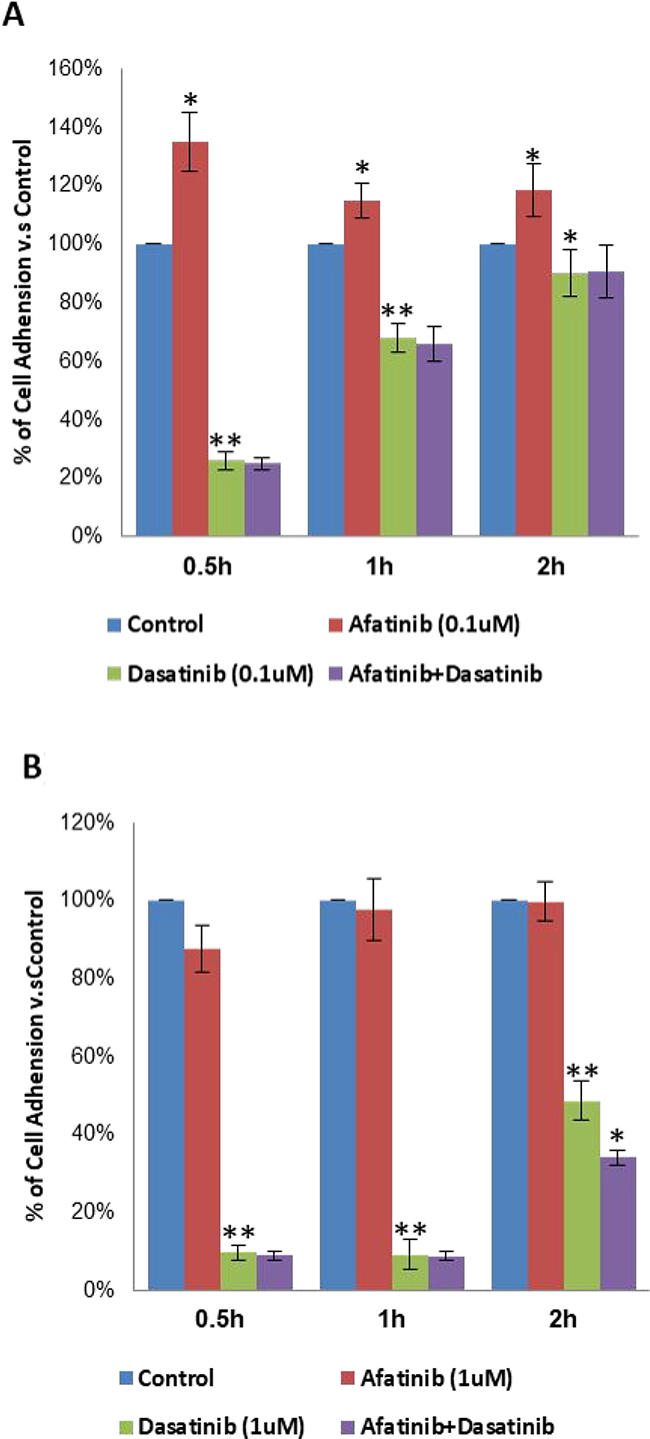
Figure 6: Inhibition of cell adhesion to collagen I by the treatment of afatinib, dasatinib, and their combination in H1650 cells. (A, B) % of cell adhesion to collagen I upon the designated treatment at different dosage. The results were mean ± SD obtained from at least three experiments. P-value was calculated by student’s t test (**p < 0.01; *p < 0.05).
An enhanced inhibition on cell migration of H1650 was shown at various time duration when treated by either afatinib, dasatinib alone or their combination (Figure 7A). Either afatinib or dasatinib alone significantly reduced cell migration to the wound area after 24 h and 48 h (p < 0.05), and the wound completely healed after 72 h. Afatinib combined with dasatinib demonstrated further deduction of cell migration after 24 h, 48 h, and 72 h comparing with each individual drug treatment alone (p < 0.01). Figure 7B showed the dose-dependent inhibition of cell migration in H1650 cells by afatinib, dasatinib alone as well as the combination. More deduction of the cell migration was induced by the treatment at higher dosage (1 μM) than that of the lower one (0.1 μM). There is no invasion observed in H1650 cells.
