
INTRODUCTION
Apoptosis is one of the best characterized forms of programmed cell death and a crucial process in physiological and pathophysiological conditions [1]. Its execution is tightly regulated by several proteins, like Inhibitor of Apoptosis (IAP) proteins [2] or cFLIP [3]. Evasion of programmed cell death is a hallmark of cancer and accomplished, for example, by aberrant expression of antiapoptotic proteins.
One major cell death regulator is cFLIP. High cFLIP expression is correlated with a poor prognosis in several tumor entities [4–6] and its downregulation is part of effective drug-mediated cell death [7]. There are two main isoforms of cFLIP expressed in human cells which control cell death in a distinct manner: The long isoform cFLIPL, a 55 kDa protein, and the short isoform cFLIPS, a 25 kDa protein [3]. cFLIPL is a caspase-8/-10 homolog with two death effector domains (DEDs), but with an inactive caspase domain. Its influence on cell death regulation is being controversially discussed, as it is reported to exert pro- or antiapoptotic effects, depending on the context. Lower levels of cFLIPL are associated with a proapoptotic function, as highly active heterodimers are formed [8]. In higher concentrations, cFLIPL prevents caspase-8 activation in the death-inducing signaling complex (DISC) [9], which is formed upon death receptor-mediated cell death and caspase-8 activation in the ripoptosome, a signaling platform formed upon IAP depletion [10, 11]. cFLIPS consists only of two DEDs and directly inhibits caspase-8 activation at the DISC [9] and the ripoptosome [10, 11] and thereby apoptosis.
Other cell death-regulating proteins are IAP proteins which are known to be prognostic factors in different tumor entities [12]. Their inhibition as therapeutic approach has been intensively studied and IAP antagonists including Smac mimetics have been developed. Smac mimetics, e.g. BV6, induce autoubiquitination and proteasomal degradation of IAP proteins and have been shown to promote cell death induced by different stimuli [13].
New treatment strategies are required for ALL, as the prognosis for relapsed patients is still poor [14]. We previously discovered that the Smac mimetic BV6 sensitizes ALL cells to glucocorticoids, such as Dexa, which are part of the standard therapy of ALL patients, by promoting the formation of the ripoptosome complex and by exerting antileukemic activity in a patient-derived xenograft model of ALL in vivo [15]. The assembly of the ripoptosome is regulated, amongst others, by the two major isoforms of cFLIP, i.e. cFLIPL and cFLIPS [10, 11]. Therefore, we studied the role of cFLIP in regulating Dexa/BV6-mediated cell death to gain new insights into the molecular mechanisms underlying the synergism of Dexa and BV6.
RESULTS
Dexa downregulates cFLIPL protein in ALL cells
Initially, we determined protein expression of the two major isoforms of cFLIP (i.e. cFLIPL and cFLIPS) in ALL cell lines. Since all analyzed ALL cell lines predominately expressed cFLIPL rather than cFLIPS protein (Supplementary Figure 1), we focused our analysis in ALL on cFLIPL. We then asked whether Dexa as single agent or in combination with BV6 affects cFLIPL levels. Interestingly, treatment with Dexa alone or together with BV6 downregulated cFLIPL protein levels already after a few hours (Figure 1A). To examine whether the loss of cFLIPL protein is due to changes in mRNA expression, we performed qRT-PCR analysis. Dexa treatment increased rather than suppressed cFLIPL mRNA levels (Figure 1B), indicating that the observed loss of cFLIPL protein by Dexa or Dexa/BV6 treatment is independent of mRNA expression. These data indicate a Dexa-mediated downregulation of cFLIPL protein independent of mRNA expression.
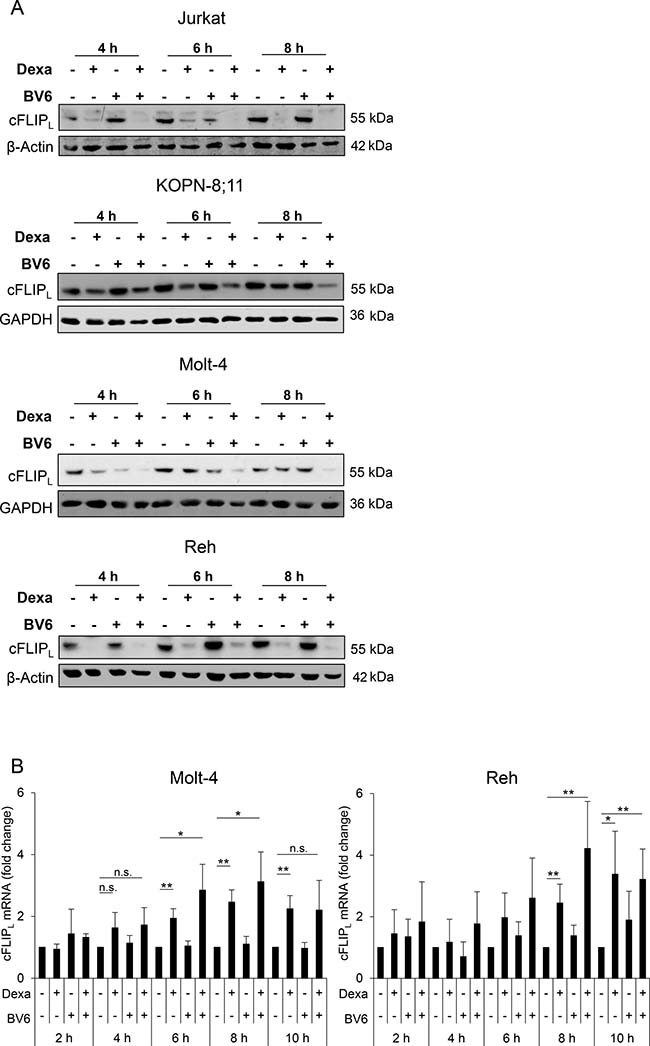
Figure 1: Dexa downregulates cFLIPL protein in ALL cells. ALL cells were treated for indicated time points with Dexa and/or BV6 (Jurkat: 300 μM Dexa, 7 μM BV6; KOPN-8;11: 150 μM Dexa, 2 μM BV6; Molt-4: 300 μM Dexa, 5 μM BV6; Reh: 300 μM Dexa, 0.3 μM BV6). (A) Protein expression of cFLIPL was analyzed by Western blotting. β-Actin or GAPDH served as loading control. (B) cFLIPL mRNA levels were analyzed using qRT-PCR. Fold change of cFLIPL mRNA as mean and standard deviation (SD) of at least three independent experiments are shown.
