
INTRODUCTION
Hepatocellular carcinoma (HCC) is the one of the most common malignancies in the world [1]. Despite the rapid progress in the development of therapeutic strategies, the prognosis of HCC is still poor due to high rate of recurrence and metastasis [2, 3]. Recently, molecular target agents, such as tyrosine kinases inhibitors, have been developed for the treatment of advanced HCC [4, 5]. To date, however, the efficacy of these drugs seems to be insufficient to achieve a satisfactory therapeutic effect. Thus, it is needed to identify the new pathways based on the detailed molecular mechanisms.
It is well known that metabolic reprogramming occurs in cancer cells and that dysregulation of energy homeostasis may contribute to the development of cancers [6, 7]. In HCC, there is increasing evidence that the mevalonate pathway responsible for cholesterol biosynthesis is implicated in its pathogenesis [8, 9]. Statins are inhibitors of 3-hydroxy-3-methylglutaryl CoA reductase (HMGCR), a rate limiting enzyme for the mevalonate pathway, and are widely used to reduce cholesterol levels, leading to the prevention of cardiovascular diseases [10]. In addition to cholesterol-lowering property, statins have been shown to reduce risk of HCC in clinical studies [11] and have anti-tumor effect on HCC cells in in vitro and animal studies [12]. Biologically, the mevalonate pathway is known to play a crucial role for protein prenylation, which is post-translational modification of small GTPases, such as Rho family proteins [13, 14], suggesting that anti-tumor effect of statins on HCC might be through the regulation of protein modification in the mevalonate pathway [15]. However, the precise molecular mechanisms underlying the interplay between the mevalonate pathway and oncogenic signaling in HCC have not been fully determined.
The Forkhead Box M1 (FoxM1) transcription factor, a member of the Fox family of proteins, is highly expressed in a variety of human cancers including HCC [16–19]. Mouse genetic approach have demonstrated that FoxM1 is associated with progression and metastasis of HCC [20–22]. Very recently, we identified FoxM1 expression in tumor tissues as an independent prognostic factor affecting recurrence of HCC and overall survival of HCC patients following surgery, indicating that FoxM1 might not only be a promising therapeutic target but also a prognostic biomarker for HCC [23]. Furthermore, it has been shown that FoxM1 promotes reprograming of glucose metabolism in pancreatic and ovarian cancers [24, 25]. These data suggest that FoxM1 might be implicated in metabolic pathways of HCC pathogenesis. However, the involvement of FoxM1 in cancer lipid metabolism, especially, in the mevalonate pathway of HCC, has not been fully elucidated.
Considering these findings, in this study, we proposed our hypothesis that FoxM1 might be involved in the mevalonate pathway in HCC. To clarify this issue, we utilized in vitro culture systems using human hepatoma cell lines along with several inhibitors or metabolites of the mevalonate pathway. Furthermore, we evaluated the gene expression of FoxM1 and that of the mevalonate pathway-related genes in surgically resected HCC tissue samples.
RESULTS
FoxM1 expression is regulated by the mevalonate pathway in human hepatoma cells
To investigate the involvement of FoxM1 in the mevalonate pathway, we examined whether the inhibition of the mevalonate pathway might affect the FoxM1 protein expression in human hepatoma cells. HepG2, Huh7 and HLF cells were treated with pitavastatin, a synthetic HMGCR inhibitor, and the FoxM1 protein expression was examined using Western blot analysis. Administration of pitavastatin significantly reduced the FoxM1 protein expression in a dose-dependent manner in these hepatoma cell lines (Figure 1A). Furthermore, re-exposure to mevalonate (MV), a product of HMGCR, restored the reduction of FoxM1 protein expression induced by pitavastatin (Figure 1A). The effect of pitavastatin or MV on FoxM1 expression was also confirmed by the quantitative real-time RT-PCR analysis (Supplementary Figure 1). Similar results were obtained when HepG2 cells were treated with simvastatin or fluvastatin (Supplementary Figure 2A and 2C). Moreover, siRNA-mediated depletion of HMGCR caused the reduction of the FoxM1 protein expression in HepG2 cells (Figure 1B). These results indicated that FoxM1 was regulated by mevalonate pathway in human hepatoma cells.