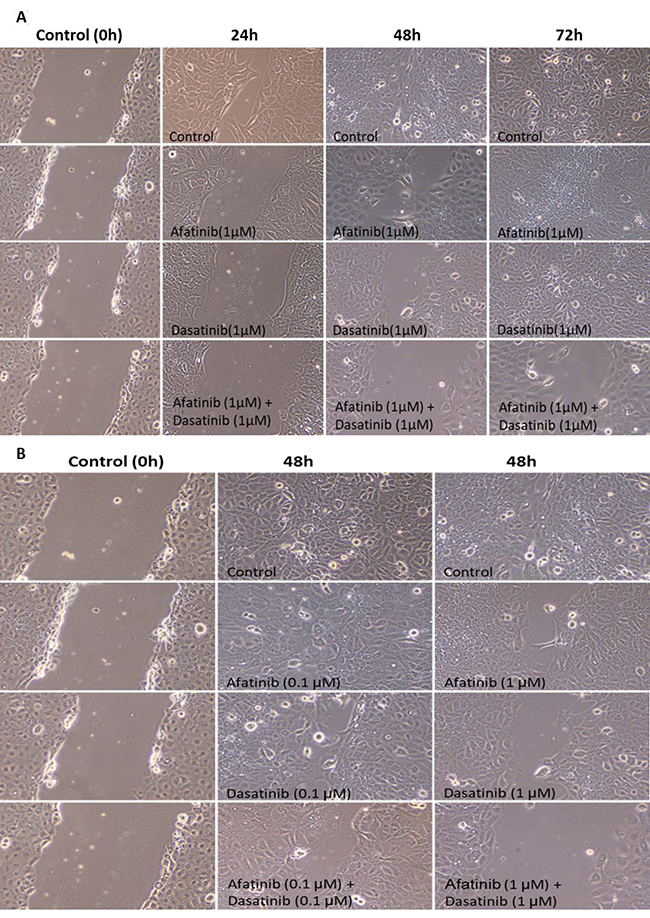
Figure 7: Time and dose-dependent inhibition of cell migration by the treatment of afatinib, dasatinib, and their combination in H1650 cells. (A) Enhanced inhibition of cell migration at various time duration by the combination of afatinib and dasatinib. (B) enhanced dose-dependent inhibition of cell migration by the combination of afatinib and dasatinib.
DISCUSSION
Both afatinib and dasatinib achieved better efficacy against cell proliferation than reversible EGFR-TKI and EGFR monoclonal antibody. However, neither afatinib nor dasatinib as a single agent would meet the demand of treatment for NSCLC in clinical practice due to resistance. Afatinib combined with dasatinib showed more synergistic effect than combined with cetuximab. Theoretically, the afatinib and dasatinib combination inhibited cell signalling in different signal pathways due to synergic interaction of Src with EGFR or their downstream factors, but the gefitinib or afatinib and cetuximab only affected the same EGFR pathway [26]. Afatinib in combination with dasatinib was unable to overcome the primary resistance with K-ras mutation since the activation of mutant K-ras is non-EGFR or Src dependent [27]. However, afatinib combined with dasatinib synergistically reduced cell proliferation of H1650 which was also resistant to either afatinib or dasatinib alone. In this study, we specifically focused on this cell line to elucidate the mechanism underlying synergistic anti-cancer effect between afatinib and dasatinib.
