
Introduction
Microtubule-targeting agents or mitotic inhibitors are one of the cornerstones of modern chemotherapy [1-5]. Despite different effects on microtubules and tubulin, a variety of structures and binding sites, all microtubule-active drugs at low concentrations kill proliferating (cycling) cells, by causing fatal mitotic arrest [6-9]. In addition to killing cancer cells, these mitotic inhibitors (MI) can kill normal cycling cells (bone marrow cells, hair follicles, mucosal and epithelial cells), thus causing certain side effects. Side effects may not only be devastating, but they also limit anticancer therapy. The goal is to protect proliferating normal cells from the cytotoxicity of MI, without protecting cancer cells.
Given that MI cannot possibly cause mitotic arrest in the cells that do not enter mitosis, a transient G1 and/or G2 arrest (protective arrest) must protect cells from MI. How can we cause a protective G1/G2 arrest selectively in normal cells but not in cancer cells? Whereas all normal cells have wt p53, this tumor suppressor is mutant or lost in 50% of cancers [10]. This absence provides a means for selective protection of normal cells without protecting cancer cells lacking wt p53 [11-13]. Thus, induction of p53 can arrest cells in G1 and G2 phases, preventing their entry into mitosis. Specifically, low concentrations of DNA damaging drugs (DDD) induce wt p53 and arrest cells in G1 and G2, thus protecting cells from mitotic inhibitors [14-17]. In paired cell lines, DDD protected cells with wt p53 but not cells lacking p53 and p21 [14]. However, the effects of DDD are poorly reversible. While mitotic arrest and polyploidization were prevented (a short term protection), the prospect on long-term cell survival and recovery of proliferation remained unclear. Also DDD can protect some cancer cell lines lacking p53 [15]. It was given consideration from the start that less toxic and more selective drugs should be found to induce p53 for the optimal protection of normal cells [14].
Nutlin-3a, an inhibitor of Mdm2, induces wt p53 without causing DNA damage [18, 19]. Also, its effect is strictly p53-dependent [20]. In paired and isogenic cancer cell lines (with and without wt p53), nutlin-3a selectively protected cells with wt p53 [16, 21, 22]. Importantly, nutlin-3a provided a long-term protection from paclitaxel to skin-derived fibroblast cell line in culture, so that cells proliferated after removal of paclitaxel [21]. This outstanding result needed to be further confirmed in other normal cell types including epithelial cells.
Nutlin-3a is an experimental therapeutic and is not approved yet for clinical use. Therefore, we also planned to investigate clinically approved drugs that might protect normal cells from MI. Cancer cells are characterized by dysregulation of the PI-3K/mTOR pathway [23-26]. It was hypothesized that the mTOR inhibitor rapamycin may protect normal cells from cycle-dependent chemotherapy [27]. Although seemingly unrelated, a remarkable study by Longo and co-workers showed that fasting protected mice from the toxicity of chemotherapy [28-30] and, most importantly, abrogated side effects of chemotherapy in patients with cancer [31]. We suggest that protective effect of fasting may in part be due to inhibition of the nutrient-sensing mTOR pathway in normal cells. Fasting decreases blood levels of nutrients (glucose, amino acids), IGF-1 and insulin, which otherwise activate mTOR in the organism [32-37].
Metformin, an anti-diabetic drug, can in part substitute for fasting because it decreases levels of glucose and insulin [38, 39]. Metformin deactivates mTOR in mice [40]. Also, metformin inhibits the mTOR pathway in cell culture [41-43]. Both metformin and rapalogs (rapamycin and its analogs) are clinically approved drugs. Rapalogs are also approved for cancer therapy [44, 45]. Metformin and rapamycin potentiate chemotherapy against cancer [46]. Furthermore, rapalogs and metformin in combination with chemotherapy (including MI) are undergoing clinical trials [47-50].
Here we investigated whether nutlin-3a, rapamycin, metformin and their combinations can protect 3 normal cell types: WI-38t fibroblasts, RPE (retinal pigment epithelial) and NKE (normal kidney epithelial) cells, without protection of MDA-MB-231 breast cancer cells with mutant p53. We investigated whether normal cells were fully protected and recovered upon removal MI. We identified effective combinations of rapamycin with both metformin and nutlin-3a for protection of normal cells from MI.
Results
Protection of WI-38t cells by nutlin-3a
To evaluate long-term protection, we determined cell numbers six days after removal of mitotic inhibitors (MI) such as nocodazole and paclitaxel (Fig. 1A). We initially used nocodazole (Noco) because it is easily removable by washing cell culture. Cells were pretreated with nutlin-3 and the next day nocodazole was added. After 3 days of treatment with nocodazole, cells were washed and cultured in the fresh medium for 6 days to allow recovery of the protected cells (Fig. 1 A). Nocodazole alone decreased cell numbers more than 50-fold compared with control in both MDA-MB-231 and WI-38t (Fig. 1B). Pretreatment with nutlin-3a did not protect MDA-MB-231 cells, but completely prevented the effects of nocodazole in WI-38t cells (Fig. 1B). Nutlin-3 caused G1 and G2 arrest (Fig. 1C). Nocodazole arrested cells with 4N DNA content, corresponding to mitotic arrest as evidenced by mitotic/round cells visualized on live microscopy (Fig. 1C). Pretreatment with nutlin-3a (Nu) completely prevented nocodazole-induced mitotic arrest (Fig. 1C, Nu+Noco). Cell cycle distributions of nutlin-pretreated cells were almost identical with (+Noco) and without nocodazole (Fig. 1C). Thus, G1 and G2 arrest caused by nutlin-3a prevented mitotic arrest caused by nocodazole.
