
INTRODUCTION
Metastatic dissemination is a fundamental process in the progression of malignant disease, and is the cause of the majority of cancer-related deaths [1]. Despite this the mechanisms underlying the acquired ability of cells to metastasize are poorly understood [2]. As a consequence, the ability to accurately predict, prevent, and treat metastatic disease remain a major challenge in prognostication and oncological treatment. However, while most studies explore primary tumors in the search for molecular answers to these challenges, identification of discordances between primary tumors and metastases have the potential to reveal key drivers of metastatic progression.
Adenoid cystic carcinoma (ACC) is among the most frequent and enigmatic malignancies of the salivary gland [3]. It is characterized by an unpredictable clinical course with frequent local recurrences, late onset of metastases mainly to the lungs and liver and, consequently, an often fatal outcome [4, 5]. The genetics of primary ACC have been extensively explored, which has resulted in the identification of recurrent gene fusions involving the MYB, MYBL1, and NFIB genes and frequent mutations in NOTCH pathway genes [6–8]. So far, these characteristic genetic profiles have provided a valuable insight into the biology of ACC but have not contributed to the understanding of metastatic dissemination in the majority of patients [9, 10]. Recently, spatial genetic differences within and between paired primary ACCs and metastases have been reported, but only few and diverse mutations were found in coding regions [11]. While this study demonstrates the relatively quiet genome of primary ACC is preserved in metastases, it provides explanations to metastatic spread in a subset of cases but at the same time raises the question as to whether additional molecular mechanisms are involved in metastatic spread of ACC.
MicroRNAs (miRNAs) are short non-coding RNAs ~22 nucleotides in length, functioning as potent post-transcriptional regulators of gene expression [12]. MiRNAs have been shown to be implicated in numerous cancers and to be of prognostic value, including in salivary gland ACC [13, 14]. Several in vitro studies have characterized the effects of miRNAs on primary and metastatic ACC cell lines, but the findings have recently been discredited due to contamination of all of these cultures with various non-ACC cell lines [15]. Hence, reliable data from human ACC samples is highly warranted.
To further explore the molecular background for ACC dissemination, we performed fluorescence in situ hybridization (FISH), targeted next-generation sequencing (NGS), and global miRNA expression profiling in paired samples of primary and metastatic ACC.
RESULTS
Clinicopathologic characteristics
Patients included six males and five females with primary ACC of various head and neck sites (Table 1, Figure 1A). Radical surgical removal of the primary lesion was performed in 4/11 primary tumors. All metastases were metachronous and were from brain (case 1), lung (cases 2–9), and liver (case 5, 10, and 11), with latency from surgery of the primary lesion to metastatic spread ranging from 7–119 months (median: 60.5) (Figure 1B–1D). Radical surgical removal of the metastatic lesion was performed in 9/11 patients. Median follow-up was 46 months (range: 2–97). At the end of follow-up, five patients had died of disseminated disease, three were alive with disease, and three had no evidence of disease. Clinicopathologic characteristics of the 25 patients without recurrence and 11 additional patients with metastatic spread are listed in Supplementary Table 1.
Table 1: Clinicopathologic characteristics at diagnosis of primary and metastatic adenoid cystic carcinoma
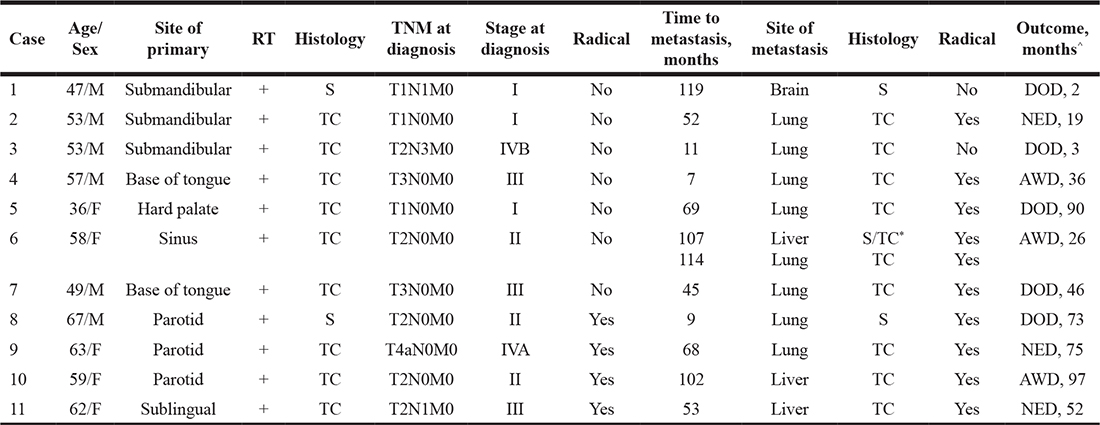
^After diagnosis of metastatic disease. *multiple metastases of which some were solid and others tubulocribriform. RT = radiotherapy; S = solid; TC = tubulocribriform; DOD = Dead of disease; NED = No evidence of disease; AWD = Alive with disease.
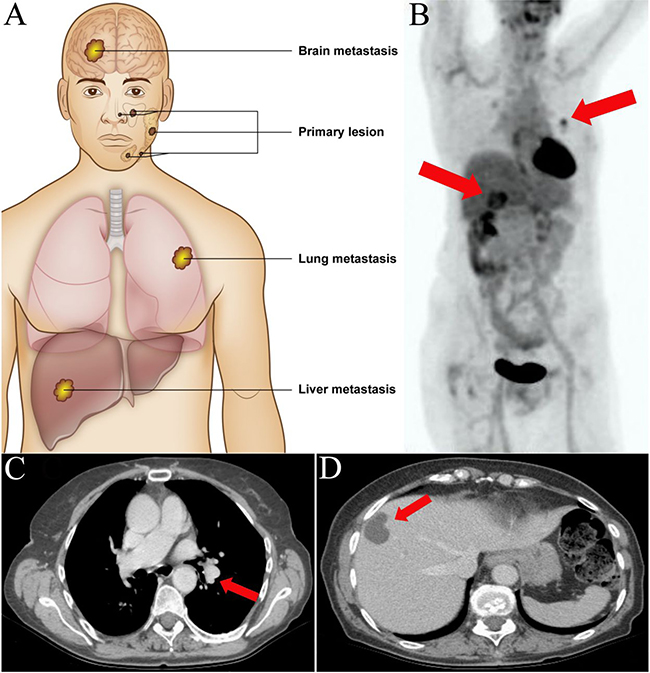
Figure 1: Anatomical localization of primary salivary gland adenoid cystic carcinomas and distant metastases. (A) Primary adenoid cystic carcinomas were located to the major and minor salivary glands of the head and neck. Distant metastases were located to the brain (n = 1), lungs (n = 8), and liver (n = 3). (B) Positron emission tomography – computerized tomography (PET-CT) scan of case 6 with metachronous metastases to the hilar region of the left lung and liver (red arrows). (C) CT-scan of the same patient showing the pulmonary and (D) hepatic metastases.