In H1650 cells, the synergistic anti-tumour activity of the combination of afatinib and dasatinib was exhibited in cell proliferation, survival, apoptosis, adhesion and migration through the interaction amongst the various signalling pathways. Previous studies have reported that depletion of PTEN protein or loss of PTEN function reversely increases PI3K/Akt activity and causes drug resistance in a variety of cell types [28–31]. Afatinib alone completely inhibited P-EGFR (Tyr1068), P-EGFR (Tyr845) and P-HER2 (1221/1222) subsequently decreased the activity of Akt and MAPK but not complete inhibition (Figure 4B), thus suggesting that afatinib alone was not sufficient to reverse the resistance to gefitinib via blocking both EGFR/PI3k/Akt and EGFR/Ras/Raf/MAPK signalling pathways, even though it showed more efficacy than gefitinib (IC50: 3.8 μM vs 22.5 μM). In contrast, dasatinib alone didn’t have any inhibition on phosphorylation of Akt, MAPK, but slightly increased P-EGFR at the point of Tyr1068, indicating that there was no suppression effect on EGFR/PI3k/Akt and EGFR/Ras/Raf/MAPK signalling pathways but dasatinib alone reduced Src activity and subsequently inhibited P-EGFR (Tyr845), thus H1650 cells could survive via the activated EGFR/PI3K/Akt / EGFR/Ras/Raf/MAPK pathway to bypass Src signalling pathway when treated by dasatinib alone. The interaction between Src and FAK has been shown to active both cell motility and invasion [32, 33]. Upon stimulation of EGF or PDGF receptors, FAK auto-phosphorylated, creating a high affinity binding site for Src, the association between Src and FAK resulted in activation of Src and phosphorylation of FAK at various tyrosine sites. The Src/FAK complex phosphorylated a number of other focal adhesion proteins and activated other intra-cellular signalling pathway including PI3K/PTEN/Akt, and Ras/Raf/MEK/ERK signalling pathways [34]. In this study, we found that dasatinib alone substantially inhibited the activity of FAK at the site of Tyr925, which enable dasatinib to produce more inhibition of cell adhesion and migration than afatinib. The inactivation of EGFR by afatinib could further inhibit the formation of FAK/Src complex, resulted in down-regulation of the activity of FAK. As a result, we observed: 1) enhanced the inhibition on both cell adhesion and migration (Figures 6 and 7); 2) reduced the binding of PI3K to FAK, and sequentially fully abolished the phosphorylation of Akt (Figure 4); 3) inhibited the binding of Gr2 to FAK at the Tyr925 and significantly reduced the activity of MAPK (Figure 4). In addition, Src-mediated pathway is also involved in activating signal transduction and transcription (Stat3) to mediate cell cycle progression and against apoptosis [35]. We found that dasatinib produced significantly more inhibition of P-Stat3 (Tyr705) than afatinib. Dasatinib might restrain the activity of Stat3 to reduce the cell survival by inactivation of Src, and it demonstrated more efficacy on Stat3 signalling pathway than that of afatinib. Previous studies found that MAPK played a direct inhibitory modification on Stat3 in tumour cells [35]. When the activity of MAPK was restricted by afatinib, Stat3 might be activated relevantly to inhibit cell apoptosis and increase cell survival. From our results, dasatinib was able to overcome this inhibitory modification by inhibiting the activity of Stat3. The small dosage of afatinib (0.1 μM) achieved more inhibition of P-MAPK than that of 1 μM. This was in accordance with that combination between small dosages (0.1 μM of afatinib + 0.1 μM of dasatinib) was more synergic than that of large dosages (1 μM of afatinib + 1 μM of dasatinib). The large dosage may trigger the inhibitory modification among the related cell signalling pathways to bypass the inhibition. We also found that afatinib demonstrated more efficacy on the cell apoptosis (Figure 5), which complemented and confirmed by the cleavage of PARP. The increased cleaved PARP by the combination of afatinib and dasatinib was further confirmed the enhanced induction of apoptosis.
According to our results, afatinib inhibited cell proliferation and induced apoptosis via affecting PI3K/PTEN/Akt, and Ras/Raf/MEK/ERK signalling pathways; dasatinib decreased cell survival, adhesion and migration, and induced apoptosis through affecting Src/FAK and JAK/Stat signalling pathways. The synergistic interaction between afatinib and dasatinib was not only due to their blockage of different signalling pathways but also by inhibition of the cross-talking among the related signalling molecules. T. Yoshida has demonstrated that afatinib combined with dasatinib produced significant in vivo tumour regression in PC9GR xenograft studies [36]. Clinical phase I trial on this combination is ongoing to discover its efficacy by Lee Moffitt Cancer Center (https://clinicaltrials.gov identifier: NCT01999985). JAK/Stat signalling pathway may play important role in the gefitinib resistant cells. So far there is no effective therapy for NSCLC patients with PTEN deletions. Our study suggests combination of afatinib and dasatinib may be effective in this particular group of patients. In our study, we didn’t test osimertinib either alone or combination with dasatinib. This should be an area of subsequent investigation in view the superior efficacy of osimertinib over erlotinib/gefitinib as the first line treatment of EGFR sensitive mutation NSCLC with less toxicities [18].