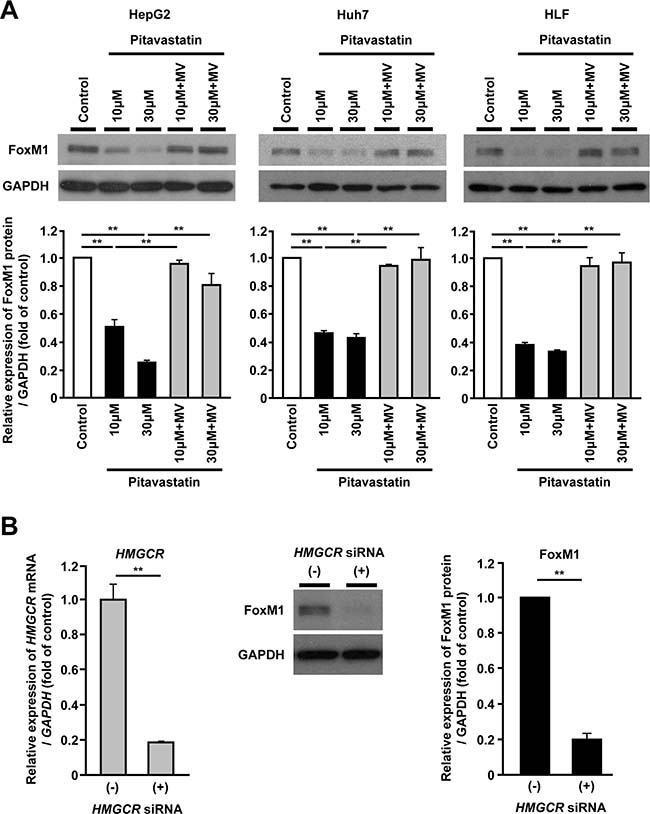
Figure 1: FoxM1 expression is regulated by the mevalonate pathway in human hepatoma cells. (A) Western blot analysis showing the protein expression of FoxM1 (upper panels) in HepG2 cells (left panel), Huh7 cells (middle panel), and HLF cells (right panel) after treatment with pitavastatin (10 μM or 30 μM), either alone or along with mevalonate (MV, 100 μM), for 24 hours. Quantification of the protein expression of FoxM1 (lower panels) in HepG2 cells (left panel), Huh7 cells (middle panel), and HLF cells (right panel) after treatment with pitavastatin (10 μM or 30 μM), either alone or along with MV (100 μM), for 24 hours. DMSO was used as control. (B) Effect of siRNA-mediated depletion of HMGCR on FoxM1 protein expression in HepG2 cells. Quantification of HMGCR gene expression in siRNA against HMGCR-treated HepG2 cells (left panel). Western blot analysis showing the protein expression of FoxM1 in siRNA against HMGCR-treated HepG2 cells (representative images: middle panel, quantification of the protein expression: right panel). The values of the protein expression were normalized to the control. Data are expressed as mean ± SEM, **p < 0.01.
Inhibition of HMGCR decreases the nuclear expression and the transcriptional activity of FoxM1 in human hepatoma cells
To confirm the regulation of FoxM1 expression via the mevalonate pathway, we immunohistochemically examined the expression of FoxM1 in HepG2 cells treated with pitavastatin. Immunofluorescence analysis showed that the administration of pitavastatin reduced nuclear expression of FoxM1 whereas its expression was detected mainly in the nucleus in control cells. Furthermore, re-exposure to MV restored the nuclear expression of FoxM1 (Figure 2A). Because the nuclear expression of FoxM1 is shown to be required for its transcriptional activity [26], we also examined whether inhibition of HMGCR might affect expression of FoxM1 target genes. The administration of pitavastatin resulted in a significant reduction of CCNB1 or BIRC5, known FoxM1 target genes [27, 28]. Re-exposure to MV restored the expression of these genes (Figure 2B and 2C). Together, these data suggested that the mevalonate pathway could regulate the transcriptional activity of FoxM1 in human hepatoma cells.