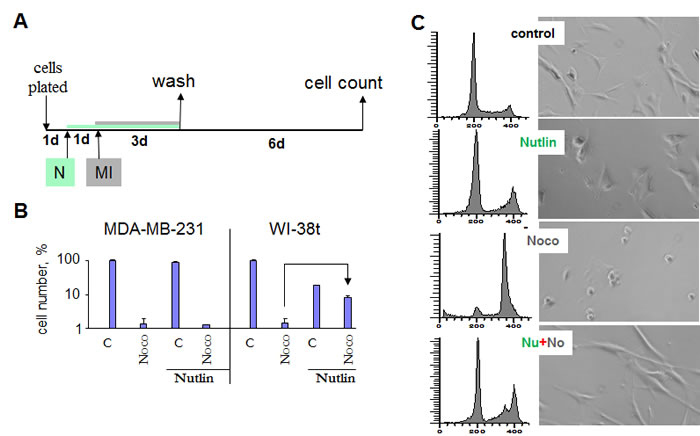
Figure 1: Nutlin-3a protects cells by preventing mitotic arrest caused by nocodazole. (A) Drug treatment schedule: MI was added 1 day after nutlin-3a. 3days later all drugs were washed off and cells were allowed to recover for 6 d and then counted. (B) Selective protection of WI-38t, but not MDA-MB-231 cells, from the cytotoxic effect of nocodazole. 10,000 cells were plated per well (in 6 well plates), treated as shown in panel A and counted after 6 days. The results are shown as % of control, in log scale. (C) Prevention of toxic mitotic arrest by non-toxic G1 and G2 arrest. WI-38-t cells were pre-treated with 2.5 µM nutlin-3a, and then were treated with 200 nM nocodazole. After 24 hours, cells were microphotographed (right panel), collected and analyzed by flow cytometry.
As expected, nutlin-3a induced p53 and p21 (Fig. 2A). Nocodazole alone induced p53 without inducing p21. This is consistent with induction of p53 during prolonged mitotic arrest [7, 51, 52]. Isolated induction of p53 (without induction of p21) by nocodazole is a consequence of mitotic arrest [7, 52]. At both concentrations of nutlin-3a (2.5 µM and 10 µM), cells were completely protected from nocodazole: cell numbers were equal after treatment with nutlin-3a alone and nutlin-3a plus nocodazole (Fig. 2B).
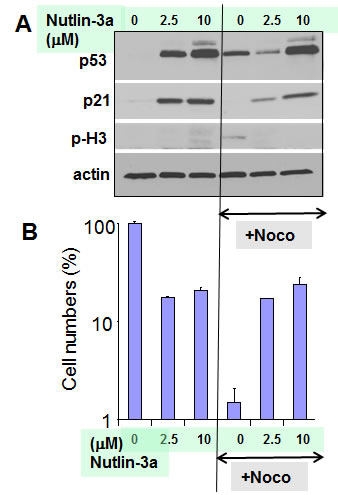
Figure 2: Protective induction of p53/p21 by nutlin-3a in WI-38t cells.
A. Immunoblot. WI-38t cells were pre-treated with indicated concentrations of nutlin-3a (2.5 µM and 10 µM), and then treated with 200 nM nocodazole. The next day, cells were lysed and immunoblot was performed as described in the Methods section.
B. 50, 000 cells were plated per well (in 6 well plates) and treated as shown in Fig. 1A. The results present cell numbers expressed as % of control in log scale. Note: Cell numbers in Nutlin+Noco exceeded plated cell numbers almost 10-fold, indicating active cell proliferation after drug removal (not shown).
Protection of RPE cells by nutlin-3a
Nutlin-3a also causes reversible arrest in RPE cells, so that cells could resume proliferation after nutlin-3a was removed [53, 54]. We next investigated whether this arrest can protect RPE cells from the toxicity of MI. Pretreatment with nutlin-3a partially abrogated the toxicity of nocodazole and paclitaxel (Taxol) in RPE cells but not in cancer MDA-MB-231 cells with mutant p53 (Fig. 3). The effect of paclitaxel (PTX) was less reversible than the effect of nocodazole because PTX is poorly washable from the cell culture.
Analysis of cell cycle distribution revealed that nutlin-3a caused G2 arrest in RPE cells (Fig. 4). As expected, nocodazole caused mitotic arrest, which was indistinguishable from G2 arrest by flow cytometry, but was evident by the appearance of mitotic cells (Fig. 4A, microphotographs). By arresting cells in G2, nutlin-3a prevented mitotic arrest, thus protecting normal cells. In contrast, nutlin-3a did not cause arrest in MDA-MB-231 cells (Fig. 4B). Therefore, nocodazole caused mitotic arrest in MDA-MB-231 cells both in the presence and the absence of nutlin-3a (Fig. 4B).