In conclusion, combination of afatinib and dasatinib had more synergistic effect than cetuximab plus afatinib against gefitinib resistant NSCLC cells. The combination reversed the resistance to EGFR-TKI-resistant H1650 cells with PTEN mutations via affecting SFK/FAK, PI3K/PTEN/Akt, Ras/Raf/MEK/ERK, and JAK/Stat pathways. The level of MAPK, Src and Stat3 may be useful biomarkers predicating synergism between afatinib and dasatinib for the treatment of gefitinib-resistant NSCLC cells. Our work also provided strong scientific support for future in vivo study as well as clinical trials.
MATERIALS AND METHODS
Drugs
Cetuximab (monoclonal antibody against EGFR) was purchased from Merck AG and stored at 4° C. Gefitinib (EGFR TKI) was purchased from Biaffin GmbH & Co KG (proteinkinase.de, Germany). Afatinib (irreversible EGFR and HER2 kinase inhibitor) was bought from LC Laboratories, USA. Dasatinib (Src kinase inhibitor) was obtained from Bristol-Myer Squibb Company. Stock solution was prepared at 10 mM in pure DMSO and stored in aliquots at −80° C. These agents were further diluted with culture medium immediately before use.
Cell lines
A549, H1975, H1650, HCC827, and H820 were obtained from American type Culture Collections (ATCC). As13, As87, and As125 NSCLC cell lines were derived from patients’ pleural effusion by us. All cell lines were cultured in RPMI culture medium with Hepes and L-glutamin (PAA laboratories cell Culture Products, Austria), containing 10% Fatal Bovine Serum (Invitrogen, USA), 1% antibiotic with 100 UI/ml Penicillin and 100ug/ml Streptomycin (Invitrogen, USA). Incubation condition was set at 37° C in a humidified atmosphere of 95% air and 5% CO2. The culture medium was changed 2 to 3 times a week and cells were subcultured using trypsin/EDTA (Invitrogen, USA).
Direct sequencing was applied for mutational analysis of the EGFR, PTEN and K-Ras genes for the three self-established cell lines. Exons 18~21 of EGFR, exon 2 of K-ras, and all 9 exons of PTEN were first amplified using intron-based primers (two PCR reactions for exon 8 of PTEN). The amplicons were then purified for sequencing analysis on ABI PRISM 3100 genetic analyzer (Applied Biosystems) [37].
Growth inhibition assay
CellTiter 96 Aqueous Non-Radioaction cell proliferation Assay Kit (Promega Corporation, USA) was used for growth viability assays. 3000–8000 cells from 8 cell lines were plated in 96-well flat-bottomed plates and cultured for 24 hours (h). Cells were exposed to serially diluted drugs in completed RPMI cell culture medium for an additional 72 hours. 20 μl MTS/PMS solution was added into each well containing 100 μl of the culture medium. Then, the cells were incubated for 3 h at 37° C before measurement of absorbance at 490 nm with a Benchmark Plus microplate spectrophotometer (Bio-RAD, USA). Absorbance values were expressed as a percentage of that for untreated cells, and the concentration of dasatinib resulting in 50% growth inhibition (IC50) was calculated for each cell line. A sensitive cell line to test agents is arbitrarily defined if IC50 is less than or equal to 1 μM or falls below the Cmax of each agent on the basis of clinical phase I pharmacokinetic data [38].