Histopathology
Histologically, the majority of primary ACCs (9/11) displayed tubulocribriform growth patterns with dual populations of luminal and abluminal cells, whereas two cases were of solid type (Figure 2A, 2B). The growth pattern between primary tumors and metastases was preserved in all but one case in which the primary tumor was tubulocribriform and some of the multiple metastases were of solid type (case 6) (Table 1) (Figure 2C, 2D). Immunohistochemically, all primary ACCs and metastases were positive for CK7, CD117, and MYB (Figure 3B, insert) and consistently negative for CK20 and neuroendocrine markers (CD56, chromogranin A). Dual TTF-1 and napsin A positivity was focal in pulmonary metastases of two cases. Ki-67 indices in primary tumors and metastases ranged from 5–60% (median: 10). All immunohistochemical results are summarized in Supplementary Table 2.
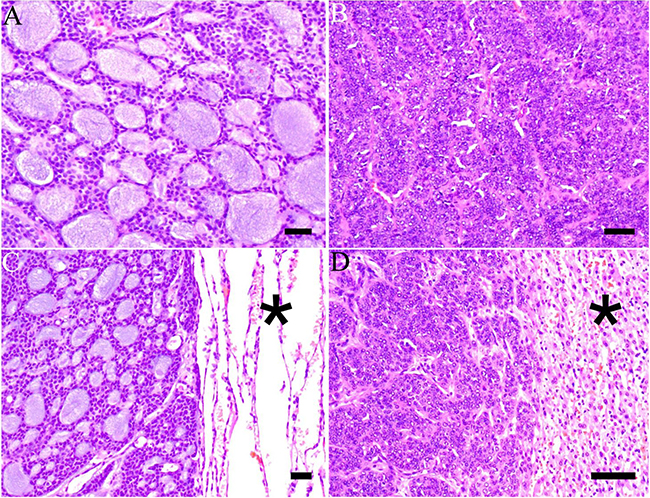
Figure 2: Histological features of primary and metastatic salivary gland adenoid cystic carcinoma. (A) Adenoid cystic carcinoma of the sinonasal minor salivary glands with tubular and cribriform areas and accumulation of basophilic basal membrane material in pseudoluminae (case 6) (hematoxylin & eosin, H&E). (B) Adenoid cystic carcinoma of the submandibular gland from case 1 with solid aggregates of basaloid tumor cells. Nuclei are large and mitoses are frequent (H&E). (C) Pulmonary metastasis of a well-differentiated tubulocribriform adenoid cystic carcinoma from the primary tumor in (A). Normal lung parenchyma is seen to the right (asterisk) (H&E). (D) Hepatic metastasis showing a solid adenoid cystic carcinoma. This specimen is from the same patient as in (A and C). Normal liver parenchyma is seen to the right (asterisk) (H&E). Scale bar = 100 μm.
FISH
In all cases, the pattern of MYB and NFIB aberrations was identical between primary ACC and metastasis (Table 2). In 4/10 patients, concurrent rearrangements of MYB and NFIB were identified (Figure 3A, 3B). In the majority of tumor cells, the MYB break-apart probe showed a green/red signal ratio of 3 in case 3 and a red/green signal ratio of 3 in case 6, consistent with amplification of the 5′ and 3′ parts of MYB, respectively (Figure 3C). Four cases with MYB protein expression did not harbor identifiable alterations in MYB (Figure 3B, insert and Table 2). MYBL1 was intact in all cases (not shown).
Table 2: Rearrangement of MYB and NFIB in primary adenoid cystic carcinoma and metastases
Case |
Primary adenoid cystic carcinoma |
Metastatic adenoid cystic carcinoma |
|---|---|---|
1 |
MYB-NFIB |
MYB-NFIB |
2 |
Normal |
Normal |
3 |
MYB amplification1 |
MYB amplification1 |
4 |
MYB-NFIB |
MYB-NFIB |
5 |
Normal |
Normal |
6 |
MYB amplification2 |
Liver |
Lung |
||
7 |
MYB-NFIB |
MYB-NFIB |
8 |
MYB-NFIB |
MYB-NFIB |
9 |
Normal |
Lung3 |
Lung3 |
||
10 |
Not available |
Not available |
11 |
Normal |
Normal |
All patients had intact MYBL1 genes.1Multiple green signals per nucleus, consistent with 5′ amplification. 2Multiple red signals per nucleus, consistent with 3′ amplification. 3Separate tubulocribriform metastases from right and left lung, respectively.
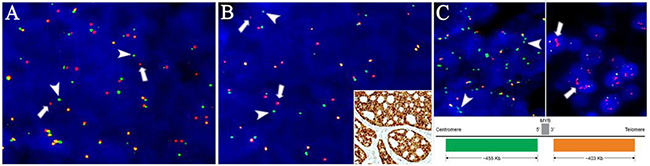
Figure 3: Rearrangements and copy number status of MYB and NFIB in primary and metastatic adenoid cystic carcinoma. Fluorescence in situ hybridization with break-apart probes showing split signals of the green centromeric (arrowheads) and red telomeric (arrows) for (A) MYB and (B) NFIB in a pulmonary metastasis. (B, insert) Representative area of the primary tumor in case 5 showing diffuse positivity for MYB protein despite being without detectable MYB abnormalities. (C, upper left) MYB break-apart probe showing isolated amplification of the green centromeric probe (arrowheads, case 3) and (C, upper right) red telomeric probe (arrows, case 6). (C, bottom) Schematic presentation of the chromosomal location of the break-apart probe. Multiple green and red signals are consistent with isolated amplification of the 5′ and 3′ parts of the MYB gene, respectively. Image is not drawn to scale.
Targeted next-generation sequencing of primary and metastatic tumor tissue
Point mutations in primary and metastatic ACC was assessed using a targeted NGS panel covering approximately 2,800 mutational hotspots in 50 cancer-related oncogenes and tumor-supressor genes. Truncal mutations present in both primary tumor and metastasis were identified in case 1 (NRAS, NOTCH1), case 4 (BRAF, TP53), and case 11 (APC). In wild-type primary tumors, metastases showed single mutations in case 3 (NRAS), case 6 (PIK3CA in lung metastasis and BRAF in liver metastasis), and case 8 (HRAS) (Table 3). Loss of mutations from primary tumor to metastases were identified in case 5 (PDGFRA) and case 9 (FGFR2). No mutations were identified in 6/11 (54.5%) of primary ACCs and three of these had no mutations in the metastatic lesions. In order to account for patient-specific single nucleotide polymorphisms (SNPs), matched normal salivary gland tissue was sequenced from the seven cases with this material available. This did not exclude any of the mutations identified in neither primary ACCs or distant metastases.
