
INTRODUCTION
Angiogenesis drives tumor progression [1], and pathways involving vascular endothelial growth factors (VEGFs) and its receptors (VEGFRs) promote tumor angiogenesis [2]. Bevacizumab (Bev) is a humanized anti-VEGF-A neutralizing monoclonal antibody, and the combination regimen of add-on Bev with pre-existing chemotherapeutic agents provide improved clinical benefits more than chemotherapy alone in several malignancy types [3–5]. Unlike chemotherapeutic agents, the anti-angiogenesis strategy should result in far less drug resistance [6, 7]. However, Bev-combined chemotherapy still could not overcome drug resistance in clinical settings [8, 9].
CD11b+/Gr-1+ cells, which include neutrophils, macrophages, and myeloid-derived suppressor cells (MDSCs), promote tumor escape from anti-VEGF therapy [10, 11]. This process likely involves at least two independent mechanisms: bypassing anti-VEGF-mediated anti-angiogenesis via secretion of the alternative angiogenic factor Bv8 [12] and a MDSC-mediated, Th17-dependent immune suppressive pathway [13]. Both mechanisms were shown to be mediated via intratumoral recruitment of myeloid cells by granulocyte-colony stimulating factor (G-CSF) [12, 13]. In the present study, we used a mouse model to investigate a possible alternative pathway that may promote tumor resistance to anti-VEGF therapy.
RESULTS
Anti-VEGF treatment increased the number of intratumoral CD11bhigh/Gr-1high cells
Shojaei et al. used B16F1 melanomas and LLC murine lung carcinomas to show resistance to anti-VEGF antibody treatment is associated with intratumoral MDSC accumulation [11]. Because Gr-1 is a cell surface marker that reflects the immune suppressive activity of MDSCs in tumor models [14–16], we used CD11b and Gr-1, rather than Ly6G/Ly6C, to identify the subpopulation of MDSCs. The anti-VEGF treatment was effective against B16F1 tumors associated with a very small amount of CD11bhigh/Gr-1high, (Figure 1A) [14–16]. In contrast, the anti-VEGF treatment accelerated intratumoral MDSC accumulation (Figure 1B).
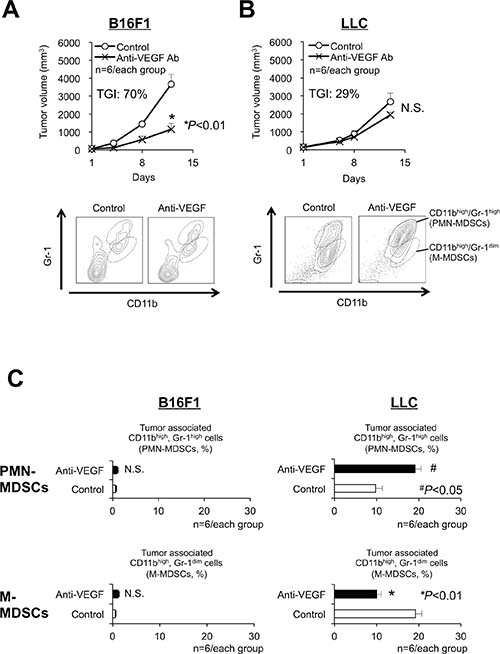
Figure 1: Antitumor responses and accumulation of myeloid-derived suppressor cells (MDSCs) related to anti-VEGF treatment in sensitive or resistant tumors. (A, B) Growth curves (upper graphs, TGI: tumor growth inhibition) and flow-cytometric analyses of the infiltration of MDSCs (bottom graphs) in anti-VEGF-sensitive (B16F1, TGI at day 12 = 70%) (A) and anti-VEGF-resistant (LLC, TGI at day 12 = 29%) (B) tumors in C57BL/6 mice. Note that 2-independent populations (CD11bhigh/Gr-1high or CD11bhigh/Gr-1dim) were identified as polymorphonuclear (PMN)- and monocytic (M)- MDSCs, respectively. (n = 6/group). (C) Quantification of the composition ratio of intratumoral CD11bhigh/Gr-1high cells (PMN-MDSCs) during anti-VEGF treatment. All data are shown as the mean ± SEM. N.S.: not significant, *P < 0.01, and #P < 0.05. (n = 6/group).
Next, because MDSCs show at least two distinct phenotypes, namely PMN- and monocytic (M)-MDSCs, we investigated the effect of anti-VEGF treatment on these populations using LLC tumors. Anti-VEGF treatment increased the ratio of CD11bhigh/Gr-1high PMN-MDSCs (Figure 1C).