In-vitro drug combination analysis
In-vitro drug combination efficacy was analysed following the method of the previous study [38]. Briefly, according to the cytotoxicity effect of single drug in NSCLC cell lines, IC25 and IC50 were calculated respectively. Two different drugs were combined in the following sequence: IC25 of first drug was combined with IC25 and IC50 of the second drug, similarly, IC50 of the first drug was also combined with IC25 and IC50 of the second drug. The NSCLC cells were treated with the combination concurrently for 72 hours. The cell growth inhibition assay was done by MTS assay. The combination index (CI) between two different drugs was evaluated by the method of Chou and Talalay. CI less than 0.90 indicates a synergistic interaction; CI between 0.90 and 1.10 indicates additive, and CI >1.10 indicates an antagonism effect.
Treatments by afatinib, dasatinib and the combination
Cells were plated at 5 × 105 per well of 6-well plates in completed RPMI culture medium for overnight. After additional 24h serum starvation cells were treated with various concentrations of afatinib, dasatinib and their combination for 3 hours prior to 100 ng/ml EGF stimulation, and 30 minutes later the cells were used for immunoblotting, cell adhesion, migration, invasion and apoptosis assay.
Western blotting analysis
The cells were lysed for protein extraction using 1× RIPA protein extraction buffer with protease inhibitor and phosphatase inhibitor (Thermo scientific, Pierce Biotechnology, USA). The total protein concentration was measured by BCA kit (Pierce Biotechnology, USA). Isolated proteins (30 μg/lane) were separated by 8% SDS-PAGE and transferred to a nitrocellulose membrane by the iblot device (Invitrogen Corporation, CA). The membranes were blocked with 5% BSA at room temperature for 1 h and then subjected to immunoblots using primary antibodies at 4° C overnight, followed by incubation with secondary goat anti-rabbit IgG conjugated to horseradish peroxidase for 1 h at room temperature. Labelled protein was visualized by chemiluminescence (Immobilon, Millipore Corporation, USA) and captured, using β-actin and β-tubulin expression as the internal standard.
Cell apoptosis assay
Cells were detached by 3 mM EDTA and washed with 1x PBS, then stained with Annexin V and 7-AAD (BD), and analysed by flow cytometry (Cyan, ADP, Beckman) for detection of apoptosis cells.
Cell adhesion assay
1.5 × 105 of pre-treated cells were seeded in collagen I coated 96-well plate and incubated for 0.5 h, 1 h, 2 h and 4 h respectively. Cells adhered to collagen I were fixed and stained with cell stain solution (Chemicon International, USA), after washing stained cells were incubated with 100 μl of extraction buffer (Chemicon International, USA), and then optical density [16] was measured at 570 nm by Benchmark Plus Micro-plate Spectrophotometer (Bio-RAD, USA).
Cell migration assay
Cells were treated following the procedure described in “Treatments by afatinib, dasatinib and the combination”. Confluent monolayer of cells were scraped with a fine 200 μl pipette tip to produce a wound. The medium containing various concentrations of drugs were replaced by completed RPMI culture medium and incubated for another 24 h, 48 h and 72 h. Migration into the wound was captured at ×10 magnification on an invert light microscope (Olympus, Japan). Three representative areas were scored and the area moved was calculated using ImageJ analysis software.
Cell invasion assay
Cell invasion assay was processed by using the cell invasion assay kit (Chemicon International, USA). A 24-well tissue culture plate with cell culture inserts which contained an 8 μm pore size polycarbonate membrane was used. 1.5 × 105 testing cells in serum free RPMI were seeded into ECM coated insert, then RPMI with 10% FBS was placed in the 24-well plate as chemo attractants. After 72 h incubation, the cells were removed from the inner surface of the insert using a cotton-tipped swab. The cells that invaded through the ECM layer and clung to the bottom of the polycarbonate membrane were fixed and stained. The number of migrating cells per insert was captured microscopically.
Statistical analysis
All the experiments were repeated at least 3 times. Data are reported as means ± SD, correlation coefficient (r) was calculated by the Pearson product-moment correlation coefficient, and statistical significance (p-value) was analysed using student’s t test.
Author contributions
Performed research: Miao wang, Designed the research study: Miao Wang, Alex Chang Yuang-chi, Analysed the data: Miao Wang, Draft the manuscript: Miao Wang, Alex Chang Yuang-chi.
