
INTRODUCTION
Pancreatic cancer (PC) remains one of the most deadly malignancies with an overall five-year survival probability less than 7% in all stages combined [1]. Moreover, it is the fourth leading cause of cancer-related death in the United States, with 53,670 new cases and estimated 43,090 deaths in 2017 [2]. In China, the estimated incidence and death of the PC is 90,100 and 79,400, respectively [3]. Surgical resection is only the main curative treatment; in any case, because of late-presenting clinical features, roughly 30 to 40 percent have the locally advanced disease and another 40 percent have a metastatic tumor at the time of diagnosis. Thus, palliative chemotherapy remains the main treatment option for most of these patients [4–6].
Recently, advances in understanding of the molecular pathology of the PC have given hope to new therapeutic approaches; however, according to recently published systematic review, lack of clinical meaningful trials in the past 25 years might be a reason behind the failure to achieve improvements in early diagnosis, management, and prolongation of overall survival of the PC [7].
With growing research, it is now well understood that the PC is a genetic disease, with complex mutation of cancer genes, and the progression of cancer is characterized by high heterogeneity [8–10]. In earlier studies, KRAS, TP53, CDKN2A, and SMAD4 have been identified as recurrently mutated genes in PC [11, 12]. These discoveries have enhanced our understanding of the molecular pathology of the PC. Recently, genomic analyses have identified different molecular subtypes of PC based on the expression of transcriptional profiles and the structural variations [8, 13–15]. Thus, it’s very crucial for early detection of mutant genes to identify PC and its subtypes, for effective management strategy of the disease. In the past decades, numerous studies have shown the potential clinical utility of liquid biopsy, such as circulating tumor cells (CTCs), circulating tumor DNA (ctDNA), and circulating tumor exosomes for various cancers, including PC [16–21]. These promising markers serve as a unique approach for early detection, monitoring and managing disease states. In recent years, noninvasive disease monitoring technology has witnessed an extraordinary explosion of research in the field of liquid biopsy since circulating cell free DNA (cfDNA) was first revealed in body fluids by Mandel and Metais in 1948 [22]. In this review, we have outlined better understanding of different components of liquid biopsy, especially CTCs, ctDNA and exosomes and their potential clinical utility for PC patients. Moreover, we have also drafted numerous of the rational challenges come across using the liquid biopsy techniques.
CURRENT STATUS IN DIAGNOSIS OF PC
Recently, studies have suggested that pancreatic cancer takes over 17 years to develop, from initiation of mutation in the gene to metastatic cancer, trailed by death roughly after 2.7 years [6, 23, 24]; therefore, if diagnosed early; it may increase overall survival dramatically, and thus, provide a window of opportunity for early detection. Currently, there is no official PC screening program, a confinement of screening for early PC is the absence of sensitive and specific markers [25]. Most commonly used blood-based tumor biomarkers in clinical practice are carbohydrate antigen (CA) 19-9 and carcinoembryonic antigen (CEA). Besides, CA19-9 is the only one currently recommended for clinical use by the NCCN guidelines for PC [26]. According to a recent meta-analysis, CA 19-9 has satisfying pooled specificity while the poor pooled sensitivity for differentiating benign from malignant pancreatic tumors, the pooled sensitivity and specificity were 0.47 (95% CI: 0.35–0.59), and 0.88 (95% CI: 0.86–0.91), respectively [27]. Additionally, it is not tumor specific and is elevated in many hepatobiliary cancers likewise in benign biliary obstruction [28]. In spite of advances in the molecular pathology of the PC, there is no dependable biomarker, the sensitivity and specificity of these currently used tumor biomarkers are definitely not adequate for the early recognition of PC [29, 30].
At present the diagnosis and staging of the PC to a great extent depends on imaging modalities, including ultrasonography (USG), computed tomography (CT), endoscopic retrograde cholangiopancreatography (ERCP), positron emission tomography (PET), magnetic resonance imaging (MRI), magnetic resonance cholangiopancreatography (MRCP), and endoscopic ultrasonography (EUS) [31–37]. In any case, little metastases are hard to detect regardless of the possibility that blends of these modalities are utilized. What’s more, these modalities require costly equipment and specialists which are more challenging [35].
Additionally, histological diagnosis often requires invasive tests before surgery. Moreover, because of the difficult anatomical position of the pancreas, a biopsy is often guided by EUS. To the date, endoscopic ultrasonography with fineneedle aspiration (EUSFNA) remains the gold standard in the workup of patients with PC for obtaining the biopsy, with the pooled sensitivity and specificity of 86.8% and 95.8%, respectively [38]. However, EUS-FNA requires sedation and is associated with the number of complications such as tumor seeding along the biopsy tract, pancreatitis, hemorrhage, bowel perforation and aspiration [39–42]. In addition, due to the dense desmoplastic reaction in a PC, the majority of the tumor mass is made up of stromal cells instead of the epithelial cancer cells. Thus, giving rise to false negative results, necessitating frequent repetitive biopsies [1, 41, 43]. Hence, the diagnosis is compellingly done on the premise of CA19-9 and radiological features only. In this manner, the current diagnostic paradigm for the diagnosis of PC experiences low diagnostic accuracy.
Consequently, it is urgent to develop new and improved strategies which can address all the above obstacles and identify primary tumors at an early and resectable stage with greater diagnostic sensitivity in vitro, whereas patients with advanced disease must be preoperatively analyzed to dodge surgical impairments and to choose appropriate treatments to enhance the nature of residual life.
MOLECULAR PATHOLOGY OF PC
Genomic analyses of cancer show that there is a complex mutational landscape and genetic stability of cancer cells are compromised, and PC is no exception to this [8, 24]. Additionally, various genetic modifications occur during the development of the PC, including an increase in duplicate chromosomal number, genetic diversification, amplifications and homozygous deletions, recapitulation of clonal expansion, clonal selection, a small subset of driver mutations and loss of heterozygosity with or without duplicate number reduction [6, 24, 44–51]. Besides, KRAS, p16, SMAD4, CDKN2A, and TP53, are most commonly mutated genes in the majority of the PC patients, particular KRAS mutations occur in almost 92% of the PC cases [8, 11, 12, 52]. In recent years, genomic analyses have an emphasis on the recognition of somatic mutations and other genetic alterations to identify different molecular subtypes of PC. As a breakthrough, some recent studies have defined subtypes of PC based on the expression of transcriptional profiles and the structural variations [8, 13–15].
Collisson et al. [13] classified 3 subtypes of PC 1. Classical- Increased expression of adhesion-associated and epithelial genes, e.g. transmembrane protein 45B (TMEM45B), trefoil factor 1 (TFF1) and mucin 13 (MUC13) 2. Quasi-mesenchymal- Increased expression of mesenchyme-associated genes, e.g. Absent in melanoma 2 (AIM2), glycoprotein m6b (GPM6B) and 5’-nucleotidase ecto (NT5E), and 3. Exocrine like- Increased expression of tumor cell-derived digestive enzyme genes, e.g. Islet-derived 1 beta (REG1B), pancreatic lipase-related protein 2 (PNLIPRP2), and cystic fibrosis transmembrane conductance regulator (CFTR).
Likewise, Moffitt et al. [14] proposed 4 subtypes of the PC: 1. Normal stroma (high expression of ACTA2, VIM, and DES) 2. Activated stroma (high expression of ITGAM, CCL13, CCL18, SPARC, WNT2, WNT5A, MMP9, and MMP11) 3. Classical (high expression of genes such as BTNL8, FAM3D, and ATAD4), and 4. Basal-like (activation of genes such as VGLL1, UCA1, and S100A2). However, basal-like and classical subtypes were also seen in both the normal stroma and activated stroma subtypes.
In addition, Waddell et al. [15] classified 4 subtypes PC based on the structural variation in the mutational landscape 1. Stable (less than 50 structural variations) 2. Locally rearranged (at the minimum of 50 somatic events in the tumor) 3. Scattered (50 to 200 structural variations), and 4. Unstable (more than 200 structural variations).
More recently, genomic expression analysis by Bailey et al. also defined 4 subtypes of the PC: 1. Squamous (TP53 and KDM6A) 2. Pancreatic progenitor (FOXA2/3, PDX1, and MNX1) 3. Immunogenic (upregulated immune networks), and 4. Aberrantly differentiated endocrine exocrine (KRAS, NR5A2, RBPJL, NEUROD1, and NKX2-2) [8].
Nonetheless, the interpretation of this molecular subtyping into the clinical setup has been questioned by conflicting outcomes between these studies. Therapeutic agents that can target these subtypes of the PC and their altered genes or their associated pathways may assume a crucial part in the success of precision medicine for the treatment of the PC [53–56].
LIQUID BIOPSY AS A GAME CHANGER
Recently, analysts at The University of Texas MD Anderson Cancer Center have indicated that PC is ready for investigation with a liquid biopsy [57]. A liquid biopsy is simple and painless, minimally invasive sampling and analysis of liquid biomarkers that can be isolated from body fluids, primarily blood [19, 58]. Moreover, liquid biopsies have turned out to be all the more clinically valuable in recent years due to the ability to pair tests on circulating tumor cells (CTCs), circulating tumor nucleic acids (ctNAs) and tumor-derived exosomes with genomic tests [58–61] (Figure 1A). Interestingly, a liquid biopsy can characterize tumor biomarkers, similar to tissue biopsy, which allows early detection of disease, real-time evaluation of metastasis, treatment monitoring, empowers examination of primary tumors, and metastases. Additionally, it enables evaluation of tumor, heterogeneity, cancer dormancy, and monitoring of tumor progression along with prognosis [58, 60, 62–66]. Recently, liquid biopsy that identifies epidermal growth factor receptor (EGFR) gene mutations in non-small cell lung cancers have been approved by FDA [67, 68].
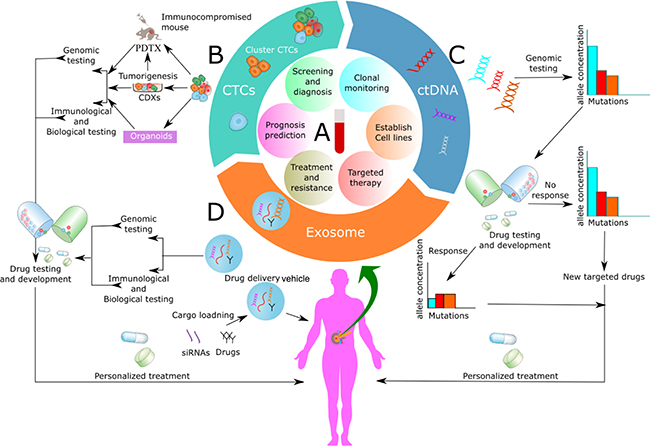
Figure 1: Application of circulating biomarkers. (A) Application of blood-based liquid biopsy analysis over the span of pancreatic cancer management, peripheral venous blood is collected from the patients for isolation of circulating tumor cells (CTCs), circulating tumor DNA (ctDNA) and exosomes. These circulating biomarkers may be applied to guide initial diagnosis, treatment monitoring or planning, prognosis prediction and developing a new targeted therapy for patients with pancreatic cancer. (B) Functional studies with CTCs and development of CTC— derived xeno-grafts (CDXs), patient-derived tumor xeno-graft (PDTX) and 3D organoids model from CTCs for dynamic monitoring of PC and development of new targeted drugs after its molecular characterization and genomic analysis. (C) Clinical application of ctDNA as a tool for therapy monitoring. ctDNA can be obtained from plasma for genomic analysis, drug testing and use in personalized medicine according to the genomic and epigenomic alteration. (D) Clinical use of exosomes for drug development after genomic and immunological testing. Moreover, use of exosome as a drug delivery vehicle where it can be loaded with drugs, siRNAs, gene etc.
CIRCULATING TUMOR CELLS (CTCS)
Circulating tumor cells (CTCs), represent tumor cells that contain a heterogeneous population of cells, including apoptotic tumor and viable tumor cells that have cast off into the circulation or lymphatic vessels from a primary or metastatic tumor and are transported around the body by undergoing phenotypic changes that are accompanied by a process called as epithelial-mesenchymal transition (EMT) [69–73]. Evidence now suggests that the tumors have ability to make their own blood vessels when they reach the size of 1–2 mm3 by inducing angiogenesis or through vasculogenic mimicry, the blood vessels composed of endothelial cells and tumor cells. However, vasculogenic mimicry forms blood vessels without endothelial cells. In fact, they are a mosaic blood vessel which allows substantial shedding of tumor cells into the circulation [74–80]. Likewise, stromal proteins like VEGF and MMP-9 have been known to stimulate angiogenesis in PC [81–83]. Consequently, it has been found that on an average metastatic cancer patient has in the vicinity of 5 and 50 CTCs for approximately every 7.5 ml of blood [84–87]. Nonetheless, the number of CTCs varies between tumor types [88]. CTCs are found in both peripheral blood and bone marrow; additionally, CTCs in the bone marrow are generally called as disseminated tumor cells (DTCs) [89–91]. Further, understanding the CTCs biological properties have demonstrated that the CTCs are involved in the distant organ colonization and metastatic spread of cancer [92–96]. What’s more, there has also been growing interest that mobilization of viable tumor cells from the site of primary tumor induced by a therapeutic or diagnostic intervention like surgery, radiation, chemotherapy or tissue biopsy may promote metastasis [97]. To comprehend this process, a team of researcher performed a tumor self-seeding mouse model experiment whereby tumor recurrence intervened by CTCs was examined utilizing human colorectal, melanoma and breast cancer cell lines, and found that tumor-derived cytokines IL-6 and IL-8 act as CTC attractants which were mediated by MMP1, FSCN1, and CXCL1 genes expressed on CTCs to promote infiltration [98]. However, it is not necessarily always associated with metastasis, as only 0.01% of CTCs takes part in metastasis [97, 99, 100]. Moreover, highly sensitive, single cell investigation showed marked heterogeneity of individual CTCs for protein expression and localization, and the CTCs reflected the character of both the primary biopsy and the transformations seen in the metastatic sites [101, 102], which corresponds with the evidence of “seed and soil” hypothesis [103–105].
Amassing proof has demonstrated that CTCs can be utilized as a biomarker to non-invasively supervise cancer progression and provide direction to monitor the treatment [106–108]. However, the American Society of Clinical Oncology (ASCO) fails to recommend the use of CTCs as a tumor marker for breast cancer, in the lack of strong evidence and conflicts of opinion between the experts [109, 110].
Nevertheless, CTCs detection, identification, enumeration, and molecular characterization are very challenging. Since, CTCs are uncommon in peripheral blood of patients (that is, 1–100 CTCs among billions of normal cells) with a half-life between 1 to 2.4 hours and due to its fragile nature, it tends to degrade when collected in standard blood collecting tubes [111–116].
CTCs being a heterogeneous population of cells, CTCs can be positively or negatively enriched on the basis of 1. Physical characteristics and 2. Immunologic or biological characteristics. Physical features include; cell diameter >15 μm, nuclear-cytoplasmic ratio >0.8, electric charges, deformability, the hyperchromatic nuclei, sunken thickened nuclear membrane, and nucleus side-shift/large nucleoli/abnormal nuclear division. Whereas, cell surface protein expression, cell surface markers, RNA, and DNA signatures are used Immunologic or biological characteristics [117–123]. The most common immunologic feature of CTCs exhibits, anti-CD45 antibody (for leukocytes), anti-vimentin antibody (mesenchymal marker), anti-CK8/18/19 antibody and the epithelial cell adhesion molecule (EpCAM) (epithelial marker) and contain a nucleus that binds to the nucleic acid dye 4’, 6-doamidino-2-phenylindole (DAPI) [117, 121]. Further, cytokeratin negative (CK-) CTCs are cancer stem cells (CSCs) that undergoes EMT. Moreover, CK-CTCs have a high potential for metastasis, and are the most resistant type of CTCs and importantly express genes associated with cancer [124, 125]. Similarly, CTCs that are undergoing apoptosis are called apoptotic CTCs, that can be detected by Epic Sciences technology that recognizes nuclear fragmentation or cytoplasmic blebbing associated with apoptosis, and measuring the ratio of viable CTCs and apoptotic CTCs may help in treatment monitoring [124, 126, 127].
Physical properties-based technologies such as ISET (isolation by size of epithelial tumor cells), ScreenCell, ApoStream™, density gradient centrifugation are used to detect, capture, and isolate CTCs. However, size-based selection strategies abuse the fact that CTCs are notably larger in size than normal blood cells [128, 129]. Importantly, nevertheless, this strategy probably results in substantial loss of CTCs which neglects the small CTCs that are cytokeratin positive and CD45 negative, and with similar sizes and shapes to white blood cells. Critically, these small CTCs have cancer-specific biomarkers that distinguish them as CTCs. Additionally, small CTCs have been found in dynamic illness and differentiation into small cell carcinomas, which often require an alternative treatment [130]. CTC cluster containing three or more CTCs is characterized as circulating tumor microemboli (CTM) [121, 129]. Moreover, CTM has high metastatic potential compared to single CTC [131–133]. Lately, researchers involved in an animal experimental model found that a thrombolytic agent like urokinase can prevent the formation of CTM and further prolong overall survival approximately by 20% compared to control. Additionally, they also concluded that CTM mobilizes at a slower rate than the single CTC due to vessel wall adhesion [133].
In the recent years, several platforms have been established for segregation of CTCs that consider both positive and negative enrichment based on physical and immunological features consolidated within the same device [112, 118, 128, 129, 134–147]. A complete outline of these strategies is beyond the scope of this article. Despite these many platforms, CellSearch™ remains only the gold standard and approved by FDA for all the CTCs detection strategies [148–150]. In particular, this CellSearch™ strategy is dependent on the expression of epithelial markers by the CTCs, more specifically the Epithelial Cell Adhesion Molecule (EpCAM) [150, 151]. On the contrary, with respect to recent finding EpCAM based strategy fails to detect CTCs with low EpCAM expression and CK-CTCs, as CTCs tend to lose their epithelial antigens by the EMT process [152–156]. In addition, it has also been recently revealed that EpCAM- negative CTCs are highly aggressive and invasive [154, 157]. Since CellSearch™ method is based on the idea that CTCs do not express the leukocyte antigen CD45, this method also neglects the fact that CTCs can directly attach to platelets and immune cells and thus accounts to be CD45-positive, which further evade immune surveillance that results in clonal expansion and metastatic [158]. In this manner, the CellSearch™ method may underestimate those CTCs that are highly aggressive and invasive [154].
Following detection and isolation of CTCs, the harvested tumor cells are studied for its genetic and biological features. Various molecular techniques, such as immunocytochemistry (ICC), fluorescence in situ hybridization (FISH), immunophenotyping, microarray, quantitative reverse transcription-polymerase chain reaction (qRT-PCR), droplet digital PCR (ddPCR), co-amplification at lower denaturation temperature-PCR (COLD-PCR), next-generation sequencing (NGS), beads, emulsion, amplification and magnetic (BEAMing), and whole genome amplification, to mention but a few have been commonly performed [159–170]. Despite advancement in these molecular techniques, a genetic study of CTCs still faces challenges of sensitivity and specificity. In particular, digital PCR-based technology is able to screen genetic variations at a very low frequency of 0.01% [171]. However, it only permits monitoring of known mutations and limited numbers of foci [172–175]. Bearing in mind, tumor cells change their mutation under the pressure of therapy or in the midst of tumor sub-clones; therefore, digital PCR-based technology may miss important information during the monitoring process. In addition to this, DNA sequencing of single CTCs for whole genome analysis requires obtaining adequate amounts of DNA and requires institutionalization [176–178]. Moreover, for RNA sequencing strategies larger blood volume is required to obtain an adequate amount of CTCs. What’s more, CTCs needs to be captured rapidly in order to avoid RNA degradation. Hence, it’s not suitable for cancer screening at the moment [163].
To enhance sensitivity and specificity despite the heterogeneity of CTCs, innovative strategies have to be developed, that can consolidate physical, immunological and genetic analyses together to ease the detection, isolation, enrichment and molecular characterization of CTCs [179].
Potential clinical utility and research model of CTCs in PC
Currently, the clinical avail of CTCs analysis remains debatable in the PC. To date, numerous analysts have attempted to identify CTCs in patients with PC and have shown its potential clinical utility utilizing different methodologies and with varying results (Supplementary Table 1) [84, 180–190]. Notably, some studies showed that CellSearch™ has a lower CTCs detection rate for PC patients with the sensitivity and specificity of 55.5 % and 100 %, respectively. Additionally, these studies also revealed that CTCs could only be found in malignant pancreatic tumor and CTCs positive patients have a significantly shorter overall survival. However, CTCs detected in these studies failed to correspond with tumor stage [84, 180, 182, 190]. Interestingly, in a study by Zhou et al. proposed that the integrated identification of c-Met, h-TERT, CK20, and CEA could be used as an indicator for CTCs in the circulation of a PC patient, which can be detected by combined use of immunomagnetic separation and RT-PCR, and thus, improving the specificity and sensitivity to 100%. Moreover, the positive expression of C-MET, CK20, and CEA was found to be closely correlated with tumor stage [189].
Ankeny et al. found that the numbers of CTCs detected from PC was able to differentiate different stages of disease as a useful biomarker and showed 100% similarity for KRAS mutation subtype between primary tumor and CTCs [187]. A meta-analysis comprising 623 patients with different stages of a PC revealed that the patients with positive CTCs had poor progress free survival (PFS) (HR=1.89, 95 %; CI=0.88–2.08, P<0.001) and overall survival (HR=1.23, 95 % CI=1.25–4.00, P<0.001) than those with the CTC-negative patients, suggesting CTCs may be a promising biomarker for the diagnosis and prognosis of a PC [191].