Capecitabine, a prodrug of 5-FU, restored the antitumor effect of anti-VEGF
Since 5-FU can selectively kill MDSCs and enhance T-lymphocyte-mediated antitumor immune responses [17], we evaluated the effect of 5-FU and the clinically available prodrug of 5-FU, capecitabine, on LLC tumor growth under anti-VEGF treatment. Capecitabine, but not 5-FU, demonstrated a combined antitumor effect with anti-VEGF (Figure 2A). In addition, capecitabine diminished the intratumoral accumulation of PMN-MDSCs (Figure 2B) and circulating PMN-MDSCs (Figure 2C, right). 5-FU only partially inhibited these same parameters (Figure 2C, left).
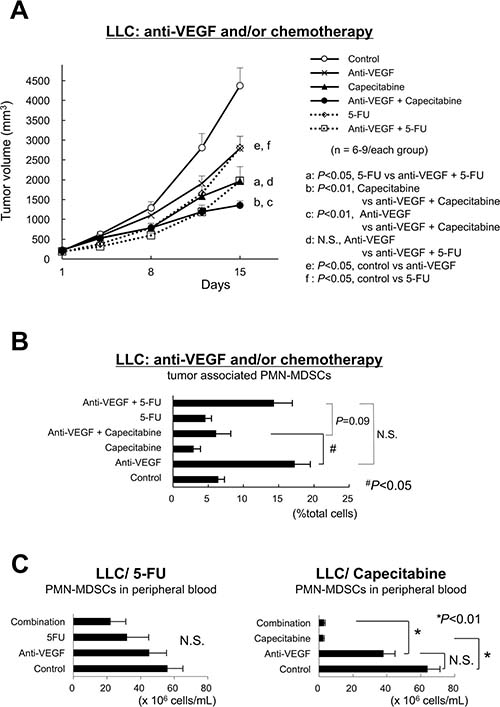
Figure 2: Capecitabine, a prodrug of 5-FU, restores restored the antitumor effect of anti-VEGF. (A) Sole and combination effects of anti-VEGF, 5-FU and capecitabine on the growth of anti-VEGF -resistant LLC tumors in vivo. Note that capecitabine, but not 5-FU, demonstrated a combined antitumor effect with anti-VEGF. (n = 6–9/group). (B) Sole and combination effects of anti-VEGF, 5-FU and capecitabine on the intratumor accumulation of PMN-MDSCs in anti-VEGF-resistant LLC tumors. Note that treatment with capecitabine, but not 5-FU, resulted in a significant reduction of the intratumor accumulation of PMN-MDSCs. Data are shown as the mean ± SEM (n = 6/group). N.S.: not significant, #P < 0.05. (n = 6/group). (C) Effect of 5-FU or capecitabine on the number of circulating PMN-MDSCs. Note that capecitabine, but not 5-FU, almost eliminated PMN-MDSCs in the peripheral blood. Data are mean ± SEM. N.S.: not significant, #P < 0.05. (n = 6/group).
G-CSF, but neither IL-17 nor Bv8, promoted intratumoral PMN-MDSC recruitment and antitumor angiogenesis in the LLC tumor model
We screened for cytokines/chemokines expressed by LLC tumors in vivo that stimulate anti-VEGF-mediated PMN-MDSC recruitment. G-CSF and CCL2, but not IL-17A or Bv8, were increased by the anti-VEGF treatment, and capecitabine reduced those upregulations (Figure 3A). The selective antagonist of the CCL2 receptor CCR2 (RS102895) at the appropriate dose [18] did not affect either tumor growth or PMN-MDSC accumulation (data not shown). Anti-mouse G-CSF-neutralizing mAb inhibited tumor growth and PMN-MDSC accumulation (Figure 3B) under anti-VEGF administration, confirming G-CSF promotes intratumoral PMN-MDSC recruitment during anti-VEGF therapy [11]. G-CSF expression increased after anti-VEGF treatment in LLC tumor lysate, while IL-17A expression was low (Figure 4A). Bv8 protein was detected, and neither anti-VEGF nor capecitabine treatment changed its protein level (Figure 4B).