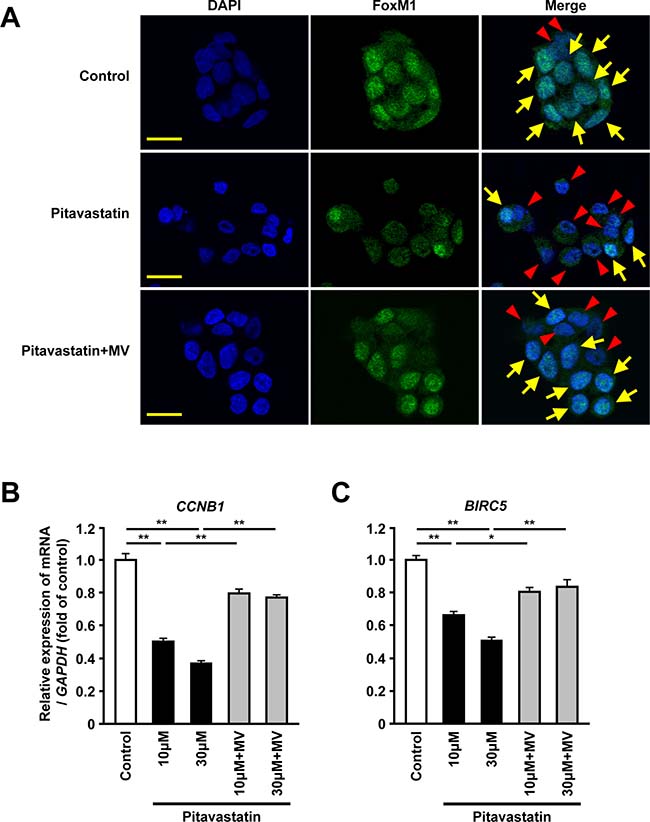
Figure 2: Inhibition of HMGCR decreases the nuclear expression and the transcriptional activity of FoxM1 in human hepatoma cells. (A) Immunofluorescence analysis showing the protein expression of FoxM1 (green) in HepG2 cells treated with pitavastatin (10 μM) or pitavastatin (10 μM) plus MV (100 μM) for 24 hours. Nuclei were stained by DAPI (blue). Scale bar: 20 μm. Arrows or arrowheads indicate the cell with or without the nuclear FoxM1 protein expression, respectively. (B–C) Quantitative real-time RT-PCR analysis showing the gene expressions of CCNB1 (B) or BIRC5 (C) in HepG2 cells after treatment with pitavastatin (10 μM or 30 μM), either alone or along with MV (100 μM) for 24 hours. DMSO was used as control. Data are expressed as mean ± SEM, *p < 0.05, **p < 0.01.
Reduced expression of FoxM1 by HMGCR-inhibition is associated with increased cell death in human hepatoma cells
Inhibition of HMGCR is shown to result in increased cell death in several cancer cells [14]. Therefore, we examined the effect of statin on cell death of human hepatoma cells. Consistent with previous reports, administration of pitavastatin induced an increase of cell death and re-exposure to MV restored these effects in HepG2, Huh7 and HLF cells (Figure 3A and 3B). Effect of pitavastatin on cell death was confirmed by the expression of cleaved PARP (Figure 3C). Furthermore, similar results were observed in case of other types of statins, such as simvastatin or fluvastatin (Supplementary Figure 2B and 2D). Furthermore, overexpression of FoxM1 resulted in loss of statin-induced cell death (Figure 3D and 3E). Collectively, these results suggest that the cell death induced by the inhibition of HMGCR might occur via FoxM1.
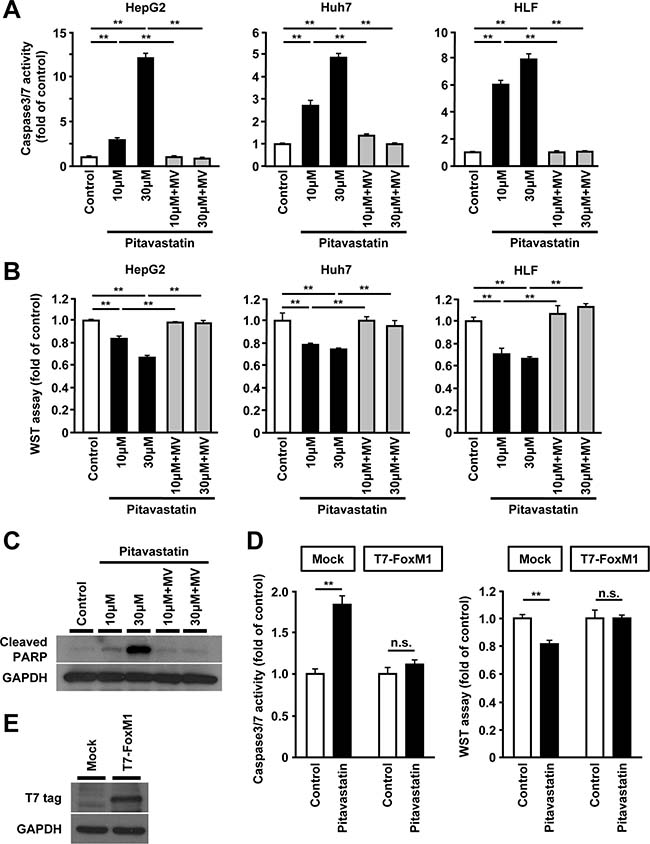
Figure 3: Reduced expression of FoxM1 by HMGCR-inhibition is associated with increased cell death in human hepatoma cells. (A) Assessment of cell death by caspase 3/7 activity in HepG2 cells (left panel), Huh7 cells (middle panel), and HLF cells (right panel) after treatment with pitavastatin (10 μM or 30 μM), either alone or along with MV (100 μM) for 48 hours. (B) Assessment of cell viability by WST assay in HepG2 cells (left panel), Huh7 cells (middle panel), and HLF cells (right panel) after treatment with pitavastatin (10 μM or 30 μM), either alone or along with MV (100 μM) for 48 hours. (C) Western blot analysis showing the protein expression of cleaved PARP in HepG2 cells after treatment with pitavastatin (10 μM or 30 μM), either alone or along with MV (100 μM) for 24 hours. (D) Effect of overexpression of FoxM1 on cell death or viability in HepG2 cells treated with pitavastatin (10 μM). (E) Western blot analysis showing the protein expression of T7-tagged FoxM1 (T7-FoxM1) using anti-T7 antibody. DMSO was used as control (for A, B, C, and D). Data are expressed as mean ± SEM, *p < 0.05, **p < 0.01, n.s. not significant.