In a study by Yu et al., CTCs was isolated by the HbCTC-Chip microfluidic device from genetically engineered KPC mice and CTCs were subjected to single molecule RNA sequencing, they identified overexpression of the WNT2 gene in CTCs, which prevent anoikis, anchorage-independent sphere formation, and surge metastatic tendency in vivo. These findings were supported in CTCs investigated from 5 of 11 patients with PC. Thus, this study proposed that the molecular study of CTCs may recognize patient drug targets [192].
In spite of the fact that evidence indicates the abundance of tumor cells in the blood of patients with PC has prognostic value, and that CTC numbers can be used as a biomarker for diagnosis, staging of a PC before treatment, and can be prescient of response to therapy after treatment and, consequently, treatment results. These results must be considered with vigilance; however, because CTC numbers are highly variable between different CTC detection platforms, and are subject to favoritism relating to the variety of detection methods used. Thus, the quantity of CTCs that can be detected is therefore highly dependent on how the platform characterizes a cell as a CTC. Nevertheless, this impediment is likely to be overcome by consolidating different technologies to enhance analysis performance. Apart from the difference in results between CTCs detection platforms, some other hypothesis has been postulated for reasons behind low CTCs in the PC 1. The blood flow in the PC is notably compromised in contrast to that of the normal pancreas [193]. Thus, less number of CTCs shed into the circulation 2. The moderately low CTCs number reported in PC may be a consequence of CTCs sequestration by the liver as blood advances through the portal circulation into the systemic circulation [190, 194].
In contemporary research, the development of cell lines from CTCs is a motivating novel field. Recently, different groups reported CTCs developed cell lines in vitro from patients with breast, mesothelioma, esophageal, bladder, lung, and colon cancer [195–200]. However, some researchers found it to be demanding and reported that CTCderived xenografts (CDXs) foresee therapeutic response conflictingly in many cancers, including the PC [195, 201–204]. Interestingly, this has been confirmed by recent research that pluripotent stem cells cultured in the lab acquire new mutations all the time, especially in TP53 gene [205], and this might be a suggesting reason that why CDXs doesn’t correspond to genetic mutations to that of the primary tumor, suggesting that research must be careful while genetic characterization of CDXs. By contrast, direct inoculation of CTCs into immune-compromised mice has met with appreciable success in lung cancer and showed to mirror the response of chemotherapy in CDXs model to that of donor patient. Additionally, it also exhibited comparable genetic profile between isolated CTCs and CDXs model [206]. CDX models have been developed for breast and prostate cancer [207–209]. More interestingly, Gao and colleagues [210] successfully cultured 3D organoids system from CTCs isolated from patients with advanced prostate cancer, which they aim to recapitulate the different subtypes prostate cancer. Likewise, some group of researchers has successfully isolated CTCs from patient-derived tumor xeno-graft (PDTX) models in some cancers, including the PC, and identified its potential clinical utility [211–213]. Perhaps, a standout amongst the most energizing utilizations of CTCs lines is that CDXs and organoids system model may bolster choice of targeted therapies, tracking cancer genetic and epigenetic modifications, and may evolve as an instrumental device for new drug development (Figure 1B). While initial studies using these models are promising, but it needs to be validated with further researches. The primary limitation of these models might be the selection method used for CTC enrichment. In a separate proof-of-concept study, Yu et al. created a pharmacogenomic (PGx) model to predict treatment response of a PC patient to chemotherapy regimens based on the genetic mutations in CTCs, and found that PGx profiling of CTCs can forecast treatment response, additionally they found clinical advantage for the patients treated with sensitive chemotherapy regimens versus insensitive chemotherapy regimens in regard to progression free (10.4 mo vs. 3.6 mo; P < 0.0001; HR, 0.14) and overall survival (17.2 mo vs. 8.3 mo; P < 0.0249; HR, 0.29) [214].
The big question remains: Can these CTCs derived xeno-graft and organoids models give a mirror image of a PC?
CIRCULATING TUMOR NUCLEIC ACIDS (CTNAS)
Circulating tumor nucleic acids (ctNAs) composed of circulating tumor DNA (ctDNA), mRNA and microRNA (miRNA) that are released and circulate in the blood of cancer patients, and changes in the levels of ctNAs in the circulation have been associated with tumor burden, tumor stage, vascularity, cellular turnover, response to therapy, and metastasis [17, 215–217]. At present, the potential clinical utility of cell free RNA (cfRNA) is debatable. A comprehensive discussion of cfRNA is beyond the scope of this article, and this topic has been well documented elsewhere [218]. Moreover, miRNAs are most copious circulating RNA and are also carried in exosomes; thus, this topic has been covered later on with regards to exosomes in this article.
CIRCULATING TUMOR DNA (CTDNA)
It has been postulated that cell free DNA (cfDNA) can originate directly from the viable tumor cells or from CTCs by apoptosis, necrosis, autophagy, micro-environmental stress, mitotic catastrophe, trauma, and treatment procedure [17, 219–226], others include viruses, such as EBV, HPV and hepatitis B virus [227–229]. Moreover, cfDNA is regarded as ‘circulating tumor DNA’ (ctDNA) after mutations in cfDNA in cancer; hence, information regarding the origination and release of ctDNA may provide insight to clinicians about their possible involvement and nature of the disease. Of these, many studies have shown that ctDNA conveys genomic and epigenomic modifications indistinguishable to those of tumor cells [230]. Studies have demonstrated that cfDNA is cleared from the circulation by means of nuclease activity and renal clearance [231–233]. Additionally, some cfDNA that are taken up by the liver and spleen are degraded by macrophages [234, 235]. Studies in both human and mice have shown that most of the apoptotic cfDNA fragments are measured in the vicinity of 166 and 200 base pairs (bp) with an observed half-life of 16 minutes to 2.5 hours [216, 236–241]. In contrast, necrosis creates higher molecular weight DNA fragments of over 10,000 bp in size due to an inadequate and irregular absorption of genomic DNA [242–245]. However, current isolation strategies are unable to capture long DNA fragments [246]. Indeed, the length of the cfDNA might be clinically valuable, utilized as a surrogate for identification of tumors as ctDNA released from necrosis represents malignant tumor’s origin, and increased in the DNA integrity index (ratio of longer fragments to shorter DNA) are seen in most of the malignant tumors [247–249]. However, there are clashing reports in the literature about the origin and composition of ctDNA, a few reports have shown that ctDNAs are shorter than that of apoptotic cfDNAs [238, 250, 251], measuring between 134 and 144 bp [250]. Withal, not all cfDNA originates from cell death; viable cells also release cfDNA as a part of homeostasis [221, 223, 252, 253]. In addition to this, it has also been seen that the activation of lymphocytes can result in the release of large numbers of cfDNA in the absence of apoptosis or necrosis [222, 252, 254]. Moreover, It has been suggested that cfDNA act as a ligand for Toll-like receptor 9 (TLR9) that may inhibit pro-apoptotic caspases by virtue of TLR9-dependent signaling [255]. This signifies a possible immunomodulatory function for cfDNA. These days cfDNA remains to be a hot topic and is widely used for a wide range of research and clinical purposes, including tumor genotyping, early cancer detection, patient prognosis, minimal residual disease monitoring, therapy evaluation, a biomarker in transplant surgery for graft injury, and prediction of allograft rejection [58, 256–268]. In recent years, multiple studies have demonstrated that patients with invasive tumors such as lung, breast, pancreas, colon, hepatocellular, ovarian, prostate, esophageal, and melanoma generally have a high level of ctDNA in their plasma than in healthy individuals [269–274]. Several genomic studies of tumor mutations have analyzed ctDNA to quantify the tumor burden and to detect therapeutic resistance conferring mutations [216, 275–277]. Moreover, a correlation has been set up between the levels of non-mutated cfDNA and mutated cfDNA in circulation and the tumor stage [278, 279]. In addition to this, some studies have also found that mutated cfDNA can lead to therapeutic resistance in cancer several months prior to detection of the tumor by imaging, helping clinicians for therapy evaluation [275, 280, 281].
The study of ctDNA in the plasma basically involves quantification of ctDNA in the circulation using various measurements, for instance mutant allele fraction or mutant allele concentration (that is, copies per milliliter) to estimate disease burden and the detection of genetic aberrations such as somatic mutations, allelic imbalances, genetic polymorphisms, microsatellite alterations, loss of heterozygosity, and methylation [17, 256, 282–288].
There are various methods and technologies used for quantitative and qualitative analysis of ctDNAs, commonly used platforms to name a few are digital PCR (dPCR) [289], droplet digital PCR (ddPCR) [290], BEAMing [291, 292], cancer personalized profiling by deep sequencing (CAPP-Seq) [293], tagged amplicon deep sequencing (TAM-Seq) [276], safe-sequencing (Safe-Seq) [294], duplex sequencing [295], integrated digital error suppression (iDES)-enhanced CAPP-Seq [296], whole-genome sequencing (WGS) [297, 298] and next-generation sequencing (NGS) [299]. Among these NGS holds great expectation for future of genomic analysis.
Regardless of these wide ranges of technologies, extraction, and analysis of ctDNA still face many challenges that need to be addressed before its regular use in a clinical setup. The major challenges are 1. Contamination of ctDNA with a large amount of wild-type cfDNA which are released from lysis of WBC of stored blood in EDTA tubes [300]. Hence, it has been proposed for the utilization of commercially available cell stabilization tubes to prevent or delay the lysis of WBC thereby decreasing the dilution impact of the ctDNA [301]. Additionally, a collection of blood at room temperature and shouldn’t freeze more than 2 hours before extracting the plasma for ctDNA analysis, avoid of use heparinised tubes, extraction of ctDNA from plasma rather than serum and a double centrifugation step to remove more cellular debris preceding DNA extraction [300–302]. 2. Low sensitivity and specificity for analysis of ctDNA [303, 304], this could be enhanced by the combination of advanced genomic approaches that have higher sensitivity to identify all ctDNA in the sample, even with small amounts of input material it has been found that multiplexed patient-specific panels in combination with targeted sequencing methods can improve the sensitivity [296, 304, 305]. 3. The expenses of NGS for liquid biopsies are high and the requirement for repeated liquid biopsies for longitudinal study may constrain its use among a substantial group of patients. The quick question emerges: Can patients bear the cost of advanced genomic approaches?
Potential clinical utility and research model of ctDNA in PC
ctDNA based liquid biopsy brings to the clinic the valuable strength for the success of targeted therapy and precision medicine. Straightaway, understanding of molecular landscapes of PC is vital to guide treatment decisions in clinical practice and with regards to clinical trials. Summary of selected studies using ctDNA for diagnosis, staging, and treatment of pancreatic cancer has been outlined in Table 1 [299, 306–312]. It was Shapiro et al. who first reported the presence of ctDNA in PC and exhibited that ctDNA is markedly elevated in PC patients contrasted with healthy controls, and further concluded that ctDNA may serve as a useful diagnostic and prognostic biomarker [269]. Moreover, PC patients with noticeable ctDNA have been appearing to have worse survival and advanced disease stage [313]. In addition to this, ctDNA can be used for real-time monitoring of tumor dynamics, because of its short half-life it can present with the true picture of tumor burden in response to different therapy [216]. Sausen et al. demonstrated that the measurement of ctDNA can be used to predict relapse and poor outcome after curative surgery, and recurrence could be detected 6.5 months before radiographic imaging [310]. Moreover, in a recent study, ctDNA was found to be an independent prognostic marker in advanced PC, and the presence of ctDNA was associated with a shorter disease-free survival (4.6 vs.17.6 months) and overall survival (19.3 vs. 32.2 months) after surgery in patients with resectable PC [299]. In a separate study, Tjensvoll et al. noted that during chemotherapy of PC patients, changes in mutant KRAS gene level in the circulation corresponded with radiological imaging data and CA19-9 level; moreover, they proposed the utility of ctDNA for monitoring treatment efficacy and tumor progression [312]. These studies demonstrate the potential clinical utility of ctDNA as a prognostic biomarker in PC and further its benefit in monitoring minimal residual disease.
Table 1: Studies of circulating tumor DNA (ctDNA) in pancreatic cancer
Study |
Pts (N) |
Stages |
Controls |
Time of Analysis |
Platform |
Markers |
Findings |
Sensitivity and Specificity |
|---|---|---|---|---|---|---|---|---|
Zill et al. 2015 [309] |
18 |
Advanced pancreatobiliary cancers |
8 biliary cancer |
Post-treatment |
NGS |
KRAS, TP53, APC, SMAD4, and FBXW7 |
Mutations were detected in 90.3% of cfDNA. The diagnostic accuracy of cfDNA sequencing was 97.7%, changes in cfDNA coordinated with tumor marker dynamics. |
92.3% and 100% |
Cheng et al. 2017 [308] |
188 |
Metastatic PC |
NA |
Pre-treatment |
NGS and ddPCR |
KRAS, BRCA2, EGFR and KDR |
The KRAS mutation was detected in 72.3% (136/188) patients. The detection of ctDNA and response to treatment as assessed by CT imaging was 76.9%, the presence of ctDNA provided the earliest measure of treatment in 60% patients. |
NA |
Berger et al. 2016 [307] |
24 |
Metastatic PC |
21 IPMN, 38 healthy controls, 26 patients with resected SCAs and 16 borderline IPMN |
NA |
ddPCR |
KRAS |
The KRAS mutation was detected in 41.7% (10/24) patients. KRAS mutation was not detected in cfDNA of controls, SCA, and IPMN. |
NA |
Sausen et al. 2015 [310] |
77 |
stage II |
NA |
Pre-treatment and Post-treatment |
NGS ddPCR |
NA |
ctDNA was detected in the 43% of patients with localized disease at diagnosis, and detection of ctDNA after resection predicts clinical relapse and poor prognosis. Moreover, ctDNA could detect recurrence 6.5 months earlier than with CT imaging. |
NA |
Henriksen et al. 2016 [311] |
95 |
NA |
27 without evidence of malignancy, 97 CP and 59 AP |
NA |
EasyMag platform, PCR |
BMP3, RASSF1A, BNC1, MESTv2, TFPI2, APC, SFRP1 and SFRP2 |
The distinction in mean number of methylated genes in the PC group (8.41 (95% CI 7.62–9.20)) versus the aggregate control group (4.74 (95% CI 4.40-5.08)) was highly significant (p < 0.001). Additionally, a diagnostic prediction model (age > 65, BMP3, RASSF1A, BNC1, MESTv2, TFPI2, APC, SFRP1 and SFRP2) had an area under the curve of 0.86 (sensitivity 76%, specificity 83%). |
NA |
Tjensvoll et al. 2016 [312] |
14 |
All stages |
29 healthy individuals |
Pre-treatment and Post-treatment |
PNA-clamp PCR |
KRAS |
KRAS mutation was detected in 71% of patients with PC. The pre-therapy ctDNA was a predictor of both progression-free and OS. Changes in ctDNA levels corresponded both with radiological follow-up data and CA19-9 levels. |
NA |
Maire et al. 2002 [306] |
47 |
NA |
31 CP |
Pre-treatment - |
PCR and allele-specific amplification |
KRAS2 |
KRAS2 mutation was detected in 22 patients (47%) with PC and in 4 controls with CP (13%) (P < 0.002). The combination of KRAS2 and CA19.9 gave a sensitivity and specificity of 98% and 77% respectively. |
47% and 87% |
Pietrasz et al. 2017 [299] |
135 |
All stages |
NA |
Pre-treatment and Post-treatment |
NGS |
NA |
ctDNA was detected in 48% of patients with advanced PC, and ctDNA emerges as an independent prognostic marker in advanced PC and indicator of shorter DFS and shorter OS when detected after surgery. |
NA |
PC: Pancreatic cancer; cfDNA: Cell-free DNA; PCR: Polymerase chain reaction; ddPCR: Droplet digital PCR; NGS: Next-generation sequencing; PNA-clamp PCR: Peptide nucleic acid clamping PCR; IPMN: Intraductal papillary mucinous neoplasm; SCAs: Serous cystadenomas; DFS: Disease-free survival OS: Overall survival; CP: Chronic pancreatitis; AP: Acute pancreatitis; CT: Computed tomography; ctDNA: Circulating tumor DNA; NA: Not available.
Since, it is known that over 90% of mutation in PC contains mutated KRAS gene and is considered to be an early event during carcinogenesis [8, 52, 314–316]. Moreover, the mutation occurs most commonly in codon G12D, G12V, and G12R [314, 317, 318]. G12V mutation is significantly associated with shorter survival contrasted to G12D, G12R, and wild-type [319]. Thus, mutated KRAS gene has been a center of surveillance for definitive diagnosis of PC. Discouragingly, chronic pancreatitis also shows the KRAS mutation in 10 to 15% of the cases in cfDNA, and to increase the sensitivity and specificity for diagnosis of PC it has additionally been proposed that combining KRAS mutation and serum CA19-9 level can enhance a sensitivity and specificity by 98% and 77%, respectively [306]. Besides, later on, some studies have shown that methylation analysis of DNA can differentiate PC from chronic pancreatitis and could be used as a potential diagnostic marker for PC [311, 320, 321]. Additionally, it has been found that ctDNA methylation analysis can also detect epigenetic alterations in different cancers, including PC that involves in tumor progression and metastasis [322–325]. Besides, these epigenetic alterations are strongly associated with patient survival [326]. Interestingly, recently it has been proposed that epigenetic alterations in a gene can be reprogrammed genetically or with a pharmacological inhibitor to reverse the epigenetic variations and inhibit their tumor-forming capacity; thus, helping in a finding of attractive therapeutic targets [327]. To the point, the essential question arises: Can ctDNA methylation analysis detect all the epigenetic alterations?
In addition to this, methylation of ctDNA has been found to conceal tissue and cell specific information that may be invaluable in cancer patients to find tissue-of-origin [251, 328], such as in the case of cancers of unknown origin. Of note, methylation analysis of ctDNA is found to be useful in determining the primary location of cancer with a specificity and sensitivity of 99.6% and 97.7%, respectively [329].
Apart from KRAS mutation, in recent years with the development of NGS, increasingly pertinent genetic aberrations have been identified, namely oncogenic BRAF V600E mutation that are observed 3% of PC patients, and that do not acquire a KRAS mutation; similarly, amplification of the MYC oncogene which is remarkably associated with poor prognosis [318]. Moreover, detection of mutations such as RBM10, MLL, MLL2, MLL3, and ARID1A is associated with longer survival [310, 318]. These studies furnish genetic indicators of prognosis and outcome in PC and have suggestions for a new period of therapeutic development (Figure 1C).
In a separate study carried by Zill et al. has demonstrated that tumor sequencing was failed in 35% cases of tissue biopsy due to inadequate a tissue sample, in addition to this all mutations were detected in ctDNA similar to that of tissue biopsy. Moreover, they proposed that ctDNA could correlate well with tumor marker dynamics in longitudinal monitoring with a diagnostic accuracy of 97.7%, and with sensitivity and specificity of 92.3% and 100%, respectively [309].
Intra- and inter-tumor heterogeneity contribute to the development of drug-resistant tumors and failure of treatment [330]. A small genetic clone carrying a drug-resistant mutation within the tumor can extend after the pressure treatment. These genetic clones can be missed by tissue biopsies due to low prevalence or the spatial partition of cells within the tumor [331]. Interestingly, ctDNA can be exploited to monitor dynamic clonal and subclonal evolution in response to the pressure of therapy [332] (Figure 2).
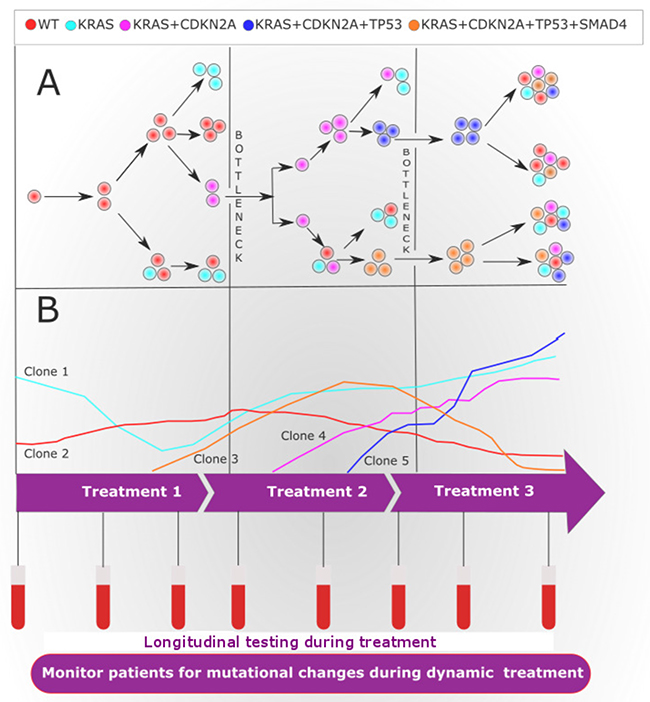
Figure 2: Tumor heterogeneity and clonal evolution during treatment. (A) Diagram showing the evolutional clonal architecture in pancreatic cancer (PC) at diagnosis and relapse. Of note, at diagnosis, the clonal and subclonal diversity evolved from a common ancestral tumor stem cell. The clonal evolution may follow linear or branched evolution, however, branched evolution is probably more likely to contribute to tumor heterogeneity. Additionally, drug treatment instigates a bottleneck effect, where resistant subclones will survive and proliferate to form a heterogeneous tumor. (B) During systemic successive targeted therapy assessed by longitudinal liquid biopsies may identify an actionable genetic alteration, therapy response or progression. In the event that progression is identified, the clinician may be able to switch treatment to target arising clones that carry additional mutations that were identified by the ctDNA analysis. At the start of targeted therapy, all cells in the patient’s with PC have actionable genetic mutations (clone 1). The administration of treatment 1 targets the clone 1. longitudinal liquid biopsy analysis demonstrates an initial decrease in the clone 1 during treatment 1, yet uncovers the evolution of new clone (clone 2 and clone 3) causing resistance to treatment 1. The clone 2 and clone 3 can be targeted with treatment 2, where longitudinal liquid biopsy analysis uncovers a decrease in the frequency of resistance clone 2 and clone 3, during this time, however, other genetic alterations clone 4 and clone 5 increases in frequency. These clones 4 and 5 are resistant to treatment 2, yet is sensitive to treatment 3. During treatment 3, the frequency of the clone 4 and clone 5 decreases, while residual earlier resistant clones may persist to give rise to therapeutic resistance.