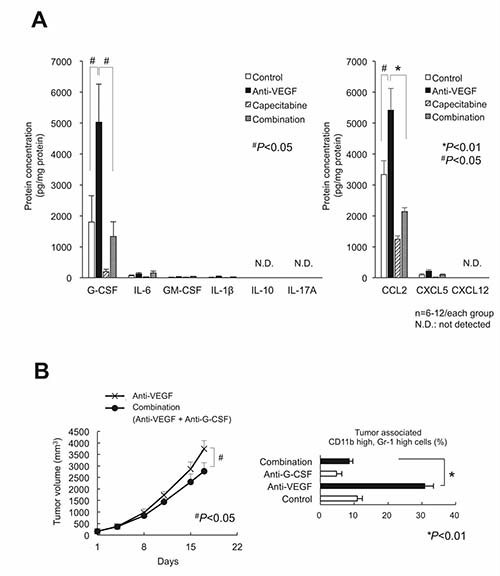
Figure 3: Screening for the cytokines/chemokines in LLC tumor lysate that were affected by anti-VEGF, capecitabine, and their combination treatments. (A) The screening results by bead array assay (left) or ELISA (right). Only G-CSF and CCL2 exhibited both the increased by anti-VEGF and decreased by capecitabine. IL-17 protein was under the detection limit. (n = 6–12/group). (B) Effect of neutralization of G-CSF on LLC tumor growth (left) and the intratumor accumulation of PMN-MDSCs (right) during anti-VEGF treatment. The increase of PMN-MDSCs by anti-VEGF was completely cancelled by anti-G-CSF treatment. Data are mean ± SEM. *P < 0.01. (n = 6/group)
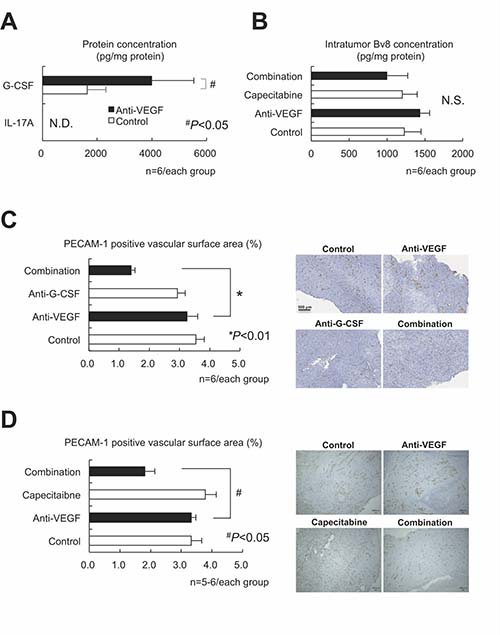
Figure 4: G-CSF, but neither IL-17 nor Bv8, was still essential to the intratumor recruitment of PMN-MDSCs and antitumor angiogenesis in the LLC tumor model. (A) Confirmation that G-CSF, but not IL-17, was involved in anti-VEGF-mediated upregulation in LLC tumor lysate assessed by ELISA assay. (n = 6/group). (B) Neither anti-VEGF nor capecitabine affected Bv8 expression as assessed by ELISA. (n = 6/group). (C) Effects of anti-VEGF, anti-G-CSF, and their combination on LLC tumor angiogenesis. The PECAM-1-positive vascular surface area was identified by immunohistochemistry. Only the combination, but not either sole therapy, could reduce the tumor angiogenesis. (n = 6/group). (D) Effects of anti-VEGF, capecitabine, and their combination on LLC tumor angiogenesis. The PECAM-1-positive vascular surface area was identified by immunohistochemistry. Note that only the combination, but not either sole therapy, could reduce the tumor angiogenesis, similarly to the findings to shown in panel (C). Data are mean ± SEM. N.S.: not significant, *P < 0.01, and #P < 0.05. (n = 5–6/group).
We focused on an Bv8-independent mechanism of tumor angiogenesis, as an IL-17-mediated immune-related mechanism appears unlikely. We assessed the platelet and endothelial cell adhesion molecule (PECAM)-1-positive vascular surface area in each tumor using an image analyzer. Anti-VEGF therapy alone did not inhibit tumor angiogenesis, whereas the addition of anti-G-CSF neutralizing antibody reduced the vascular surface area (Figure 4C). Tumor sections treated with anti-VEGF combined with capecitabine displayed similar results (Figure 4D), suggesting that an anti-VEGF/PMN-MDSCs/G-CSF axis may contribute to the Bv8-independent angiogenic escape induced by anti-VEGF treatment.