Table 3: Hotspot mutations in paired primary and metastatic salivary gland adenoid cystic carcinoma
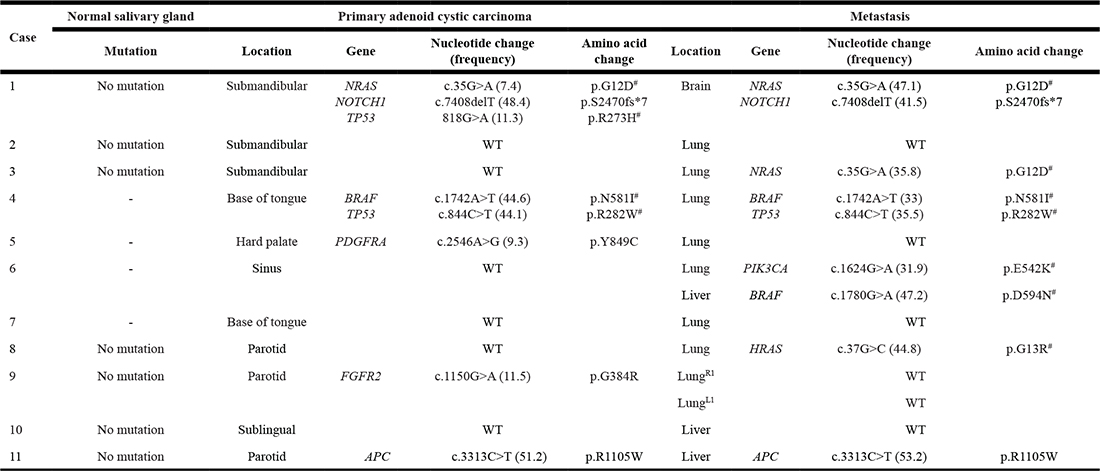
1Separate tubulocribriform metastases from right and left lung, respectively. #Recurrent oncogenic hotspot mutation.
MicroRNA expression profiling of primary and metastatic tumor tissue
Comparing miRNA expressions of primary ACC and normal salivary gland tissue identified one upregulated (hsa-miR-1271-5p) and 7 downregulated (hsa-miR-1199-3p; hsa-mir-6865; hsa-miR-4717-5p; hsa-mir-610; hsa-miR-6878-5p; hsa-mir-519e; hsa-miR-5572) miRNAs (false discovery rate [FDR] < 0.05). Comparing metastases and normal salivary gland tissue, we identified 4 upregulated (hsa-miR-922; hsa-miR-1271-3p; hsa-mir-6790; hsa-miR-6894-3p) and 7 downregulated (hsa-mir-6865; hsa-miR-1199-3p; hsa-miR-4717-5p; hsa-miR-499a-5p; hsa-miR-4790-5p; hsa-mir-3936; hsa-mir-127) miRNAs (FDR < 0.05) (Figure 4). Among these, hsa-miR-1199-3p, hsa-mir-6865, and hsa-miR-4717-5p were downregulated in primary tumors as well as metastases as compared to normal salivary gland tissue. Functional annotation of the differentially expressed miRNAs between primary ACC and normal salivary gland tissue and between metastases and normal salivary gland tissue identified their involvement in regulating genes in several Kyoto Encyclopedia of Genes and Genomes (KEGG) categories, including metabolism, migration, and signal transduction pathways (Supplementary Tables 3 and 4). When comparing primary ACCs and metastases as groups, there were no significantly differentially expressed miRNAs. Hierarchical clustering including primary and metastatic ACC as well as normal salivary gland tissue placed normal salivary gland together, with the dendrogram including all but one specimen (Figure 5). Primary and metastatic lesions were scattered in the left main cluster and only in one patient (case 7) did the primary tumor and metastasis cluster together (Figure 5). Repeating the analysis including only primary ACC and metastases and re-normalizing data, there was still no difference (FDR > 0.05) but hierarchical clustering by a p < 0.01 cut-off for differential expression placed all metastases in one main cluster of the dendrogram and 10/11 primary ACCs in the other main cluster (Figure 6). Resampling did not identify significant differences between the clusters with or without the inclusion of normal salivary gland tissue, but normal salivary gland tissue separated with the highest values (Supplementary Figures 1 and 2).
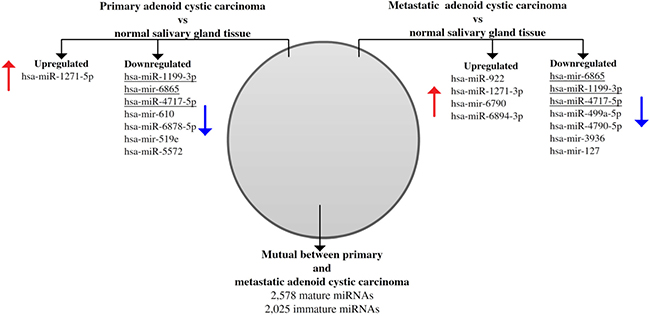
Figure 4: Venn diagram showing differentially expressed microRNAs between normal salivary gland tissue as compared to primary and metastatic adenoid cystic carcinoma. Eight miRNAs were differentially expressed between normal salivary gland tissue and primary adenoid cystic carcinoma, and 11 miRNAs were differentially expressed between normal salivary gland tissue and distant metastases. Three miRNAs were downregulated in both tumor tissues (underlined). There was no difference between primary tumor and metastases, and therefore all miRNAs investigated were mutual.
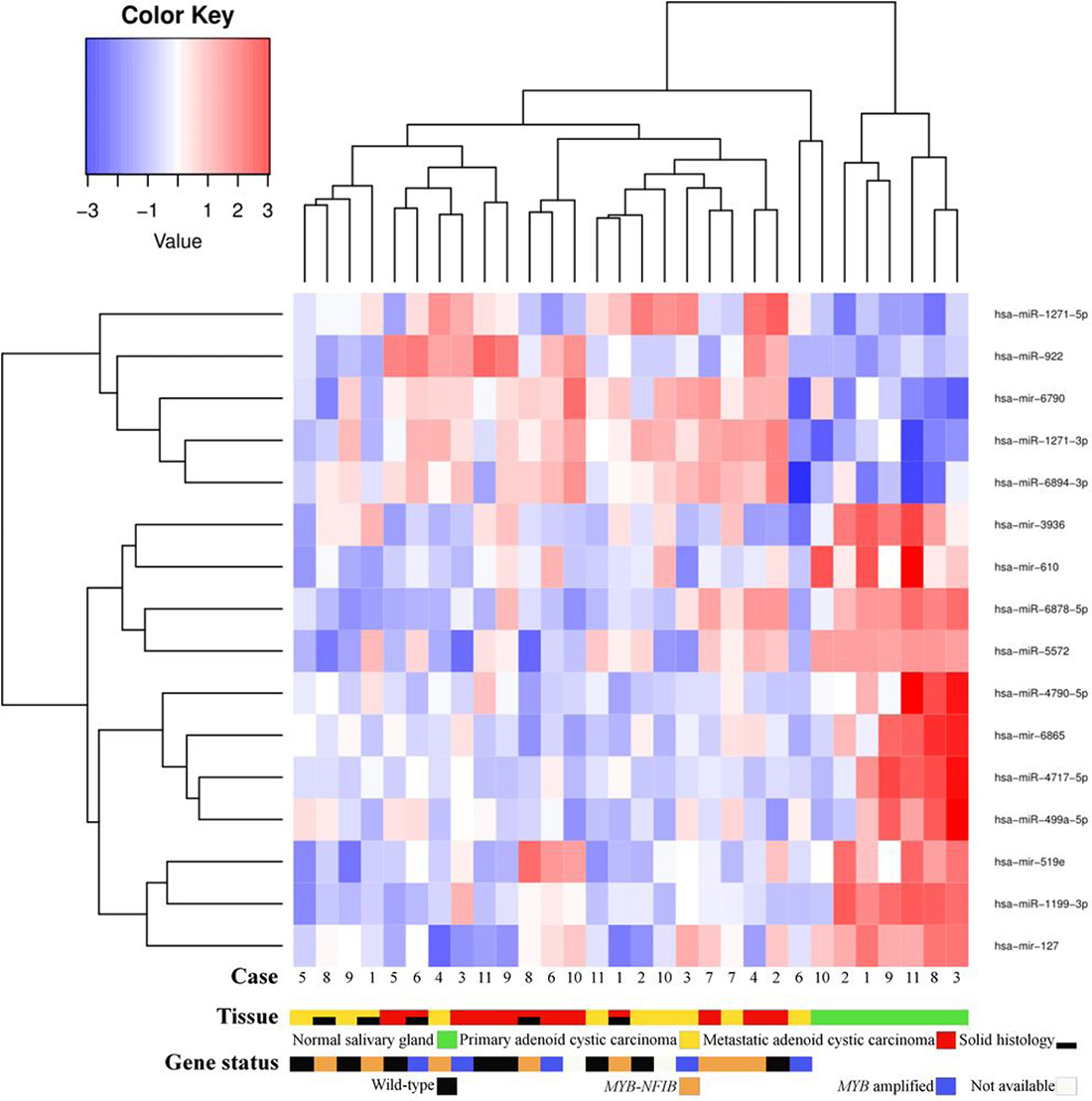
Figure 5: Heatmap of microRNA expression in primary adenoid cystic carcinoma, metastatic adenoid cystic carcinoma, and normal salivary gland tissue. Normal salivary gland tissue is clustered to the right. No difference in microRNA expression was found between primary tumors and metastases. In the dendrogram, primary tumors and corresponding metastases were only highly similar in case 7. There was no clustering of cases according to gene fusion status.