EXOSOMES
Exosomes are very stable, small cup-shaped, lipid bilayer microvesicles of endocytic origin with a size of 50–150 nm in diameter and density of 1.12–1.19 g/ml [60, 333, 334]. These microvesicles are discharged by all cells, including tumor cells, and originally thought to be that they are like cellular garbage bags [335–337]. However, recent research suggests that exosomes are involved in many physiological and pathological functions and processes such as intracellular communication, inflammation, cell proliferation and regeneration following injury, immune response, lactation, neuronal function, immunothrombosis, diabetes, atherosclerosis, development, and progression of liver disease, neurodegenerative diseases and more recently in cancer [338–352]. Evidence demonstrates that exosomes are available in numerous biologic body fluids; exosomes may in this way be viewed as open indicative biomarkers that hold incredible potential for recognition of numerous disease conditions, including cancer [353, 354]. Interestingly, exosomes are enriched with DNA, proteins, lipids, RNAs, and metabolites that are reflective of the cell types of origin [352, 355–357]. Nonetheless, whole RNA sequence can’t be bundled inside one exosome, because of its small size, which was contrasted and retrovirus particles of a comparable size that can just pack 10 kb transcriptome, subsequently, single exosome conveys just a predetermined number of transcripts [358]. Still, exosomes are remarkably abundant in plasma and when segregating the vesicle portion, the vast majority of the RNA sequence can be identified [359, 360]. Recent data from various cell type uncovers that exosomes contain 4,563 proteins, 194 lipids, 1639 mRNA and 764 microRNA [361]. Among the main 20 regularly recognized exosomal proteins, a significant number of the proteins, including CD9, ACTB, CD63, CD81, HSPA8, PKM2, ANXA2, HSP90A1, SDCBP, YWHAE, LDHA, MSN, PDCP6IP, ANXA5, FASN, ACTN4, LDHB, ANXA1, HSPA1A, and YWHA are known to be mutated in multiple cancer types [362]. Additionally, it has been found that some exosomes reveal major histocompatibility complex MHC I and MHC II on their surface, suggesting that they are derived from antigen-presenting cells and might have a regulatory immunological part in cancer biology [363, 364]. Considering, exosomes convey genomic and proteomic materials, thus it has been hypothesized that exosomes secreted by tumor cells take part in the tumor growth, invasion, pre-metastatic niches (PMNs) and metastasis through intracellular communication and escape from immunosurveillance [338, 365–373]. Biogenesis and secretion of exosome within the cell is a complex process which requires different factors like molecular motors (cytoskeleton- kinesins and myosins, polymerisation- actin, dynamin, and microtubules), molecular switches (GTPases, annexins, and flotillin) and the fusion proteins (SNARE proteins and tethering factors), cargo sorting proteins complex (ALIX and TSG101) and finally exocytotic released is promoted by cellular stress or hypoxia in cancer cells [333, 374–377]. Studies from Thery and colleagues reveal that Rab27a and Rab27b act as key downregulators of the exosome secretion pathway, which inhibit secretion of exosomes [378]. These Rab family proteins are thought to be involved in cancer progression and tumor advancement, which provided clues that exosomes have something to do with tumor biology [379]. Moreover, p53 protein, and a p53-regulated gene, TSAP6, have shown to increase production of exosome [380, 381]. Emerging evidence suggested that breast and pancreatic tumor-derived exosomes express integrins (ITGs) on their surface which direct organ-specific colonization by fusing with targeted stromal and cancer cells, thereby forming PMNs within the cancer microenvironment to transmit signals and their cargo, that includes genetic material (that is, DNA, mRNA, and miRNA), metabolites and proteins, by that determining organotropic metastasis [338, 366–368, 373, 382–384]. Subsequently, PMNs requires S100 family proteins for homing of tumor-derived exosomes in targeted organs [373]. Furthermore, tumor-derived exosomal miRNA and protein have a tendency to reprogram and instruct target cells that it fuse with towards pro-inflammatory and pro-metastatic phenotype leading to metastasis [382, 385].
Till the date, numerous of technologies and methods have been used for extraction of exosomes from body fluids which have been well documented elsewhere [386]. Commonly used methods are ultracentrifugation-based isolation, precipitation-based isolation, size-based isolation, immunoaffinity-based isolation, and microfluidics-based isolation [386]. The segregation of tumor-derived exosomes from patients; however, remains challenging due to some of the reasons: 1. Lack of a standardized method for segregation and the absence of specific markers that can differentiate tumor-derived and non-tumor derived exosomes [387]. 2. Failure in isolating large concentration of exosomes, due to contamination from other extracellular vesicles and cellular debris [386]. 3. Time-consuming technology that is hard to implement in routine clinical setup [386].
To overcome these challenges, an institutionalized technique for exosome isolation should be developed sooner rather than later, thus amplifying the significance of research facility based investigations of exosomes in the clinical setting. It is important to reliably approve each of these strategies as per meticulous definitions of exosomes, those laid out by the International Society for Extracellular Vesicles [388].
Potential clinical utility and research model of exosomes in PC
As the content of exosomes is cell-type specific with an extensive variety of molecular information carried forth from parent cells to secondary cells, exosomes may provide an idiosyncratic ‘signature’ of tumor development and metastatic progression, as well as the metabolic status of the tumor. In spite of the fact, that the mechanism of packaging is yet to be completely comprehended, it has been seen that the metastatic tumor cells show the high ability of packing and cargo secretion (that is, protein, RNA, DNA, and metabolites) in exosome [382, 385]. To date, numerous studies have outlined clinical utility of exosomes as a diagnostic, prognostic and therapeutic tool in PC patients (Table 2) [387, 389–393].
Table 2: Studies of circulating tumor exosomes in pancreatic cancer
Study |
Specimen type |
Platform |
Markers |
Findings and Conclusion |
|---|---|---|---|---|
Que et al. 2013 [389] |
Serum |
Filtration, Ultracentrifugation, and RT-PCR |
miR-17-5p, miR-21, miR-155, and miR-196a |
There were low expressions of exosomal miR-155 and miR-196a in PC patients. Moreover, there were high expressions of serum exosomal miR-17-5p and miR-21 in PC patients than control groups and high expression of miR-17-5p was significantly correlated with advanced stage of PC. |
Kahlert et al. 2014 [390] |
Serum |
Filtration, Ultracentrifugation and WGS |
KRAS, p53 |
Exosomes from PC patients contain >10-kb fragments of double-stranded genomic DNA with detectable mutations in KRAS and p53. In addition, WGS of exosomal DNA can determine genomic DNA mutations for cancer prediction, treatment, and therapy resistance. |
Madhavan et al. 2015 [391] |
Serum |
Ultracentrifugation, qRT-PCR, and Flow cytometry |
CD44v6, Tspan8, EpCAM, c-Met, CD104, miR-1246, miR-4644, miR-3976, and miR-4306 |
Serum exosomal miR-1246, miR-4644, miR-3976 and miR-4306 were significantly upregulated in 83% of PC serum-exosomes, but rarely in control groups. Additionally, It was found that a combination of five proteins (CD44v6, Tspan8, EpCAM, MET and CD104) and four miRNAs (miR-1246, miR-4644, miR-3976 and miR-4306) in circulating tumor exosomes could recognize PC from healthy control, chronic pancreatitis and benign pancreatic disease with a sensitivity and specificity of 100% and 80% respectively. |
Melo et al. 2015 [387] |
Serum |
Filtration, Ultracentrifugation, qRT-PCR And Mass spectrometry analyses |
Glypican-1 |
Expression of glypican 1 (GPC1) a membrane-bound protein on circulating exosomes of mice and humans with PC can differentiate healthy control and patients with a benign pancreatic disease. Notably, GPC1+ exosomes level correlated with tumor burden and the survival of PC patients before and after the surgery with utter sensitivity and specificity. Additionally, circulating GPC1+ exosomes of PC patients bear KRAS mutations, |
Kanwar et al. 2014 [392] |
Serum |
ExoChip (antigen based) |
CD63 |
Significantly higher exosome capture in PC patients, compared to controls. |
Allenson et al. 2017 [393] |
Whole blood |
Ultracentrifugation, Flow cytometry, and ddPCR |
KRAS |
Exosomal DNA posses KRAS mutations and was detected localized, locally advanced, and metastatic PC patients, respectively. Higher exosomal DNA KRAS mutations were associated with decreased disease-free survival in patients with localized disease. |
PC: pancreatic cancer; GPC1: glypican-1; miR: microRNA; ddPCR: Droplet digital polymerase chain reaction; qRT-PCR: Quantitative reverse transcription polymerase chain reaction; WGS: Whole-genome sequencing; EpCAM: Epithelial cellular adhesion molecule; RT-PCR: Reverse transcription polymerase chain reaction.
In a seminal research, Melo et al. [387] demonstrated an increased amount of glypican 1 (GPC1) a membrane-bound protein on circulating exosomes of mice and humans with PC can differentiate healthy control and patients with a benign pancreatic disease. Notably, GPC1+ exosomes level correlated with tumor burden and the survival of PC patients before and after the surgery with utter sensitivity and specificity. Additionally, circulating GPC1+ exosomes of PC patients bear KRAS mutations, and were able to identify pancreatic intraepithelial neoplasia (PanIN) in mice from healthy control even before detectable pancreatic lesion on MRI. Of note, the main limitation of this study was a small sample size. Undoubtedly, these findings should be verified with a larger series of the sample, but the striking evidence provided by Melo and colleagues suggest that GPC1+ exosomes may serve as a potential diagnostic and screening biomarker to detect early stages of PC for possible curative surgery. In earlier studies, overexpression of surviving [394], and mislocalization of plectin [395] in exosomes were also proposed as biomarkers for PC. Moreover, it has also recently been found that a higher rate of patients with localized PC showed noticeable KRAS mutations in exosomal DNA than previously revealed for cfDNA, and thus exosomal DNA may act as a complementary DNA source to liquid biopsy [393]. In a research, Madhavan et al. outlined that a combination of five proteins (CD44v6, Tspan8, EpCAM, MET and CD104) and four miRNAs (miR-1246, miR-4644, miR-3976 and miR-4306) in circulating tumor exosomes could recognize PC from healthy control, chronic pancreatitis, and benign pancreatic disease with a sensitivity and specificity of 100% and 80%, respectively [391].
Exosomal micro-RNAs (miRNAs) have additionally increased generous consideration in later past years. From the recent studies, the number of exosomal miRNAs including miR-21, miR-17-5p, miR-155, miR-34, miR-196a, miR-181a, miR-181b, miR-138-5p, miR-494, miR-542-3p, miR-31, and miR-205 has been identified and upregulation of these miRNAs has been shown to increase cellular proliferation, angiogenesis promotion, disease progression, metastasis, and chemo-resistance in PC patients [389, 396–403]. Moreover, these studies highlight the potential use of exosomal miRNAs as a diagnostic and prognostic biomarker. Likewise, targeting the exosomal miRNAs might be a potential therapy for PC.
Additionally, it has been found that miRNAs in circulating exosomes are representative of those increased in the primary tumor cells [21]. In a separate study, Ohuchida et al. distinguished 24 miRNAs with altered expression in gemcitabine-resistant cells, and furthermore found that patients with high miR-142-5p and miR-204 expression had significantly longer survival times than those with low miR-142-5p and miR-204 expression in the gemcitabine-treated group [404]. Despite the fact that the miRNA levels were determined in paraffin-embedded tissue, this highlights the potential use of tumor-derived exosomal miRNAs as predictors of response to chemotherapy and future use of miRNAs for targeted immune therapy in PC. Moreover, it had been proposed that exosomes miRNAs are derived from living cells, while circulating free miRNAs usually originates from apoptotic or necrotic cells [398], and thus, exosomes miRNAs might have advantages over circulating free miRNAs for monitoring therapy or late stage of PC.
Lyden and colleagues [373] in their recent paper proposed that tumor-derived exosomes integrins (ITGs) can determine organotropic metastasis as discussed in the earlier section of this paper. The consequent analysis demonstrated that liver-tropic pancreatic exosomes expressing ITGαvβ5 could communicate with F4/80+ macrophages and fuse with Kupffer cells in fibronectin rich liver niches. Besides, inhibiting ITGβ5 expression through short hairpin RNAs or hindering their binding by HYD-1/RGD peptides particularly reduced exosome uptake and additionally liver metastasis. This study explains why the liver is the most common site for PC metastasis. Moreover, these outcomes showed that exosomal ITGs may be used as organotropic biomarkers to anticipate organ-specific metastasis in PC patients, and expands our understanding of the organ-specific metastasis mechanisms involvement of exosomes in advancing tumor metastasis. In addition to Lyden results, a study by Costa-Silva et al. [385] demonstrated that the PC exosomes can expand liver metastatic burden by transferring macrophage migration inhibitory factor (MIF) to Kupffer cells and by recruiting immune cells to initiate PMNs development in the liver. It is thus proposed that the presence of MIF in exosomes may be a biomarker that can show the likelihood of PC metastasis to the liver and blockage of MIF could prevent liver metastases and may prove to be clinically relevant for the development of new targeted therapies.
The utilization of exosomes as a nucleic acid, gene or drug delivery vehicles (Figure 1D) has increased significant enthusiasm because of their phenomenal biodistribution and biocompatibility [405]. Moreover, the advantage of utilizing exosomes as a drug delivery system lies in the fact that they can be particularly targeted to a specific cell type by engineering exosome-producer cells [406]. Interestingly, past studies have demonstrated that tumor cells secrete more exosomes compared to normal cells [407]. Furthermore, malignant pancreatic tumor cells, with oncogenic RAS have also founded to uptake exosomes more readily through the active induction of macropinocytosis [408, 409], and this could strengthen the use of exosome as an ideal drug delivery vehicle. Recently, Kamerkar et al. demonstrated treatment efficacy of engineered exosome (iExosome), where iExosome was able to suppress the PC progression in genetically engineered KTC and KPC mouse models, this study exhibited an approach for direct and specific targeting of KRAS mutation in tumors using engineered exosomes [410]. However, it still needs to be verified in the clinical setting.
CONCLUSIONS
We have accomplished enormous progress in our understanding of the complex molecular and genetic mechanisms of PC, yet key inquiries stay unanswered for its early diagnosis, staging, treatment monitoring, and management. Taking everything into account, the up and coming era of ‘ liquid biopsy’ will be vital to conclusively build up the clinical relevance of bloodbased genomic profiling. Liquid biopsy methodologies will most likely give enhanced diagnostic and therapeutic outcome. However, a few issues stay to be tackled before application in a clinical setting: 1. Institutionalization of the sample collection methodology in pre-analytical setup, subsequently decreasing pre-analytical errors 2. Institutionalized and strict definition of CTCs, ctDNA, and exosomes for their segregation and analysis is required. 3. Improvement in the sensitivity and specificity of the detection methods by integrating CTCs, ctDNA, and exosomes in one platform. 4. Universal signature from CTCs, ctDNA, and exosomes for differentiating benign from malignant disease and that can cover all phases of cancer along with their subtypes, tumor characteristics, and mutations for the success of precision medicine. 5. Substantial forthcoming clinical trials, including multicenter studies, are expected to approve the clinical essentials for diagnosis, treatment monitoring, and prognosis. The comparison of CTCs, ctDNA, and exosomes is outlined in Table 3.
Table 3: Comparison between CTCs, ctDNA and exosomes as liquid biopsy
Comparison |
CTCs |
ctDNA |
Exosomes |
|---|---|---|---|
Origin |
Includes apoptotic tumor and viable tumor cells from a primary or metastatic tumor [71, 73]. |
Includes cfDNA, from the viable tumor cells or from CTCs [17, 219, 221, 224, 225]. |
Includes DNA, proteins, lipids, RNAs and metabolites and are discharged by all cells including tumor cells [333, 336, 352, 355–357]. |
Bio-banked samples used for study |
Frozen plasma, urine and other biofluids cannot be used for study of CTCs [116]. |
Frozen plasma, urine and other biofluids can be used for study of cfDNA [116, 260]. |
Frozen plasma, urine and other biofluids can be used for study of exosomes [260]. |
Stability |
Stable [116] |
Very stable [334] |
|
Genetic materials |
|||
Analytic Techniques |
CellSearch [84, 148–150, 180, 182, 190], Microfluidic [187, 181], SE-iFISH [186], MetaCell [184], Immunofluorescence [181, 185, 188], ScreenCell [185], ISET Test [188, 190] etc. |
dPCR [289], CAPP-Seq [293], TAM-Seq [276], ddPCR [290, 307, 308, 310], COLD-PCR [167], Safe-Seq [294], NGS [299, 308–310], BEAMing [291, 292], WGA [297, 298] etc. |
Ultracentrifugation [387, 386, 389–391, 393], ExoChip [392], Precipitation [386], Size-based isolation [386], Immunoaffinity-based isolation [386], Microfluidics-based isolation etc [386]. |
Morphological study and functional study of tumor cells ex vivo |
No |
No |
|
Analysis of protein location on tumor cells |
No |
No |
|
Identification of mutations |
|||
Identification of epigenetic changes |
Yes [357] |
||
Analysis of RNA transcription profiles |
No |
||
Proteomics Analysis |
Yes [157] |
No |
|
Analysis tumor heterogeneity |
Yes [206] |
Yes [332] |
No |
Use as drug delivery vehicle |
No |
No |
DNA: Deoxyribonucleic acid; RNA: Ribonucleic acid; CTCs: Circulating tumor cells; ctDNA: Circulating tumor DNA; cfDNA: Cell free DNA; ISET: Size of Epithelial Tumor; SE-iFISH: Immunostaining-fluorescence in situ hybridization; ddPCR: Droplet digital PCR; COLD-PCR: Co-amplifcation at lower denaturation temperature-PCR; NGS: Next-generation sequencing; BEAMing: Beads, Emulsion, Amplifcation and Magnetic; WGA: Whole genome amplifications, messenger RNAs (mRNAs); dPCR: digital PCR; CAPP-Seq: Cancer personalized profiling by deep sequencin; TAM-Seq: Tagged amplicon deep sequencing; Safe-Seq: Safe-sequencing.
In spite of the presence of various challenges, liquid biopsy seems to be ideal diagnostic and therapeutic strategies for PC. So far, in June 2016, a liquid biopsy was approved by the FDA for use in the USA to detect EGFR mutations in plasma ctDNA and entered clinical practice for the management of non-small cell lung cancer (NSCLC) [68]. After its approval, it represents key milestones towards the application of liquid biopsies in personalized clinical oncology.
CTCs seem to have enormous potential for PC, and can be exploited to understand the development of the distant organ colonization and metastatic spread of cancer. Moreover, CTCs can be used to understand the phenotypic changes, plasticity of tumor biology and mutational landscape of tumor by development of PDTX [213], PGx [214], CDXs [206], and 3D organoids [210] models and guide treatment decisions for complex disease like PC [184, 190, 411–413]. However, methylation analysis of CTCs remains largely unexplored, except few studies which have been reported in recent years [414, 415]. Methylation study of CTCs holds a promising future with an exciting result; this may give a new direction to upcoming research.
In the ctDNA arena, ctDNA has offered more an inclusive understanding of a patient’s disease. For instance, the total ctDNA concentration can be used for real-time monitoring of tumor dynamics and predicts relapse, poor outcome and shorter disease-free survival after curative surgery [299, 310, 313]. Methylation analysis of ctDNA can detect epigenetic alterations that involve in tumor progression and metastasis [322, 325]. Moreover, it can also differentiate PC from chronic pancreatitis and could be used as a potential diagnostic marker for PC [320, 321]. However, the sensitivity and specificity of ctDNA analysis are struggling and it can be increased by adopting a multi-marker strategy along with integrating it with other biomarkers.
Exosomes provide an enormous understanding about organ-specific colonization and PMNs [373, 385]. Moreover, exosomes can serve as a potential biomarker as its contents are largely derived from the tumors, which are enriched with DNA, proteins, lipids, RNAs, and metabolites. In addition to this, exosomes as drug delivery vehicles offer an important perspective because of its cell-specific nature, excellent biodistribution and biocompatibility [405]. Before these drug delivery systems become a therapeutic reality, it needs to be validated with further researches and large clinical trials.
In general, a liquid biopsy can possibly be used to diagnose PC at an early stage, predict prognosis, monitor PC stage, therapeutic efficacy or resistance, and provide optimal, personalized treatment strategies for patients with PC. This review has endeavored to organize the present advances in liquid biopsy for PC into a solitary idea to establish an effective management plan and implementation of these understandings to bolster energizing zones of research. But the fundamental question remains: Can liquid biopsy become a screening reality for pancreatic cancer?
CONFLICTS OF INTEREST
Authors declare no competing interests.
FUNDING
This work was supported by grants from - The National High Technology Research and Development Program of China (SS2015AA020405); Training Program of the Key Program of the National Natural Science Foundation of China (91442115); The National Natural Science Foundation of China (No.81672337); Zhejiang Provincial Key Innovation Team of Pancreatic Cancer Diagnosis & Treatment (2013TD06); The Key Program of the National Natural Science Foundation of China (81530079); Key research and development Project of Zhejiang Province (2015C03044) (Sponsored by Zhejiang Provincial Program for the Cultivation of High-level Innovative Health talents); Zhejiang Provincial Key Laboratory of Pancreatic Disease; and National High Technology Research and Development Program of China (2015AA020405).