FoxM1 expression is regulated via protein geranylgeranylation in human hepatoma cells
To investigate the specific metabolic intermediates responsible for the regulation of FoxM1 expression, we used several inhibitors of enzymes or products in the mevalonate pathway [29] (Figure 4A). Farnesyl pyrophosphate (FPP) synthase inhibitor (zoledronic acid), or geranylgeranyl transferase inhibitor (GGTI-298), had almost the same effect as pitavastatin on FoxM1 expression, whereas farnesyl transferase inhibitor (FTI-277) or squalene synthase inhibitor (YM-53601) had less effect (Figure 4B). Re-exposure to geranylgeranyl pyrophosphate (GGPP) restored pitavastatin-induced reduction of FoxM1 expression, whereas FPP or squalene had less effect (Figure 4C). Taken together, these results indicated that the regulation of FoxM1 expression in the mevalonate pathway might be mainly through protein geranylgeranylation in human hepatoma cells.
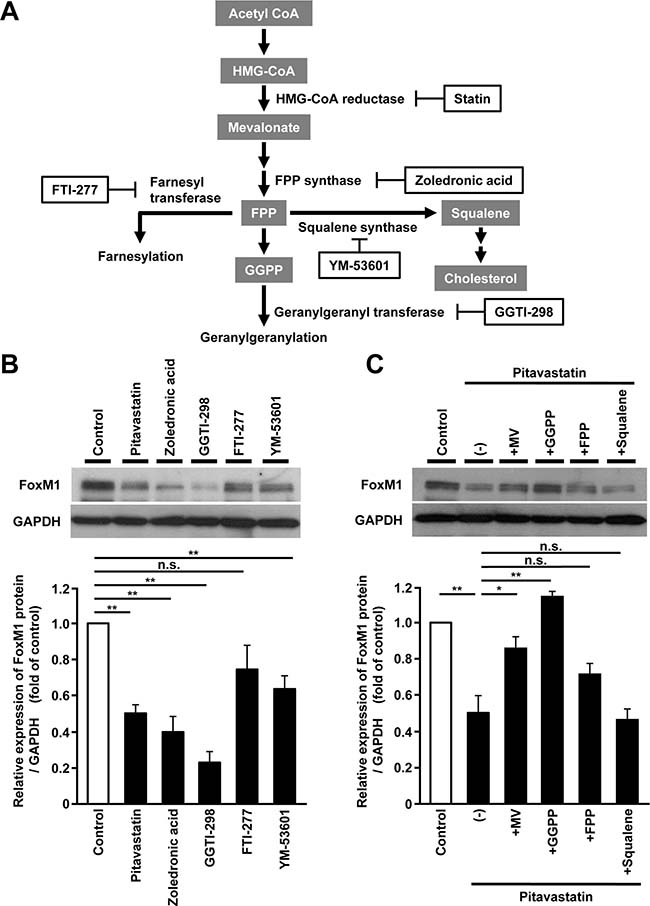
Figure 4: FoxM1 expression is regulated via protein geranylgeranylation in human hepatoma cells. (A) Schematic overview of the mevalonate pathway. The inhibitors or the products for the mevalonate pathway are shown in a frame or in a gray box, respectively. (B) Western blot analysis showing the protein expression of FoxM1 in HepG2 cells treated with the inhibitors for the mevalonate pathway, including pitavastatin (10 μM), zoledronic acid (200 μM), GGTI-298 (10 μM), FTI-277 (10 μM), or YM-53601 (5 μM), for 24 hours (upper panel). Quantification of the protein expression of FoxM1 in HepG2 cells treated with these inhibitors (lower panel). (C) Western blot analysis showing the protein expression of FoxM1 in HepG2 cells treated with pitavastatin (10 μM), either alone or along with the products for the mevalonate pathway, including MV (100 μM), geranylgeranyl pyrophosphate (GGPP, 10 μM), farnesyl pyrophosphate (FPP, 10 μM), or squalene (10 μM) for 24 hours (upper panel). Quantification of the protein expression of FoxM1 in HepG2 cells treated with pitavastatin (10 μM) alone or along with these products (lower panel). The values of the protein expression were normalized to the control. Data are expressed as mean ± SEM, *p < 0.05, **p < 0.01, n.s. not significant.