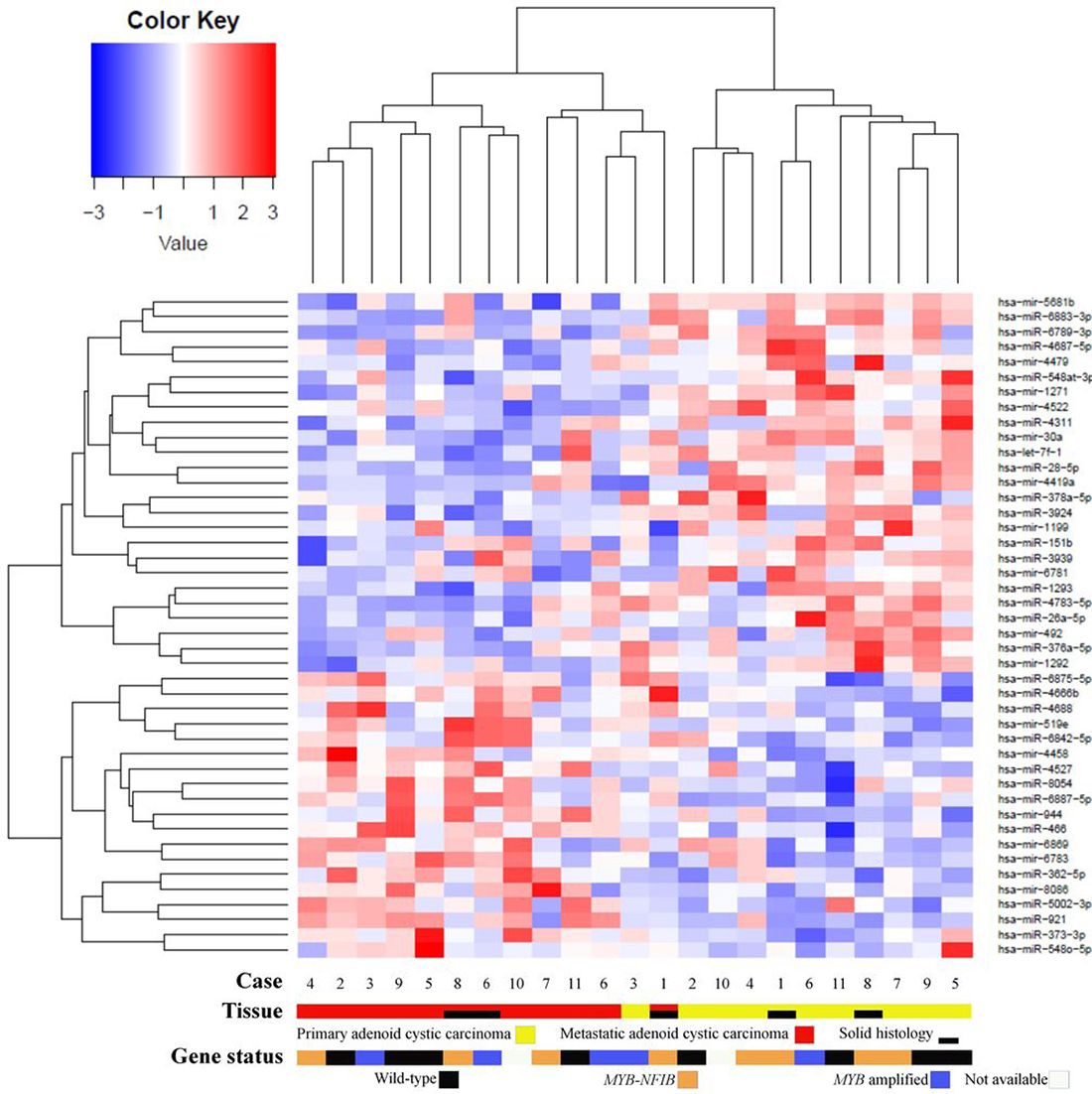
Figure 6: Heatmap of microRNA expression in primary adenoid cystic carcinoma and metastatic adenoid cystic carcinoma. The right main cluster consisted of 10/11 primary salivary gland adenoid cystic carcinoma. The left main cluster included 11/11 metastases and the remaining primary tumor. P-value < 0.01, FDR > 0.05.
MicroRNA expression profiling of primary tumors from patients with and without subsequent recurrence
When comparing the miRNA expression of the primary ACCs along with 11 additional patients who subsequently developed metastases (total n = 22) and primary ACCs from patients who remained free of metastases (n = 25), no miRNA was differentially expressed at FDR < 0.05.
DISCUSSION
The clinical course of patients with ACC is unpredictable and once metastatic disease presents, the treatment options are limited as surgical resection is far from always curative and effective targeted treatments are lacking [16, 17]. These features are illustrated in the series presented here as 3/11 patients with subsequent metastatic disease were initially diagnosed with only stage I disease, 5/11 patients died of metastatic disease within the follow-up period, and 3/11 experienced additional distant recurrence following otherwise complete metastasectomies (Table 1).