Dexa impedes BV6-stimulated NF-κB activation
Since Smac mimetics have been described to deplete IAP proteins [16, 17], we determined expression levels of cellular IAP (cIAP)1, cIAP2 and x-linked IAP (XIAP) upon treatment with Dexa and BV6. As expected, treatment with either BV6 alone or in combination with Dexa caused a loss of cIAP1 and cIAP2 in all four cell lines and XIAP expression slightly decreased by Dexa/BV6 cotreatment (Figure 2A). In KOPN-8;11 cells, we did not detect cIAP2 protein (Figure 2A).
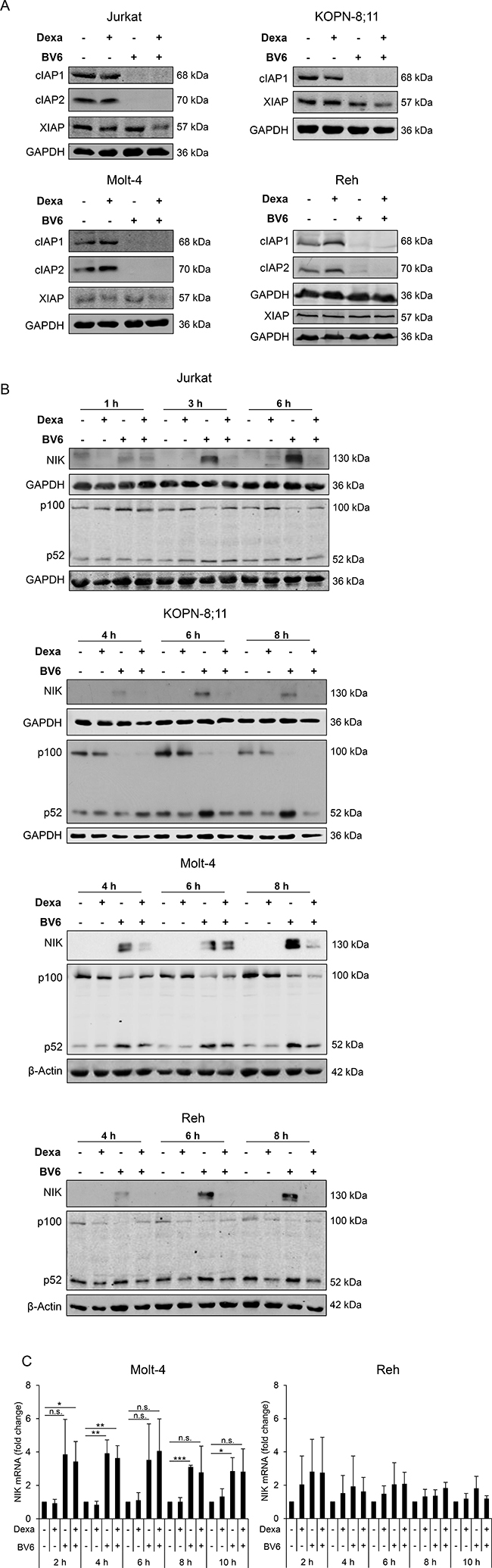
Figure 2: Dexa impedes BV6-stimulated NF-κB activation. ALL cells were treated for four hours (A) or indicated time points (B, C) with Dexa and/or BV6 (Jurkat: 300 μM Dexa, 7 μM BV6; KOPN-8;11: 150 μM Dexa, 2 μM BV6; Molt-4: 300 μM Dexa, 5 μM BV6; Reh: 300 μM Dexa, 0.3 μM BV6). (A) Protein expression of cIAP1, cIAP2 and XIAP was analyzed by Western blotting. GAPDH served as loading control. (B) Protein expression of NIK and p100/p52 was analyzed by Western blotting. β-Actin or GAPDH served as loading control. (C) NIK mRNA levels were analyzed using qRT-PCR. Fold change of NIK mRNA as mean and SD values of at least three independent experiments are shown.
As loss of cIAP proteins can lead to activation of the non-canonical NF-κB pathway [16, 17], we next assessed expression of NF-κB-inducing kinase (NIK) and p100/p52 as key components of non-canonical NF-κB signaling. Treatment with BV6 caused accumulation of NIK protein (Figure 2B), in line with the observed BV6-imposed depletion of cIAP proteins (Figure 2A), which serve as E3 ligases of NIK [16]. In addition, BV6 increased NIK mRNA expression (Figure 2C). Interestingly, addition of Dexa abolished the BV6-mediated accumulation of NIK protein (Jurkat, KOPN-8;11, Reh) or reduced it (Molt-4) (Figure 2B). Consistently, addition of Dexa slightly diminished the BV6-mediated processing of the NF-κB precursor p100 to p52 in all four cell lines (Figure 2B). Altogether these data indicate that Dexa impedes the BV6-triggered activation of NF-κB.
Dexa-stimulated downregulation of cFLIPL protein occurs largely independent of the proteasome, lysosomal enzymes and caspases
Since our data point to a transcription-independent regulation of cFLIPL, we investigated whether Dexa-stimulated downregulation of cFLIPL is due to changes in cellular protein degradation pathways. cFLIPL is described as short-lived protein, which is primarily regulated by the ubiquitin-proteasomal pathway [18]. To determine the half-life of cFLIPL in ALL cell lines, we performed cycloheximide (CHX) chase assays to assess cFLIPL levels upon inhibition of protein synthesis by CHX. CHX treatment caused a rapid decrease in cFLIPL protein (Figure 3A). To analyze the role of the proteasomal pathway, we blocked the proteasome by the specific inhibitor Bortezomib. The addition of Bortezomib delayed loss of cFLIPL protein upon protein synthesis inhibition, in particular in Reh cells, but did not completely rescue it, whereas loss of Noxa protein, a known target of the proteasome, was partially restored in both cell lines (Figure 3A), indicating that cFLIPL is not strictly regulated by the proteasome in the analyzed cell lines. To explore whether loss of cFLIPL protein upon Dexa treatment is mediated via the proteasome, we added Bortezomib to Dexa-treated cells. Interestingly, Bortezomib failed to rescue Dexa-mediated loss of cFLIPL protein (Figure 3B). To assess whether Dexa directly impairs proteasome activity, we performed a proteasome activity assay. Dexa alone or in combination with BV6 did not alter 20S proteasome activity (Figure 3C). By comparison, Dexa treatment had little effects on other short-lived proteins at early time points, such as Noxa or Mcl-1 (Supplementary Figure 2).