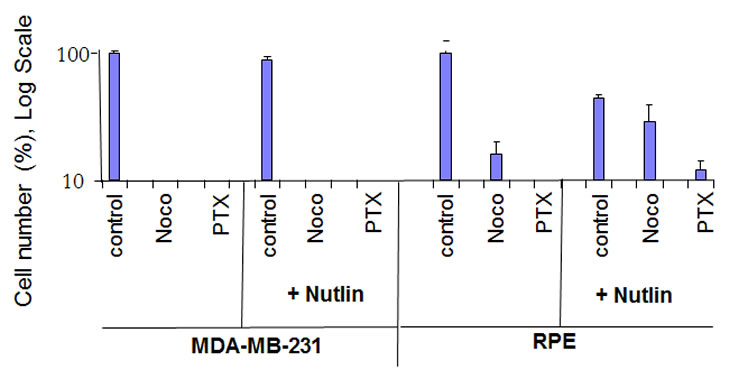
Figure 3: Nutlin-3a protects normal retinal pigment epithelial (RPE-19) cells but not MDA-MB-231 cells. 10,000 cells were plated and treated the next day with 2.5 µM nutlin-3a or left untreated. The next day, cells were treated with either 200 nM nocodazole, 50 nM Taxol (paclitaxel) or left untreated. Cells were counted 6 days after wash because control cells reached confluence at that time. The results are shown as % of control, in log scale. Note: Nutlin+Noco, final cell numbers exceeded plated cell numbers, indicating cell proliferation after drug removal.
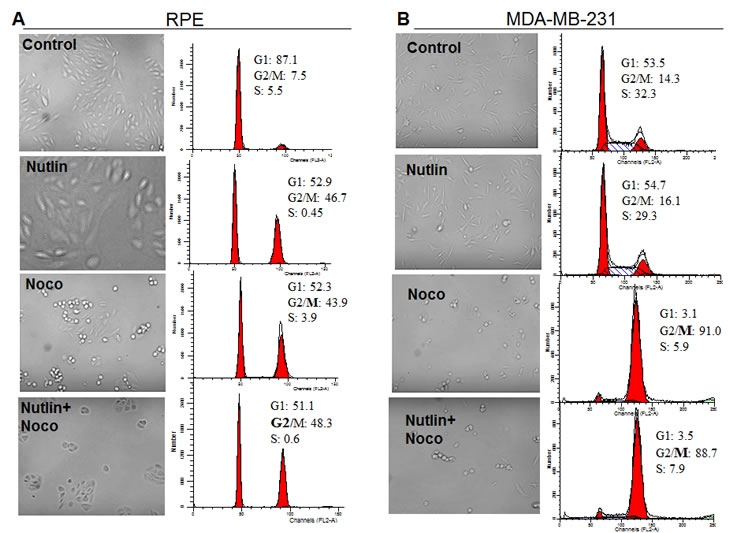
Figure 4: Nutlin-3a prevents toxic mitotic arrest by causing non-toxic G2 arrest in RPE cells. RPE (A) and MDA-MB-231 (B) cells were plated in 6-well plates at 50,000/well. Cells were either pretreated with 2.5 µM nutlin-3a or left untreated before addition of 200 nM nocodazole. After 24 hours treatment with nocodazole, cells were microphotographed (left panels), collected and analyzed by flow cytometry (right panels).
Protection of RPE by drug combinations
Noteworthy, protection of RPE by nutlin-3a was potentiated by rapamycin, which was slightly cytoprotective by itself (Fig. 5 A). Metformin alone was marginally protective but its effect was also potentiated by rapamycin. A combination of metformin plus rapamycin and a combination of nutlin-3a plus rapamycin afforded comparable protection. Similar results were obtained in low glucose medium (Fig. 5 B).
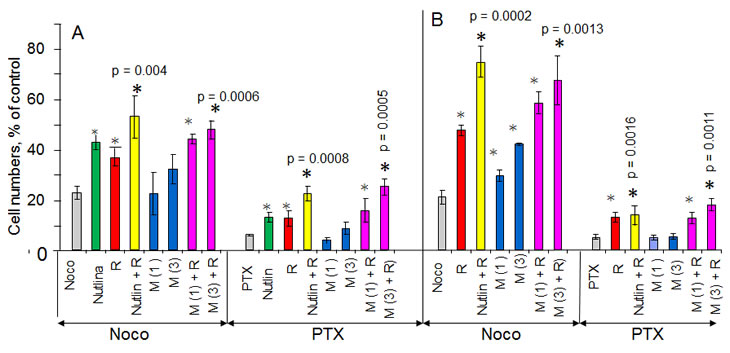
Figure 5: Cytoprotection of RPE cells with nutlin-3a, rapamycin and metformin 10,000 RPE cells were plated in normal (1g/L) glucose medium (panel A) and low (0.5 g/L) glucose medium (panel B). The next day, cells were treated with either 2.5 µM nutlin-3a (Nutlin), 1 and 3 mM metformin (M 1 and M 3), 1 nM rapamycin (R) or left untreated. The next day, cells were treated with either 200 nM nocodazole (Noco) or 50 nM paclitaxel (PTX). 3 days later, the cells were washed and cultured in normal glucose for an additional 10 days and then were counted. The results are shown as % of control (no treatment). Note: in control cells reached confluence during the experiment.
Protection of WI-38t by a combination of rapamycin and metformin
Next we investigated whether a combination of rapamycin and metformin (R+M) can protect WI-38t cells. This combination (M + R) afforded 2-3 fold protection in WI-38t, but not in MDA-MB-231 cells (Suppl. Fig. 1). Individually, 3 mM metformin and 1 nM-100 nM rapamycin were slightly protective. Combinations of metformin with 1 nM rapamycin were more effective (Fig. 6). We next investigated the mechanism of cytoprotection by rapamycin plus metformin (R+M). The combination (R+M) caused G1/G2 arrest and this partially prevented mitotic arrest caused by nocodazole (Fig. 7). In contrast, R+M did not protect MDA-MB-231 cells (Fig. 7). Noteworthy, the R+M combination was toxic to MDA-MB-231 cells cultured at low glucose (Suppl. Fig. 2).