REFERENCES
1. Court CM, Ankeny JS, Hou S, Tseng HR, Tomlinson JS. Improving pancreatic cancer diagnosis using circulating tumor cells: prospects for staging and single-cell analysis. Expert Rev Mol Diagn. 2015; 15:1491–504. https://doi.org/10.1586/14737159.2015.1091311.
2. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67:7–30. https://doi.org/10.3322/caac.21387.
3. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016; 66:115–32. https://doi.org/10.3322/caac.21338.
4. Assifi MM, Lu X, Eibl G, Reber HA, Li G, Hines OJ. Neoadjuvant therapy in pancreatic adenocarcinoma: a meta-analysis of phase II trials. Surgery. 2011; 150:466–73. https://doi.org/10.1016/j.surg.2011.07.006.
5. Shrikhande SV, Kleeff J, Reiser C, Weitz J, Hinz U, Esposito I, Schmidt J, Friess H, Buchler MW. Pancreatic resection for M1 pancreatic ductal adenocarcinoma. Ann Surg Oncol. 2007; 14:118–27. https://doi.org/10.1245/s10434-006-9131-8.
6. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, Velculescu VE, Kinzler KW, Vogelstein B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010; 467:1114–7. https://doi.org/10.1038/nature09515.
7. Rahib L, Fleshman JM, Matrisian LM, Berlin JD. Evaluation of Pancreatic Cancer Clinical Trials and Benchmarks for Clinically Meaningful Future Trials: A Systematic Review. JAMA Oncol. 2016; 2:1209–16. https://doi.org/10.1001/jamaoncol.2016.0585.
8. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, Nourse C, Murtaugh LC, Harliwong I, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016; 531:47–52. https://doi.org/10.1038/nature16965.
9. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74. https://doi.org/10.1016/j.cell.2011.02.013.
10. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57–70.
11. Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008; 3:157–88. https://doi.org/10.1146/annurev.pathmechdis.3.121806.154305.
12. Mimeault M, Brand RE, Sasson AA, Batra SK. Recent advances on the molecular mechanisms involved in pancreatic cancer progression and therapies. Pancreas. 2005; 31:301–16.
13. Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, Feiler HS, Ko AH, Olshen AB, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011; 17:500–3. https://doi.org/10.1038/nm.2344.
14. Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, Smyla JK, Anderson JM, Kim HJ, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015; 47:1168–78. https://doi.org/10.1038/ng.3398.
15. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, Quinn MC, Robertson AJ, Fadlullah MZ, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015; 518:495–501. https://doi.org/10.1038/nature14169.
16. Alix-Panabieres C, Pantel K. Circulating tumor cells: liquid biopsy of cancer. Clin Chem. 2013; 59:110–8. https://doi.org/10.1373/clinchem.2012.194258.
17. Schwarzenbach H, Hoon DSB, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011; 11:426–37.
18. van de Stolpe A, Pantel K, Sleijfer S, Terstappen LW, den Toonder JM. Circulating tumor cell isolation and diagnostics: toward routine clinical use. Cancer Res. 2011; 71:5955–60. https://doi.org/10.1158/0008-5472.can-11-1254.
19. Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013; 10:472–84. https://doi.org/10.1038/nrclinonc.2013.110.
20. Gold B, Cankovic M, Furtado LV, Meier F, Gocke CD. Do Circulating Tumor Cells, Exosomes, and Circulating Tumor Nucleic Acids Have Clinical Utility?: A Report of the Association for Molecular Pathology. J Mol Diagn. 2015; 17:209–24. https://doi.org/10.1016/j.jmoldx.2015.02.001.
21. Nedaeinia R, Manian M, Jazayeri MH, Ranjbar M, Salehi R, Sharifi M, Mohaghegh F, Goli M, Jahednia SH, Avan A, Ghayour-Mobarhan M. Circulating exosomes and exosomal microRNAs as biomarkers in gastrointestinal cancer. Cancer Gene Ther. 2017; 24:48–56. https://doi.org/10.1038/cgt.2016.77.
22. Anker P, Mulcahy H, Qi Chen X, Stroun M. Detection of Circulating Tumour DNA in the Blood (Plasma/Serum) of Cancer Patients. Cancer Metastasis Rev. 1999; 18:65–73. https://doi.org/10.1023/a:1006260319913.
23. Kim MS, Zhong Y, Yachida S, Rajeshkumar NV, Abel ML, Marimuthu A, Mudgal K, Hruban RH, Poling JS, Tyner JW, Maitra A, Iacobuzio-Donahue CA, Pandey A. Heterogeneity of pancreatic cancer metastases in a single patient revealed by quantitative proteomics. Mol Cell Proteomics. 2014; 13:2803–11. https://doi.org/10.1074/mcp.M114.038547.
24. Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal SA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010; 467:1109–13. https://doi.org/10.1038/nature09460.
25. Lin QJ, Yang F, Jin C, Fu DL. Current status and progress of pancreatic cancer in China. World J Gastroenterol. 2015; 21:7988–8003. https://doi.org/10.3748/wjg.v21.i26.7988.
26. Pancreatic adenocarcinoma guidelines. National Cancer Comprehensive Network. 2017; Version 2.2016.
27. Cao S, Hu Y, Gao X, Liao Q, Zhao Y. Serum Carbohydrate Antigen 19-9 in Differential Diagnosis of Benign and Malignant Pancreatic Cystic Neoplasms: A Meta-Analysis. PLoS One. 2016; 11:e0166406. https://doi.org/10.1371/journal.pone.0166406.
28. Winter JM, Cameron JL, Campbell KA, Arnold MA, Chang DC, Coleman J, Hodgin MB, Sauter PK, Hruban RH, Riall TS, Schulick RD, Choti MA, Lillemoe KD, et al. 1423 pancreaticoduodenectomies for pancreatic cancer: A single-institution experience. J Gastrointest Surg. 2006; 10:1199–210. https://doi.org/10.1016/j.gassur.2006.08.018.
29. Kim JE, Lee KT, Lee JK, Paik SW, Rhee JC, Choi KW. Clinical usefulness of carbohydrate antigen 19-9 as a screening test for pancreatic cancer in an asymptomatic population. J Gastroenterol Hepatol. 2004; 19:182–6.
30. Homma T, Tsuchiya R. The study of the mass screening of persons without symptoms and of the screening of outpatients with gastrointestinal complaints or icterus for pancreatic cancer in Japan, using CA19-9 and elastase-1 or ultrasonography. Int J Pancreatol. 1991; 9:119–24.
31. Karmazanovsky G, Fedorov V, Kubyshkin V, Kotchatkov A. Pancreatic head cancer: accuracy of CT in determination of resectability. Abdom Imaging. 2005; 30:488–500. https://doi.org/10.1007/s00261-004-0279-z.
32. Appel BL, Tolat P, Evans DB, Tsai S. Current staging systems for pancreatic cancer. Cancer J. 2012; 18:539–49. https://doi.org/10.1097/PPO.0b013e318278c5b5.
33. Fusaroli P, Kypraios D, Caletti G, Eloubeidi MA. Pancreatico-biliary endoscopic ultrasound: a systematic review of the levels of evidence, performance and outcomes. World J Gastroenterol. 2012; 18:4243–56. https://doi.org/10.3748/wjg.v18.i32.4243.
34. Conrad C, Fernandez-Del Castillo C. Preoperative evaluation and management of the pancreatic head mass. J Surg Oncol. 2013; 107:23–32. https://doi.org/10.1002/jso.23165.
35. Lee ES, Lee JM. Imaging diagnosis of pancreatic cancer: A state-of-the-art review. World J Gastroenterol. 2014; 20:7864–77. https://doi.org/10.3748/wjg.v20.i24.7864.
36. Mohammad Alizadeh AH, Shahrokh S, Hadizadeh M, Padashi M, Zali MR. Diagnostic potency of EUS-guided FNA for the evaluation of pancreatic mass lesions. Endosc Ultrasound. 2016; 5:30–4. https://doi.org/10.4103/2303-9027.175879.
37. Shulik O, Cavanagh Y, Grossman M. Pancreatic Lesion: Malignancy or Abscess? Am J Case Rep. 2016; 17:337–9.
38. Puli SR, Bechtold ML, Buxbaum JL, Eloubeidi MA. How good is endoscopic ultrasound-guided fine-needle aspiration in diagnosing the correct etiology for a solid pancreatic mass?: A meta-analysis and systematic review. Pancreas. 2013; 42:20–6. https://doi.org/10.1097/MPA.0b013e3182546e79.
39. Ducreux M, Cuhna AS, Caramella C, Hollebecque A, Burtin P, Goéré D, Seufferlein T, Haustermans K, Van Laethem JL, Conroy T, Arnold D. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology. 2015; 26:v56–v68. https://doi.org/10.1093/annonc/mdv295.
40. Rimbaş M, Deaconu M, Croitoru A, Haidar A. Sudden appearance of free fluid during endoscopic ultrasound-guided fine-needle aspiration. Endosc Ultrasound. 2016; 5:55–7. https://doi.org/10.4103/2303-9027.175900.
41. Fujii LL, Levy MJ. Basic techniques in endoscopic ultrasound-guided fine needle aspiration for solid lesions: Adverse events and avoiding them. Endosc Ultrasound. 2014; 3:35–45. https://doi.org/10.4103/2303-9027.123006.
42. Chertoff J, Khullar V, Burke L. Duodenal perforation following esophagogastroduodenoscopy (EGD) with cautery and epinephrine injection for peptic ulcer disease: An interesting case of nonoperative management in the medical intensive care unit (MICU). Int J Surg Case Rep. 2015; 10:121–5. https://doi.org/10.1016/j.ijscr.2015.03.011.
43. Bournet B, Vignolle-Vidoni A, Grand D, Roques C, Breibach F, Cros J, Muscari F, Carrère N, Selves J, Cordelier P, Buscail L. Endoscopic ultrasound-guided fine-needle aspiration plus KRAS and GNAS mutation in malignant intraductal papillary mucinous neoplasm of the pancreas. Endosc Int Open. 2016; 4:E1228–35. https://doi.org/10.1055/s-0042-117216.
44. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008; 321:1801–6. https://doi.org/10.1126/science.1164368.
45. Hahn SA, Kern SE. Molecular genetics of exocrine pancreatic neoplasms. Surg Clin North Am. 1995; 75:857–69.
46. Karhu R, Mahlamaki E, Kallioniemi A. Pancreatic adenocarcinoma — genetic portrait from chromosomes to microarrays. Genes Chromosomes Cancer. 2006; 45:721–30. https://doi.org/10.1002/gcc.20337.
47. Calhoun ES, Hucl T, Gallmeier E, West KM, Arking DE, Maitra A, Iacobuzio-Donahue CA, Chakravarti A, Hruban RH, Kern SE. Identifying allelic loss and homozygous deletions in pancreatic cancer without matched normals using high-density single-nucleotide polymorphism arrays. Cancer Res. 2006; 66:7920–8. https://doi.org/10.1158/0008-5472.can-06-0721.
48. Harada T, Chelala C, Bhakta V, Chaplin T, Caulee K, Baril P, Young BD, Lemoine NR. Genome-wide DNA copy number analysis in pancreatic cancer using high-density single nucleotide polymorphism arrays. Oncogene. 2008; 27:1951–60. https://doi.org/10.1038/sj.onc.1210832.
49. Beroukhim R, Lin M, Park Y, Hao K, Zhao X, Garraway LA, Fox EA, Hochberg EP, Mellinghoff IK, Hofer MD, Descazeaud A, Rubin MA, Meyerson M, et al. Inferring loss-of-heterozygosity from unpaired tumors using high-density oligonucleotide SNP arrays. PLoS Comput Biol. 2006; 2:e41. https://doi.org/10.1371/journal.pcbi.0020041.
50. Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012; 481:306–13. http://www.nature.com/nature/journal/v481/n7381/abs/nature10762.html#supplementary-information.
51. Makohon-Moore A, Iacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016; 16:553–65. https://doi.org/10.1038/nrc.2016.66.
52. Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002; 2:897–909. https://doi.org/10.1038/nrc949.
53. Chantrill LA, Nagrial AM, Watson C, Johns AL, Martyn-Smith M, Simpson S, Mead S, Jones MD, Samra JS, Gill AJ, Watson N, Chin VT, Humphris JL, et al. Precision Medicine for Advanced Pancreas Cancer: The Individualized Molecular Pancreatic Cancer Therapy (IMPaCT) Trial. Clin Cancer Res. 2015; 21:2029–37. https://doi.org/10.1158/1078-0432.ccr-15-0426.
54. Yu J, Liu SH, Sanchez R, Nemunaitis J, Rozengurt E, Brunicardi FC. Pancreatic cancer actionable genes in precision medicine and personalized surgery. Surgeon. 2017; 15:24–9. https://doi.org/10.1016/j.surge.2016.05.002.
55. Narayanan V, Weekes CD. Molecular therapeutics in pancreas cancer. World J Gastrointest Oncol. 2016; 8:366–79. https://doi.org/10.4251/wjgo.v8.i4.366.
56. Wong K, Qian Z, Le Y. The Role of Precision Medicine in Pancreatic Cancer: Challenges for Targeted Therapy, Immune Modulating Treatment, Early Detection, and Less Invasive Operations. Cancer Translational Medicine. 2016; 2:41–7. https://doi.org/10.4103/2395-3977.181434.
57. San Lucas FA, Allenson K, Bernard V, Castillo J, Kim DU, Ellis K, Ehli EA, Davies GE, Petersen JL, Li D, Wolff R, Katz M, Varadhachary G, et al. Minimally invasive genomic and transcriptomic profiling of visceral cancers by next-generation sequencing of circulating exosomes. Ann Oncol. 2016; 27:635–41. https://doi.org/10.1093/annonc/mdv604.
58. Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014; 32:579–86. https://doi.org/10.1200/JCO.2012.45.2011.
59. Siravegna G, Bardelli A. Blood circulating tumor DNA for non-invasive genotyping of colon cancer patients. Mol Oncol. 2016; 10:475–80. https://doi.org/10.1016/j.molonc.2015.12.005.
60. Rahbari M, Rahbari N, Reissfelder C, Weitz J, Kahlert C. Exosomes: novel implications in diagnosis and treatment of gastrointestinal cancer. Langenbecks Arch Surg. 2016; 401:1097–110. https://doi.org/10.1007/s00423-016-1468-2.
61. Cheng F, Su L, Qian C. Circulating tumor DNA: a promising biomarker in the liquid biopsy of cancer. Oncotarget. 2016; 7:48832–41. https://doi.org/10.18632/oncotarget.9453.
62. Thiele JA, Bethel K, Kralickova M, Kuhn P. Circulating Tumor Cells: Fluid Surrogates of Solid Tumors. Annu Rev Pathol. 2017; 12:419–47. https://doi.org/10.1146/annurev-pathol-052016-100256.
63. Shigeyasu K, Toden S, Zumwalt TJ, Okugawa Y, Goel A. Emerging Role of MicroRNAs as Liquid Biopsy Biomarkers in Gastrointestinal Cancers. Clin Cancer Res. 2017; 23:2391–2399. https://doi.org/10.1158/1078-0432.ccr-16-1676.
64. Vendrell JA, Mau-Them FT, Beganton B, Godreuil S, Coopman P, Solassol J. Circulating Cell Free Tumor DNA Detection as a Routine Tool forLung Cancer Patient Management. Int J Mol Sci. 2017; 18. https://doi.org/10.3390/ijms18020264.
65. Lianidou ES, Mavroudis D, Sotiropoulou G, Agelaki S, Pantel K. What’s new on circulating tumor cells? A meeting report. Breast Cancer Res. 2010; 12:307. https://doi.org/10.1186/bcr2601.
66. Lim SH, Becker TM, Chua W, Caixeiro NJ, Ng WL, Kienzle N, Tognela A, Lumba S, Rasko JE, de Souza P, Spring KJ. Circulating tumour cells and circulating free nucleic acid as prognostic and predictive biomarkers in colorectal cancer. Cancer Lett. 2014; 346:24–33. https://doi.org/10.1016/j.canlet.2013.12.019.
67. Voelker R. Liquid biopsy receives approval. JAMA. 2016; 316:260. https://doi.org/10.1001/jama.2016.8833.
68. cobas EGFR Mutation Test v2. Available online. 2016.
69. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006; 127:679–95. https://doi.org/10.1016/j.cell.2006.11.001.
70. Riquet M, Rivera C, Gibault L, Pricopi C, Mordant P, Badia A, Arame A, Le Pimpec Barthes F. [Lymphatic spread of lung cancer: anatomical lymph node chains unchained in zones]. [Article in French]. Rev Pneumol Clin. 2014; 70:16–25. https://doi.org/10.1016/j.pneumo.2013.07.001.
71. Pantel K, Speicher MR. The biology of circulating tumor cells. Oncogene. 2016; 35:1216–24. https://doi.org/10.1038/onc.2015.192.
72. Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004; 4:448–56. https://doi.org/10.1038/nrc1370.
73. Książkiewicz M, Markiewicz A, Żaczek AJ. Epithelial-Mesenchymal Transition: A Hallmark in Metastasis Formation Linking Circulating Tumor Cells and Cancer Stem Cells. Pathobiology. 2012; 79:195–208.
74. Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010; 468:829–33. https://doi.org/10.1038/nature09624.
75. Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM, De Maria R. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010; 468:824–8. https://doi.org/10.1038/nature09557.
76. Katsnelson A. Tumours grow their own blood vessels. Nature News. 2010. https://doi.org/doi:10.1038/news.2010.623.
77. Folberg R, Hendrix MJ, Maniotis AJ. Vasculogenic mimicry and tumor angiogenesis. Am J Pathol. 2000; 156:361–81. https://doi.org/10.1016/s0002-9440(10)64739-6.
78. Folberg R, Maniotis AJ. Vasculogenic mimicry. APMIS. 2004; 112:508–25. https://doi.org/10.1111/j.1600-0463.2004.apm11207-0810.x.
79. McDougall SR, Anderson AR, Chaplain MA. Mathematical modelling of dynamic adaptive tumour-induced angiogenesis: clinical implications and therapeutic targeting strategies. J Theor Biol. 2006; 241:564–89. https://doi.org/10.1016/j.jtbi.2005.12.022.
80. Spill F, Guerrero P, Alarcon T, Maini PK, Byrne HM. Mesoscopic and continuum modelling of angiogenesis. Journal of Mathematical Biology. 2015; 70:485–532. https://doi.org/10.1007/s00285-014-0771-1.
81. Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006; 103:12493–8. https://doi.org/10.1073/pnas.0601807103.
82. Inoue M, Hager JH, Ferrara N, Gerber HP, Hanahan D. VEGF-A has a critical, nonredundant role in angiogenic switching and pancreatic beta cell carcinogenesis. Cancer Cell. 2002; 1:193–202.
83. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000; 2:737–44. https://doi.org/10.1038/35036374.
84. Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AG, Uhr JW, Terstappen LW. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res. 2004; 10:6897–904. https://doi.org/10.1158/1078-0432.ccr-04-0378.
85. Cristofanilli M, Broglio KR, Guarneri V, Jackson S, Fritsche HA, Islam R, Dawood S, Reuben JM, Kau SW, Lara JM, Krishnamurthy S, Ueno NT, Hortobagyi GN, et al. Circulating tumor cells in metastatic breast cancer: biologic staging beyond tumor burden. Clin Breast Cancer. 2007; 7:471–9.
86. Ross JS, Slodkowska EA. Circulating and disseminated tumor cells in the management of breast cancer. Am J Clin Pathol. 2009; 132:237–45. https://doi.org/10.1309/ajcpji7deolkcs6f.
87. Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, Dinulescu DM, Connolly D, Foster R, Dombkowski D, Preffer F, Maclaughlin DT, Donahoe PK. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci U S A. 2006; 103:11154–9. https://doi.org/10.1073/pnas.0603672103.
88. Haber DA, Velculescu VE. Blood-based analyses of cancer: circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014; 4:650–61. https://doi.org/10.1158/2159-8290.CD-13-1014.
89. Masuda T, Hayashi N, Iguchi T, Ito S, Eguchi H, Mimori K. Clinical and biological significance of circulating tumor cells in cancer. Mol Oncol. 2016; 10:408–17. https://doi.org/10.1016/j.molonc.2016.01.010.
90. Braun S, Naume B. Circulating and Disseminated Tumor Cells. Journal of Clinical Oncology. 2005; 23:1623–6. https://doi.org/doi:10.1200/JCO.2005.10.073.
91. Mohme M, Riethdorf S, Pantel K. Circulating and disseminated tumour cells - mechanisms of immune surveillance and escape. Nat Rev Clin Oncol. 2016; 14:155–167. https://doi.org/10.1038/nrclinonc.2016.144.
92. Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003; 3:453–8. https://doi.org/10.1038/nrc1098.
93. Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016; 529:298–306. https://doi.org/10.1038/nature17038.
94. Giuliano M, Giordano A, Jackson S, De Giorgi U, Mego M, Cohen EN, Gao H, Anfossi S, Handy BC, Ueno NT, Alvarez RH, De Placido S, Valero V, et al. Circulating tumor cells as early predictors of metastatic spread in breast cancer patients with limited metastatic dissemination. Breast Cancer Research. 2014; 16:440. https://doi.org/10.1186/s13058-014-0440-8.
95. Xue J, Xie VK, Wang P, Cui J, Gao Y, Lu Z. Interrelationships of Circulating Tumor Cells with Metastasis and Thrombosis: Role of MicroRNAs. Curr Pharm Des. 2014; 20:5298–308.
96. Fabisiewicz A, Grzybowska E. CTC clusters in cancer progression and metastasis. Med Oncol. 2017; 34:12. https://doi.org/10.1007/s12032-016-0875-0.
97. Martin OA, Anderson RL, Narayan K, MacManus MP. Does the mobilization of circulating tumour cells during cancer therapy cause metastasis? Nat Rev Clin Oncol. 2017; 14:32–44. https://doi.org/10.1038/nrclinonc.2016.128.
98. Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XHF, Norton L, Massagué J. Tumor Self-Seeding by Circulating Cancer Cells. Cell. 2009; 139:1315–26. https://doi.org/10.1016/j.cell.2009.11.025.
99. Zhe X, Cher ML, Bonfil RD. Circulating tumor cells: finding the needle in the haystack. Am J Cancer Res. 2011; 1:740–51.
100. Sergeant G, Roskams T, van Pelt J, Houtmeyers F, Aerts R, Topal B. Perioperative cancer cell dissemination detected with a real-time RT-PCR assay for EpCAM is not associated with worse prognosis in pancreatic ductal adenocarcinoma. BMC Cancer. 2011; 11:47. https://doi.org/10.1186/1471-2407-11-47.
101. Dittamore R, Louw J, Krupa R, Anand A, Danila DC, Arslan Z, Bales N, Marrinucci D, Scher HI. Molecular characterization of circulating tumor cells (CTC) and CTC subpopulations in progressive metastatic castration resistant prostate cancer (mCRPC). Journal of Clinical Oncology. 2014; 32:132. https://doi.org/doi:10.1200/jco.2014.32.4_suppl.132.
102. Punnoose EA, Ferraldeschi R, Szafer-Glusman E, Tucker EK, Mohan S, Flohr P, Riisnaes R, Miranda S, Figueiredo I, Rodrigues DN, Omlin A, Pezaro C, Zhu J, et al. PTEN loss in circulating tumour cells correlates with PTEN loss in fresh tumour tissue from castration-resistant prostate cancer patients. Br J Cancer. 2015; 113:1225–33. https://doi.org/10.1038/bjc.2015.332.
103. Hart IR. ‘Seed and soil’ revisited: mechanisms of site-specific metastasis. Cancer Metastasis Rev. 1982; 1:5–16. https://doi.org/10.1007/bf00049477.
104. Scott J, Kuhn P, Anderson AR. Unifying metastasis--integrating intravasation, circulation and end-organ colonization. Nat Rev Cancer. 2012; 12:445–6. https://doi.org/10.1038/nrc3287.
105. Qian CN, Teh BT. “Seed and Soil” Theory of Metastasis. In: Schwab M, ed. Encyclopedia of Cancer. (Berlin, Heidelberg: Springer Berlin Heidelberg). 2011; 3354–5.
106. Hou JM, Krebs M, Ward T, Sloane R, Priest L, Hughes A, Clack G, Ranson M, Blackhall F, Dive C. Circulating tumor cells as a window on metastasis biology in lung cancer. Am J Pathol. 2011; 178:989–96. https://doi.org/10.1016/j.ajpath.2010.12.003.
107. Matsusaka S, Chin K, Ogura M, Suenaga M, Shinozaki E, Mishima Y, Terui Y, Mizunuma N, Hatake K. Circulating tumor cells as a surrogate marker for determining response to chemotherapy in patients with advanced gastric cancer. Cancer Sci. 2010; 101:1067–71. https://doi.org/10.1111/j.1349-7006.2010.01492.x.
108. Cohen SJ, Punt CJ, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, Picus J, Morse MA, Mitchell E, Miller MC, Doyle GV, Tissing H, Terstappen LW, et al. Prognostic significance of circulating tumor cells in patients with metastatic colorectal cancer. Ann Oncol. 2009; 20:1223–9. https://doi.org/10.1093/annonc/mdn786.
109. Moussavi-Harami SF, Wisinski KB, Beebe DJ. Circulating Tumor Cells in Metastatic Breast Cancer: A Prognostic and Predictive Marker. J Patient Cent Res Rev. 2014; 1:85–92. https://doi.org/10.17294/2330-0698.1017.
110. Harris L, Fritsche H, Mennel R, Norton L, Ravdin P, Taube S, Somerfield MR, Hayes DF, Bast RC Jr. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J Clin Oncol. 2007; 25:5287–312. https://doi.org/10.1200/jco.2007.14.2364.
111. Danova M, Torchio M, Mazzini G. Isolation of rare circulating tumor cells in cancer patients: technical aspects and clinical implications. Expert Rev Mol Diagn. 2011; 11:473–85. https://doi.org/10.1586/erm.11.33.
112. Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007; 450:1235–9. https://doi.org/10.1038/nature06385.
113. Barriere G, Fici P, Gallerani G, Fabbri F, Zoli W, Rigaud M. Circulating tumor cells and epithelial, mesenchymal and stemness markers: characterization of cell subpopulations. Ann Transl Med. 2014; 2. https://doi.org/10.3978/j.issn.2305-5839.2014.10.04.
114. Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, Beitsch PD, Leitch M, Hoover S, Euhus D, Haley B, Morrison L, Fleming TP, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004; 10:8152–62. https://doi.org/10.1158/1078-0432.ccr-04-1110.
115. Sollier E, Go DE, Che J, Gossett DR, O’Byrne S, Weaver WM, Kummer N, Rettig M, Goldman J, Nickols N, McCloskey S, Kulkarni RP, Di Carlo D. Size-selective collection of circulating tumor cells using Vortex technology. Lab Chip. 2014; 14:63–77. https://doi.org/10.1039/c3lc50689d.
116. Qin J, Alt JR, Hunsley BA, Williams TL, Fernando MR. Stabilization of circulating tumor cells in blood using a collection device with a preservative reagent. Cancer Cell Int. 2014; 14:23. https://doi.org/10.1186/1475-2867-14-23.
117. Toss A, Mu Z, Fernandez S, Cristofanilli M. CTC enumeration and characterization: moving toward personalized medicine. Ann Transl Med. 2014; 2. https://doi.org/10.3978/j.issn.2305-5839.2014.09.06.
118. Vona G, Sabile A, Louha M, Sitruk V, Romana S, Schutze K, Capron F, Franco D, Pazzagli M, Vekemans M, Lacour B, Brechot C, Paterlini-Brechot P. Isolation by size of epithelial tumor cells : a new method for the immunomorphological and molecular characterization of circulatingtumor cells. Am J Pathol. 2000; 156:57–63. https://doi.org/10.1016/s0002-9440(10)64706-2.
119. Hofman V, Long E, Ilie M, Bonnetaud C, Vignaud JM, Flejou JF, Lantuejoul S, Piaton E, Mourad N, Butori C, Selva E, Marquette CH, Poudenx M, et al. Morphological analysis of circulating tumour cells in patients undergoing surgery for non-small cell lung carcinoma using the isolation by size of epithelial tumour cell (ISET) method. Cytopathology. 2012; 23:30–8. https://doi.org/10.1111/j.1365-2303.2010.00835.x.
120. Hofman VJ, Ilie MI, Bonnetaud C, Selva E, Long E, Molina T, Vignaud JM, Flejou JF, Lantuejoul S, Piaton E, Butori C, Mourad N, Poudenx M, et al. Cytopathologic detection of circulating tumor cells using the isolation by size of epithelial tumor cell method: promises and pitfalls. Am J Clin Pathol. 2011; 135:146–56. https://doi.org/10.1309/ajcp9x8ozbeiqvvi.
121. Zhang D, Zhao L, Zhou P, Ma H, Huang F, Jin M, Dai X, Zheng X, Huang S, Zhang T. Circulating tumor microemboli (CTM) and vimentin+ circulating tumor cells (CTCs) detected by a size-based platform predict worse prognosis in advanced colorectal cancer patients during chemotherapy. Cancer Cell International. 2017; 17:6. https://doi.org/10.1186/s12935-016-0373-7.
122. Kanwar N, Hu P, Bedard P, Clemons M, McCready D, Done SJ. Identification of genomic signatures in circulating tumor cells from breast cancer. International Journal of Cancer. 2015; 137:332–44. https://doi.org/10.1002/ijc.29399.
123. Kalinich M, Bhan I, Kwan TT, Miyamoto DT, Javaid S, LiCausi JA, Milner JD, Hong X, Goyal L, Sil S, Choz M, Ho U, Kapur R, et al. An RNA-based signature enables high specificity detection of circulating tumor cells in hepatocellular carcinoma. Proceedings of the National Academy of Sciences. 2017; 114:1123–8. https://doi.org/10.1073/pnas.1617032114.
124. Marrinucci D, Bethel K, Kolatkar A, Luttgen MS, Malchiodi M, Baehring F, Voigt K, Lazar D, Nieva J, Bazhenova L, Ko AH, Korn WM, Schram E, et al. Fluid biopsy in patients with metastatic prostate, pancreatic and breast cancers. Phys Biol. 2012; 9:016003. https://doi.org/10.1088/1478-3975/9/1/016003.
125. Schneck H, Gierke B, Uppenkamp F, Behrens B, Niederacher D, Stoecklein NH, Templin MF, Pawlak M, Fehm T, Neubauer H; Disseminated Cancer Cell Network (DCC Net) Duesseldorf. EpCAM-Independent Enrichment of Circulating Tumor Cells in Metastatic Breast Cancer. PLoS One. 2015; 10:e0144535. https://doi.org/10.1371/journal.pone.0144535.
126. Spiliotaki M, Mavroudis D, Kapranou K, Markomanolaki H, Kallergi G, Koinis F, Kalbakis K, Georgoulias V, Agelaki S. Evaluation of proliferation and apoptosis markers in circulating tumor cells of women with early breast cancer who are candidates for tumor dormancy. Breast Cancer Res. 2014; 16. https://doi.org/10.1186/s13058-014-0485-8.
127. Bidard FC, Proudhon C, Pierga JY. Circulating tumor cells in breast cancer. Molecular Oncology. 2016; 10:418–30. https://doi.org/http://dx.doi.org/10.1016/j.molonc.2016.01.001.
128. Lin HK, Zheng S, Williams AJ, Balic M, Groshen S, Scher HI, Fleisher M, Stadler W, Datar RH, Tai YC, Cote RJ. Portable filter-based microdevice for detection and characterization of circulating tumor cells. Clin Cancer Res. 2010; 16:5011–8. https://doi.org/10.1158/1078-0432.ccr-10-1105.
129. Paterlini-Brechot P, Benali NL. Circulating tumor cells (CTC) detection: clinical impact and future directions. Cancer Lett. 2007; 253:180–204. https://doi.org/10.1016/j.canlet.2006.12.014.
130. Ferraldeschi R, McDaniel A, Krupa R, Louw J, Tucker E, Bales N, Marrinucci D, Riisnaes R, Mateo J, Dittamore R, Bono JSD, Tomlins SA, Attard G. CK- and small nuclear size circulating tumor cell (CTCs) phenotypes in metastatic castration-resistant prostate cancer (mCRPC). Journal of Clinical Oncology. 2014; 32:209. https://doi.org/10.1200/jco.2014.32.4_suppl.209.
131. Aceto N. Circulating Tumor Cell Clusters are Oligoclonal Precursors of Breast Cancer Metastasis. Cell. 2014; 158:1110–22. https://doi.org/10.1016/j.cell.2014.07.013.
132. Hong Y, Li Z, Zhang Q. A circulating tumor cell cluster-based model for tumor metastasis (Hypothesis). Oncol Lett. 2016; 12:4891–5. https://doi.org/10.3892/ol.2016.5358.
133. Choi JW, Kim JK, Yang YJ, Kim P, Yoon KH, Yun SH. Urokinase Exerts Antimetastatic Effects by Dissociating Clusters of Circulating Tumor Cells. Cancer Research. 2015; 75:4474–82. https://doi.org/10.1158/0008-5472.can-15-0684.
134. Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK, Floyd FP Jr, Gilman AJ, Lord JB, et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci U S A. 2010; 107:18392–7. https://doi.org/10.1073/pnas.1012539107.
135. Riahi R, Gogoi P, Sepehri S, Zhou Y, Handique K, Godsey J, Wang Y. A novel microchannel-based device to capture and analyze circulating tumor cells (CTCs) of breast cancer. Int J Oncol. 2014; 44:1870–8. https://doi.org/10.3892/ijo.2014.2353.
136. Beije N, Jager A, Sleijfer S. Circulating tumor cell enumeration by the CellSearch system: the clinician’s guide to breast cancer treatment? Cancer Treat Rev. 2015; 41:144–50. https://doi.org/10.1016/j.ctrv.2014.12.008.
137. Banerjee SS, Khobragade V, Khandare J. Designing Multicomponent Nanosystems for Rapid Detection of Circulating Tumor Cells. Methods Mol Biol. 2017; 1530:271–81. https://doi.org/10.1007/978-1-4939-6646-2_16.
138. Sheng Y, Wang T, Li H, Zhang Z, Chen J, He C, Li Y, Lv Y, Zhang J, Xu C, Wang Z, Huang C, Wang L. Comparison of analytic performances of Cellsearch and iFISH approach in detecting circulating tumor cells. Oncotarget. 2017; 8:8801–6. https://doi.org/10.18632/oncotarget.6688.
139. Ming Y, Li Y, Xing H, Luo M, Li Z, Chen J, Mo J, Shi S. Circulating Tumor Cells: From Theory to Nanotechnology-Based Detection. Front Pharmacol. 2017; 8:35. https://doi.org/10.3389/fphar.2017.00035.
140. Went PT, Lugli A, Meier S, Bundi M, Mirlacher M, Sauter G, Dirnhofer S. Frequent EpCam protein expression in human carcinomas. Hum Pathol. 2004; 35:122–8.
141. Hyun KA, Jung HI. Advances and critical concerns with the microfluidic enrichments of circulating tumor cells. Lab Chip. 2014; 14:45–56. https://doi.org/10.1039/c3lc50582k.
142. Launiere C, Gaskill M, Czaplewski G, Myung JH, Hong S, Eddington DT. Channel surface patterning of alternating biomimetic protein combinations for enhanced microfluidic tumor cell isolation. Anal Chem. 2012; 84:4022–8. https://doi.org/10.1021/ac2033408.
143. Yoon HJ, Kim TH, Zhang Z, Azizi E, Pham TM, Paoletti C, Lin J, Ramnath N, Wicha MS, Hayes DF, Simeone DM, Nagrath S. Sensitive capture of circulating tumour cells by functionalized graphene oxide nanosheets. Nat Nanotechnol. 2013; 8:735–41. https://doi.org/10.1038/nnano.2013.194.
144. Li N, Xiao T, Zhang Z, He R, Wen D, Cao Y, Zhang W, Chen Y. A 3D graphene oxide microchip and a Au-enwrapped silica nanocomposite-based supersandwich cytosensor toward capture and analysis of circulating tumor cells. Nanoscale. 2015; 7:16354–60. https://doi.org/10.1039/c5nr04798f.
145. Murlidhar V, Zeinali M, Grabauskiene S, Ghannad-Rezaie M, Wicha MS, Simeone DM, Ramnath N, Reddy RM, Nagrath S. A radial flow microfluidic device for ultra-high-throughput affinity-based isolation of circulating tumor cells. Small. 2014; 10:4895–904. https://doi.org/10.1002/smll.201400719.
146. Yoon HJ, Shanker A, Wang Y, Kozminsky M, Jin Q, Palanisamy N, Burness ML, Azizi E, Simeone DM, Wicha MS, Kim J, Nagrath S. Tunable Thermal-Sensitive Polymer–Graphene Oxide Composite for Efficient Capture and Release of Viable Circulating Tumor Cells. Advanced Materials. 2016; 28:4891–7. https://doi.org/10.1002/adma.201600658.
147. Chen H, Cao B, Sun B, Cao Y, Yang K, Lin YS, Chen H. Highly-sensitive capture of circulating tumor cells using micro-ellipse filters. Sci Rep. 2017; 7:610. https://doi.org/10.1038/s41598-017-00232-6.
148. Kuske A, Gorges TM, Tennstedt P, Tiebel AK, Pompe R, Preißer F, Prues S, Mazel M, Markou A, Lianidou E, Peine S, Alix-Panabières C, Riethdorf S, et al. Improved detection of circulating tumor cells in non-metastatic high-risk prostate cancer patients. Scientific Reports. 2016; 6:39736. https://doi.org/10.1038/srep39736.
149. Müller V, Riethdorf S, Rack B, Janni W, Fasching PA, Solomayer E, Aktas B, Kasimir-Bauer S, Pantel K, Fehm T. Prognostic impact of circulating tumor cells assessed with the CellSearch System™ and AdnaTest Breast™ in metastatic breast cancer patients: the DETECT study. Breast Cancer Research. 2012; 14:R118. https://doi.org/10.1186/bcr3243.
150. Satelli A, Brownlee Z, Mitra A, Meng QH, Li S. Circulating Tumor Cell Enumeration with a Combination of Epithelial Cell Adhesion Molecule– and Cell-Surface Vimentin–Based Methods for Monitoring Breast Cancer Therapeutic Response. Clinical Chemistry. 2014; 61:259.
151. Grover PK, Cummins AG, Price TJ, Roberts-Thomson IC, Hardingham JE. Circulating tumour cells: the evolving concept and the inadequacy of their enrichment by EpCAM-based methodology for basic and clinical cancer research. Annals of Oncology. 2014; 25:1506–16. https://doi.org/10.1093/annonc/mdu018.
152. Wu S, Liu S, Liu Z, Huang J, Pu X, Li J, Yang D, Deng H, Yang N, Xu J. Classification of Circulating Tumor Cells by Epithelial-Mesenchymal Transition Markers. PLoS One. 2015; 10:e0123976. https://doi.org/10.1371/journal.pone.0123976.
153. Krawczyk N, Meier-Stiegen F, Banys M, Neubauer H, Ruckhaeberle E, Fehm T. Expression of Stem Cell and Epithelial-Mesenchymal Transition Markers in Circulating Tumor Cells of Breast Cancer Patients. Biomed Res Int. 2014; 2014:415721. https://doi.org/10.1155/2014/415721.
154. Gorges TM, Tinhofer I, Drosch M, Rose L, Zollner TM, Krahn T, von Ahsen O. Circulating tumour cells escape from EpCAM-based detection due to epithelial-to-mesenchymal transition. BMC Cancer. 2012; 12:178. https://doi.org/10.1186/1471-2407-12-178.
155. Punnoose EA, Atwal SK, Spoerke JM, Savage H, Pandita A, Yeh RF, Pirzkall A, Fine BM, Amler LC, Chen DS, Lackner MR. Molecular biomarker analyses using circulating tumor cells. PLoS One. 2010; 5:e12517. https://doi.org/10.1371/journal.pone.0012517.
156. Konigsberg R, Obermayr E, Bises G, Pfeiler G, Gneist M, Wrba F, de Santis M, Zeillinger R, Hudec M, Dittrich C. Detection of EpCAM positive and negative circulating tumor cells in metastatic breast cancer patients. Acta Oncol. 2011; 50:700–10. https://doi.org/10.3109/0284186x.2010.549151.
157. Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, Concannon KF, Donaldson MC, Sequist LV, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013; 339:580–4. https://doi.org/10.1126/science.1228522.
158. Liu Q, Liao Q, Zhao Y. Myeloid-derived suppressor cells (MDSC) facilitate distant metastasis of malignancies by shielding circulating tumor cells (CTC) from immune surveillance. Med Hypotheses. 2016; 87:34–9. https://doi.org/10.1016/j.mehy.2015.12.007.
159. Balic M, Lin H, Williams A, Datar RH, Cote RJ. Progress in circulating tumor cell capture and analysis: implications for cancer management. Expert Review of Molecular Diagnostics. 2012; 12:303–12. https://doi.org/10.1586/erm.12.12.
160. Jueckstock J, Rack B, Friedl TW, Scholz C, Steidl J, Trapp E, Tesch H, Forstbauer H, Lorenz R, Rezai M, Häberle L, Alunni-Fabbroni M, Schneeweiss A, et al. Detection of circulating tumor cells using manually performed immunocytochemistry (MICC) does not correlate with outcome in patients with early breast cancer – Results of the German SUCCESS-A- trial. BMC Cancer. 2016; 16:401. https://doi.org/10.1186/s12885-016-2454-3.
161. Lowes LE, Allan AL. Recent Advances in the Molecular Characterization of Circulating Tumor Cells. Cancers. 2014; 6:595–624. https://doi.org/10.3390/cancers6010595.
162. Oxnard GR, Paweletz CP, Kuang Y, Mach SL, O’Connell A, Messineo MM, Luke JJ, Butaney M, Kirschmeier P, Jackman DM, Jänne PA. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014; 20:1698–705. https://doi.org/10.1158/1078-0432.CCR-13-2482.
163. Pfitzner C, Schröder I, Scheungraber C, Dogan A, Runnebaum IB, Dürst M, Häfner N. Digital-Direct-RT-PCR: a sensitive and specific method for quantification of CTC in patients with cervical carcinoma. Scientific Reports. 2014; 4:3970. https://doi.org/10.1038/srep03970.
164. Diamond E, Lee GY, Akhtar NH, Kirby BJ, Giannakakou P, Tagawa ST, Nanus DM. Isolation and characterization of circulating tumor cells in prostate cancer. Frontiers in Oncology. 2012; 2:131. https://doi.org/10.3389/fonc.2012.00131.
165. Denis JA, Patroni A, Guillerm E, Pepin D, Benali-Furet N, Wechsler J, Manceau G, Bernard M, Coulet F, Larsen AK, Karoui M, Lacorte JM. Droplet digital PCR of circulating tumor cells from colorectal cancer patients can predict KRAS mutations before surgery. Mol Oncol. 2016; 10:1221–31. https://doi.org/10.1016/j.molonc.2016.05.009.
166. Freidin MB, Freydina DV, Leung M, Montero Fernandez A, Nicholson AG, Lim E. Circulating tumor DNA outperforms circulating tumor cells for KRAS mutation detection in thoracic malignancies. Clin Chem. 2015; 61:1299–304. https://doi.org/10.1373/clinchem.2015.242453.
167. Castellanos-Rizaldos E, Milbury CA, Guha M, Makrigiorgos GM. COLD-PCR enriches low-level variant DNA sequences and increases the sensitivity of genetic testing. Methods Mol Biol. 2014; 1102:623–39. https://doi.org/10.1007/978-1-62703-727-3_33.