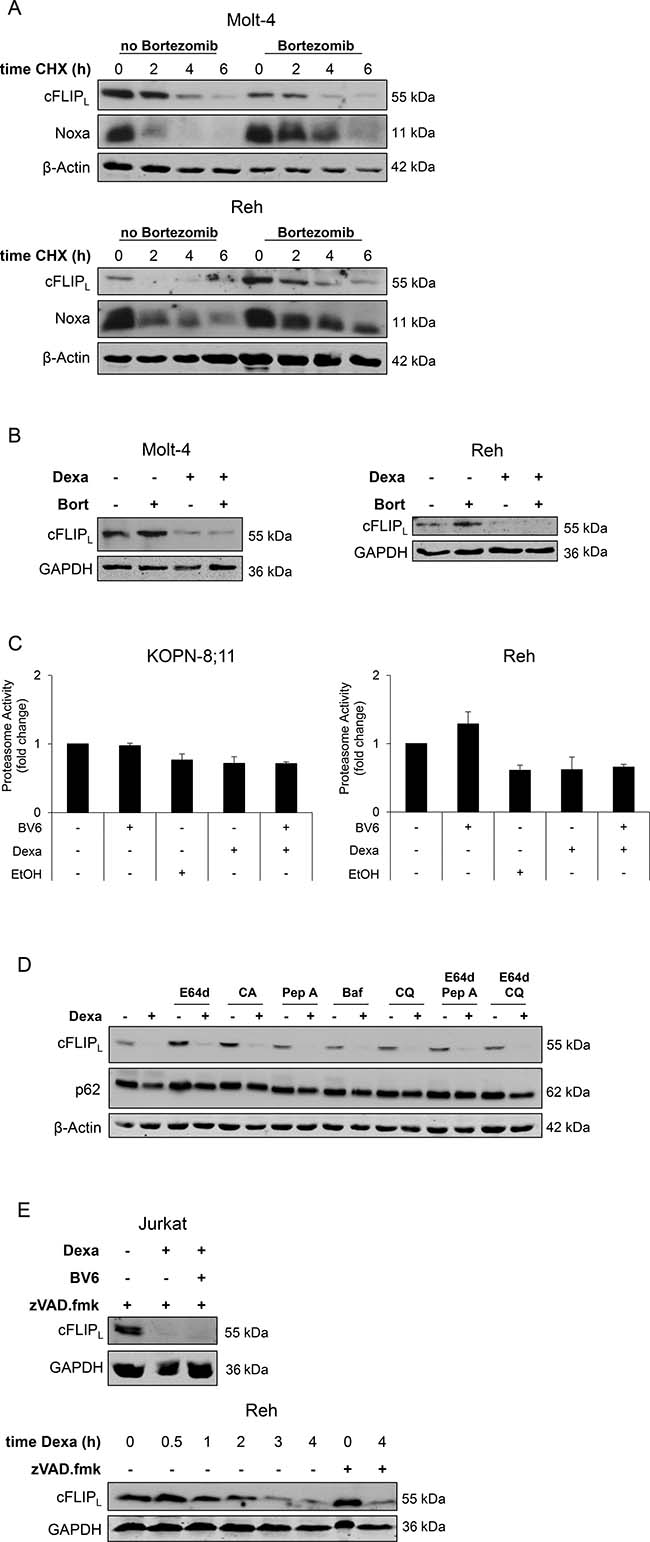
Figure 3: Dexa-stimulated downregulation of cFLIPL protein occurs largely independent of the proteasome, lysosomal enzymes and caspases. (A) Molt-4 and Reh cells were treated with 30 nM Bortezomib to block proteasomal activity, after one hour cells were treated with 10 ng/ml CHX for indicated time points. Expression of cFLIPL and Noxa was analyzed by Western blotting. β-Actin served as loading control. (B) Molt-4 and Reh cells were treated with 30 nM Bortezomib (Bort) to block proteasomal activity, after one hour cells were treated with 300 μM Dexa for four hours. Expression of cFLIPL was analyzed by Western blotting. GAPDH served as loading control. (C) Proteasome activity was analyzed using Chemicon 20S Proteasome Activity Assay. Fold change of proteasome activity of two independent experiments performed in duplicates is shown. EtOH was used as solvent for Dexa. (D) Reh cells were incubated with inhibitors of lysosomal enzymes (10 μg/ml E64d, 10 μg/ml CA-074 methyl ester (CA), 10 μg/ml Pepstatin A (Pep A), 50 nM Bafilomycin A (Baf), 25 μM Chloroquine (CQ)) for one hour, followed by treatment with 300 μM Dexa for four hours. cFLIPL protein expression was analyzed using Western blotting. β-Actin served as loading control. (E) Jurkat and Reh cells were treated with 20 μM zVAD.fmk and 300 μM Dexa for four hours or indicated time points. cFLIPL expression was analyzed by Western blotting. GAPDH served as loading control.