Over recent decades, genetics has continued to provide insights into the causes of human cancer through the identification of genetic aberrations underlying different malignant phenotypes. But while some cancers are heterogeneous in their genetic background, the identification of recurrent genetic alterations in several types of salivary gland tumors has greatly increased the understanding of these relatively rare entities [18]. In ACC, NOTCH1 mutations have been shown to be enriched in recurrent and metastatic tumors and mutations in NOTCH pathway genes characterize a small subset of cases with solid histology and a particularly aggressive clinical course [19, 20]. Aside from this, genetics has otherwise not contributed to an understanding of the main cause of ACC mortality, i.e. metastatic spread. Recurrent gene fusions involving MYB, MYBL1, and NFIB are hallmarks of ACC, and we demonstrate the preservation of MYB and NFIB rearrangements between the primary ACC and the metastases, consistent with the fundamental role of these genes in ACC formation [11]. Interestingly, we found amplification of the 3′ as well as 5′ parts of MYB (case 6 and 3, respectively) which were retained in the metastatic cells. While the 5′ part of the MYB gene hosts the DNA binding and transactivation domains, the 3′ part of MYB hosts the negative regulatory domain that is consistently lost in the MYB-NFIB fusion [21]. Therefore, amplification of the 5′ part of MYB in case 3 goes well in hand with the oncogenic properties of this part of the MYB gene, while the significance of isolated amplification of the 3′ part of MYB in case 6 is unclear. Also, we found diffuse expression of MYB protein in all cases, including those without abnormalities of MYB as characterized with FISH, which further underline the importance of this protein in ACC biology (Figure 3B). While elucidating the mechanism responsible for MYB overexpression is beyond the scope of this study, others have shown the translocation of various super-enhancer sequences to result in MYB overexpression [8].
Targeted treatment directed by cancer genomics face substantial complexity with the concept of intratumoral heterogeneity, and a study by Liu et al. demonstrated this phenomenon in ACC, although to a low extent as compared to other carcinomas [11, 22]. Additional complexity arises when also considering the metastatic compartment of malignant disease, but accounting for this is of paramount importance, since eradication of this cell population is a prerequisite for a sustainable effect of targeted treatment [22]. Liu et al. demonstrated that mutations exclusive for metastatic lesions (mainly lymph node metastases) were indeed present in ACC, and to a lesser extent in synchronous as compared to metachronous metastases [11]. In the present study, all metastases were metachronous and most were located at the clinically most relevant anatomical sites for ACC patients, i.e. lung and liver. Using targeted next-generation sequencing, we identified mutations exclusive for the metastatic lesions in 3/11 patients but also mutations exclusive for the primary lesions in another three patients (Table 3). While mutations found exclusively in the metastatic lesions of case 3, 6, and 8 could represent mutations involved in the metastatic phenotype, the significance of the mutations found exclusively in the primary lesions of case 1, 5, and 9 are more likely to be passenger mutations (Table 3). This latter statement is supported by the low frequencies of these mutations (9.3–11.5%). Although the degree of intratumoral heterogeneity in ACC has been shown to be present only to a minor extent, it must be noted that our results rests on the repertoire of mutations identified in 1mm tumor biopsies and not the entire tumor. Therefore, it is possible that the frequencies of different mutations could differ in other areas of the same tumor. In the cases with mutations found exclusively in the metastatic lesions these mutations were all present in high frequencies, but it is not unlikely that the same mutation could be found in the primary lesion, although probably at a lower frequency, if another region had been sampled. Therefore, although we employed a targeted sequencing panel, we confirmed the heterogeneous genomic landscape of paired primary and metastatic ACC. However, in contrast to the tumor-defining oncogenic driver function of ACC gene fusions, interpreting the significance of point mutations in ACC is not an easy task. This is exemplified by a comparison of our findings with datasets from 207 primary salivary gland ACCs and 7 ACC metastases from cBioPortal [23]. In this dataset, mutations in APC, FGFR2, HRAS, PIK3CA, and TP53 were previously reported but were all different from the mutations we found in our material (Supplementary Table 5). However, in accordance with previous studies, mutations in NOTCH1 tended to cluster in the proline-glutamic acid-serine-threonine (PEST) domain, similar to the activating mutations in T-cell acute lymphoblastic leukaemia (Supplementary Table 5) [11, 19, 20, 24]. Furthermore, demonstrating the diversity of the mutational repertoire involved in the metastatic process is the identification of distinctly different oncogenic hotspot mutations in the lung and liver metastases in case 6. Importantly, the PIK3CA in case 6 and NRAS in case 1 and 3 are rational candidates for off-label targeted treatments and the NOTCH1 mutation in case 1 is eligible for inclusion in an ongoing clinical trial for advanced salivary gland cancers (clinicaltrials.gov identifier: NCT020697309) (Table 3). Collectively, this underscores the need for extensive sampling of ACC patients in the context of clinical trials.
While genetics explains the events leading to ACC formation mainly in the form of fusion genes, the low rate of mutations exclusive for the metastatic lesions presented here makes alternative biological mechanisms likely to be involved. As previous in vitro studies on the involvement of miRNA in the metastatic progression of ACC has been hampered by cross-contamination, data from human samples has not been available until now [15]. By global miRNA expression profiling we identified miRNAs differentially expressed between primary ACC and metastases as compared to normal salivary gland tissue but no difference between primary and metastatic lesions (Figure 4). Some of these miRNAs were exclusive for primary and metastatic ACCs, but three poorly characterized miRNAs were downregulated in both groups (Figure 4). The significance of these three miRNAs is currently uncertain. Interestingly, functional annotation showed the miRNAs dysregulated in metastases to target genes involved in functions of key importance for the metastatic phenotype, with the most significant function being extracellular molecule interaction (Supplementary Table 4). Normal salivary gland tissue separated from ACC in hierarchical clustering but primary ACCs and metastases were intermingled, with only one case having primary tumor and metastasis placed adjacently (Figure 5 and Supplementary Figure 1). Although this gives an impression of primary tumors and metastases being similar, the inclusion of three different groups with a relatively low number of samples could obscure possible patterns. Although not reaching statistical significance, there was a near complete separation between primary ACC and metastases when normal salivary gland tissue was left out of the clustering analysis (Figure 6). Functional annotation of the miRNAs in Figure 6 identify these to be involved in a large number of metabolic functions, signaling pathways, and as being involved in other types of cancer (Supplementary Table 6). While this finding is intriguing, it must be interpreted with caution as it still relies on non-significant differences and needs further validation. Alternatively, given the highly diverse spectrum of somatic mutations in ACC shown here as well as in other studies, it is possible that ACC is a mainly fusion-driven disease with otherwise diverse molecular changes separating primary and metastatic lesions, including miRNA expression [7, 11, 18, 19, 22].
The acquirement of different somatic mutations on top of the MYB-NFIB fusion has been demonstrated to occur during high-grade transformation of breast ACC, which also included mutations in NOTCH1 and FGFR1 [25]. Also in breast cancer, remarkably similar miRNA and global gene expression profiles have been found between paired primary breast carcinoma and metastases [26, 27]. In one study these findings extended so far that primary tumors were as similar to their metastases as repeated sampling from the primary tumor, suggesting that the metastatic capability was an inherent feature of the primary tumor and not something unique for the metastatic lesion [27–29]. If this was also the case in salivary gland ACC, this could imply that patients developing metastases had primary tumors that were inherently more “metastatis-like”. Since this would make our comparison of paired primary ACC and metastases futile, we compared the miRNA expression profile of primary ACCs with and without subsequent development of recurrent disease and found no difference in their miRNA expression. This implies that primary ACCs are similar in miRNA expression irrespective of later development of metastases. In direct extension of this consideration it is noteworthy that only the present material of ACC metastases has undergone miRNA expressional profiling, and that comprehensive genetic and miRNA expressional profiling of additional metastatic lesions will extend the understanding of the biological underpinnings of this relatively rare disease.