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
REFERENCES
1. Scagliotti GV, Selvaggi G, Novello S, Hirsch FR. The biology of epidermal growth factor receptor in lung cancer. Clin Cancer Res. 2004; 10:4227s–32s.
2. Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004; 304:1497–500.
3. Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005; 352:786–92.
4. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science. 2007; 316:1039–43.
5. Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, Liu DD, Kurie JM, Mao L, Khuri FR. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res. 2002; 8:1178–84.
6. Forgacs E, Biesterveld EJ, Sekido Y, Fong K, Muneer S, Wistuba II, Milchgrub S, Brezinschek R, Virmani A, Gazdar AF, Minna JD. Mutation analysis of the PTEN/MMAC1 gene in lung cancer. Oncogene. 1998; 17:1557–65.
7. Jin G, Kim MJ, Jeon HS, Choi JE, Kim DS, Lee EB, Cha SI, Yoon GS, Kim CH, Jung TH, Park JY. PTEN mutations and relationship to EGFR, ERBB2, KRAS, and TP53 mutations in non-small cell lung cancers. Lung Cancer. 2010; 69:279–83.
8. Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P, Michel K, Peifer M, Mermel C, et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009; 69:3256–61.
9. Yamamoto C, Basaki Y, Kawahara A, Nakashima K, Kage M, Izumi H, Kohno K, Uramoto H, Yasumoto K, Kuwano M, Ono M. Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib-resistant lung cancer cells harboring epidermal growth factor receptor-activating mutations. Cancer Res. 2010; 70:8715–25.
10. Giaccone G, Zucali PA. Src as a potential therapeutic target in non-small-cell lung cancer. Ann Oncol. 2008; 19:1219–23.
11. Mazurenko NN, Kogan EA, Zborovskaya IB, Kisseljov FL. Expression of pp60c-src in human small cell and non-small cell lung carcinomas. Eur J Cancer. 1992; 28:372–77.
12. Song L, Morris M, Bagui T, Lee FY, Jove R, Haura EB. Dasatinib (BMS-354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Res. 2006; 66:5542–48.
13. Johnson FM, Saigal B, Talpaz M, Donato NJ. Dasatinib (BMS-354825) tyrosine kinase inhibitor suppresses invasion and induces cell cycle arrest and apoptosis of head and neck squamous cell carcinoma and non-small cell lung cancer cells. Clin Cancer Res. 2005; 11:6924–32.
14. Kruser TJ, Traynor AM, Wheeler DL. The use of single-agent dasatinib in molecularly unselected non-small-cell lung cancer patients. Expert Opin Investig Drugs. 2011; 20:305–07.
15. Johnson FM, Bekele BN, Feng L, Wistuba I, Tang XM, Tran HT, Erasmus JJ, Hwang LL, Takebe N, Blumenschein GR, Lippman SM, Stewart DJ. Phase II study of dasatinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010; 28:4609–15.
16. Sos ML, Rode HB, Heynck S, Peifer M, Fischer F, Klüter S, Pawar VG, Reuter C, Heuckmann JM, Weiss J, Ruddigkeit L, Rabiller M, Koker M, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. J Cancer Res. 2010; 70:868–74.
17. Subramaniam DS, Hwang J. BIBW 2992 in non-small cell lung cancer. Expert Opin Investig Drugs. 2011; 20:415–22.
18. Jänne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015; 372:1689–99.
19. Arulananda S, Do H, Musafer A, Mitchell P, Dobrovic A, John T. Combination Osimertinib and Gefitinib in C797S and T790M EGFR-mutated non-small cell lung cancer. J Thorac Oncol. 2017; 12:1728–32.
20. Murakami Y, Sonoda K, Abe H, Watari K, Kusakabe D, Azuma K, Kawahara A, Akiba J, Oneyama C, Pachter JA, Sakai K, Nishio K, Kuwano M, Ono M. The activation of SRC family kinases and focal adhesion kinase with the loss of the amplified, mutatedEGFRgene contributes to the resistance to afatinib, erlotinib and osimertinib in human lung cancer cells. Oncotarget. 2017; 8:70736–51. https://doi.org/10.18632/oncotarget.19982.