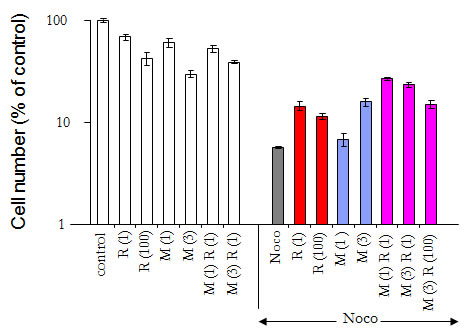
Figure 6: Cytoprotection of WI-38t cells by combining rapamycin and metformin 10,000 WI-38t cells were plated in normal glucose medium. The next day, cells were treated with either 1 mM or 3 mM metformin (M 1 and M 3) and 1 nM or 100 nM rapamycin (R 1 and R 100) or left untreated (control). The next day, cells were treated with 200 nM nocodazole (Noco) as indicated. After the 3 days, the cells were washed and cultured for an additional 10 days and then were counted. The results are shown as % of control (no treatment). Note: in control cells reached confluence during the experiment.
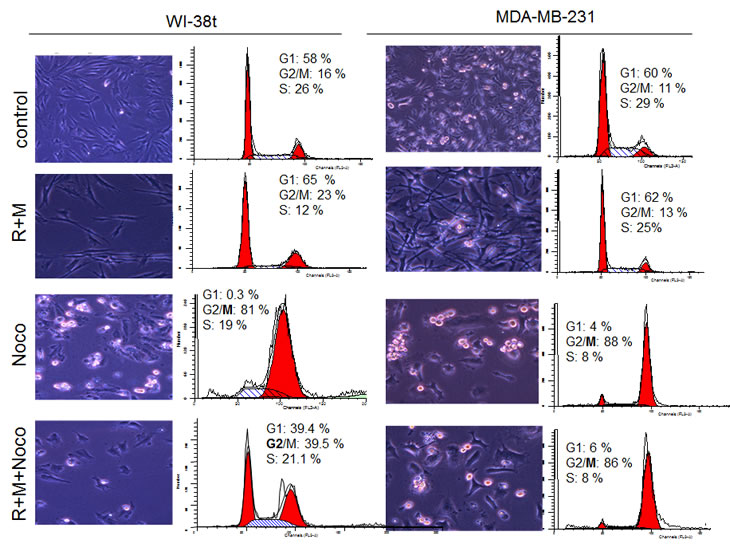
Figure 7: A combination of rapamycin and metformin prevents mitotic arrest in WI-38t cells. 50,000 WI-38t and MDA-MB-231 cells were plated in 6-well plates and pre-treated with a combination of 100 nM rapamycin and 3 mM metformin (R+M). The next day, cells were treated with 200 nM nocodazole. After 24 hours, cells were microphotographed (left panels), collected and flow cytometry was performed (right panels).
Comparison of M+R and nutlin-3a in NKE cells
We next tested whether these combinations were protective in a third type of normal cells. Normal kidney epithelial (NKE) cells were partially protected from nocodazole and paclitaxel (PTX) in both normal and low glucose media. Combinations of nutlin-3a with rapamycin (N+R), even though rapamycin alone was not protective, were the most effective in NKE cells (Fig. 8). Combinations of metformin with rapamycin (R+M) were moderately protective (Fig. 8).
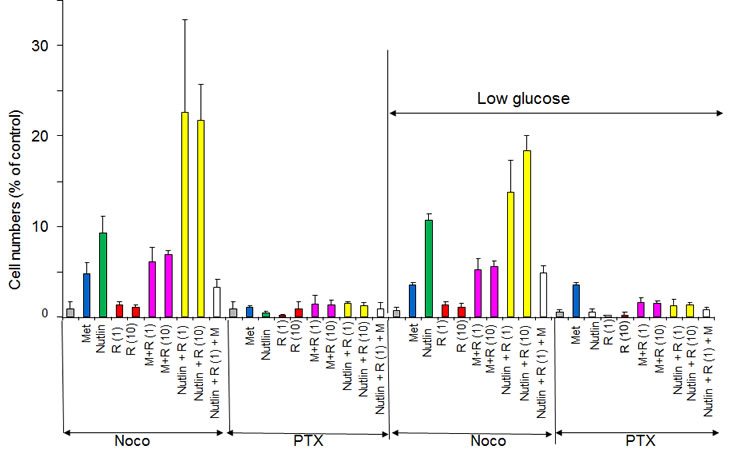
Figure 8: Protection of NKE cells from nocodazole and paclitaxel in normal glucose and low glucose. 10,000 NKE cells were plated in either 1g/L glucose or 0.5g/L glucose (low). Cells were pre-treated for 24 hrs with either 2.5 µM nutlin-3a, 1 and 10 nM rapamycin (R1 and R10), 3 mM metformin (M) alone or in combination and then were treated with either 200 nM nocodazole (Noco) or 50 nM Taxol (PTX). After 3 days, cells were washed and cultured for 6 days in fresh medium before counting.