To investigate whether cFLIPL protein is degraded upon Dexa treatment via the lysosomal pathway, we blocked lysosomal enzymes by different pharmacological inhibitors. All of them (alone or in combination) failed to prevent loss of cFLIPL protein after Dexa treatment (Figure 3D). Since cFLIPL is a known target of caspase-8, we analyzed whether caspase-mediated cleavage of cFLIPL is responsible for its loss upon Dexa treatment. But the addition of N-benzyloxycarbonyl-Val-Ala-Asp-(OMe)-fluoromethylketone (zVAD.fmk), a pan-caspase inhibitor, did not prevent Dexa-mediated downregulation of cFLIPL (Figure 3E). This set of experiments indicates that Dexa-mediated loss of cFLIPL protein in ALL cell lines is not primarily mediated via the proteasome, lysosomal enzymes or caspases.
High cFLIPL levels impair Dexa/BV6-mediated cell death in Reh cells
To explore the functional relevance of cFLIPL in Dexa/BV6-mediated cell death, we created cell lines stably overexpressing cFLIPL (Figure 4A, 4C, 4E). Of note, cFLIPL overexpression significantly reduced Dexa/BV6- as well as BV6-mediated cell death in Reh cells (Figure 4B), in which the ectopically expressed cFLIPL protein was resistant to Dexa/BV6-imposed downregulation (Figure 4G). In Jurkat and Molt-4 cells, Dexa/BV6 downregulated ectopically expressed cFLIPL in addition to endogenous cFLIPL protein (Figure 4G), consistent with the failure of cFLIPL overexpression to rescue Dexa/BV6-induced cell death in these cell lines (Figure 4D, 4F). Control experiments confirmed that cFLIPL overexpression significantly reduced TNFα/BV6-mediated cell death in Jurkat and Molt-4 cells (Supplementary Figure 3). In line with the cFLIPL-conferred protection from Dexa/BV6-induced cell death (Figure 4B), cFLIPL overexpression reduced Dexa/BV6-imposed cleavage of caspase-8, caspase-9 and caspase-3 compared to Reh control cells expressing empty vector (EV) (Figure 4H), whereas overexpression of cFLIPL had little effects on Dexa/BV6-induced caspase cleavage in Jurkat cells (Figure 4H). These data show that high cFLIPL levels impair Dexa/BV6-mediated cell death in a cell line-dependent manner.
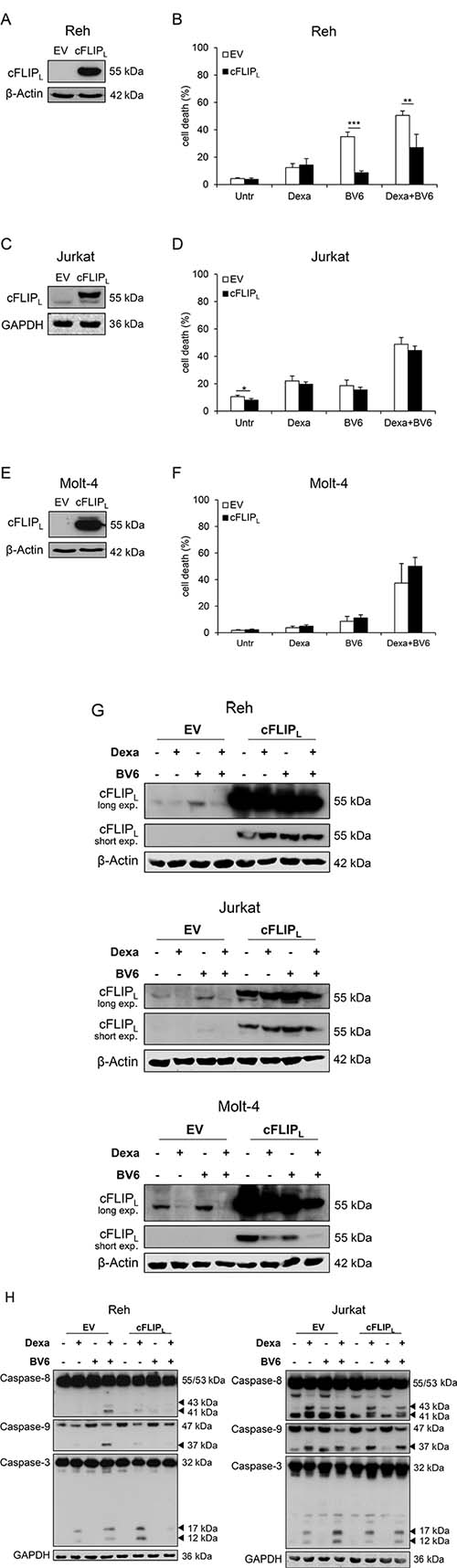
Figure 4: High cFLIPL levels impair Dexa/BV6-mediated cell death. (A, C, E) Protein levels of cFLIPL were assessed by Western blotting in EV or cFLIPL overexpressing (cFLIPL) cells. GAPDH served as loading control. (B, D, F) cFLIPL overexpressing (OE) cells were treated with Dexa and/or BV6 (Jurkat: 300 μM Dexa; 7 μM BV6; Molt-4: 300 μM Dexa, 5 μM BV6; Reh: 300 μM Dexa, 0.3 μM BV6) for 24 hours (Reh, Molt-4) or 15 hours (Jurkat), respectively, and cell death was determined by FSC/SSC analysis and flow cytometry. Mean and SD of at least three independent experiments performed in triplicate are shown; **p < 0.01, ***p < 0.001. (G) OE cells were treated with 300 μM Dexa and/or BV6 (Jurkat: 7 μM BV6; Molt-4: 5 μM BV6; Reh: 0.3 μM BV6) for four hours. cFLIPL expression was assessed by Western blotting. β-Actin served as loading control. (H) Reh and Jurkat OE cells were treated with 300 μM Dexa and/or BV6 (Jurkat: 7 μM BV6; Reh: 0.3 μM BV6) for six hours. Pro-caspase expression and caspase cleavage were analyzed by Western blotting. GAPDH served as loading control.