The complexity in providing a complete molecular landscape of the metastatic process in ACC gives rise to some limitations in our study. Sampling of multiple tumor regions would allow for more solid statements to be made regarding tumor heterogeneity, which would be further strengthened by the use of a more comprehensive sequencing platform for the detection of mutational diversity. Due to the lack of significance between primary and metastatic lesions, the clustering based on miRNA expression may not necessarily reflect the underlying biology, but the rarity of ACCs with available primary and metastatic tissue dictated the sample size which is an inherent limitation to the study of rare diseases.
In conclusion, we present a series of paired primary and metastatic salivary gland ACCs and demonstrate the preservation of MYB and NFIB aberrations during the course of metastatic progression. Contrasting with the fundamental role of these two genes in ACC, no general pattern of somatic mutations or significant changes in miRNA expression was found in metastatic lesions. Although larger materials could assist in delineating these issues further, our findings support the metastatic process in ACC as being a complex and heterogeneous biological phenomenon.
MATERIALS AND METHODS
Patient material
Patients surgically treated for salivary gland ACC and resected metastatic lesion were identified in the Danish Pathology Data Bank (Patobank). Eleven patients with both tissues available were identified, and formalin-fixed and paraffin-embedded (FFPE) specimens were collected from pathology departments all over Denmark. In seven cases, triplet material from primary ACC, metastatic ACC, and normal salivary gland tissue was available. In the remaining four cases only the primary and metastatic tumors were available. For the analysis of miRNA expression in primary tumors only, 11 additional ACCs with subsequent metastases were included along with 25 ACCs that remained free of recurrence. Median follow-up periods for patients with and without subsequent distant failure were 78 months (range: 14–233) and 154 months (range: 72–322), respectively (Supplementary Table 1). None of the patients had received radiotherapy or chemotherapy prior to surgery or biopsy. All cases were reviewed by a head and neck pathologist (TKA) [30–32]. The Regional Ethics Committee (H-6-2014-086) and the Danish Data Protection Agency (REG-94-2014) approved this study.
Immunohistochemistry
FFPE blocks were sectioned and mounted on coated slides (Dako, Glostrup, Denmark) and stained with H&E according to standard protocols. Tumor sections were deparaffinized using EZ-prep (Ventana Medical Systems®, Tucson, AZ, USA) and immunohistochemistry was performed using the Ventana Benchmark Ultra platform (Ventana Medical Systems) as previously described [33]. The following primary antibodies were employed: CD117 (polyclonal, 1:100, Dako (Glostrup, Denmark)), CD56 (clone 1B6, 1:50, Novocastra (Newcastle, UK)), chromogranin A (polyclonal, 1:2000, Dako), CK7 (clone OV-TL 12/30),1:1000, Dako), CK20 (clone KS20.8, 1:400, Dako), ki-67 (clone MIB1, 1:100, Dako), MYB (clone EP769Y 1:150 (AbCam, Cambridge, UK), napsin A (clone IP64, 1:400, Novocastra), and TTF-1(clone SPT24, 1:100, Novocastra). MYB was considered positive when nuclear staining was observed in at least 5% of tumor cells [31]. Positive controls as suggested on datasheets were used. Negative controls omitting the primary antibody were performed for all antibodies.
Fluorescence in situ hybridization
Primary tumor and metastatic lesion were evaluated for rearrangements of MYB (Empire Genomics, Buffalo, NY, USA), MYBL1 (Empire Genomics), and NFIB (Empire Genomics) genes with break-apart probes according to the manufacturer’s instructions using the HYBrite platform (Abbott Molecular) and counterstained with DAPI II (ZytoVision GmbH, Bremerhaven, Germany) as previously described [31]. One hundred nuclei were counted, and only nuclei where the entire nuclear membrane could be visualized were scored. Break-apart signals in ≥ 10% of cells was considered to represent rearrangement [31]. Amplification was defined as a red/green or green/red signal ratios ≥ 2 signals for 3′ and 5′ amplification, respectively. Amplification was defined as a red/green or green/red signal ratio of ≥ 3 in in more than 10% of tumor cells for 3′ and 5′ amplification, respectively.
DNA extraction and targeted next-generation sequencing
DNA was extracted from one 1 mm core obtained from the original FFPE block after identification of a representative tumor area from an H&E slide in order to minimize admixture of stromal cells and consequently a high tumor cell content, as previously described [34]. Briefly, targeted NGS was carried out on the Ion PGM™ System (Thermo Fisher Scientific, Walham, MA, USA) with the Ion AmpliSeq Cancer Hotspot Panel version 2 (Thermo Fisher Scientific) according to the manufacturer’s instructions. The Ion AmpliSeq Cancer Hotspot Panel v.2 covers approximately 2,800 mutational hotspots in 50 cancer-related oncogenes and tumor-supressor genes. Library preparation was performed from 10 ng of purified DNA using Ion AmpliSeq Library kit 2.0 according to manufacturer’s instructions (Thermo Fisher Scientific) and using the Ion PGM Hi-Q view kit (Thermo Fisher Scientific) as template kit for the IonChef (Thermo Fisher Scientific). Sequencing was carried out using Ion PGM Hi-Q view Sequencing kit with the Ion 316™ Chip v2 (Thermo Fisher Scientific). Median number of mapped reads in normal salivary gland, primary tumor, and metastasis were 296,709, 215,455, and 468,226, respectively. Median allele coverage in normal salivary gland, primary tumor, and metastasis were 1,286, 735, and 2,097, respectively. Sequencing statistics are given for each sample in Supplementary Table 7. Variant calling was conducted using the Torrent Variant Caller v4.6 of the Torrent Suite™ Software (Thermo Fisher Scientific) using default settings. Variants were evaluated according to the American College of Medical Genetics and Genomics (ACMG) guidelines using the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/docs/acmg/), COSMIC (http://cancer.sanger.ac.uk/cosmic), and by literature search (https://www.ncbi.nlm.nih.gov/pubmed) [35]. Mutant reads were not allowed in the normal tissue. A mutation frequency of 5%, with equal ± strand distribution, was regarded as mutation positive given that DNA was extracted from cores and not whole sections.
RNA extraction
RNA was extracted from one 1mm core as described above. In case 9, only one pulmonary metastasis had sufficient tissue left. The QiaCube (Qiagen, Valencia, CA, USA) was used for automated isolation of miRNA with the miRNeasy FFPE Kit (Qiagen) according to the manufacturer’s instructions [36]. Total RNA concentration was measured using the NanoDrop ND-1000 (Thermo Scientific, Wilmington, DE, USA) and ranged from 366–1536 ng/μl with A260/A280 ratio ranging from 1.8–2.6, indicating high nucleic acid purity.
MicroRNA array
The Affymetrix miRNA 4.1 array platform (Affymetrix, Santa Clara, CA, USA) was used, covering all entries in Sanger miRbase database v.20, including 2,578 mature and 2,025 immature human miRNAs with a dynamic range of 4 logs. For miRNA analysis, 300 μg of total RNA was labelled with the FlashTag Biotin HSR RNA Labelling kit (Affymetrix) according to the manufacturer’s instructions. All cases were run in a single batch to avoid batch variation. Array plates were washed, stained, and scanned on the GeneTitan Instrument (Affymetrix). Raw data are available from the journal website as Supplementary Table 8.