21. Watanabe S, Yoshida T, Kawakami H, Takegawa N, Tanizaki J, Hayashi H, Takeda M, Yonesaka K, Tsurutani J, Nakagawa K. T790M-selective EGFR-TKI combined with Dasatinib as an optimal strategy for overcoming EGFR-TKI resistance in T790M-positive non-small cell lung cancer. Mol Cancer Ther. 2017; 16:2563–71.
22. Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, Ince WL, Jänne PA, Januario T, Johnson DH, Klein P, Miller VA, Ostland MA, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005; 23:5900–09.
23. Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005; 2:e17.
24. Rodenhuis S, Slebos RJ, Boot AJ, Evers SG, Mooi WJ, Wagenaar SS, van Bodegom PC, Bos JL. Incidence and possible clinical significance of K-ras oncogene activation in adenocarcinoma of the human lung. Cancer Res. 1988; 48:5738–41.
25. Cho JY, Kim JH, Lee YH, Chung KY, Kim SK, Gong SJ, You NC, Chung HC, Roh JK, Kim BS. Correlation between K-ras gene mutation and prognosis of patients with nonsmall cell lung carcinoma. Cancer. 1997; 79:462–67.
26. Janjigian YY, Groen HJ, Horn L, Smit EF, Fu Y, Wang F, Shahidi M, Denis LJ, Pao W, Miller VA. Activity and tolerability of afatinib (BIBW 2992) and cetuximab in NSCLC patients with acquired resistance to erlotinib or gefitinib. J Clin Oncol. 2011; 29:7525.
27. Rodenhuis S, van de Wetering ML, Mooi WJ, Evers SG, van Zandwijk N, Bos JL. Mutational activation of the K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. N Engl J Med. 1987; 317:929–35.
28. McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA, D’Assoro AB, Salisbury JL, Mazzarino MC, Stivala F, Libra M. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul. 2006; 46:249–79.
29. Kandasamy K, Srivastava RK. Role of the phosphatidylinositol 3′-kinase/PTEN/Akt kinase pathway in tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in non-small cell lung cancer cells. Cancer Res. 2002; 62:4929–37.
30. Bianco R, Shin I, Ritter CA, Yakes FM, Basso A, Rosen N, Tsurutani J, Dennis PA, Mills GB, Arteaga CL. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene. 2003; 22:2812–22.
31. Kokubo Y, Gemma A, Noro R, Seike M, Kataoka K, Matsuda K, Okano T, Minegishi Y, Yoshimura A, Shibuya M, Kudoh S. Reduction of PTEN protein and loss of epidermal growth factor receptor gene mutation in lung cancer with natural resistance to gefitinib (IRESSA). Br J Cancer. 2005; 92:1711–19.
32. Ishizawar R, Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell. 2004; 6:209–14.
33. Chang AY, Wang M. Molecular mechanisms of action and potential biomarkers of growth inhibition of dasatinib (BMS-354825) on hepatocellular carcinoma cells. BMC Cancer. 2013; 13:267.
34. Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003; 116:1409–16.
35. Quesnelle KM, Boehm AL, Grandis JR. STAT-mediated EGFR signaling in cancer. J Cell Biochem. 2007; 102:311–19.
36. Yoshida T, Zhang G, Bai Y, Fang B, Rawal B, Fisher K, Chen A, Okabe T, Okamoto I, Nakagawa K. MIS4. Molecular networks of the EGFR-TKI resistant non-small cell lung cancer: how to apply new technologies to bench-to-bed research. Ann Oncol. 2012; 23:xi47–xi48.
37. Zhang X, Zhao Y, Wang M, Yap WS, Chang AY. Detection and comparison of epidermal growth factor receptor mutations in cells and fluid of malignant pleural effusion in non-small cell lung cancer. Lung Cancer. 2008; 60:175–82.
38. Chang AY, Wang M. In-vitro growth inhibition of chemotherapy and molecular targeted agents in hepatocellular carcinoma. Anticancer Drugs. 2013; 24:251–59.