Discussion
The goal of chemotherapeutic cyclotherapy is selective protection of normal cells by exploiting aberrations in cancer cell cycle such as loss of p53 [13, 55-57]. By causing G1 and G2 arrest, nutlin-3a protects cells with wt p53 from mitotic inhibitors. As a proof of principle in paired cell lines, nutlin-3a prevented mitotic arrest, cell death or polyploidization (a short-term protection) [16, 17, 21, 22]. Furthermore, pretreatment with nutlin-3a allowed normal skin-derived fibroblasts to recover from MI treatment, as measured by an increase in cell numbers after 6 days (a long-term protection) in drug-free medium [21]. Here we focused on a long-term protection and extended this observation to WI-38 fibroblasts and two epithelial cell lines: RPE and NKE. As expected, nutlin-3a arrested normal cells in both G1 and/or G2 (depending on cell type) and thus protected them from mitotic arrest caused by MI. While affording a long-term protection to all 3 normal cell types, nutlin-3a did not protect MDA-MB-231 cancer cells in any experiments. Similar results in normal cell lines of different origin indicate that nutlin-3a might also protect relevant normal cells in the organism. Remarkably, Sur et al demonstrated that oral administration of Nutlin-3 (200 mg/kg) efficiently protected mice from neutropenia caused by a mitotic inhibitor [20]. This important result indicates that protection in cell culture is indeed applicable to the organism.
We found that rapamycin further potentiated the protective effect of nutlin-3a in three cell lines, most robustly in NKE cells. There are two potential mechanisms. First, rapamycin, as a mild cytostatic agent, may increase the durability of arrest, thus potentiating the protection. Second, to be protective in a long-term, the arrest must be reversible. Nutlin-3-induced arrest is mostly reversible in most cell lines [58, 59]. But nutlin-3a can also induce irreversible senescence, depending on the activity of mTOR, p53 levels and the duration of the arrest [53, 54, 60]. Rapamycin may improve the reversibility of arrest caused by p53 [53]. We are currently investigating the mechanism of potentiation of nutlin-3a by rapamycin (Leontieva et al, MS in preparation).
Since nutlin-3a does not cause arrest in cancer cells lacking p53, the addition of rapamycin cannot possibly potentiate nutlin-3a in cells with mutant p53. Therefore, like nutlin-3a alone, a combination of nutlin-3a plus rapamycin (N+R) does not protect cancer cells with mutant p53. Therefore, the combination of nutlin-3a plus rapamycin can be used for protection of normal cells in patients having tumors with mutant p53. In heterogeneous tumors with co-existing cells having wt and mutant p53, the N+R combination may spare cancer cells with wt p53. This will revert the tumor to a less aggressive phenotype, which is more sensitive to conventional chemotherapy. Therefore, a combination of N+R+MI may be promising in heterogeneous cancers too.
It was shown that fasting, which decreases levels of glucose and insulin and inactivates nutrient-sensing signaling pathways, decreases side-effects of chemotherapy [29]. Like fasting, metformin, an anti-diabetic drug, decreases glucose levels. Also, metformin lowers levels of insulin in women with early breast cancer [38]. Furthermore, metformin decreases tumor growth in mice fed by high-calorie diet [61, 62]. Metformin and rapamycin differently affects nutrient-sensing pathways [63]. Therefore, 2 drugs may in theory potentiate each other. A combination of temsirolimus (an analog of rapamycin) and metformin showed promising results in phase I clinical trial as cancer therapy [43, 64].
Here we demonstrated that a combination of rapamycin and metformin (R+M) caused protective G1 and G2 arrest in normal cells. Furthermore, this combination provided a long-term protection against MI in all three normal cell types. Not only did the R+M combination not protect cancer cells but, in contrast, it was toxic by itself to MDA-MB-231 cells in low glucose conditions. Low glucose levels are toxic to cancer cells, which depend on glucose as substrates for their glycolytic phenotype [65-72]. Noteworthy, metformin decreases glucose levels in the organism.
Metformin enhances susceptibility of p53-/- cells to apoptosis upon nutrient deprivation [73]. In contrast, p53-proficient cells can undergo cell cycle arrest (instead of apoptosis) in low glucose [74]. This explains selective toxicity of metformin to p53-deficient cells in low glucose [73, 74] observed in our study too. Thus a combination of metformin, rapamycin and fasting may be toxic to cancer cells, while protecting normal cells from chemotherapy with MI (Fig. 9), thus increasing therapeutic window. Concentrations of metformin (1 mM) and rapamycin (1 nM) are lower than those achievable in patients [75-80]. We suggest that metformin and rapamycin should be administrated for a 1-2-day period prior and simultaneously with MI such as the Vinca drugs and Taxanes (Fig. 10). In conclusion, based on cell culture data and analysis of the literature, we suggest that nutlin-3a, nutlin-3a plus rapamycin and rapamycin plus metformin may extend the therapeutic window of microtubule-active chemotherapy. Low concentrations of rapamycin and metformin, especially combined with fasting, may be used in the clinic today.
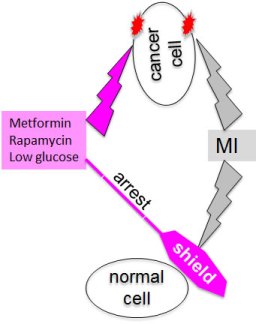
Figure 9: Cyclotherapy: protection of normal cells and unshielding of cancer cells.