MicroRNA data analysis
The raw miRNA expression data was normalized using the quantile normalization using the free R package preprocessCore [37]. The normalized data was log transformed with base 2 to ensure normal or approximately normal distribution before statistical modelling. Considering the fact that multiple samples (normal salivary gland tissue, primary ACC and metastases) were taken from each patient, we fitted the linear mixed-effect models with miRNA expression levels as the dependent variable, sample grouping (normal, primary, metastasis) as the fixed effect variable, and patient ID as the random effect variable. In fitting the models, normal tissue was defined as the reference group. The mixed-effect models were fitted using the R package lmerTest. Differential expression analysis of primary tumors from patients with and without subsequent metastases was done by fitting a regression model that regresses miRNA expression on metastasis status equivalent to a student t-test. P values from the statistical analysis were adjusted for multiple testing by calculating the FDR using the R function p.adjust() defining method=”fdr” [38]. The R scripts used for data analysis are available upon request. We applied clustering analysis on the significant miRNAs using the heatmap.2 function in the R package gplots. The default distance algorithm set as “Euclidean” distance was used to compute the distances between clusters. The miRNAs with FDR < 0.05 were defined as statistically significant. Bootstrap resampling was used to generate probability values as implemented in pvclust [39].
Pathway analysis and functional annotation
Pathway analysis of miRNAs was performed with DIANA-miRPath v3.0 software available from http://snf-515788.vm.okeanos.grnet.gr/ [40]. The DIANA-TarBase v7.0 was selected as the miRNAtarget prediction algorithm [41]. For functional annotation, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were used and enrichment score for KEGG pathways presented by –ln (p value) [42].
Abbreviations
ACC: adenoid cystic carcinoma; miRNA: microRNA; FISH: fluorescence in situ hybridization; NGS: next-generation sequencing; PEST domain: proline-glutamic acid-serine-threonine domain; FFPE: formalin-fixed and paraffin-embedded; FDR: false discovery rate; KEGG: Kyoto Encyclopedia of Genes and Genomes.
Author contributions
SA, SH, IW, and PH conceptualized the study and drafted the manuscript. TKA, DE, SRL, and BPU reviewed slides and provided specimens for analysis. LCM performed targeted NGS. KB provided clinical information. QT performed statistical analysis of miRNA data. All authors approved the manuscript.
ACKNOWLEDGMENTS
The authors thank Heidi Ugleholdt, Camilla Cordua Mortensen, Sanni Pedersen and Tine Adelfest for their invaluable assistance in the laboratories. Thank you for your patience and always positive attitudes. The study was funded by the Einar Willumsen Foundation, Merchant Kristjan Kjær and wife Margrethe Kjær’s Foundation, The Danielsen Foundation, Region Zealand’s Research Fund, and Hans Skouby and wife Emma Skouby’s Fundation.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer 2006; 6:449–458. https://doi.org/10.1038/nrc1886.
2. Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet 2007; 369:1742–1757. https://doi.org/10.1016/S0140-6736(07)60781-8.
3. Bjørndal K, Krogdahl A, Therkildsen MH, Overgaard J, Johansen J, Kristensen CA, Homøe P, Sørensen CH, Andersen E, Bundgaard T, Primdahl H, Lambertsen K, Andersen LJ, Godballe C. Salivary gland carcinoma in Denmark 1990-2005: a national study of incidence, site and histology. Results of the Danish Head and Neck Cancer Group (DAHANCA). Oral Oncol. 2011; 47:677–682. https://doi.org/10.1016/j.oraloncology.2011.04.020.
4. Bjørndal K, Krogdahl A, Therkildsen MH, Charabi B, Kristensen CA, Andersen E, Schytte S, Primdahl H, Johansen J, Pedersen HB, Andersen LJ, Godballe C. Salivary adenoid cystic carcinoma in Denmark 1990–2005: Outcome and independent prognostic factors including the benefit of radiotherapy. Results of the Danish Head and Neck Cancer Group (DAHANCA). Oral Oncol. 2015; 51:1138–1142. https://doi.org/10.1016/j.oraloncology.2015.10.002.
5. Spiro RH. Distant metastasis in adenoid cystic carcinoma of salivary origin. Am J Surg. 1997; 174:495–458.
6. Persson M, Andrén Y, Mark J, Horlings HM, Persson F, Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci USA. 2009; 106:18740–18744. https://doi.org/10.1073/pnas.0909114106.
7. Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, Ramaswami D. Walsh LA, Eng S, Huse JT, Zhang J, Dolgalev I, Huberman K, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013; 45:791–798. https://doi.org/10.1038/ng.2643.
8. Drier Y, Cotton MJ, Williamson KE, Gillespie SM, Ryan RJ, Kluk MJ, Carey CD, Rodig SJ, Sholl LM, Afrogheh AH, Faquin WC, Queimado L, Qi J, et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet. 2016; 48:265–272. https://doi.org/10.1038/ng.3502.
9. Mitani Y, Liu B, Rao PH, Borra VJ, Zafereo M, Weber RS, Kies M, Lozano G, Futreal PA, Caulin C, El-Naggar AK. Novel MYBL1 gene rearrangements with recurrent MYBL1-NFIB fusions in salivary adenoid cystic carcinomas lacking t(6;9) translocations. Clin Cancer Res. 2016; 22:725–733. https://doi.org/10.1158/1078-0432.CCR-15-2867-T.
10. Mitani Y, Li J, Rao PH, Zhao YJ, Bell D, Lippman SM, Weber RS, Caulin C, El-Naggar AK. Comprehensive analysis of the MYB-NFIB gene fusion in salivary adenoid cystic carcinoma: Incidence, variability, and clinicopathologic significance. Clin Cancer Res. 2010; 16:4722–4731. https://doi.org/10.1158/1078-0432.CCR-10-0463.
11. Liu B, Mitani Y, Rao X, Zafereo M, Zhang J, Zhang J, Futreal A, Lozano G, El-Naggar AK. Spatio-Temporal Genomic Heterogeneity, Phylogeny, and Metastatic Evolution in Salivary Adenoid Cystic Carcinoma. J Natl Cancer Inst. 2017; 109. https://doi.org/10.1093/jnci/djx033.
12. Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008; 9:102–114. https://doi.org/10.1038/nrg2290.
13. Pencheva N, Tavazoie SF. Control of Metastatic Progression by microRNA Regulatory Networks. Nat Cell Biol. 2013; 15:546–554. https://doi.org/10.1038/ncb2769.
14. Mitani Y, Roberts DB, Fatani H, Weber RS, Kies MS, Lippman SM, El-Naggar AK. MicroRNA Profiling of Salivary Adenoid Cystic Carcinoma: Association of miR-17-92 Upregulation with Poor Outcome. PLoS One 2013; 8:e66778. https://doi.org/10.1371/journal.pone.0066778.
15. Phuchareon J, Ohta Y, Woo JM, Eisele DW, Tetsu O. Genetic profiling reveals cross-contamination and misidentification of 6 adenoid cystic carcinoma cell lines: ACC2, ACC3, ACCM, ACCNS, ACCS and CAC2. PLoS One. 2009; 4:e6040. https://doi.org/10.1371/journal.pone.0006040.