Intratumoral MDSCs have PyNPase activities and the proangiogenic factor TP
Pyrimidine nucleotide phosphorylases (PyNPases), composed of thymidine phosphorylase (TP) and uridine phosphorylase (UP), are proangiogenic factors [19–21] and capecitabine converting enzymes [22, 23]. We investigated PyNPase activity in various cells, including MDSCs. The capecitabine-sensitive human colorectal cancer cell line HCT116 [22] demonstrated high PyNPase activity as a positive control, whereas CD45+ cells derived from murine spleen cells showed no activity (Figure 5A). The LLC cells and PMN- and M-MDSCs sorted from the LLC tumors had high PyNPase activity. The in vivo capecitabine treatment reduced the intratumoral content of TP independent of anti-VEGF treatment (Figure 5B), indicating that capecitabine could eliminate PyNPases in tumors.
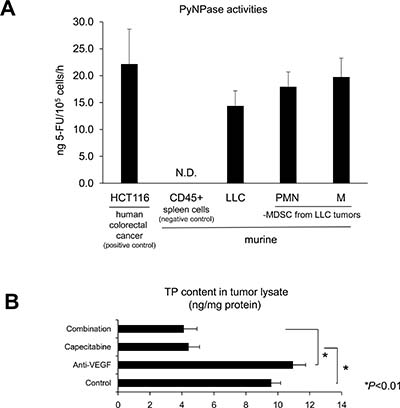
Figure 5: Intratumoral MDSCs have PyNPase activities and thymidine phosphorylase (TP), which is known as a proangiogenic factor. (A) High PyNPase activity in LLC cells as well as both PMN-/M-MDSCs isolated from LLC tumors in vivo. PyNPase activity was assessed by converting efficiency to 5-FU. All data are shown as the mean ± SEM (n = 3–5/group). (B) Capecitabine reduced TP contents in LLC tumor lysate assessed by ELISA. All data are shown as the mean ± SEM. N.S.: not significant, *P < 0.01. (n = 6/group).
Impairment of capecitabine effects under anti-VEGF in PyNPases TP−/−/UP−/− double-deficient mice
We assessed TP activity in host-originated PMN-MDSCs, using LLC tumors bearing TP−/−/UP−/− double-deficient mice [24] under anti-VEGF treatment. The capecitabine-dependent reduction of intratumoral PMN-MDSC recruitment in LLC tumors of the wild-type mice was suppressed in the LLC tumors of the TP−/−/UP−/− double-deficient mice (Figure 6A). Capecitabine’s enhanced antitumor effect and restoration of antitumor angiogenesis under anti-VEGF therapy observed in the wild-type mice was reduced in the TP−/−/UP−/− mice (Figure 6B and 6C, respectively).
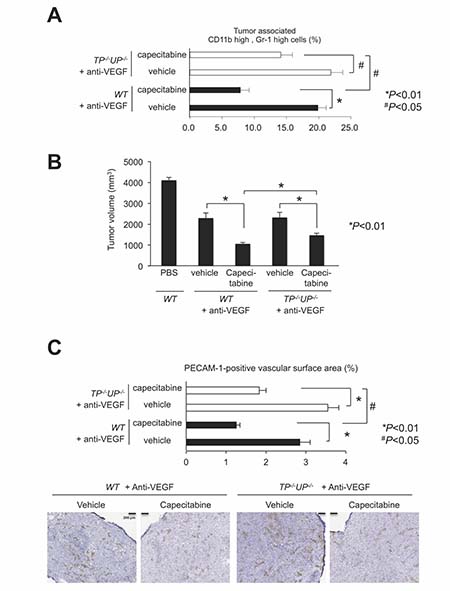
Figure 6: Impairment of capecitabine’s effects in PyNPases TP−/−/UP−/− double deficient mice. (A) Capecitabine-mediated reduction of PMN-MDSC infiltration during anti-VEGF therapy was impaired in PyNPases TP−/−/UP−/− double deficient mice. All data are shown as the mean ± SEM. N.S.: not significant, *P < 0.01 and #P < 0.05. (n = 6/group). (B) Capecitabine-mediated reduction of tumor volume during anti-VEGF therapy was also impaired in PyNPases TP−/−/UP−/− double deficient mice. All data are shown as the mean ± SEM. N.S.: not significant, *P < 0.01 (n = 6/group). (C) Capecitabine-mediated reduction of tumor angiogenesis during anti-VEGF therapy was also impaired in PyNPases TP−/−/UP−/− double deficient mice. All data are shown as the mean ± SEM. N.S.: not significant, *P < 0.01 and #P < 0.05. (n = 6/group).