Figure 10: Proposal for potential clinical trials
Methods and Materials
Cell Culture
Cells were cultured in RPMI media purchased from Cellgro (Manassas, VA) supplemented with 5% (v/v) FBS. Rapamycin was purchased from LC Laboratory (Woburn, MA), and metformin, nutlin-3a, nocodazole and paclitaxel were from Sigma-Aldrich (St. Louis, MO, USA). Glucose was added to glucose-free RPMI to desired concentrations or mixed in ratios using RPMI with glucose (2 g/L) and RPMI without glucose. WI-38, WI-38t fibroblasts immortalized by telomerase and retinal pigment epithelial (RPE) ARPE-19 cells were described previously [81], [59]. MDA-MB-231 was from ATCC (Manassas, VA). Normal Kidney Epithelial (NKE) cells were provided by Dr. Katerina V. Gurova (RPCI).
Flow cytometry
Cells were fixed in 70% ethanol and stained with 25 μg/ml propidium iodide (PI), 0.2% Triton X- 100 and 40 μg/ml RNase in PBS. Flow cytometry was performed using FACScan, Becton Dickinson, San Jose, CA with 10,000-20,000 events per sample. Cell cycle distribution was analyzed using Modfit LT software (Verity Software House Inc., Topsham, ME) with histograms of DNA content profiles on the X-axis and cell numbers on the y-axis.
Mitotic Index
Following flow cytometry, propidium iodine stained nuclei were visualized on coverslips and mitotic cells were identified by morphological characteristics allowing G2 phase verses mitotic cells to be distinguished. Cells were photographed on a Zeiss Axioplan 2 microscope (Thornwood, NY).
Cell Counting
Cells were counted on Vicell Series Cell Viability Analyzer (Beckman Coulter, Inc., Brea, CA) using Trypan Blue exclusion method to provide a proportion of live and dead cells.
Immunoblot analysis
Immunoblot was performed as described previously [53]. The following antibodies were used: mouse anti-p53 (Ab-6) from Oncogene, mouse anti-p21 from BD Biosciences (San Jose, CA); rabbit anti-actin from Sigma-Aldrich (St. Louis, MO); secondary goat anti-rabbit and goat antimouse HRP conjugated antibodies were from Chemicon (Billerica, MA) and Bio-Rad (St. Louis, MO), respectively.
Author Contributions
MVB and FL conceptualized the project and designed the experiments. PA, OL and ZD performed the experiments. MVB wrote the manuscript. All authors reviewed the manuscript and gave their feedback. MVB oversaw the project.
FUNDING
Roswell Park Cancer Institute, Buffalo, NY 14203.
CONFLICTS OF INTEREST
No conflicts to declare.
References
1. Blagosklonny MV. Analysis of FDA approved anticancer drugs reveals the future of cancer therapy. Cell Cycle. 2004; 3:1035-1042.
2. Jordan MA. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr Med Chem Anticancer Agents. 2002; 2:1-17.
3. Rowinsky EK, Calvo E. Novel agents that target tublin and related elements. Semin Oncol. 2006; 33:421-435.
4. Morris PG, Fornier MN. Microtubule active agents: beyond the taxane frontier. Clin Cancer Res. 2008; 14:7167-7172.
5. Perez EA. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol Cancer Ther. 2009; 8:2086-2095.
6. Jordan MA, Thrower D, Wilson L. Mechanism of inhibition of cell proliferation by Vinca alkaloids. Cancer Res. 1991; 51:2212-2222.
7. Demidenko ZN, Kalurupalle S, Hanko C, Lim CU, Broude E, Blagosklonny MV. Mechanism of G1-like arrest by low concentrations of paclitaxel: next cell cycle p53-dependent arrest with sub G1 DNA content mediated by prolonged mitosis. Oncogene. 2008; 27:4402-4410.
8. Blagosklonny MV. Mitotic arrest and cell fate: why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle. 2007; 6:70-74.
9. Bekier ME, Fischbach R, Lee J, Taylor WR. Length of mitotic arrest induced by microtubule-stabilizing drugs determines cell death after mitotic exit. Mol Cancer Ther. 2009; 8:1646-1654.
10. Vogelstein B, Lane DP, Levine AJ. Surfing the p53 network. Nature. 2000; 408:307-310.
11. Blagosklonny MV, Pardee AB. Exploiting cancer cell cycling for selective protection of normal cells. Cancer Res. 2001; 61:4301-4305.
12. Blagosklonny MV. Matching targets for selective cancer therapy. Drug Discov Today. 2003; 8:1104-1107.
13. Blagosklonny MV. “Targeting the absence” and therapeutic engineering for cancer therapy. Cell Cycle. 2008; 7:1307-1312.
14. Blagosklonny MV, Robey R, Bates S, Fojo T. Pretreatment with DNA-damaging agents permits selective killing of checkpoint-deficient cells by microtubule-active drugs. J Clin Invest. 2000; 105:533-539.
15. Blagosklonny MV. Sequential activation and inactivation of G2 checkpoints for selective killing of p53-deficient cells by microtubule-active drugs. Oncogene. 2002; 21:6249-6254.
16. Choong ML, Yang H, Lee MA, Lane DP. Specific activation of the p53 pathway by low dose actinomycin D: A new route to p53 based cyclotherapy. Cell Cycle. 2009; 8:2810-8
17. Rao B, van Leeuwen IMM, Higgins M, Campbel J, Thompson AM, Lane DP, Lain S, 639-650 p. Evaluation of an Actinomycin D/VX-680 aurora kinase inhibitor combination in p53-based cyclotherapy. Oncotarget. 2010; 1:639-650.
18. Vassilev LT. Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle. 2004; 3:419-421.
19. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004; 303:844-848.
20. Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA, Jr., Kinzler KW, Vogelstein B, Papadopoulos N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A. 2009; 106:3964-3969.
21. Carvajal D, Tovar C, Yang H, Vu BT, Heimbrook DC, Vassilev LT. Activation of p53 by MDM2 antagonists can protect proliferating cells from mitotic inhibitors. Cancer Res. 2005; 65:1918-1924.
22. Cheok CF, Kua N, Kaldis P, Lane DP. Combination of nutlin-3 and VX-680 selectively targets p53 mutant cells with reversible effects on cells expressing wild-type p53. Cell Death Differ. 2010:1486-1500.
23. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004; 10:789-799.
24. Kang S, Bader AG, Zhao L, Vogt PK. Mutated PI 3-kinases: cancer targets on a silver platter. Cell Cycle. 2005; 4:578-581.
25. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006; 441:424-430.
26. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007; 12:9-22.
27. Blagosklonny MV, Darzynkiewicz Z. Four birds with one stone: RAPA as potential anticancer therapy. Cancer Biol Ther. 2002; 1:359-361.
28. Raffaghello L, Lee C, Safdie FM, Wei M, Madia F, Bianchi G, Longo VD. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci U S A. 2008; 105:8215-8220.
29. Raffaghello L, Safdie F, Bianchi G, Dorff T, Fontana L, Longo VD. Fasting and differential chemotherapy protection in patients. Cell Cycle. 2010; 9:4474-4476.
30. Lee C, Safdie FM, Raffaghello L, Wei M, Madia F, Parrella E, Hwang D, Cohen P, Bianchi G, Longo VD. Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res. 2010; 70:1564-1572.
31. Safdie FM, Dorff T, Quinn D, Fontana L, Wei M, Lee C, Cohen P, Longo VD. Fasting and cancer treatment in humans: A case series report. Aging. 2009; 1:988-1007.
32. Tremblay F, Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem. 2001; 276:38052-38060.
33. Tremblay F, Krebs M, Dombrowski L, Brehm A, Bernroider E, Roth E, Nowotny P, WaldhŠusl W, Marette A, Roden M. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes. 2005; 54:2674-2684.
34. Shah OJ, Hunter T. Tuberous sclerosis and insulin resistance. Unlikely bedfellows reveal a TORrid affair. Cell Cycle. 2005; 4:46-51.
35. Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. 2005; 146:1473-1481.
36. Tzatsos A, Kandror KV. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol. 2006; 26:63-76.
37. Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, Roth E, FŸrnsinn C, Promintzer M, Anderwald C, Bischof M, Roden M. The Mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes. 2007; 56:1600-1607.
38. Goodwin PJ, Pritchard KI, Ennis M, Clemons M, Graham M, Fantus IG. Insulin-lowering effects of metformin in women with early breast cancer. Clin Breast Cancer. 2008; 8:501-505.
39. Berstein LM. Modern approach to metabolic rehabilitation of cancer patients: biguanides (phenformin and metformin) and beyond. Future Oncol. 2010; 6:1313-1323.
40. Erdemoglu E, Guney M, Giray SG, Take G, Mungan T. Effects of metformin on mammalian target of rapamycin in a mouse model of endometrial hyperplasia. Eur J Obstet Gynecol Reprod Biol. 2009; 145:195-199.
41. Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007; 67:10804-10812.
42. Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle. 2009; 8:88-96.
43. Vazquez-Martin A, Oliveras-Ferraros C, del Barco S, Martin-Castillo B, Menendez JA. mTOR inhibitors and the anti-diabetic biguanide metformin: new insights into the molecular management of breast cancer resistance to the HER2 tyrosine kinase inhibitor lapatinib (Tykerb). Clin Transl Oncol. 2009; 11:455-459.
44. Hudes GR. Targeting mTOR in renal cell carcinoma. Cancer. 2009; 115:2313-2320.
45. Janes MR, Fruman DA. Targeting TOR dependence in cancer. Oncotarget. 2010; 1:69-76.
46. Marimpietri D, Brignole C, Nico B, Pastorino F, Pezzolo A, Piccardi F, Cilli M, Di Paolo D, Pagnan G, Longo L, Perri P, Ribatti D, Ponzoni M. Combined therapeutic effects of vinblastine and rapamycin on human neuroblastoma growth, apoptosis, and angiogenesis. Clin Cancer Res. 2007; 13:3977-3988.
47. Ramalingam SS, Harvey RD, Saba N, Owonikoko TK, Kauh J, Shin DM, Sun SY, Strychor S, Tighiouart M, Egorin MJ, Fu H, Khuri FR. Phase 1 and pharmacokinetic study of everolimus, a mammalian target of rapamycin inhibitor, in combination with docetaxel for recurrent/refractory nonsmall cell lung cancer. Cancer. 2010; 116:3903-3909.
48. Andre F, Campone M, O’Regan R, Manlius C, Massacesi C, Sahmoud T, Mukhopadhyay P, Soria JC, Naughton M, Hurvitz SA. Phase I study of everolimus plus weekly paclitaxel and trastuzumab in patients with metastatic breast cancer pretreated with trastuzumab. J Clin Oncol. 28:5110-5115.
49. Perotti A, Locatelli A, Sessa C, Hess D, Vigano L, Capri G, Maur M, Cerny T, Cresta S, Rojo F, Albanell J, Marsoni S, Corradino I, Berk L, Rivera VM, Haluska F et al. Phase IB study of the mTOR inhibitor ridaforolimus with capecitabine. J Clin Oncol. 28:4554-4561.