16. Girelli L, Locati L, Galeone C, Scanagatta P, Duranti L, Licitra L, Pastorino U. Lung metastasectomy in adenoid cystic cancer: Is it worth it? Oral Oncol. 2017; 65:114–8. https://doi.org/10.1016/j.oraloncology.2016.10.018.
17. Dillon PM, Chakraborty S, Moskaluk CA, Joshi PJ, Thomas CY. Adenoid cystic carcinoma: A review of recent advances, molecular targets, and clinical trials. Head Neck. 2016; 38:620–627. https://doi.org/10.1002/hed.23925.
18. Andersson MK, Stenman G. The landscape of gene fusions and somatic mutations in salivary gland neoplasms – Implications for diagnosis and therapy. Oral Oncol. 2016; 57:63–69. https://doi.org/10.1016/j.oraloncology.2016.04.002.
19. Morris LG, Chandramohan R, West L, Zehir A, Chakravarty D, Pfister DG, Wong RJ, Lee NY, Sherman EJ, Baxi SS, Ganly I, Singh B, Shah JP, et al. The Molecular Landscape of Recurrent and Metastatic Head and Neck Cancers: Insights From a Precision Oncology Sequencing Platform. JAMA Oncol. 2016 Jul 21. [Epub ahead of print] https://doi.org/10.1001/jamaoncol.2016.1790.
20. Ferrarotto R, Mitani Y, Diao L, Guijarro I, Wang J, Zweidler-McKay P, Bell D, William WN Jr, Glisson BS, Wick MJ, Kapoun AM, Patnaik A, Eckhardt G, et al. Activating NOTCH1 Mutations Define a Distinct Subgroup of Patients With Adenoid Cystic Carcinoma Who Have Poor Prognosis, Propensity to Bone and Liver Metastasis, and Potential Responsiveness to Notch1 Inhibitors. J Clin Oncol. 2017; 35:352–60. https://doi.org/10.1200/JCO.2016.67.5264.
21. Stenman G, Andersson MK, Andrén Y. New tricks from an old oncogene: Gene fusion and copy number alterations of MYB in human cancer. Cell Cycle. 2010; 9:2986–2995. https://doi.org/10.4161/cc.9.15.12515.
22. Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013; 108:479–485. https://doi.org/10.1038/bjc.2012.581.
23. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404. https://doi.org/10.1158/2159-8290.CD-12-0095.
24. Ferrando AA. The role of NOTCH1 signaling in T-ALL. Hematology (Am Soc Hematol Educ Program). 2009; 2009:353–61. https://doi.org/10.1182/asheducation-2009.1.353.
25. Fusco N, Geyer FC, De Filippo MR, Martelotto LG, Ng CK, Piscuoglio S, Guerini-Rocco E, Schultheis AM, Fuhrmann L, Wang L, Jungbluth AA, Burke KA, Lim RS, et al. Genetic events in the progression of adenoid cystic carcinoma of the breast to high-grade triple-negative breast cancer. Mod Pathol. 2016; 29:1292–305. https://doi.org/10.1038/modpathol.2016.134.
26. Gravgaard KH, Lyng MB, Laenkholm AV, Søkilde R, Nielsen BS, Litman T, Ditzel HJ. The miRNA-200 family and miRNA-9 exhibit differential expression in primary versus corresponding metastatic tissue in breast cancer. Breast Cancer Res Treat. 2012; 134:207–217. https://doi.org/10.1007/s10549-012-1969-9.
27. Weigelt B, Glas AM, Wessels LF, Witteveen AT, Peterse JL, van’t Veer LJ. Gene expression profiles of primary breast tumors maintained in distant metastases. Proc Natl Acad Sci USA. 2003; 100:15901–15905. https://doi.org/10.1073/pnas.2634067100.
28. Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, et al. Molecular portraits of human breast tumours. Nature. 2000; 406:747–752. https://doi.org/10.1038/35021093.
29. Bernards R, Weinberg RA. A progression puzzle. Nature. 2002; 418:823. https://doi.org/10.1038/418823a.
30. Andreasen S, Melchior LC, Kiss K, Bishop JA, Høgdall E, Grauslund M, Wessel I, Homøe P, Agander TK. The PRKD1 E710D hotspot mutation is highly specific in polymorphous adenocarcinoma of the palate from adenoid cystic carcinoma and pleomorphic adenoma on FNA. Cancer Cytopathol. 2017 Dec 20. https://doi.org/10.1002/cncy.21959. [Epub ahead of print].
31. Andreasen S, Bishop JA, Hansen TV, Westra WH, Bilde A, von Buchwald C, Kiss K. Human Papillomavirus-related Carcinoma with Adenoid Cystic-like Features of the Sinonasal Tract: Clinical and Morphological Characterization of 6 New Cases. Histopathology. 2017; 70:880–888. https://doi.org/10.1111/his.13162.
32. Andreasen S, Bjørndal K, Agander TK, Wessel I, Homøe P. Tumors of the sublingual gland: a national clinicopathologic study of 29 cases. Eur Arch Otorhinolaryngol. 2016; 273:3847–3856. https://doi.org/10.1007/s00405-016-4000-y.
33. Andreasen S, Therkildsen MH, Grauslund M, Friis-Hansen L, Wessel I, Homøe P. Activation of the interleukin-6/Janus kinase/STAT3 pathway in pleomorphic adenoma of the parotid gland. APMIS. 2015; 123:706–715. https://doi.org/10.1111/apm.12407.
34. Andreasen S, Grauslund M, Heegaard S. Lacrimal gland ductal carcinomas: Clinical, Morphological and Genetic characterization and implications for targeted treatment. Acta Ophthalmol. 2017; 95:299–306. https://doi.org/10.1111/aos.13310.
35. Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O’Daniel JM, Ormond KE, Rehm HL, Watson MS, Williams MS, Biesecker LG, and American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013; 15:565–74. https://doi.org/10.1038/gim.2013.73.
36. Howe K. Extraction of miRNAs from Formalin-Fixed Paraffin-Embedded (FFPE) Tissues. Methods Mol Biol. 2017; 1509:17–24. https://doi.org/10.1007/978-1-4939-6524-3_3.
37. Garmire LX, Subramaniam S. Evaluation of normalization methods in mammalian microRNA-Seq data. RNA 2012; 18:1279–1288. https://doi.org/10.1261/rna.030916.111.
38. Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc B. 1995; 57:289–300.
39. Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006; 22:1540–42. https://doi.org/10.1093/bioinformatics/btl117.
40. Vlachos IS, Zagganas K, Paraskevopoulou MD, Georgakilas G, Karagkouni D, Vergoulis T, Dalamagas T, Hatzigeorgiou AG. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015; 43:W460–466. https://doi.org/10.1093/nar/gkv403.
41. Vlachos IS, Paraskevopoulou MD, Karagkouni D, Georgakilas G, Vergoulis T, Kanellos I, Anastasopoulos IL, Maniou S, Karathanou K, Kalfakakou D, Fevgas A, Dalamagas T, Hatzigeorgiou AG. DIANA-TarBase v7.0: Indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015; 43:D153–159. https://doi.org/10.1093/nar/gku1215.
42. Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999; 27:29–34.