168. Yee SS, Lieberman DB, Blanchard T, Rader J, Zhao J, Troxel AB, DeSloover D, Fox AJ, Daber RD, Kakrecha B, Sukhadia S, Belka GK, DeMichele AM, et al. A novel approach for next-generation sequencing of circulating tumor cells. Mol Genet Genomic Med. 2016; 4:395–406. https://doi.org/10.1002/mgg3.210.
169. Perakis S, Speicher MR. Emerging concepts in liquid biopsies. BMC Medicine. 2017; 15:75. https://doi.org/10.1186/s12916-017-0840-6.
170. Lohr JG, Adalsteinsson VA, Cibulskis K, Choudhury AD, Rosenberg M, Cruz-Gordillo P, Francis JM, Zhang CZ, Shalek AK, Satija R, Trombetta JJ, Lu D, Tallapragada N, et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol. 2014; 32:479–84. https://doi.org/10.1038/nbt.2892.
171. Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011; 83:8604–10. https://doi.org/10.1021/ac202028g.
172. Tellez-Gabriel M, Ory B, Lamoureux F, Heymann MF, Heymann D. Tumour Heterogeneity: The Key Advantages of Single-Cell Analysis. International Journal of Molecular Sciences. 2016; 17:2142. https://doi.org/10.3390/ijms17122142.
173. Bi W, Stambrook PJ. Detection of known mutation by proof-reading PCR. Nucleic Acids Research. 1998; 26:3073–5.
174. Barnard R, Futo V, Pecheniuk N, Slattery M, Walsh T. PCR bias toward the wild-type k-ras and p53 sequences: implications for PCR detection of mutations and cancer diagnosis. Biotechniques. 1998; 25:684–91.
175. Li J, Makrigiorgos GM. COLD-PCR: a new platform for highly improved mutation detection in cancer and genetic testing. Biochem Soc Trans. 2009; 37:427–32. https://doi.org/10.1042/bst0370427.
176. Babayan A, Alawi M, Gormley M, Muller V, Wikman H, McMullin RP, Smirnov DA, Li W, Geffken M, Pantel K, Joosse SA. Comparative study of whole genome amplification and next generation sequencing performance of single cancer cells. Oncotarget. 2016; 8:56066–56080. https://doi.org/10.18632/oncotarget.10701.
177. El Messaoudi S, Rolet F, Mouliere F, Thierry AR. Circulating cell free DNA: Preanalytical considerations. Clin Chim Acta. 2013; 424:222–30. https://doi.org/10.1016/j.cca.2013.05.022.
178. Malentacchi F, Pizzamiglio S, Verderio P, Pazzagli M, Orlando C, Ciniselli CM, Gunther K, Gelmini S. Influence of storage conditions and extraction methods on the quantity and quality of circulating cell-free DNA (ccfDNA): the SPIDIA-DNAplas External Quality Assessment experience. Clin Chem Lab Med. 2015; 53:1935–42. https://doi.org/10.1515/cclm-2014-1161.
179. Pantel K, Brakenhoff RH, Brandt B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat Rev Cancer. 2008; 8:329–40. https://doi.org/10.1038/nrc2375.
180. Iwanicki-Caron I, Basile P, Toure E, Antonietti M, Lecleire S, Di Fiore A, Oden-Gangloff A, Blanchard F, Lemoine F, Di Fiore F, Sabourin JC, Michel P. Usefulness of circulating tumor cell detection in pancreatic adenocarcinoma diagnosis. Am J Gastroenterol. 2013; 108:152–5. https://doi.org/10.1038/ajg.2012.367.
181. Rhim AD, Thege FI, Santana SM, Lannin TB, Saha TN, Tsai S, Maggs LR, Kochman ML, Ginsberg GG, Lieb JG, Chandrasekhara V, Drebin JA, Ahmad N, et al. Detection of Circulating Pancreas Epithelial Cells in Patients with Pancreatic Cystic Lesions. Gastroenterology. 2014; 146:647–51. https://doi.org/10.1053/j.gastro.2013.12.007.
182. Earl J, Garcia-Nieto S, Martinez-Avila JC, Montans J, Sanjuanbenito A, Rodriguez-Garrote M, Lisa E, Mendia E, Lobo E, Malats N, Carrato A, Guillen-Ponce C. Circulating tumor cells (Ctc) and kras mutant circulating free Dna (cfdna) detection in peripheral blood as biomarkers in patients diagnosed with exocrine pancreatic cancer. BMC Cancer. 2015; 15:797. https://doi.org/10.1186/s12885-015-1779-7.
183. Zhang Y, Wang F, Ning N, Chen Q, Yang Z, Guo Y, Xu D, Zhang D, Zhan T, Cui W. Patterns of circulating tumor cells identified by CEP8, CK and CD45 in pancreatic cancer. Int J Cancer. 2015; 136:1228–33. https://doi.org/10.1002/ijc.29070.
184. Bobek V, Gurlich R, Eliasova P, Kolostova K. Circulating tumor cells in pancreatic cancer patients: Enrichment and cultivation. World J Gastroenterol. 2014; 20:17163–70. https://doi.org/10.3748/wjg.v20.i45.17163.
185. Kulemann B, Liss AS, Warshaw AL, Seifert S, Bronsert P, Glatz T, Pitman MB, Hoeppner J. KRAS mutations in pancreatic circulating tumor cells: a pilot study. Tumour Biol. 2016; 37:7547–54. https://doi.org/10.1007/s13277-015-4589-2.
186. Gao Y, Zhu Y, Zhang Z, Zhang C, Huang X, Yuan Z. Clinical significance of pancreatic circulating tumor cells using combined negative enrichment and immunostaining-fluorescence in situ hybridization. J Exp Clin Cancer Res. 2016; 35:66. https://doi.org/10.1186/s13046-016-0340-0.
187. Ankeny JS, Court CM, Hou S, Li Q, Song M, Wu D, Chen JF, Lee T, Lin M, Sho S, Rochefort MM, Girgis MD, Yao J, et al. Circulating tumour cells as a biomarker for diagnosis and staging in pancreatic cancer. Br J Cancer. 2016; 114:1367–75. https://doi.org/10.1038/bjc.2016.121.
188. Poruk KE, Blackford AL, Weiss MJ, Cameron JL, He J, Goggins M, Rasheed ZA, Wolfgang CL, Wood LD. Circulating Tumor Cells Expressing Markers of Tumor-Initiating Cells Predict Poor Survival and Cancer Recurrence in Patients with Pancreatic Ductal Adenocarcinoma. Clin Cancer Res. 2017; 23:2681–2690. https://doi.org/10.1158/1078-0432.ccr-16-1467.
189. Zhou J, Hu L, Yu Z, Zheng J, Yang D, Bouvet M, Hoffman RM. Marker expression in circulating cancer cells of pancreatic cancer patients. J Surg Res. 2011; 171:631–6. https://doi.org/10.1016/j.jss.2010.05.007.
190. Khoja L, Backen A, Sloane R, Menasce L, Ryder D, Krebs M, Board R, Clack G, Hughes A, Blackhall F, Valle JW, Dive C. A pilot study to explore circulating tumour cells in pancreatic cancer as a novel biomarker. Br J Cancer. 2012; 106:508–16. https://doi.org/10.1038/bjc.2011.545.
191. Han L, Chen W, Zhao Q. Prognostic value of circulating tumor cells in patients with pancreatic cancer: a meta-analysis. Tumour Biol. 2014; 35:2473–80. https://doi.org/10.1007/s13277-013-1327-5.
192. Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, Ciciliano JC, Smas ME, Winokur D, Gilman AJ, Ulman MJ, Xega K, Contino G, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signaling in metastasis. Nature. 2012; 487:510–3. https://doi.org/10.1038/nature11217.
193. Komar G, Kauhanen S, Liukko K, Seppanen M, Kajander S, Ovaska J, Nuutila P, Minn H. Decreased blood flow with increased metabolic activity: a novel sign of pancreatic tumor aggressiveness. Clin Cancer Res. 2009; 15:5511–7. https://doi.org/10.1158/1078-0432.ccr-09-0414.
194. Catenacci DVT, Chapman CG, Xu P, Koons A, Konda VJ, Siddiqui UD, Waxman I. Acquisition of Portal Venous Circulating Tumor Cells From Patients With Pancreaticobiliary Cancers by Endoscopic Ultrasound. Gastroenterology. 2015; 149:1794–803.e4. https://doi.org/10.1053/j.gastro.2015.08.050.
195. Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, Desai R, Zhu H, Comaills V, Zheng Z, Wittner BS, Stojanov P, Brachtel E, et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014; 345:216–20. https://doi.org/10.1126/science.1253533.
196. Bobek V, Kacprzak G, Rzechonek A, Kolostova K. Detection and cultivation of circulating tumor cells in malignant pleural mesothelioma. Anticancer Res. 2014; 34:2565–9.
197. Bobek V, Matkowski R, Gurlich R, Grabowski K, Szelachowska J, Lischke R, Schutzner J, Harustiak T, Pazdro A, Rzechonek A, Kolostova K. Cultivation of circulating tumor cells in esophageal cancer. Folia Histochem Cytobiol. 2014; 52:171–7. https://doi.org/10.5603/fhc.2014.0020.
198. Cegan M, Kolostova K, Matkowski R, Broul M, Schraml J, Fiutowski M, Bobek V. In vitro culturing of viable circulating tumor cells of urinary bladder cancer. Int J Clin Exp Pathol. 2014; 7:7164–71.
199. Zhang Z, Shiratsuchi H, Lin J, Chen G, Reddy RM, Azizi E, Fouladdel S, Chang AC, Lin L, Jiang H, Waghray M, Luker G, Simeone DM, et al. Expansion of CTCs from early stage lung cancer patients using a microfluidic co-culture model. Oncotarget. 2014; 5:12383–97. https://doi.org/10.18632/oncotarget.2592.
200. Cayrefourcq L, Mazard T, Joosse S, Solassol J, Ramos J, Assenat E, Schumacher U, Costes V, Maudelonde T, Pantel K, Alix-Panabieres C. Establishment and characterization of a cell line from human circulating colon cancer cells. Cancer Res. 2015; 75:892–901. https://doi.org/10.1158/0008-5472.can-14-2613.
201. Voskoglou-Nomikos T, Pater JL, Seymour L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin Cancer Res. 2003; 9:4227–39.
202. Johnson JI, Decker S, Zaharevitz D, Rubinstein LV, Venditti JM, Schepartz S, Kalyandrug S, Christian M, Arbuck S, Hollingshead M, Sausville EA. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. 2001; 84:1424–31. https://doi.org/10.1054/bjoc.2001.1796.
203. End DW, Smets G, Todd AV, Applegate TL, Fuery CJ, Angibaud P, Venet M, Sanz G, Poignet H, Skrzat S, Devine A, Wouters W, Bowden C. Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro. Cancer Res. 2001; 61:131–7.
204. Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, Schoffski P, Post S, Verslype C, Neumann H, Safran H, Humblet Y, Perez Ruixo J, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004; 22:1430–8. https://doi.org/10.1200/jco.2004.10.112.
205. Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, Kashin S, Mekhoubad S, Ilic D, Charlton M, Saphier G, Handsaker RE, Genovese G, et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature. 2017; 545:229–33. https://doi.org/10.1038/nature22312.
206. Hodgkinson CL, Morrow CJ, Li Y, Metcalf RL, Rothwell DG, Trapani F, Polanski R, Burt DJ, Simpson KL, Morris K, Pepper SD, Nonaka D, Greystoke A, et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med. 2014; 20:897–903. https://doi.org/10.1038/nm.3600.
207. Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V, Klein C, Saini M, Bauerle T, Wallwiener M, Holland-Letz T, Hofner T, Sprick M, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol. 2013; 31:539–44. https://doi.org/10.1038/nbt.2576.
208. Rossi E, Rugge M, Facchinetti A, Pizzi M, Nardo G, Barbieri V, Manicone M, De Faveri S, Chiara Scaini M, Basso U, Amadori A, Zamarchi R. Retaining the long-survive capacity of Circulating Tumor Cells (CTCs) followed by xeno-transplantation: not only from metastatic cancer of the breast but also of prostate cancer patients. Oncoscience. 2014; 1:49–56. https://doi.org/10.18632/oncoscience.8.
209. Williams ES, Rodriguez-Bravo V, Chippada-Venkata U, De Ia Iglesia-Vicente J, Gong Y, Galsky M, Oh W, Cordon-Cardo C, Domingo-Domenech J. Generation of Prostate Cancer Patient Derived Xenograft Models from Circulating Tumor Cells. J Vis Exp. 2015; 53182. https://doi.org/10.3791/53182.
210. Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, Wongvipat J, Kossai M, Ramazanoglu S, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014; 159:176–87. https://doi.org/10.1016/j.cell.2014.08.016.
211. Giuliano M, Herrera S, Christiny P, Shaw C, Creighton CJ, Mitchell T, Bhat R, Zhang X, Mao S, Dobrolecki LE, Al-rawi A, Chen F, Veneziani BM, et al. Circulating and disseminated tumor cells from breast cancer patient-derived xenograft-bearing mice as a novel model to study metastasis. Breast Cancer Research. 2015; 17:3. https://doi.org/10.1186/s13058-014-0508-5.
212. Khoo BL, Grenci G. Liquid biopsy and therapeutic response: Circulating tumor cell cultures for evaluation of anticancer treatment. Sci Adv. 2016; 2:e1600274. https://doi.org/10.1126/sciadv.1600274.
213. Torphy RJ, Tignanelli CJ, Kamande JW, Moffitt RA, Herrera Loeza SG, Soper SA, Yeh JJ. Circulating tumor cells as a biomarker of response to treatment in patient-derived xenograft mouse models of pancreatic adenocarcinoma. PLoS One. 2014; 9:e89474. https://doi.org/10.1371/journal.pone.0089474.
214. Yu KH, Ricigliano M, Hidalgo M, Abou-Alfa GK, Lowery MA, Saltz LB, Crotty JF, Gary K, Cooper B, Lapidus R, Sadowska M, O’Reilly EM. Pharmacogenomic Modeling of Circulating Tumor and Invasive Cells for Prediction of Chemotherapy Response and Resistance in Pancreatic Cancer. Clinical Cancer Research. 2014; 20:5281–9. https://doi.org/10.1158/1078-0432.ccr-14-0531.
215. Mansour H. Cell-free nucleic acids as noninvasive biomarkers for colorectal cancer detection. Front Genet. 2014; 5. https://doi.org/10.3389/fgene.2014.00182.
216. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA Jr. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008; 14:985–90. https://doi.org/10.1038/nm.1789.
217. Kohler C, Barekati Z, Radpour R, Zhong XY. Cell-free DNA in the circulation as a potential cancer biomarker. Anticancer Res. 2011; 31:2623–8.
218. Schwarzenbach H, Nishida N, Calin GA, Pantel K. Clinical relevance of circulating cell-free microRNAs in cancer. Nat Rev Clin Oncol. 2014; 11:145–56. https://doi.org/10.1038/nrclinonc.2014.5.
219. Akca H, Demiray A, Yaren A, Bir F, Koseler A, Iwakawa R, Bagci G, Yokota J. Utility of serum DNA and pyrosequencing for the detection of EGFR mutations in non-small cell lung cancer. Cancer Genet. 2013; 206:73–80. https://doi.org/10.1016/j.cancergen.2013.01.005.
220. Stroun M, Anker P. Nucleic acids spontaneously released by living frog auricles. Biochem J. 1972; 128:100p-1p.
221. Stroun M, Lyautey J, Lederrey C, Olson-Sand A, Anker P. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta. 2001; 313:139–42.
222. Rogers JC, Boldt D, Kornfeld S, Skinner A, Valeri CR. Excretion of deoxyribonucleic acid by lymphocytes stimulated with phytohemagglutinin or antigen. Proc Natl Acad Sci U S A. 1972; 69:1685–9.
223. Anker P, Stroun M, Maurice PA. Spontaneous release of DNA by human blood lymphocytes as shown in an in vitro system. Cancer Res. 1975; 35:2375–82.
224. Delgado PO, Alves BC, Gehrke Fde S, Kuniyoshi RK, Wroclavski ML, Del Giglio A, Fonseca FL. Characterization of cell-free circulating DNA in plasma in patients with prostate cancer. Tumour Biol. 2013; 34:983–6. https://doi.org/10.1007/s13277-012-0634-6.
225. Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat. 2001; 4:303–13. https://doi.org/10.1054/drup.2001.0213.
226. Laktionov PP, Tamkovich SN, Rykova EY, Bryzgunova OE, Starikov AV, Kuznetsova NP, Sumarokov SV, Kolomiets SA, Sevostianova NV, Vlassov VV. Extracellular circulating nucleic acids in human plasma in health and disease. Nucleosides Nucleotides Nucleic Acids. 2004; 23:879–83. https://doi.org/10.1081/ncn-200026035.
227. Lo YM, Chan LY, Lo KW, Leung SF, Zhang J, Chan AT, Lee JC, Hjelm NM, Johnson PJ, Huang DP. Quantitative analysis of cell-free Epstein-Barr virus DNA in plasma of patients with nasopharyngeal carcinoma. Cancer Res. 1999; 59:1188–91.
228. Yang HJ, Liu VW, Tsang PC, Yip AM, Tam KF, Wong LC, Ng TY, Ngan HY. Quantification of human papillomavirus DNA in the plasma of patients with cervical cancer. Int J Gynecol Cancer. 2004; 14:903–10. https://doi.org/10.1111/j.1048-891X.2004.014528.x.
229. Ono A, Fujimoto A, Yamamoto Y, Akamatsu S, Hiraga N, Imamura M, Kawaoka T, Tsuge M, Abe H, Hayes CN, Miki D, Furuta M, Tsunoda T, et al. Circulating Tumor DNA Analysis for Liver Cancers and Its Usefulness as a Liquid Biopsy. Cell Mol Gastroenterol Hepatol. 2015; 1:516–34. https://doi.org/10.1016/j.jcmgh.2015.06.009.
230. Huang X, O’Connor R, Kwizera EA. Gold Nanoparticle Based Platforms for Circulating Cancer Marker Detection. Nanotheranostics. 2017; 1:80–102. https://doi.org/10.7150/ntno.18216.
231. Tamkovich SN, Cherepanova AV, Kolesnikova EV, Rykova EY, Pyshnyi DV, Vlassov VV, Laktionov PP. Circulating DNA and DNase activity in human blood. Ann N Y Acad Sci. 2006; 1075:191–6. https://doi.org/10.1196/annals.1368.026.
232. Tsui NB, Jiang P, Chow KC, Su X, Leung TY, Sun H, Chan KC, Chiu RW, Lo YM. High resolution size analysis of fetal DNA in the urine of pregnant women by paired-end massively parallel sequencing. PLoS One. 2012; 7:e48319. https://doi.org/10.1371/journal.pone.0048319.
233. Reckamp KL, Melnikova VO, Karlovich C, Sequist LV, Camidge DR, Wakelee H, Perol M, Oxnard GR, Kosco K, Croucher P, Samuelsz E, Vibat CR, Guerrero S, et al. A Highly Sensitive and Quantitative Test Platform for Detection of NSCLC EGFR Mutations in Urine and Plasma. J Thorac Oncol. 2016; 11:1690–700. https://doi.org/10.1016/j.jtho.2016.05.035.
234. Chan KC, Jiang P, Chan CW, Sun K, Wong J, Hui EP, Chan SL, Chan WC, Hui DS, Ng SS, Chan HL, Wong CS, Ma BB, et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc Natl Acad Sci U S A. 2013; 110:18761–8. https://doi.org/10.1073/pnas.1313995110.
235. Chused TM, Steinberg AD, Talal N. The clearance and localization of nucleic acids by New Zealand and normal mice. Clinical and Experimental Immunology. 1972; 12:465–76.
236. Heitzer E, Auer M, Hoffmann EM, Pichler M, Gasch C, Ulz P, Lax S, Waldispuehl-Geigl J, Mauermann O, Mohan S, Pristauz G, Lackner C, Höfler G, et al. Establishment of tumor-specific copy number alterations from plasma DNA of patients with cancer. Int J Cancer. 2013; 133:346–56. https://doi.org/10.1002/ijc.28030.
237. Thierry AR, Mouliere F, Gongora C, Ollier J, Robert B, Ychou M, Del Rio M, Molina F. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 2010; 38:6159–75. https://doi.org/10.1093/nar/gkq421.
238. Mouliere F, Robert B, Arnau Peyrotte E, Del Rio M, Ychou M, Molina F, Gongora C, Thierry AR. High fragmentation characterizes tumour-derived circulating DNA. PLoS One. 2011; 6:e23418. https://doi.org/10.1371/journal.pone.0023418.
239. Chan KC, Zhang J, Hui AB, Wong N, Lau TK, Leung TN, Lo KW, Huang DW, Lo YM. Size distributions of maternal and fetal DNA in maternal plasma. Clin Chem. 2004; 50:88–92. https://doi.org/10.1373/clinchem.2003.024893.
240. To EW, Chan KC, Leung SF, Chan LY, To KF, Chan AT, Johnson PJ, Lo YM. Rapid clearance of plasma Epstein-Barr virus DNA after surgical treatment of nasopharyngeal carcinoma. Clin Cancer Res. 2003; 9:3254–9.
241. Yao W, Mei C, Nan X, Hui L. Evaluation and comparison of in vitro degradation kinetics of DNA in serum, urine and saliva: A qualitative study. Gene. 2016; 590:142–8. https://doi.org/10.1016/j.gene.2016.06.033.
242. Jiang P, Lo YMD. The Long and Short of Circulating Cell-Free DNA and the Ins and Outs of Molecular Diagnostics. Trends Genet. 2016; 32:360–71. https://doi.org/10.1016/j.tig.2016.03.009.