Knockdown of cFLIP increases BV6-mediated cell death
Next, we tested the functional relevance of Dexa-mediated loss of cFLIP protein to sensitize ALL cells to BV6 by siRNA-mediated knockdown of cFLIP to mimic its depletion by Dexa. In all tested cell lines, knockdown of cFLIP by using two independent siRNA sequences (Figure 5A, 5C, 5E, 5G) caused an increase in TNFα/BV6-mediated cell death that served as positive control (Figure 5B, 5D, 5F, 5H). Importantly, cFLIP knockdown significantly increased BV6-mediated cell death in KOPN-8;11 cells (Figure 5D). In Jurkat and Reh cells, cFLIP knockdown by using sequence #1 significantly enhanced cell death in BV6-treated cells (Figure 5B, 5G), while cFLIP silencing did not alter BV6-induced cell death in Molt-4 cells (Figure 5F). These results demonstrate that cFLIP silencing increases BV6-mediated cell death in a cell line-dependent manner.
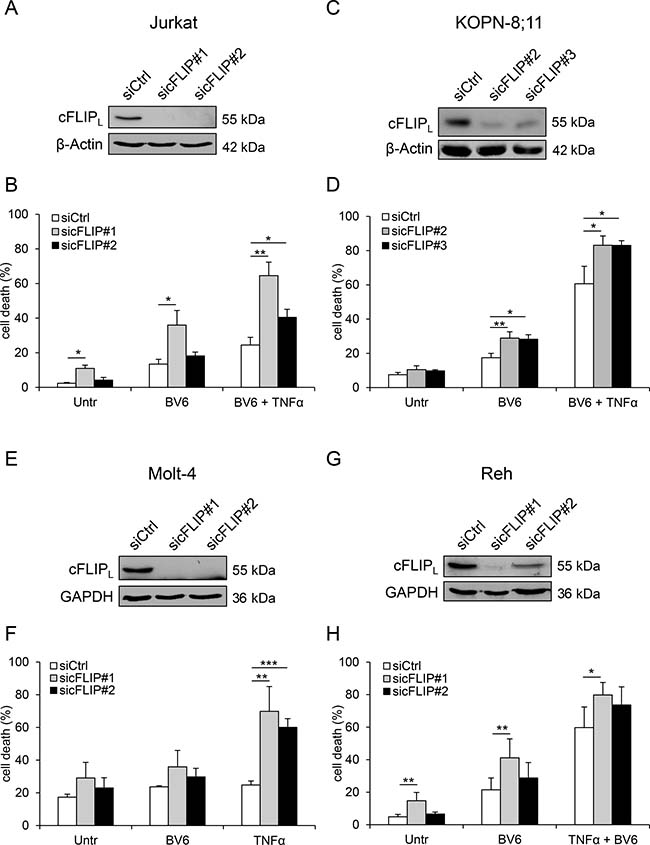
Figure 5: Knockdown of cFLIP increases BV6-mediated cell death. Cells were transiently transfected with two distinct siRNAs targeting cFLIP or control siRNA. (A, C, E, G) Protein expression of cFLIPL was analyzed by Western blotting. β-Actin or GAPDH served as loading control. (B, D, F, H) Cells were treated with BV6 and/or TNFα (Jurkat: 7 μM BV6, 1 ng/ml TNFα; KOPN-8;11 μM BV6, 1 ng/ml TNFα; Molt-4: 5 μM BV6, 100 ng/ml TNFα; Reh: 0.1 μM BV6, 0.05 ng/ml TNFα) for 24 hours or 15 hours (Jurkat), respectively. Cell death was determined by FSC/SSC analysis and flow cytometry. Mean and SD of at least three independent experiments performed in triplicate are shown; *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
Regulation of Smac mimetic-induced cell death by cFLIP
In the present study, we discover that Dexa-imposed suppression of cFLIPL contributes to Dexa/BV6-induced cell death. The conclusion that cFLIPL negatively regulates Dexa/BV6-induced cell death is supported by our data showing that overexpression of cFLIPL to an extent that is no longer subject to Dexa-mediated downregulation rescues Dexa/BV6-mediated cell death. In addition, knockdown of cFLIP mimics Dexa treatment in ALL, as it increases BV6-mediated cell death. All these findings underscore the functional relevance of cFLIP in Dexa/BV6-mediated cell death. Since we previously demonstrated that Dexa/BV6-induced depletion of cIAP proteins is followed by ripoptosome formation [15], which is known to be negatively regulated by cFLIP [10, 11, 19], reduction of cFLIP protein levels by Dexa/BV6 treatment may well promote ripoptosome formation and thereby cell death. Thus, in addition to downregulating cIAP proteins, Dexa/BV6-induced suppression of cFLIP contributes to Dexa/BV6-mediated cell death.
cFLIP is a well-described negative regulator of cell death in many tumor entities and treatment strategies. cFLIP has been shown to protect from Smac mimetic-induced cell death [20, 21] and silencing of cFLIP was found to sensitize different tumor cell lines to Smac mimetics [21, 22]. Also, there are several reports showing that cFLIP can protect cancer cells from TRAIL-, CD95- or chemotherapy-induced cell death [23–26].
Molecular mechanisms of cFLIP downregulation
Furthermore, our study provides new insights into the molecular mechanisms that are responsible for Dexa-imposed downregulation of cFLIP protein. cFLIP expression is tightly regulated by various transcriptional and post-transcriptional mechanisms. While cFLIP is known to be regulated by different transcription factors including NF-κB [27, 28], our data point to a transcription-independent regulation of cFLIP by Dexa, as Dexa suppressed cFLIPL protein but not mRNA levels. Nevertheless, Dexa attenuated BV6-stimulated non-canonical NF-κB activation in ALL cell lines, which is consistent with other reports showing that glucocorticoids such as Dexa can block NF-κB [29–31].
cFLIP is known as a short-lived protein and its turnover has been shown to determine sensitivity to cell death, e.g. to death receptor signals [32]. Several E3 ubiquitin ligases, for example Itch [33, 34], have been identified that polyubiquitinate cFLIP to induce its proteasome-mediated proteolysis, and proteasome inhibitors have been described to rescue the degradation of cFLIP protein [35, 36]. In addition, increased proteasome activity is associated with downregulation of cFLIP protein [37]. However, our findings suggest that Dexa-induced loss of cFLIPL protein is not primarily due to increased proteasomal degradation, since i) addition of the proteasome inhibitor Bortezomib failed to fully protect from Dexa-induced loss of cFLIP protein and since ii) treatment with Dexa alone or in combination with BV6 did not alter 20S proteasomal activity.