DISCUSSION
Our study produced three key observations. (i) Anti-VEGF therapy accelerated intratumoral CD11bhigh/Gr-1high PMN-MDSC accumulation in anti-VEGF-resistant LLC tumors, but capecitabine diminished this effect and restored the antitumor activity of anti-VEGF treatment. (ii) The beneficial effect of capecitabine on LLC with anti-VEGF therapy is likely promoted by G-CSF expression, not IL-17 or Bv8. (iii) PyNPases, expressed by PMN-MDSCs, stimulated tumor resistance to anti-VEGF treatment.
The potential role of TP (platelet-derived endothelial cell growth factor [PD-ECGF]) in tumor angiogenesis in VEGF-negative tumor tissue was first suggested for colon cancer [19, 25, 26]. Since then, a number of studies have reported various tumor types and tumor infiltrating macrophages (TAMs) and lymphocytes to be the cellular sources of TP [27]. The present study is the first to determine that MDSCs, in particular the PMN type, are the functionally essential TP-expressing cells in tumors during anti-VEGF therapy.
Considering our present findings and the good safety and efficacy profiles of the combination of capecitabine and bevacizumab in phase III trials [4, 28], the observed partial effect of capecitabine on the restoration of anti-angiogenesis and inhibition of tumor growth during anti-VEGF treatment suggests more study may enable further optimization of anti-VEGF therapy to provide greater clinical benefit.
The capecitabine dose used in this study almost eliminated PMN-MDSCs in peripheral blood, but this effect was partially reduced in intratumoral areas (Figure 2). This may be due to (i) an insufficient distribution of capecitabine to the whole tumor tissue even though the systemic dose of it was sufficient, and/or (ii) the recruitment of other cells that are insensitive to capecitabine/5-FU and express TP/UP to tumor microenvironment. The latter might be likely, because CD68-positive tumor-associated macrophages (TAMs) were identified as the dominant cell source of TP/PD-ECGF in human colorectal cancer tissue [25]. Capecitabine reversed tumor escape from anti-VEGF, and may be a favorable chemotherapeutic agent that should be combined with bevacizumab in clinical settings (Supplementary Figure 1).
MATERIALS AND METHODS
Cell lines and culture conditions
The murine lung cancer (LLC) and murine melanoma (B16F1) cell lines were obtained in 2004 from the American Type Culture Collection (ATCC; Rockville, MD). The human colorectal cancer cell line HCT116 was obtained in 1990 from the ATCC. The LLC and B16F1 cells were cultured in high-glucose Dulbecco’s Modified Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 4 mM glutamine, and maintained at 37° C in 5% CO2. The HCT116 was cultured in McCoy’s 5A Medium supplemented with 10% FBS and maintained at 37° C in a 5% CO2. All cell lines were passaged up to 20 times. All cell lines were authenticated using STR analyses, and were routinely tested for mycoplasma contamination using PCR-based detection methods in Central Institute for Experimental Animals (Kanagawa, Japan).
Mice
Male 5- to 9-week-old C57BL/6 mice were obtained from KBT Oriental (Charles River Grade, Tosu, Saga, Japan). Male 7-week-old thymidine phosphorylase and uridine phosphorylase double-deficient mice (TP−/−/UP−/−) on the C57BL/6 background were provided by Prof. Furukawa (Department of Cancer Chemotherapy, Kagoshima University, Kagoshima) [24]. The mice were kept under specific pathogen-free and humane conditions, and the animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee and by the Biosafety Committee for Recombinant DNA Experiments of Kyushu University (approval ID: A26–240–0). These experiments were also done in accordance with the recommendations for the proper care and use of laboratory animals and according to The Law (No. 105) and Notification (No. 6) of the Japanese Government, and the U.S. National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Tumor models
LLC and B16-F1 tumor cells (5.0 × 106) in 200 μL of DMEM were subcutaneously injected in the left flank of C57BL/6 mice or TP−/−/UP−/− mice. The administration of anticancer agents was started when the tumor volumes reached 50 to 300 mm3. Anti-mouse VEGF-A monoclonal antibody (mAb) B20-4.1.1 (Genentech, Oceanside, CA) was intraperitoneally administered to the mice at the dose of 5 mg/kg weekly. Anti-mouse G-CSF mAb (clone 67604, rat IgG1, R&D Systems, Minneapolis, MN) was administered intraperitoneally at the dose of 10 μg/head daily. Capecitabine (pentylN-[1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-methyloxolan-2-yl]-5-fluoro-2- oxopyrimidin-4-yl]carbamate, Chugai Pharmaceutical, Tokyo) and CCR2 antagonist (1′-[2-[4-(trifluoromethyl)phenyl]ethyl]spiro[1H-3,1-benzoxazine-4,4′-piperidine]-2-one, RS102895, Sigma-Aldrich, St. Louis, MO) were orally administered daily at the dose of 718 mg/kg and 10 mg/kg, respectively. 5-Fluorouracil (5-fluoro-1H-pyrimidine- 2,4-dione, 5-FU, Invitrogen, Carlsbad, CA) was administered intraperitoneally at the dose of 50 mg/kg twice a week. All animal experiments were conducted in accord with the institutional Animal Care and Use Committee. Tumor volume was estimated from the equation (L × W2 × 0.5); L = length and W = width.