243. Galluzzi L, Kroemer G. Secondary Necrosis: Accidental No More. Trends Cancer. 2017; 3:1–2. https://doi.org/10.1016/j.trecan.2016.12.001.
244. Wang BG, Huang HY, Chen YC, Bristow RE, Kassauei K, Cheng CC, Roden R, Sokoll LJ, Chan DW, Shih Ie M. Increased plasma DNA integrity in cancer patients. Cancer Res. 2003; 63:3966–8.
245. Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, Knippers R. DNA Fragments in the Blood Plasma of Cancer Patients: Quantitations and Evidence for Their Origin from Apoptotic and Necrotic Cells. Cancer Research. 2001; 61:1659–65.
246. Raymond CK, Hernandez J, Karr R, Hill K, Li M. Collection of cell-free DNA for genomic analysis of solid tumors in a clinical laboratory setting. PLoS One. 2017; 12:e0176241. https://doi.org/10.1371/journal.pone.0176241.
247. Jiang WW, Zahurak M, Goldenberg D, Milman Y, Park HL, Westra WH, Koch W, Sidransky D, Califano J. Increased plasma DNA integrity index in head and neck cancer patients. Int J Cancer. 2006; 119:2673–6. https://doi.org/10.1002/ijc.22250.
248. Zaher ER, Anwar MM, Kohail HMA, El-Zoghby SM, Abo-El-Eneen MS. Value of circulating DNA concentration and integrity as a screening test for detection of cancer in an Egyptian cohort. Alexandria Journal of Medicine. 2012; 48:187–96. https://doi.org/http://dx.doi.org/10.1016/j.ajme.2012.03.003.
249. Kamel AM, Teama S, Fawzy A, El Deftar M. Plasma DNA integrity index as a potential molecular diagnostic marker for breast cancer. Tumour Biol. 2016; 37:7565–72. https://doi.org/10.1007/s13277-015-4624-3.
250. Underhill HR, Kitzman JO, Hellwig S, Welker NC, Daza R, Baker DN, Gligorich KM, Rostomily RC, Bronner MP, Shendure J. Fragment Length of Circulating Tumor DNA. PLoS Genetics. 2016; 12:e1006162. https://doi.org/10.1371/journal.pgen.1006162.
251. Sun K, Jiang P, Chan KC, Wong J, Cheng YK, Liang RH, Chan WK, Ma ES, Chan SL, Cheng SH, Chan RW, Tong YK, Ng SS, et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci U S A. 2015; 112:E5503–12. https://doi.org/10.1073/pnas.1508736112.
252. Stroun M, Maurice P, Vasioukhin V, Lyautey J, Lederrey C, Lefort F, Rossier A, Chen XQ, Anker P. The origin and mechanism of circulating DNA. Ann N Y Acad Sci. 2000; 906:161–8.
253. Stroun M, Lyautey J, Lederrey C, Mulcahy HE, Anker P. Alu repeat sequences are present in increased proportions compared to a unique gene in plasma/serum DNA: evidence for a preferential release from viable cells? Ann N Y Acad Sci. 2001; 945:258–64.
254. Rogers JC. Identification of an intracellular precursor to DNA excreted by human lymphocytes. Proc Natl Acad Sci U S A. 1976; 73:3211–5.
255. Kostiuk SV, Malinovskaia EM, Ermakov AV, Smirnova TD, Kameneva LV, Chvartatskaia OV, Loseva PA, Ershova ES, Liubchenko LN, Veiko NN. [Cell-free DNA fragments increase transcription in human mesenchymal stem cells, activate TLR-dependent signal pathway and supress apoptosis]. [Article in Russian]. Biomed Khim. 2012; 58:673–83.
256. Fleischhacker M, Schmidt B. Circulating nucleic acids (CNAs) and cancer—a survey. Biochim Biophys Acta. 2007; 1775:181–232. https://doi.org/10.1016/j.bbcan.2006.10.001.
257. Elshimali YI, Khaddour H, Sarkissyan M, Wu Y, Vadgama JV. The Clinical Utilization of Circulating Cell Free DNA (CCFDNA) in Blood of Cancer Patients. Int J Mol Sci. 2013; 14:18925–58. https://doi.org/10.3390/ijms140918925.
258. Marzese DM, Hirose H, Hoon DS. Diagnostic and prognostic value of circulating tumor-related DNA in cancer patients. Expert Rev Mol Diagn. 2013; 13:827–44. https://doi.org/10.1586/14737159.2013.845088.
259. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci Transl Med. 2014; 6:224ra24. https://doi.org/10.1126/scitranslmed.3007094.
260. Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol. 2017; 14:531–548. https://doi.org/10.1038/nrclinonc.2017.14.
261. Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem. 2015; 61:112–23. https://doi.org/10.1373/clinchem.2014.222679.
262. Qin Z, Ljubimov VA, Zhou C, Tong Y, Liang J. Cell-free circulating tumor DNA in cancer. Chin J Cancer. 2016; 35:36. https://doi.org/10.1186/s40880-016-0092-4.
263. Beck J, Oellerich M, Schulz U, Schauerte V, Reinhard L, Fuchs U, Knabbe C, Zittermann A, Olbricht C, Gummert JF, Shipkova M, Birschmann I, Wieland E, et al. Donor-Derived Cell-Free DNA Is a Novel Universal Biomarker for Allograft Rejection in Solid Organ Transplantation. Transplant Proc. 2015; 47:2400–3. https://doi.org/10.1016/j.transproceed.2015.08.035.
264. Beck J, Bierau S, Balzer S, Andag R, Kanzow P, Schmitz J, Gaedcke J, Moerer O, Slotta JE, Walson P, Kollmar O, Oellerich M, Schutz E. Digital droplet PCR for rapid quantification of donor DNA in the circulation of transplant recipients as a potential universal biomarker of graft injury. Clin Chem. 2013; 59:1732–41. https://doi.org/10.1373/clinchem.2013.210328.
265. Zou J, Duffy B, Slade M, Young AL, Steward N, Hachem R, Mohanakumar T. Rapid detection of donor cell free DNA in lung transplant recipients with rejections using donor-recipient HLA mismatch. Hum Immunol. 2017; 78:342–349. https://doi.org/10.1016/j.humimm.2017.03.002.
266. Adamek M, Opelz G, Klein K, Morath C, Tran TH. A fast and simple method for detecting and quantifying donor-derived cell-free DNA in sera of solid organ transplant recipients as a biomarker for graft function. Clin Chem Lab Med. 2016; 54:1147–55. https://doi.org/10.1515/cclm-2015-0622.
267. Grskovic M, Hiller DJ, Eubank LA, Sninsky JJ, Christopherson C, Collins JP, Thompson K, Song M, Wang YS, Ross D, Nelles MJ, Yee JP, Wilber JC, et al. Validation of a Clinical-Grade Assay to Measure Donor-Derived Cell-Free DNA in Solid Organ Transplant Recipients. J Mol Diagn. 2016; 18:890–902. https://doi.org/10.1016/j.jmoldx.2016.07.003.
268. Bloom RD, Bromberg JS, Poggio ED, Bunnapradist S, Langone AJ, Sood P, Matas AJ, Mehta S, Mannon RB, Sharfuddin A, Fischbach B, Narayanan M, Jordan SC, et al. Cell-Free DNA and Active Rejection in Kidney Allografts. J Am Soc Nephrol. 2017; 28:2221–2232. https://doi.org/10.1681/asn.2016091034.
269. Shapiro B, Chakrabarty M, Cohn EM, Leon SA. Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer. 1983; 51:2116–20.
270. Silva JM, Garcia JM, Dominguez G, Silva J, Miralles C, Cantos B, Coca S, Provencio M, Espana P, Bonilla F. Persistence of tumor DNA in plasma of breast cancer patients after mastectomy. Ann Surg Oncol. 2002; 9:71–6.
271. Sozzi G, Conte D, Leon M, Ciricione R, Roz L, Ratcliffe C, Roz E, Cirenei N, Bellomi M, Pelosi G, Pierotti MA, Pastorino U. Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol. 2003; 21:3902–8. https://doi.org/10.1200/jco.2003.02.006.
272. Ren N, Ye QH, Qin LX, Zhang BH, Liu YK, Tang ZY. Circulating DNA level is negatively associated with the long-term survival of hepatocellular carcinoma patients. World J Gastroenterol. 2006; 12:3911–4.
273. Boddy JL, Gal S, Malone PR, Harris AL, Wainscoat JS. Prospective study of quantitation of plasma DNA levels in the diagnosis of malignant versus benign prostate disease. Clin Cancer Res. 2005; 11:1394–9. https://doi.org/10.1158/1078-0432.ccr-04-1237.
274. Banki F, Mason RJ, Oh D, Hagen JA, DeMeester SR, Lipham JC, Tanaka K, Danenberg KD, Yacoub WN, Danenberg PV, DeMeester TR. Plasma DNA as a molecular marker for completeness of resection and recurrent disease in patients with esophageal cancer. Arch Surg. 2007; 142:533–8. https://doi.org/10.1001/archsurg.142.6.533.
275. Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012; 486:537–40. https://doi.org/10.1038/nature11219.
276. Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, Dawson SJ, Piskorz AM, Jimenez-Linan M, Bentley D, Hadfield J, May AP, Caldas C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012; 4:136ra68. https://doi.org/10.1126/scitranslmed.3003726.
277. McBride DJ, Orpana AK, Sotiriou C, Joensuu H, Stephens PJ, Mudie LJ, Hamalainen E, Stebbings LA, Andersson LC, Flanagan AM, Durbecq V, Ignatiadis M, Kallioniemi O, et al. Use of cancer-specific genomic rearrangements to quantify disease burden in plasma from patients with solid tumors. Genes Chromosomes Cancer. 2010; 49:1062–9. https://doi.org/10.1002/gcc.20815.
278. Garcia-Olmo DC, Samos J, Picazo MG, Asensio AI, Toboso I, Garcia-Olmo D. Release of cell-free DNA into the bloodstream leads to high levels of non-tumor plasma DNA during tumor progression in rats. Cancer Lett. 2008; 272:133–40. https://doi.org/10.1016/j.canlet.2008.07.003.
279. Gonzalez-Masia JA, Garcia-Olmo D, Garcia-Olmo DC. Circulating nucleic acids in plasma and serum (CNAPS): applications in oncology. Onco Targets Ther. 2013; 6:819–32. https://doi.org/10.2147/ott.s44668.
280. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012; 486:532–6. https://doi.org/10.1038/nature11156.
281. Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, Marass F, Humphray S, Hadfield J, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013; 497:108–12. https://doi.org/10.1038/nature12065.
282. Salvi S, Gurioli G, De Giorgi U, Conteduca V, Tedaldi G, Calistri D, Casadio V. Cell-free DNA as a diagnostic marker for cancer: current insights. Onco Targets Ther. 2016; 9:6549–59. https://doi.org/10.2147/ott.s100901.
283. van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002; 415:530–6. https://doi.org/10.1038/415530a.
284. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, Rajan S, Humphray S, Becq J, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013; 368:1199–209. https://doi.org/10.1056/NEJMoa1213261.
285. Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz LA Jr, Goodman SN, David KA, Juhl H, Kinzler KW, Vogelstein B. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005; 102:16368–73. https://doi.org/10.1073/pnas.0507904102.
286. Chan KC, Jiang P, Zheng YW, Liao GJ, Sun H, Wong J, Siu SS, Chan WC, Chan SL, Chan AT, Lai PB, Chiu RW, Lo YM. Cancer genome scanning in plasma: detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin Chem. 2013; 59:211–24. https://doi.org/10.1373/clinchem.2012.196014.
287. Bohers E, Viailly PJ, Dubois S, Bertrand P, Maingonnat C, Mareschal S, Ruminy P, Picquenot JM, Bastard C, Desmots F, Fest T, Leroy K, Tilly H, et al. Somatic mutations of cell-free circulating DNA detected by next-generation sequencing reflect the genetic changes in both germinal center B-cell-like and activated B-cell-like diffuse large B-cell lymphomas at the time of diagnosis. Haematologica. 2015; 100:e280–4. https://doi.org/10.3324/haematol.2015.123612.
288. Parkinson CA, Gale D, Piskorz AM, Biggs H, Hodgkin C, Addley H, Freeman S, Moyle P, Sala E, Sayal K, Hosking K, Gounaris I, Jimenez-Linan M, et al. Exploratory Analysis of TP53 Mutations in Circulating Tumour DNA as Biomarkers of Treatment Response for Patients with Relapsed High-Grade Serous Ovarian Carcinoma: A Retrospective Study. PLoS Medicine. 2016; 13:e1002198. https://doi.org/10.1371/journal.pmed.1002198.
289. Taly V, Pekin D, Benhaim L, Kotsopoulos SK, Le Corre D, Li X, Atochin I, Link DR, Griffiths AD, Pallier K, Blons H, Bouche O, Landi B, et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin Chem. 2013; 59:1722–31. https://doi.org/10.1373/clinchem.2013.206359.
290. Huang A, Zhang X, Zhou SL, Cao Y, Huang XW, Fan J, Yang XR, Zhou J. Detecting Circulating Tumor DNA in Hepatocellular Carcinoma Patients Using Droplet Digital PCR Is Feasible and Reflects Intratumoral Heterogeneity. J Cancer. 2016; 7:1907–14. https://doi.org/10.7150/jca.15823.
291. Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A. 2003; 100:8817–22. https://doi.org/10.1073/pnas.1133470100.
292. Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D. BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods. 2006; 3:551–9. https://doi.org/10.1038/nmeth898.
293. Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, Shrager JB, Loo BW Jr. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014; 20:548–54. https://doi.org/10.1038/nm.3519.
294. Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 2011; 108:9530–5. https://doi.org/10.1073/pnas.1105422108.
295. Kennedy SR. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat Protoc. 2014; 9:2586–606. https://doi.org/10.1038/nprot.2014.170.
296. Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, Stehr H, Liu CL, Bratman SV, Say C, Zhou L, Carter JN, West RB, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016; 34:547–55. https://doi.org/10.1038/nbt.3520.
297. Heitzer E, Ulz P, Belic J, Gutschi S, Quehenberger F, Fischereder K, Benezeder T, Auer M, Pischler C, Mannweiler S. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013; 5. https://doi.org/10.1186/gm434.
298. Leary RJ, Sausen M, Kinde I, Papadopoulos N, Carpten JD, Craig D, O’Shaughnessy J, Kinzler KW, Parmigiani G, Vogelstein B. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med. 2012; 4. https://doi.org/10.1126/scitranslmed.3004742.
299. Pietrasz D, Pecuchet N, Garlan F, Didelot A, Dubreuil O, Doat S, Imbert-Bismut F, Karoui M, Vaillant JC, Taly V, Laurent-Puig P, Bachet JB. Plasma Circulating Tumor DNA in Pancreatic Cancer Patients Is a Prognostic Marker. Clin Cancer Res. 2017; 23:116–23. https://doi.org/10.1158/1078-0432.ccr-16-0806.
300. Xue X, Teare MD, Holen I, Zhu YM, Woll PJ. Optimizing the yield and utility of circulating cell-free DNA from plasma and serum. Clin Chim Acta. 2009; 404:100–4. https://doi.org/10.1016/j.cca.2009.02.018.
301. Sherwood JL, Corcoran C, Brown H, Sharpe AD, Musilova M, Kohlmann A. Optimised Pre-Analytical Methods Improve KRAS Mutation Detection in Circulating Tumour DNA (ctDNA) from Patients with Non-Small Cell Lung Cancer (NSCLC). PLoS One. 2016; 11. https://doi.org/10.1371/journal.pone.0150197.
302. Vallee A, Marcq M, Bizieux A, Kouri CE, Lacroix H, Bennouna J, Douillard JY, Denis MG. Plasma is a better source of tumor-derived circulating cell-free DNA than serum for the detection of EGFR alterations in lung tumor patients. Lung Cancer. 2013; 82:373–4. https://doi.org/10.1016/j.lungcan.2013.08.014.
303. Ma M, Zhu H, Zhang C, Sun X, Gao X, Chen G. “Liquid biopsy”—ctDNA detection with great potential and challenges. Ann Transl Med. 2015; 3. https://doi.org/10.3978/j.issn.2305-5839.2015.09.29.
304. Yi X, Ma J, Guan Y, Chen R, Yang L, Xia X. The feasibility of using mutation detection in ctDNA to assess tumor dynamics. Int J Cancer. 2017; 140:2642–7. https://doi.org/10.1002/ijc.30620.
305. Ng SB, Chua C, Ng M, Gan A, Poon PS, Teo M, Fu C, Leow WQ, Lim KH, Chung A, Koo SL, Choo SP, Ho D, et al. Individualised multiplexed circulating tumour DNA assays for monitoring of tumour presence in patients after colorectal cancer surgery. Sci Rep. 2017; 7. https://doi.org/10.1038/srep40737.
306. Maire F, Micard S, Hammel P, Voitot H, Lévy P, Cugnenc P, Ruszniewski P, Puig PL. Differential diagnosis between chronic pancreatitis and pancreatic cancer: value of the detection of KRAS2 mutations in circulating DNA. Br J Cancer. 2002; 87:551–4. https://doi.org/10.1038/sj.bjc.6600475.
307. Berger AW, Schwerdel D, Costa IG, Hackert T, Strobel O, Lam S, Barth TF, Schroppel B, Meining A, Buchler MW, Zenke M, Hermann PC, Seufferlein T, et al. Detection of Hot-Spot Mutations in Circulating Cell-Free DNA From Patients With Intraductal Papillary Mucinous Neoplasms of the Pancreas. Gastroenterology. 2016; 151:267–70. https://doi.org/10.1053/j.gastro.2016.04.034.
308. Cheng H, Liu C, Jiang J, Luo G, Lu Y, Jin K, Guo M, Zhang Z, Xu J, Liu L, Ni Q, Yu X. Analysis of ctDNA to predict prognosis and monitor treatment responses in metastatic pancreatic cancer patients. Int J Cancer. 2017; 140:2344–50. https://doi.org/10.1002/ijc.30650.
309. Zill OA, Greene C, Sebisanovic D, Siew LM, Leng J, Vu M, Hendifar AE, Wang Z, Atreya CE, Kelley RK, Van Loon K, Ko AH, Tempero MA, et al. Cell-Free DNA Next-Generation Sequencing in Pancreatobiliary Carcinomas. Cancer Discov. 2015; 5:1040–8. https://doi.org/10.1158/2159-8290.cd-15-0274.
310. Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, Anagnostou V, Parpart-Li S, Murphy D, Kay Li Q, Hruban CA, Scharpf R, White JR, et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun. 2015; 6:7686. https://doi.org/10.1038/ncomms8686.
311. Henriksen SD, Madsen PH, Larsen AC, Johansen MB, Drewes AM, Pedersen IS, Krarup H, Thorlacius-Ussing O. Cell-free DNA promoter hypermethylation in plasma as a diagnostic marker for pancreatic adenocarcinoma. Clin Epigenetics. 2016; 8:117. https://doi.org/10.1186/s13148-016-0286-2.
312. Tjensvoll K, Lapin M, Buhl T, Oltedal S, Steen-Ottosen Berry K, Gilje B, Soreide JA, Javle M, Nordgard O, Smaaland R. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol Oncol. 2016; 10:635–43. https://doi.org/10.1016/j.molonc.2015.11.012.
313. Singh N, Gupta S, Pandey RM, Chauhan SS, Saraya A. High levels of cell-free circulating nucleic acids in pancreatic cancer are associated with vascular encasement, metastasis and poor survival. Cancer Invest. 2015; 33:78–85. https://doi.org/10.3109/07357907.2014.1001894.
314. Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014; 39:91–100. https://doi.org/10.1016/j.tibs.2013.12.004.
315. Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer. 2014; 111:817–22. https://doi.org/10.1038/bjc.2014.215.
316. Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, Hruban RH, Maitra A, Kinzler K, Vogelstein B, Goggins M. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012; 142:730–3.e9. https://doi.org/10.1053/j.gastro.2011.12.042.
317. Yachida S, White CM, Naito Y, Zhong Y, Brosnan JA, Macgregor-Das AM, Morgan RA, Saunders T, Laheru DA, Herman JM, Hruban RH, Klein AP, Jones S, et al. Clinical Significance of the Genetic Landscape of Pancreatic Cancer and Implications for Identification of Potential Long Term Survivors. Clin Cancer Res. 2012; 18:6339–47. https://doi.org/10.1158/1078-0432.CCR-12-1215.
318. Witkiewicz AK, McMillan EA, Balaji U, Baek GH, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, Choti MA, Yeo CJ, McCue P, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015; 6:6744. https://doi.org/10.1038/ncomms7744.
319. Kinugasa H, Nouso K, Miyahara K, Morimoto Y, Dohi C, Tsutsumi K, Kato H, Matsubara T, Okada H, Yamamoto K. Detection of K-ras gene mutation by liquid biopsy in patients with pancreatic cancer. Cancer. 2015; 121:2271–80. https://doi.org/10.1002/cncr.29364.
320. Matsubayashi H, Canto M, Sato N, Klein A, Abe T, Yamashita K, Yeo CJ, Kalloo A, Hruban R, Goggins M. DNA methylation alterations in the pancreatic juice of patients with suspected pancreatic disease. Cancer Res. 2006; 66:1208–17. https://doi.org/10.1158/0008-5472.can-05-2664.
321. Liggett T, Melnikov A, Yi QL, Replogle C, Brand R, Kaul K, Talamonti M, Abrams RA, Levenson V. Differential methylation of cell-free circulating DNA among patients with pancreatic cancer versus chronic pancreatitis. Cancer. 2010; 116:1674–80. https://doi.org/10.1002/cncr.24893.
322. Nogueira da Costa A, Herceg Z. Detection of cancer-specific epigenomic changes in biofluids: powerful tools in biomarker discovery and application. Mol Oncol. 2012; 6:704–15. https://doi.org/10.1016/j.molonc.2012.07.005.