Besides proteasome-mediated proteolysis, tumor necrosis factor (TNF) receptor-associated factor (TRAF)7, another E3 ubiquitin ligase, has been described to polyubiquitinate cFLIP and to induce its lysosomal degradation [38]. However, our findings suggest a lysosome-independent downregulation of cFLIPL, as inhibition of several lysosomal enzymes, either alone or in combination, failed to rescue Dexa-stimulated cFLIPL degradation. It is also unlikely that caspase-8-triggered cleavage of cFLIPL is responsible for its downregulation by Dexa, since the pan-caspase inhibitor zVAD.fmk did not prevent Dexa-mediated loss of cFLIPL.
As transcriptional or post-translational regulation, e.g. by caspases or the proteasome, are not primarily responsible for downregulation of cFLIPL, it might be that translational processes are affected by Dexa. While in ALL cells glucocorticoids have been shown to repress genes involved in RNA, protein and nucleotide synthesis [39], which is in line with studies on other tumor entities [40] or tissues [41, 42], Dexa has recently been reported to not alter mRNA translation in ALL cell lines [43]. While the rapid kinetic of Dexa-mediated loss of cFLIPL protein similar to inhibition of protein synthesis by CHX is consistent with a block of translation or ribosomal proteins upon Dexa treatment, further studies are required to fully understand the mechanisms underlying Dexa-stimulated decrease of cFLIPL.
By showing that Dexa-mediated downregulation of cFLIPL contributes to its sensitization to BV6-induced cell death, our study provides new insights into the molecular mechanisms of the cooperative induction of cell death by Dexa/BV6. As glucocorticoids are part of treatment regimens for ALL patients, our findings showing that Dexa and BV6 cooperate to induce cell death provide new concepts to enhance glucocorticoid sensitivity.
MATERIALS AND METHODS
Cell culture and chemicals
ALL cell lines were obtained from DSMZ (Braunschweig, Germany). Cells were cultured in RPMI 1640 medium (Life Technologies/Thermo Fisher Scientific, Darmstadt, Germany). Media were supplemented with 10% FCS (fetal calf serum) (Life Technologies/Thermo Fisher Scientific), 1 mM pyruvate (Life Technologies/Thermo Fisher Scientific), 25 mM HEPES (Life Technologies/Thermo Fisher Scientific) and 1% penicillin/streptomycin (Life Technologies/Thermo Fisher Scientific). The Smac mimetic BV6 was kindly provided by Genentech, Inc. (South San Francisco, CA, USA). The glucocorticoid Dexa was purchased from Sigma-Aldrich (Steinheim, Germany), the caspase inhibitor zVAD.fmk from Bachem (Heidelberg, Germany), the proteasome inhibitor Bortezomib from Selleckchem (Houston, TX, USA) and cycloheximide (CHX), E64d, CA-074 methyl ester, chloroquine, pepstatin A and Bafilomycin A from Sigma-Aldrich. All other chemicals were purchased from Sigma-Aldrich (Steinheim, Germany) or Carl Roth (Karlsruhe, Germany) unless indicated otherwise.
Determination of cell death and proteasome activity
Cell death was assessed by forward/side scatter (FSC/SSC) analysis and flow cytometry (FACS Canto II; BD Biosciences). 20S Proteasome activity was analyzed using CHEMICON 20S Proteasome Activity Assay Kit according to the manufacturer’s instructions (Merck, Darmstadt, Germany).
Western blot analysis
Western blot analysis was performed as described previously [44] using the following antibodies: Mouse anti-cFLIP (Enzo Life Sciences, Lörrach, Germany), goat anti-cIAP1 (R&D Systems, Wiesbaden, Germany), rat anti-cIAP2 (Enzo Life Sciences), mouse anti-XIAP (BD Biosciences, Heidelberg, Germany), rabbit anti-NIK (Cell Signaling Technologies, Beverly, MA USA), mouse anti-p100/p52 (Merck Millipore, Darmstadt, Germany), rabbit anti-p62 (MBL International, Woburn, MA, USA), mouse anti-Noxa (Enzo Life Sciences), rabbit anti-Mcl-1 (Enzo Life Sciences), mouse anti-caspase-8 (Enzo Life Sciences), rabbit anti-caspase-3 (Cell Signaling Technologies), rabbit anti-caspase-9 (Cell Signaling Technologies), mouse anti-β-actin (Sigma-Aldrich), mouse anti-GAPDH (HyTest, Turku, Finland), mouse anti-tubulin (Calbiochem, Merck Millipore, Darmstadt, Germany). Secondary antibodies conjugated to horseradish peroxidase (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used for enhanced chemiluminescence detection (Amersham Bioscience, Freiburg, Germany). Alternatively, secondary antibodies labeled with IRDye infrared dyes were used for fluorescence detection (Odyssey Imaging System, LI-COR Bioscience, Bad Homburg, Germany). All Western blots shown are representative of two or three independent experiments.
qRT-PCR analysis
Total RNA was isolated using peqGOLD Total RNA Kit (peqlab/VWR, Darmstadt, Germany), cDNA synthesis was performed with 1 μg RNA using RevertAid H Minus First Strand cDNA Synthesis Kit (Molecular Biology/Thermo Fisher Scientific). To quantify gene expression qRT-PCR analysis was performed using SYBR Green Reaction Mix (Applied Biosystems, Darmstadt, Germany) with 7900HT Fast Real-Time PCR System (Applied Biosystems). The following primers were used: 28S (forward: 5′-TTGAAAATCCGGGGGAGAG-3′; reverse: 5′-ACATTGTTCCAACATGCCAG-3′), 18S (forward: 5′-C GCAAATTACCCACTCCCG-3′; reverse: 5′-TTCCAAT TACAGGGCCTCGAA-3′), glucose 6-phosphate dehydrogenase (G6PDH) (forward: 5′-ATCGACCAC TACCTGGGCAA-3′; reverse: 5′-AGGCCCTGCACTACG TCTT-3′), RNA polymerase II (forward: 5′-GCACCACG TCCAATGACAT-3′; reverse: 5′-AATACCTTCGTCGG CGTG-3′), cFLIP (forward: 5′-GCTCACCATCCCTGTA CCTG-3′; reverse: 5′-CAGGAGTGGGCGTTTTCTT-3′), NIK (forward: 5′- CCAGCTGCCATCTCTATCATC-3′; reverse: 5′-AAAAAGTGGGGCTGAACTCT-3′). mRNA expression levels of genes of interest were normalized to three housekeeping genes and relative expression of target gene transcript and reference gene transcripts was calculated as ΔΔCt.