Flow cytometric analysis
Tumors were excised from control- and anti-VEGF-treated mice, and single cell suspensions were obtained by mincing tumors and homogenizing by disruption and digestion with a gentle MACS™ Dissociator and Tumor Dissociation Kit for mouse (Miltenyi Biotec, Bergisch Gladbach, Germany). Peripheral blood was pretreated with VersaLyse™ lysing solution for red blood cells lysis (Beckman Coulter, Indianapolis, IN).
Cells were stained with the following FITC-, PE-, PE-Cy5-, BV785, or Alexa647-conjugated monoclonal antibodies: mouse CD11b, Gr1, CD45 (BioLegend, San Diego, CA), G-CSF (eBioscience, San Diego, CA), and TP (Proteintech, Chicago, IL). The appropriate conjugated isotype-matched Immunoglobulin Gs (IgGs) were used for control. Intracellular cytokine staining was performed with the Cell Fixation/Permeabilization kit (BD Biosciences, Franklin Lakes, NJ). Cells were analyzed using a FACSAria™ cell sorter and an LSRFortessa™ cell analyzer (BD Biosciences) and FlowJo 7.6 software (Tree Star, San Carlos, CA).
Immunohistochemistry
We evaluated microvessel density in the tumor tissue by performing immunohistochemical staining of PECAM-1 (rat anti-mouse PECAM-1 mAb, clone MEC 13.3; BD Biosciences). Tumor samples were collected at the end of the study. Immunohistochemistry was performed as described previously [29]. The microvessel density (%) was calculated from the ratio of the PECAM-1-positive staining area to the total observation area in the viable region. Positive staining areas were calculated using the imaging analysis software Tissue Studio® (Definiens, Munich, Germany).
Immunoassays
Tumor homogenates collected at the end of the study were analyzed with the BD™ Cytometric Bead Array (BD Biosciences). The concentrations of mouse IL-17A, G-CSF, CXCL12, CXCL5, and CCL2 were measured by a Quantikine ELISA kit (R&D Systems). Bv8 was quantified using the mouse prokineticin-2 (Bv8) ELISA kit (Cusabio Biotech, Selangor, Malaysia). The protein amount of TP was quantified using a specific ELISA kit (Cloud-Clone, Katy, TX).
PyNPase enzymatic activity assay
Cells were homogenized in 10 mM Tris buffer (pH 7.4), containing 15 mM NaCl, 1.5 mM MgCl2, and 50 μM potassium phosphate. The homogenate was then centrifuged at 105,000 g for 90 min. The supernatant was dialyzed overnight against 20 mM potassium phosphate buffer (pH 7.4) containing 1 mM β-mercaptoethanol and used as a source of crude enzyme. All procedures were carried out below 4° C. The reaction mixture (120 μl) for the assay of the enzyme activity contained 183 mM potassium phosphate (pH 7.4), 10 mM 5′-dFUrd, and the crude enzyme from cells. The reaction was carried out at 37° C for 60 min, and then terminated by adding 360 μl of methanol. After removal of the precipitate by centrifugation, the amount of 5-FUra produced in the supernatant was measured with the My5-FU assay (Saladax Biomedical, Bethlehem, PA). The dThdPase and UPase activities are expressed as ng of 5-FUra converted/105 cells/h.
Statistical analysis
All data are expressed as the mean ± standard error of the mean (SEM). The Wilcoxon test was used, and P-values < 0.05 were accepted as significant. Statistical analyses were carried out using JMP, version 10 (SAS Institute, Cary, NC).
Abbreviations
Bev: bevacizumab; FDA: U.S. Food and Drug Administration; VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor; MDSCs: myeloid-derived suppressor cells; PyNPase: pyrimidine nucleotide phosphorylase; TP: thymidine phosphorylase; UP: uridine phosphorylase; PMN: polymorphonuclear; LLC: Lewis lung carcinoma; 5-FU: 5- fluorouracil; G-CSF: granulocyte-colony stimulating factor; IL-17: interleukin-17; PFS: progression-free survival; OS: overall survival; CCL2: chemokine (C-C motif) ligand 2; CCR2: C-C chemokine receptor type 2; PECAM-1: platelet and endothelial cell adhesion molecule-1; PD-ECGF platelet-derived endothelial cell growth factor; TAMs: tumor infiltrating macrophages.