323. Sato N, Goggins M. The role of epigenetic alterations in pancreatic cancer. J Hepatobiliary Pancreat Surg. 2006; 13:286–95. https://doi.org/10.1007/s00534-005-1057-1.
324. Tan AC, Jimeno A, Lin SH, Wheelhouse J, Chan F, Solomon A, Rajeshkumar NV, Rubio-Viqueira B, Hidalgo M. Characterizing DNA methylation patterns in pancreatic cancer genome. Molecular Oncology. 2009; 3:425–38. https://doi.org/10.1016/j.molonc.2009.03.004.
325. Warton K, Mahon KL, Samimi G. Methylated circulating tumor DNA in blood: power in cancer prognosis and response. Endocrine-Related Cancer. 2016; 23:R157-R71. https://doi.org/10.1530/ERC-15-0369.
326. Thompson MJ, Rubbi L, Dawson DW, Donahue TR, Pellegrini M. Pancreatic Cancer Patient Survival Correlates with DNA Methylation of Pancreas Development Genes. PLoS One. 2015; 10:e0128814. https://doi.org/10.1371/journal.pone.0128814.
327. McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, Word AE, Carrer A, Salz TH, Natsume S, Stauffer KM, Makohon-Moore A, Zhong Y, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet. 2017; 49:367–76. https://doi.org/10.1038/ng.3753.
328. Lehmann-Werman R, Neiman D, Zemmour H, Moss J, Magenheim J, Vaknin-Dembinsky A, Rubertsson S, Nellgård B, Blennow K, Zetterberg H, Spalding K, Haller MJ, Wasserfall CH, et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci U S A. 2016; 113:E1826-E34. https://doi.org/10.1073/pnas.1519286113.
329. Moran S, Martinez-Cardus A, Sayols S, Musulen E, Balana C, Estival-Gonzalez A, Moutinho C, Heyn H, Diaz-Lagares A, de Moura MC, Stella GM, Comoglio PM, Ruiz-Miro M, et al. Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol. 2016; 17:1386–95. https://doi.org/10.1016/s1470-2045(16)30297-2.
330. Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010; 1805:105–17. https://doi.org/10.1016/j.bbcan.2009.11.002.
331. Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature. 2013; 501:355–64. https://doi.org/10.1038/nature12627.
332. Murtaza M, Dawson SJ, Pogrebniak K, Rueda OM, Provenzano E, Grant J, Chin SF, Tsui DW, Marass F, Gale D, Ali HR, Shah P, Contente-Cuomo T, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun. 2015; 6:8760. https://doi.org/10.1038/ncomms9760.
333. Willms E, Johansson HJ, Mäger I, Lee Y, Blomberg KE, Sadik M, Alaarg A, Smith CI, Lehtiö J, El Andaloussi S, Wood MJ, Vader P. Cells release subpopulations of exosomes with distinct molecular and biological properties. Scientific Reports. 2016; 6:22519. https://doi.org/10.1038/srep22519.
334. Boukouris S, Mathivanan S. Exosomes in bodily fluids are a highly stable resource of disease biomarkers. Proteomics Clin Appl. 2015; 9:358–67. https://doi.org/10.1002/prca.201400114.
335. Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem. 1987; 262:9412–20.
336. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002; 2:569–79. https://doi.org/10.1038/nri855.
337. Pan BT, Teng K, Wu C, Adam M, Johnstone RM. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J Cell Biol. 1985; 101:942–8.
338. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007; 9:654–9. https://doi.org/10.1038/ncb1596.
339. Alexander M, Hu R, Runtsch MC, Kagele DA, Mosbruger TL, Tolmachova T, Seabra MC, Round JL, Ward DM, O’Connell RM. Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nature Communications. 2015; 6:7321. https://doi.org/10.1038/ncomms8321.
340. Tan CY, Lai RC, Wong W, Dan YY, Lim SK, Ho HK. Mesenchymal stem cell-derived exosomes promote hepatic regeneration in drug-induced liver injury models. Stem Cell Research & Therapy. 2014; 5:76. https://doi.org/10.1186/scrt465.
341. Keller S, Sanderson MP, Stoeck A, Altevogt P. Exosomes: from biogenesis and secretion to biological function. Immunol Lett. 2006; 107:102–8. https://doi.org/10.1016/j.imlet.2006.09.005.
342. Admyre C, Johansson SM, Qazi KR, Filen JJ, Lahesmaa R, Norman M, Neve EP, Scheynius A, Gabrielsson S. Exosomes with immune modulatory features are present in human breast milk. J Immunol. 2007; 179:1969–78.
343. Simhadri VR, Reiners KS, Hansen HP, Topolar D, Simhadri VL, Nohroudi K, Kufer TA, Engert A, Pogge von Strandmann E. Dendritic cells release HLA-B-associated transcript-3 positive exosomes to regulate natural killer function. PLoS One. 2008; 3:e3377. https://doi.org/10.1371/journal.pone.0003377.
344. Bahrini I, Song JH, Diez D, Hanayama R. Neuronal exosomes facilitate synaptic pruning by up-regulating complement factors in microglia. Scientific Reports. 2015; 5:7989. https://doi.org/10.1038/srep07989.
345. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013; 13:34–45. https://doi.org/10.1038/nri3345.
346. Guay C, Regazzi R. Circulating microRNAs as novel biomarkers for diabetes mellitus. Nat Rev Endocrinol. 2013; 9:513–21. https://doi.org/10.1038/nrendo.2013.86.
347. Lee MJ, Park DH, Kang JH. Exosomes as the source of biomarkers of metabolic diseases. Ann Pediatr Endocrinol Metab. 2016; 21:119–25. https://doi.org/10.6065/apem.2016.21.3.119.
348. Boulanger CM, Loyer X, Rautou PE, Amabile N. Extracellular vesicles in coronary artery disease. Nat Rev Cardiol. 2017; 14:259–72. https://doi.org/10.1038/nrcardio.2017.7.
349. Perrotta I, Aquila S. Exosomes in human atherosclerosis: An ultrastructural analysis study. Ultrastruct Pathol. 2016; 40:101–6. https://doi.org/10.3109/01913123.2016.1154912.
350. Masyuk AI, Masyuk TV, Larusso NF. Exosomes in the pathogenesis, diagnostics and therapeutics of liver diseases. J Hepatol. 2013; 59:621–5. https://doi.org/10.1016/j.jhep.2013.03.028.
351. Vella LJ, Sharples RA, Nisbet RM, Cappai R, Hill AF. The role of exosomes in the processing of proteins associated with neurodegenerative diseases. Eur Biophys J. 2008; 37:323–32. https://doi.org/10.1007/s00249-007-0246-z.
352. Azmi AS, Bao B, Sarkar FH. Exosomes in Cancer Development, Metastasis and Drug Resistance: A Comprehensive Review. Cancer Metastasis Rev. 2013; 32:10.1007/s10555-013-9441-9. https://doi.org/10.1007/s10555-013-9441-9.
353. Herring JM, McMichael MA, Smith SA. Microparticles in health and disease. J Vet Intern Med. 2013; 27:1020–33. https://doi.org/10.1111/jvim.12128.
354. Ciardiello C, Cavallini L, Spinelli C, Yang J, Reis-Sobreiro M, de Candia P, Minciacchi VR, Di Vizio D. Focus on Extracellular Vesicles: New Frontiers of Cell-to-Cell Communication in Cancer. Int J Mol Sci. 2016; 17:175. https://doi.org/10.3390/ijms17020175.
355. Kalluri R, LeBleu VS. Discovery of Double-Stranded Genomic DNA in Circulating Exosomes. Cold Spring Harb Symp Quant Biol. 2016; 81:275–280. https://doi.org/10.1101/sqb.2016.81.030932.
356. Beach A, Zhang HG, Ratajczak MZ, Kakar SS. Exosomes: an overview of biogenesis, composition and role in ovarian cancer. Journal of Ovarian Research. 2014; 7:14. https://doi.org/10.1186/1757-2215-7-14.
357. Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, Zheng Y, Hoshino A, Brazier H, Xiang J, Williams C, Rodriguez-Barrueco R, Silva JM, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014; 24:766–9. https://doi.org/10.1038/cr.2014.44.
358. Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003; 4:346–58. https://doi.org/10.1038/nrg1066.
359. Freedman JE, Gerstein M, Mick E, Rozowsky J, Levy D, Kitchen R, Das S, Shah R, Danielson K, Beaulieu L, Navarro FC, Wang Y, Galeev TR, et al. Diverse human extracellular RNAs are widely detected in human plasma. Nature Communications. 2016; 7:11106. https://doi.org/10.1038/ncomms11106.
360. Huang X, Yuan T, Tschannen M, Sun Z, Jacob H, Du M, Liang M, Dittmar RL, Liu Y, Liang M, Kohli M, Thibodeau SN, Boardman L, et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genomics. 2013; 14:319. https://doi.org/10.1186/1471-2164-14-319.
361. Mathivanan S, Fahner CJ, Reid GE, Simpson RJ. ExoCarta 2012: database of exosomal proteins, RNA and lipids. Nucleic Acids Res. 2012; 40:D1241–4. https://doi.org/10.1093/nar/gkr828.
362. Keerthikumar S, Chisanga D, Ariyaratne D, Saffar HA, Anand S, Zhao K, Samuel M, Pathan M, Jois M, Chilamkurti N, Gangoda L, Mathivanan S. ExoCarta: A web-based compendium of exosomal cargo. J Mol Biol. 2016; 428:688–92. https://doi.org/10.1016/j.jmb.2015.09.019.
363. Admyre C, Grunewald J, Thyberg J, Gripenback S, Tornling G, Eklund A, Scheynius A, Gabrielsson S. Exosomes with major histocompatibility complex class II and co-stimulatory molecules are present in human BAL fluid. Eur Respir J. 2003; 22:578–83.
364. Zhang HG, Grizzle WE. Exosomes and cancer: a newly described pathway of immune suppression. Clin Cancer Res. 2011; 17:959–64. https://doi.org/10.1158/1078-0432.ccr-10-1489.
365. Lee Y, El Andaloussi S, Wood MJ. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum Mol Genet. 2012; 21:R125-34. https://doi.org/10.1093/hmg/dds317.
366. Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008; 10:619–24. https://doi.org/10.1038/ncb1725.
367. Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008; 10:1470–6. https://doi.org/10.1038/ncb1800.
368. Ding G, Zhou L, Qian Y, Fu M, Chen J, Chen J, Xiang J, Wu Z, Jiang G, Cao L. Pancreatic cancer-derived exosomes transfer miRNAs to dendritic cells and inhibit RFXAP expression via miR-212-3p. Oncotarget. 2015; 6:29877–88. https://doi.org/10.18632/oncotarget.4924.
369. Syn N, Wang L, Sethi G, Thiery JP, Goh BC. Exosome-Mediated Metastasis: From Epithelial-Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol Sci. 2016; 37:606–17. https://doi.org/10.1016/j.tips.2016.04.006.
370. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009; 9:285–93. https://doi.org/10.1038/nrc2621.
371. Ordonez-Moran P, Huelsken J. Complex metastatic niches: already a target for therapy? Curr Opin Cell Biol. 2014; 31:29–38. https://doi.org/10.1016/j.ceb.2014.06.012.
372. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013; 19:1423–37. https://doi.org/10.1038/nm.3394.
373. Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, Singh S, Williams C, Soplop N, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015; 527:329–35. https://doi.org/10.1038/nature15756.
374. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014; 30:255–89. https://doi.org/10.1146/annurev-cellbio-101512-122326.
375. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013; 200:373–83. https://doi.org/10.1083/jcb.201211138.
376. Yu IM, Hughson FM. Tethering factors as organizers of intracellular vesicular traffic. Annu Rev Cell Dev Biol. 2010; 26:137–56. https://doi.org/10.1146/annurev.cellbio.042308.113327.
377. King HW, Michael MZ, Gleadle JM. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer. 2012; 12:421. https://doi.org/10.1186/1471-2407-12-421.
378. Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, Moita CF, Schauer K, Hume AN, Freitas RP, Goud B, Benaroch P, Hacohen N, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010; 12:19–30. https://doi.org/10.1038/ncb2000.
379. Bobrie A, Krumeich S, Reyal F, Recchi C, Moita LF, Seabra MC, Ostrowski M, Thery C. Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 2012; 72:4920–30. https://doi.org/10.1158/0008-5472.can-12-0925.
380. Yu X, Harris SL, Levine AJ. The regulation of exosome secretion: a novel function of the p53 protein. Cancer Res. 2006; 66:4795–801. https://doi.org/10.1158/0008-5472.can-05-4579.
381. Lespagnol A, Duflaut D, Beekman C, Blanc L, Fiucci G, Marine JC, Vidal M, Amson R, Telerman A. Exosome secretion, including the DNA damage-induced p53-dependent secretory pathway, is severely compromised in TSAP6//Steap3-null mice. Cell Death Differ. 2008; 15:1723–33. http://www.nature.com/cdd/journal/v15/n11/suppinfo/cdd2008104s1.html.
382. Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, Nitadori-Hoshino A, Hoffman C, Badal K, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012; 18:883–91. https://doi.org/10.1038/nm.2753.
383. Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO, Skog J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat Commun. 2011; 2:180. https://doi.org/10.1038/ncomms1180.
384. Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, Ratajczak MZ. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia. 2006; 20:847–56. https://doi.org/10.1038/sj.leu.2404132.
385. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, Becker A, Hoshino A, Mark MT, Molina H. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015; 17. https://doi.org/10.1038/ncb3169.
386. Li P, Kaslan M, Lee SH, Yao J, Gao Z. Progress in Exosome Isolation Techniques. Theranostics. 2017; 7:789–804. https://doi.org/10.7150/thno.18133.
387. Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, LeBleu VS, Mittendorf EA, Weitz J, Rahbari N. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015; 523. https://doi.org/10.1038/nature14581.
388. Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, Gho YS, Kurochkin IV, Mathivanan S, Quesenberry P, Sahoo S, Tahara H, Wauben MH, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. Journal of Extracellular Vesicles. 2014; 3:10.3402/jev.v3.26913. https://doi.org/10.3402/jev.v3.26913.
389. Que R, Ding G, Chen J, Cao L. Analysis of serum exosomal microRNAs and clinicopathologic features of patients with pancreatic adenocarcinoma. World J Surg Oncol. 2013; 11:219. https://doi.org/10.1186/1477-7819-11-219.
390. Kahlert C, Melo SA, Protopopov A, Tang J, Seth S, Koch M, Zhang J, Weitz J, Chin L, Futreal A, Kalluri R. Identification of Double-stranded Genomic DNA Spanning All Chromosomes with Mutated KRAS and p53 DNA in the Serum Exosomes of Patients with Pancreatic Cancer. J Biol Chem. 2014; 289:3869–75. https://doi.org/10.1074/jbc.C113.532267.
391. Madhavan B, Yue S, Galli U, Rana S, Gross W, Muller M, Giese NA, Kalthoff H, Becker T, Buchler MW, Zoller M. Combined evaluation of a panel of protein and miRNA serum-exosome biomarkers for pancreatic cancer diagnosis increases sensitivity and specificity. Int J Cancer. 2015; 136:2616–27. https://doi.org/10.1002/ijc.29324.
392. Kanwar SS, Dunlay CJ, Simeone DM, Nagrath S. Microfluidic device (ExoChip) for on-chip isolation, quantification and characterization of circulating exosomes. Lab Chip. 2014; 14:1891–900. https://doi.org/10.1039/c4lc00136b.
393. Allenson K, Castillo J, San Lucas FA, Scelo G, Kim DU, Bernard V, Davis G, Kumar T, Katz M, Overman MJ, Foretova L, Fabianova E, Holcatova I, et al. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann Oncol. 2017; 28:741–7. https://doi.org/10.1093/annonc/mdx004.
394. Khan S, Jutzy JMS, Aspe JR, Wall NR. Abstract B97: Exosomal survivin, a potential tool for early detection of pancreatic cancer health disparity? Cancer Research. 2012; 72:B97–B. https://doi.org/10.1158/1538-7445.panca2012-b97.
395. Shin SJ, Smith JA, Rezniczek GA, Pan S, Chen R, Brentnall TA, Wiche G, Kelly KA. Unexpected gain of function for the scaffolding protein plectin due to mislocalization in pancreatic cancer. Proc Natl Acad Sci U S A. 2013; 110:19414–9. https://doi.org/10.1073/pnas.1309720110.
396. Pang W, Su J, Wang Y, Feng H, Dai X, Yuan Y, Chen X, Yao W. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci. 2015; 106:1362–9. https://doi.org/10.1111/cas.12747.
397. Taucher V, Mangge H, Haybaeck J. Non-coding RNAs in pancreatic cancer: challenges and opportunities for clinical application. Cell Oncol (Dordr). 2016; 39:295–318. https://doi.org/10.1007/s13402-016-0275-7.
398. Zöller M. Pancreatic cancer diagnosis by free and exosomal miRNA. World Journal of Gastrointestinal Pathophysiology. 2013; 4:74–90. https://doi.org/10.4291/wjgp.v4.i4.74.
399. Yu C, Wang M, Li Z, Xiao J, Peng F, Guo X, Deng Y, Jiang J, Sun C. MicroRNA-138-5p regulates pancreatic cancer cell growth through targeting FOXC1. Cell Oncol (Dordr). 2015; 38:173–81. https://doi.org/10.1007/s13402-014-0200-x.
400. Tian S, Guo X, Yu C, Sun C, Jiang J. miR-138-5p suppresses autophagy in pancreatic cancer by targeting SIRT1. Oncotarget. 2017; 8:11071–82. https://doi.org/10.18632/oncotarget.14360.
401. Rana S, Malinowska K, Zoller M. Exosomal tumor microRNA modulates premetastatic organ cells. Neoplasia. 2013; 15:281–95.
402. Ali S, Dubaybo H, Brand RE, Sarkar FH. Differential Expression of MicroRNAs in Tissues and Plasma Co-exists as a Biomarker for Pancreatic Cancer. J Cancer Sci Ther. 2015; 7:336–46. https://doi.org/10.4172/1948-5956.1000372.
403. Mikamori M, Yamada D, Eguchi H, Hasegawa S, Kishimoto T, Tomimaru Y, Asaoka T, Noda T, Wada H, Kawamoto K, Gotoh K, Takeda Y, Tanemura M, et al. MicroRNA-155 Controls Exosome Synthesis and Promotes Gemcitabine Resistance in Pancreatic Ductal Adenocarcinoma. Scientific Reports. 2017; 7:42339. https://doi.org/10.1038/srep42339.
404. Ohuchida K, Mizumoto K, Kayashima T, Fujita H, Moriyama T, Ohtsuka T, Ueda J, Nagai E, Hashizume M, Tanaka M. MicroRNA Expression as a Predictive Marker for Gemcitabine Response after Surgical Resection of Pancreatic Cancer. Annals of Surgical Oncology. 2011; 18:2381–7. https://doi.org/10.1245/s10434-011-1602-x.
405. van den Boorn JG, Dassler J, Coch C, Schlee M, Hartmann G. Exosomes as nucleic acid nanocarriers. Adv Drug Deliv Rev. 2013; 65:331–5. https://doi.org/10.1016/j.addr.2012.06.011.
406. Bellavia D, Raimondo S, Calabrese G, Forte S, Cristaldi M, Patinella A, Memeo L, Manno M, Raccosta S, Diana P, Cirrincione G, Giavaresi G, Monteleone F, et al. Interleukin 3- receptor targeted exosomes inhibit in vitro and in vivo Chronic Myelogenous Leukemia cell growth. Theranostics. 2017; 7:1333–45. https://doi.org/10.7150/thno.17092.
407. Kharaziha P, Ceder S, Li Q, Panaretakis T. Tumor cell-derived exosomes: a message in a bottle. Biochim Biophys Acta. 2012; 1826:103–11. https://doi.org/10.1016/j.bbcan.2012.03.006.
408. Nakase I, Kobayashi NB, Takatani-Nakase T, Yoshida T. Active macropinocytosis induction by stimulation of epidermal growth factor receptor and oncogenic Ras expression potentiates cellular uptake efficacy of exosomes. Sci Rep. 2015; 5:10300. https://doi.org/10.1038/srep10300.
409. Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, Rabinowitz JD, Metallo CM, Vander Heiden MG, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013; 497:633–7. https://doi.org/10.1038/nature12138.
410. Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, Lee JJ, Kalluri R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017; 546:498–503. https://doi.org/10.1038/nature22341. http://www.nature.com/nature/journal/vaop/ncurrent/abs/nature22341.html#supplementary-information.
411. Kowalik A, Kowalewska M, Góźdź S. Current approaches for avoiding the limitations of circulating tumor cells detection methods—implications for diagnosis and treatment of patients with solid tumors. Translational Research. 2017; 185:58–84.e15. https://doi.org/10.1016/j.trsl.2017.04.002.
412. Meacham CE, Morrison SJ. Tumor heterogeneity and cancer cell plasticity. Nature. 2013; 501:328–37. https://doi.org/10.1038/nature12624.
413. Correnti M, Raggi C. Stem-like plasticity and heterogeneity of circulating tumor cells: current status and prospect challenges in liver cancer. Oncotarget. 2017; 8:7094–115. https://doi.org/10.18632/oncotarget.12569.
414. Pixberg CF, Schulz WA, Stoecklein NH, Neves RP. Characterization of DNA Methylation in Circulating Tumor Cells. Genes (Basel). 2015; 6:1053–75. https://doi.org/10.3390/genes6041053.
415. Pixberg CF, Raba K, Muller F, Behrens B, Honisch E, Niederacher D, Neubauer H, Fehm T, Goering W, Schulz WA, Flohr P, Boysen G, Lambros M, et al. Analysis of DNA methylation in single circulating tumor cells. Oncogene. 2017; 36:3223–3231. https://doi.org/10.1038/onc.2016.480.