Gene silencing and transduction
Knockdown experiments with small interfering RNA (siRNA) were performed using Neon Transfection System (Invitrogen, Karlsruhe, Germany) according to the manufacturer’s instructions [45], using 80 nM Silencer®Select siRNAs (Thermo Fisher Scientific) against cFLIP (#1: s16864, #2: s16865, #3: s229912), or non-targeting control siRNA (no. 4390843). Jurkat, KOPN-8;11 and Molt-4 cells were treated 48 hours after transfection. Reh cells were transfected twice at intervals of 48 hours and treated 24 hours after the second transfection. Overexpression of cFLIP was performed by retroviral transduction using cFLIPL and the pBABE-puro retroviral vector system. For virus production, Phoenix cells were transfected with 20 μg DNA using calcium phosphate method. Virus-containing supernatant of Phoenix cells was filtered and added via spin transduction in the presence of 4 μg/ml protamine sulfate (Sigma-Aldrich) to Jurkat, Molt-4 or Reh cells. Transduced cells were selected with 5 μg/ml puromycin (Sigma-Aldrich).
Statistical analysis
Statistical significance was assessed using Student’s t-Test (two-tailed distribution, equal variance) calculated with Microsoft Excel (Munich, Germany).
ACKNOWLEDGMENTS
We thank C. Hugenberg for expert secretarial assistance.
CONFLICTS OF INTEREST
None to declare.
FUNDING
This work has been partially supported by grants from the BMBF (to S.F.).
REFERENCES
1. Lockshin RA, Zakeri Z. Cell death in health and disease. J Cell Mol Med. 2007; 11:1214–1224.
2. LaCasse EC, Baird S, Korneluk RG, MacKenzie AE. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene. 1998; 17:3247–3259.
3. Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997; 388:190–195.
4. Valnet-Rabier MB, Challier B, Thiebault S, Angonin R, Margueritte G, Mougin C, Kantelip B, Deconinck E, Cahn JY, Fest T. c-Flip protein expression in Burkitt’s lymphomas is associated with a poor clinical outcome. Br J Haematol. 2005; 128:767–773.
5. Harris J, Ibrahim H, Amen F, Karadimitris A, Naresh KN, Macdonald DH. Cellular (FLICE) like inhibitory protein (cFLIP) expression in diffuse large B-cell lymphoma identifies a poor prognostic subset, but fails to predict the molecular subtype. Hematol Oncol. 2012; 30:8–12.
6. McLornan D, Hay J, McLaughlin K, Holohan C, Burnett AK, Hills RK, Johnston PG, Mills KI, McMullin MF, Longley DB, Gilkes A. Prognostic and therapeutic relevance of c-FLIP in acute myeloid leukaemia. Br J Haematol. 2013; 160:188–198.
7. Du X, Bao G, He X, Zhao H, Yu F, Qiao Q, Lu J, Ma Q. Expression and biological significance of c-FLIP in human hepatocellular carcinomas. J Exp Clin Cancer Res. 2009; 28:24.
8. Boatright KM, Deis C, Denault JB, Sutherlin DP, Salvesen GS. Activation of caspases-8 and -10 by FLIP(L). Biochem J. 2004; 382:651–657.
9. Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem. 1999; 274:1541–1548.
10. Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Hacker G, Leverkus M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011; 43:449–463.
11. Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, Meier P. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011; 43:432–448.
12. Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012; 11:109–124.
13. Fulda S. Promises and challenges of Smac mimetics as cancer therapeutics. Clin Cancer Res. 2015; 21:5030–5036.
14. Tallen G, Ratei R, Mann G, Kaspers G, Niggli F, Karachunsky A, Ebell W, Escherich G, Schrappe M, Klingebiel T, Fengler R, Henze G, von Stackelberg A. Long-term outcome in children with relapsed acute lymphoblastic leukemia after time-point and site-of-relapse stratification and intensified short-course multidrug chemotherapy: results of trial ALL-REZ BFM 90. J Clin Oncol. 2010; 28:2339–2347.
15. Belz K, Schoeneberger H, Wehner S, Weigert A, Bonig H, Klingebiel T, Fichtner I, Fulda S. Smac mimetic and glucocorticoids synergize to induce apoptosis in childhood ALL by promoting ripoptosome assembly. Blood. 2014; 124:240–250.
16. Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, Flygare JA, Fairbrother WJ, Deshayes K, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007; 131:669–681.
17. Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M, Brink R, Leverkus M, Tergaonkar V, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007; 131:682–693.
18. Noh HJ, Lee SJ, Sung EG, Song IH, Kim JY, Woo CH, Kwon TK, Lee TJ. CHOP down-regulates cFLIP(L) expression by promoting ubiquitin/proteasome-mediated cFLIP(L) degradation. J Cell Biochem. 2012; 113:3692–3700.
19. Geserick P, Hupe M, Moulin M, Wong WW, Feoktistova M, Kellert B, Gollnick H, Silke J, Leverkus M. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J Cell Biol. 2009; 187:1037–1054.
20. Zhao L, Yang G, Bai H, Zhang M, Mou D. NCTD promotes Birinapant-mediated anticancer activity in breast cancer cells by downregulation of c-FLIP. Oncotarget. 2017; 8:26886–26895. https://doi.org/10.18632/oncotarget.15848.
21. Crawford N, Stasik I, Holohan C, Majkut J, McGrath M, Johnston PG, Chessari G, Ward GA, Waugh DJ, Fennell DA, Longley DB. SAHA overcomes FLIP-mediated inhibition of SMAC mimetic-induced apoptosis in mesothelioma. Cell Death Dis. 2013; 4:e733.