Author contributions
Conception and design: T. Iwai, Y. Harada, and Y. Yonemitsu, Development of methodology: T. Iwai, Y. Harada, H. Saeki, E. Oki, and Y. Yonemitsu, Acquisition of data: T. Iwai, Y. Harada, H. Saeki, E. Oki, and Y. Yonemitsu, Analysis and interpretation of data (statistical analysis, etc.): T. Iwai, Y. Harada, and Y. Yonemitsu, Writing, review, and/or revision of the manuscript: T. Iwai, Y. Harada, and Y. Yonemitsu, Administrative, technical, or material support (i.e., reporting or organizing data, etc.): T. Iwai, and Y. Harada, Study supervision: Y. Maehara, and Y. Yonemitsu.
ACKNOWLEDGMENTS
We thank Mitsue Kurasawa, Keigo Yorozu (Product Research Department, Chugai Pharmaceutical) and Hiroshi Fujii (Department of Pathology, Kyushu University) for help with the immunohistochemistry, and Kaname Yamamoto, Naoki Harada, Kaori Fujimoto-Ouchi and Kazushige Mori (Product Research Department, Chugai Pharmaceutical) for the helpful discussion and comments.
CONFLICTS OF INTEREST
This study was a basic collaboration study with Kyushu University (Professors Y. Maehara and Y. Yonemitsu) and Chugai Pharmaceutical Co. Ltd., and the study was partly supported by Chugai Pharmaceutical Co. Ltd.
FUNDING
This study was supported in part by grants-in-aid from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (15638738 and 16690166 to Y.M., and 16690165 to Y.Y.), and in part by Chugai Pharmaceutical Co. Ltd.
REFERENCES
1. Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005; 438:967–74. https://doi.org/10.1038/nature04483.
2. Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008; 8:579–91. https://doi.org/10.1038/nrc2403.
3. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004; 350:2335–42. https://doi.org/10.1056/NEJMoa032691.
4. Miller KD, Chap LI, Holmes FA, Cobleigh MA, Marcom PK, Fehrenbacher L, Dickler M, Overmoyer BA, Reimann JD, Sing AP, Langmuir V, Rugo HS. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol. 2005; 23:792–9. https://doi.org/10.1200/jco.2005.05.098.
5. Saltz LB, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS, Rivera F, Couture F, Sirzen F, Cassidy J. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008; 26:2013–9. https://doi.org/10.1200/jco.2007.14.9930.
6. Klement G, Baruchel S, Rak J, Man S, Clark K, Hicklin DJ, Bohlen P, Kerbel RS. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest. 2000; 105:R15–24. https://doi.org/10.1172/jci8829.
7. Klement G, Huang P, Mayer B, Green SK, Man S, Bohlen P, Hicklin D, Kerbel RS. Differences in therapeutic indexes of combination metronomic chemotherapy and an anti-VEGFR-2 antibody in multidrug-resistant human breast cancer xenografts. Clin Cancer Res. 2002; 8:221–32.
8. Miles DW, Chan A, Dirix LY, Cortes J, Pivot X, Tomczak P, Delozier T, Sohn JH, Provencher L, Puglisi F, Harbeck N, Steger GG, Schneeweiss A, et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2010; 28:3239–47. https://doi.org/10.1200/jco.2008.21.6457.
9. Pujade-Lauraine E, Hilpert F, Weber B, Reuss A, Poveda A, Kristensen G, Sorio R, Vergote I, Witteveen P, Bamias A, Pereira D, Wimberger P, Oaknin A, et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The AURELIA open-label randomized phase III trial. J Clin Oncol. 2014; 32:1302–8. https://doi.org/10.1200/jco.2013.51.4489.
10. Shojaei F, Ferrara N. Refractoriness to antivascular endothelial growth factor treatment: role of myeloid cells. Cancer Res. 2008; 68:5501–4. https://doi.org/10.1158/0008-5472.can-08-0925.
11. Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP, Ferrara N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007; 25:911–20. https://doi.org/10.1038/nbt1323.
12. Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, Blanchard D, Bais C, Peale FV, van Bruggen N, Ho C, Ross J, Tan M, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007; 450:825–31. https://doi.org/10.1038/nature06348.
13. Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, Vernes JM, Jiang Z, Meng YG, Peale FV, Ouyang W, Ferrara N. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med. 2013; 19:1114–23. https://doi.org/10.1038/nm.3291.
14. Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, Geilich M, Winkels G, Traggiai E, Casati A, Grassi F, Bronte V. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. 2010; 40:22–35. https://doi.org/10.1002/eji.200939903.
15. Youn JI, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010; 40:2969–75. https://doi.org/10.1002/eji.201040895.
16. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008; 181:5791–802.
17. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rebe C, Ghiringhelli F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010; 70: 3052–61. https://doi.org/10.1158/0008-5472.can-09-3690.
18. Cerri C, Genovesi S, Allegra M, Pistillo F, Puntener U, Guglielmotti A, Perry VH, Bozzi Y, Caleo M. The Chemokine CCL2 Mediates the Seizure-enhancing Effects of Systemic Inflammation. J Neurosci. 2016; 36:3777–88. https://doi.org/10.1523/jneurosci.0451-15.2016.
19. Folkman J. What is the role of thymidine phosphorylase in tumor angiogenesis. J Natl Cancer Inst. 1996; 88:1091–2.
20. Ishikawa F, Miyazono K, Hellman U, Drexler H, Wernstedt C, Hagiwara K, Usuki K, Takaku F, Risau W, Heldin CH. Identification of angiogenic activity and the cloning and expression of platelet-derived endothelial cell growth factor. Nature. 1989; 338:557–62. https://doi.org/10.1038/338557a0.
21. Sumizawa T, Furukawa T, Haraguchi M, Yoshimura A, Takeyasu A, Ishizawa M, Yamada Y, Akiyama S. Thymidine phosphorylase activity associated with platelet-derived endothelial cell growth factor. J Biochem. 1993; 114:9–14.
22. Ishikawa T, Utoh M, Sawada N, Nishida M, Fukase Y, Sekiguchi F, Ishitsuka H. Tumor selective delivery of 5-fluorouracil by capecitabine, a new oral fluoropyrimidine carbamate, in human cancer xenografts. Biochem Pharmacol. 1998; 55:1091–7.
23. Miwa M, Ura M, Nishida M, Sawada N, Ishikawa T, Mori K, Shimma N, Umeda I, Ishitsuka H. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer. 1998; 34:1274–81.
24. Haraguchi M, Tsujimoto H, Fukushima M, Higuchi I, Kuribayashi H, Utsumi H, Nakayama A, Hashizume Y, Hirato J, Yoshida H, Hara H, Hamano S, Kawaguchi H, et al. Targeted deletion of both thymidine phosphorylase and uridine phosphorylase and consequent disorders in mice. Mol Cell Biol. 2002; 22:5212–21.
25. Takahashi Y, Bucana CD, Liu W, Yoneda J, Kitadai Y, Cleary KR, Ellis LM. Platelet-derived endothelial cell growth factor in human colon cancer angiogenesis: role of infiltrating cells. J Natl Cancer Inst. 1996; 88:1146–51.
26. Takebayashi Y, Akiyama S, Akiba S, Yamada K, Miyadera K, Sumizawa T, Yamada Y, Murata F, Aikou T. Clinicopathologic and prognostic significance of an angiogenic factor, thymidine phosphorylase, in human colorectal carcinoma. J Natl Cancer Inst. 1996; 88:1110–7.
27. Elamin YY, Rafee S, Osman N, O Byrne KJ, Gately K. Thymidine Phosphorylase in Cancer; Enemy or Friend? Cancer Microenviron. 2016; 9:33–43. https://doi.org/10.1007/s12307-015-0173-y.
28. Simkens LH, van Tinteren H, May A, ten Tije AJ, Creemers GJ, Loosveld OJ, de Jongh FE, Erdkamp FL, Erjavec Z, van der Torren AM, Tol J, Braun HJ, Nieboer P, et al. Maintenance treatment with capecitabine and bevacizumab in metastatic colorectal cancer (CAIRO3): a phase 3 randomised controlled trial of the Dutch Colorectal Cancer Group. Lancet. 2015; 385:1843–52. https://doi.org/10.1016/s0140-6736(14)62004-3.
29. Yanagisawa M, Yorozu K, Kurasawa M, Nakano K, Furugaki K, Yamashita Y, Mori K, Fujimoto-Ouchi K. Bevacizumab improves the delivery and efficacy of paclitaxel. Anticancer Drugs. 2010; 21:687–94. https://doi.org/10.1097/CAD.0b013e32833b7598.