50. Sessa C, Tosi D, Vigano L, Albanell J, Hess D, Maur M, Cresta S, Locatelli A, Angst R, Rojo F, Coceani N, Rivera VM, Berk L, Haluska F, Gianni L. Phase Ib study of weekly mammalian target of rapamycin inhibitor ridaforolimus (AP23573; MK-8669) with weekly paclitaxel. Ann Oncol. 21:1315-1322.
51. Giannakakou P, Robey R, Fojo T, Blagosklonny MV. Low concentrations of paclitaxel induce cell type-dependent p53, p21 and G1/G2 arrest instead of mitotic arrest: molecular determinants of paclitaxel-induced cytotoxicity. Oncogene. 2001; 20:3806-3813.
52. Blagosklonny MV. Prolonged mitosis versus tetraploid checkpoint: how p53 measures the duration of mitosis. Cell Cycle. 2006; 5:971-975.
53. Leontieva O, Gudkov A, Blagosklonny M. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010; 9:4323-4327.
54. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY). 2010; 2:344-352.
55. Blagosklonny MV, Darzynkiewicz Z. Cyclotherapy: Protection of normal cells and unshielding of cancer cells. Cell Cycle. 2002; 1:375-382.
56. Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009; 9:862-873.
57. Lane DP, Cheok CF, Lain S. p53-based cancer therapy. Cold Spring Harb Perspect Biol. 2010; 2:a001222.
58. Huang B, Deo D, Xia M, Vassilev LT. Pharmacologic p53 Activation Blocks Cell Cycle Progression but Fails to Induce Senescence in Epithelial Cancer Cells. Mol Cancer Res. 2009; 7:1497-1509.
59. Korotchkina LG, Demidenko ZN, Gudkov AV, Blagosklonny MV. Cellular quiescence caused by the Mdm2 inhibitor nutlin-3a. Cell Cycle. 2009; 8:3777-3781.
60. Efeyan A, Ortega-Molina A, Velasco-Miguel S, Herranz D, Vassilev LT, Serrano M. Induction of p53-dependent senescence by the MDM2 antagonist nutlin-3a in mouse cells of fibroblast origin. Cancer Res. 2007; 67:7350-7357.
61. Algire C, Zakikhani M, Blouin MJ, Shuai JH, Pollak M. Metformin attenuates the stimulatory effect of a high-energy diet on in vivo LLC1 carcinoma growth. Endocr Relat Cancer. 2008; 15:833-839.
62. Algire C, Amrein L, Zakikhani M, Panasci L, Pollak M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010; 17:351-360.
63. Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat. 2010; 123:271-279.
64. Mackenzie MJ, Ernst S, Johnson C, Winquist E. A phase I study of temsirolimus and metformin in advanced solid tumours. Invest New Drugs. in press
65. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004; 101:3329-3335.
66. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004; 4:891-899.
67. Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008; 134:703-707.
68. Yeluri S, Madhok B, Prasad KR, Quirke P, Jayne DG. Cancer’s craving for sugar: an opportunity for clinical exploitation. J Cancer Res Clin Oncol. 2009; 135:867-877.
69. Young CD, Anderson SM. Sugar and fat - that’s where it’s at: metabolic changes in tumors. Breast Cancer Res. 2008; 10:202.
70. Diaz-Ruiz R, Uribe-Carvajal S, Devin A, Rigoulet M. Tumor cell energy metabolism and its common features with yeast metabolism. Biochim Biophys Acta. 2009; 1796:252-265.
71. Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006; 66:8927-8930.
72. Bui T, Thompson CB. Cancer’s sweet tooth. Cancer Cell. 2006; 9:419-420.
73. Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007; 67:6745-6752.
74. Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005; 18:283-293.
75. Dell’Aglio DM, Perino LJ, Kazzi Z, Abramson J, Schwartz MD, Morgan BW. Acute metformin overdose: examining serum pH, lactate level, and metformin concentrations in survivors versus nonsurvivors: a systematic review of the literature. Ann Emerg Med. 2009; 54:818-823.
76. Mahalati K, Kahan BD. Clinical pharmacokinetics of sirolimus. Clin Pharmacokinet. 2001; 40:573-585.
77. Rodriguez AE, Rodriguez Alemparte M, Vigo CF, Fernandez Pereira C, Llaurado C, Vetcher D, Pocovi A, Ambrose J. Role of oral rapamycin to prevent restenosis in patients with de novo lesions undergoing coronary stenting: results of the Argentina single centre study (ORAR trial). Heart. 2005; 91:1433-1437.
78. Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, Liau L, Martin N, Becker D, Bergsneider M, Lai A, Green R et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008; 5:e8.
79. Boehm A, Mayerhofer M, Herndlhofer S, Knoebl P, Sillaber C, Sperr WR, Jaeger U, Valent P. Evaluation of in vivo antineoplastic effects of rapamycin in patients with chemotherapy-refractory AML. Eur J Intern Med. 2009; 20:775-778.
80. Armstrong AJ, Netto GJ, Rudek MA, Halabi S, Wood DP, Creel PA, Mundy K, Davis SL, Wang T, Albadine R, Schultz L, Partin AW, Jimeno A, Fedor H, Febbo PG, George DJ et al. A pharmacodynamic study of rapamycin in men with intermediate- to high-risk localized prostate cancer. Clin Cancer Res. 2010; 16:3057-3066.
81. Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7:3355-3361.