22. Cheung HH, Mahoney DJ, Lacasse EC, Korneluk RG. Down-regulation of c-FLIP Enhances death of cancer cells by smac mimetic compound. Cancer Res. 2009; 69:7729–7738.
23. Palacios C, Yerbes R, Lopez-Rivas A. Flavopiridol induces cellular FLICE-inhibitory protein degradation by the proteasome and promotes TRAIL-induced early signaling and apoptosis in breast tumor cells. Cancer Res. 2006; 66:8858–8869.
24. Grund K, Ahmadi R, Jung F, Funke V, Gdynia G, Benner A, Sykora J, Walczak H, Joos S, Felsberg J, Reife nberger G, Wiestler OD, Herold-Mende C, Roth W. Troglitazone-mediated sensitization to TRAIL-induced apoptosis is regulated by proteasome-dependent degradation of FLIP and ERK1/2-dependent phosphorylation of BAD. Cancer Biol Ther. 2008; 7:1982–90.
25. Kim Y, Suh N, Sporn M, Reed JC. An inducible pathway for degradation of FLIP protein sensitizes tumor cells to TRAIL-induced apoptosis. J Biol Chem. 2002; 277:22320–22329.
26. Haag C, Stadel D, Zhou S, Bachem MG, Moller P, Debatin KM, Fulda S. Identification of c-FLIP(L) and c-FLIP(S) as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut. 2011; 60:225–237.
27. Kreuz S, Siegmund D, Scheurich P, Wajant H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol. 2001; 21:3964–3973.
28. Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001; 21:5299–5305.
29. Heck S, Bender K, Kullmann M, Göttlicher M, Herrlich P, Cato AC. I kappaB alpha-independent downregulation of NF-kappaB activity by glucocorticoid receptor. EMBO J. 1997; 16:4698–707.
30. Machuca C, Mendoza-Milla C, Cordova E, Mejia S, Covarrubias L, Ventura J, Zentella A. Dexamethasone protection from TNF-alpha-induced cell death in MCF-7 cells requires NF-kappaB and is independent from AKT. BMC Cell Biol. 2006; 7:9.
31. Beck IM, Vanden Berghe W, Vermeulen L, Bougarne N, Vander Cruyssen B, Haegeman G, De Bosscher K. Altered subcellular distribution of MSK1 induced by glucocorticoids contributes to NF-kappaB inhibition. EMBO J. 2008; 27:1682–93.
32. Toivonen HT, Meinander A, Asaoka T, Westerlund M, Pettersson F, Mikhailov A, Eriksson JE, Saxen H. Modeling reveals that dynamic regulation of c-FLIP levels determines cell-to-cell distribution of CD95-mediated apoptosis. J Biol Chem. 2011; 286:18375–18382.
33. Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006; 124:601–613.
34. Zhang G, Liu J, Zhang Y, Qu J, Xu L, Zheng H, Liu Y, Qu X. Cbl-b-dependent degradation of FLIP(L) is involved in ATO-induced autophagy in leukemic K562 and gastric cancer cells. FEBS Lett. 2012; 586:3104–3110.
35. Fukazawa T, Fujiwara T, Uno F, Teraishi F, Kadowaki Y, Itoshima T, Takata Y, Kagawa S, Roth JA, Tschopp J, Tanaka N. Accelerated degradation of cellular FLIP protein through the ubiquitin-proteasome pathway in p53-mediated apoptosis of human cancer cells. Oncogene. 2001; 20:5225–5231.
36. Liu X, Yue P, Schonthal AH, Khuri FR, Sun SY. Cellular FLICE-inhibitory protein down-regulation contributes to celecoxib-induced apoptosis in human lung cancer cells. Cancer Res. 2006; 66:11115–11119.
37. Seo SU, Cho HK, Min KJ, Woo SM, Kim S, Park JW, Kim SH, Choi YH, Keum YS, Hyun JW, Park HH, Lee SH, Kim DE, Kwon TK. Thioridazine enhances sensitivity to carboplatin in human head and neck cancer cells through downregulation of c-FLIP and Mcl-1 expression . Cell Death Dis. 2017; 8:e2599.
38. Scudiero I, Zotti T, Ferravante A, Vessichelli M, Reale C, Masone MC, Leonardi A, Vito P, Stilo R. Tumor necrosis factor (TNF) receptor-associated factor 7 is required for TNFalpha-induced Jun NH2-terminal kinase activation and promotes cell death by regulating polyubiquitination and lysosomal degradation of c-FLIP protein. J Biol Chem. 2012; 287:6053–6061.
39. Obexer P, Certa U, Kofler R, Helmberg A. Expression profiling of glucocorticoid-treated T-ALL cell lines: rapid repression of multiple genes involved in RNA-, protein- and nucleotide synthesis. Oncogene. 2001; 20:4324–4336.
40. Meyuhas O, Thompson EA Jr, Perry RP. Glucocorticoids selectively inhibit translation of ribosomal protein mRNAs in P1798 lymphosarcoma cells. Mol Cell Biol. 1987; 7:2691–99.
41. Fussell JC, Kelly FJ. Effects of dexamethasone on lung protein turnover. Biochem J. 1991; 273:93–97.
42. Burrin DG, Wester TJ, Davis TA, Fiorotto ML, Chang X. Dexamethasone inhibits small intestinal growth via increased protein catabolism in neonatal pigs. Am J Physiol. 1999; 276:E269–277.
43. Aneichyk T, Bindreither D, Mantinger C, Grazio D, Goetsch K, Kofler R, Rainer J. Translational profiling in childhood acute lymphoblastic leukemia: no evidence for glucocorticoid regulation of mRNA translation. BMC Genomics. 2013; 14:844.
44. Fulda S, Strauss G, Meyer E, Debatin KM. Functional CD95 ligand and CD95 death-inducing signaling complex in activation-induced cell death and doxorubicin-induced apoptosis in leukemic T cells. Blood. 2000; 95:301–308.
45. Schenk B, Fulda S. Reactive oxygen species regulate Smac mimetic/TNFalpha-induced necroptotic signaling and cell death. Oncogene. 2015; 34:5796–5806.