Rho family proteins are involved in the mevalonate pathway-dependent regulation of FoxM1
To investigate which specific factors regulate FoxM1 expression in the mevalonate pathway, we focused on the Rho family proteins as they require geranylgeranylation when they are activated [13]. To explore the requirement of Rho family proteins in regulating FoxM1 expression, we used the inhibitors for major Rho family proteins, such as RhoA, Rac1 and Cdc42. Administration of each inhibitor significantly reduced FoxM1 expression (Figure 5A–5C). Additionally, siRNA-mediated depletion of RhoA, Rac1 and Cdc42 also reduced FoxM1 expression (Figure 5D–5F). These results suggested that the mevalonate pathway-dependent FoxM1 expression could be regulated via Rho family proteins.
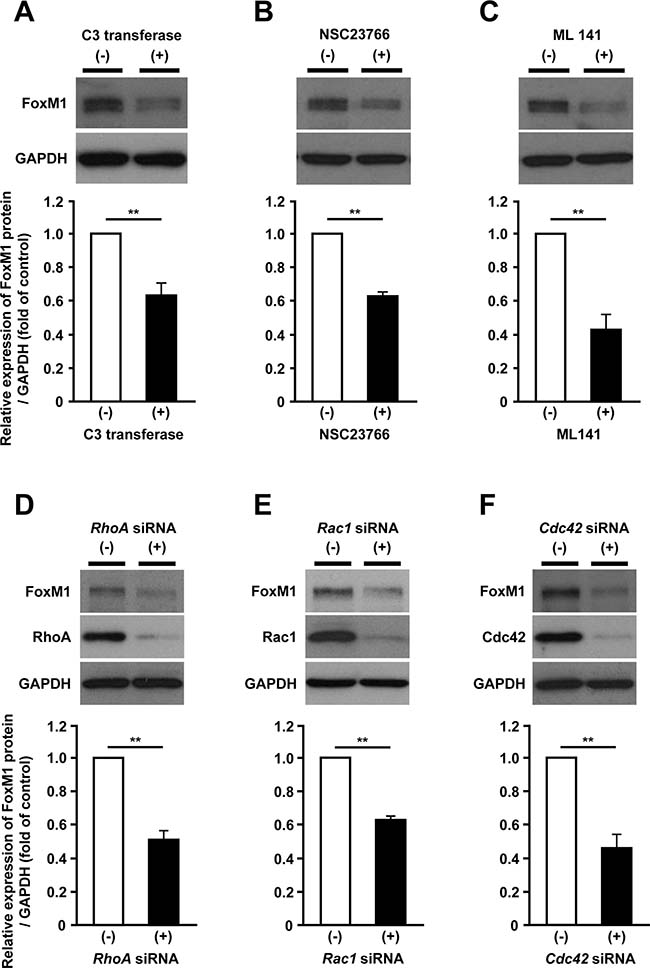
Figure 5: Rho family proteins are involved in the mevalonate pathway-dependent regulation of FoxM1. (A–C) Western blot analysis showing the protein expression of FoxM1 in HepG2 cells treated with 2 μg/ml of RhoA inhibitor C3 transferase (A, upper panel), 100 μM of Rac1 inhibitor NSC23766 (B, upper panel), or 20 μM of Cdc42 inhibitor ML141 (C, upper panel). Quantification of the protein expression of FoxM1 in HepG2 cells with these inhibitors (lower panels). (D–F) Western blot analysis showing the protein expression of FoxM1 transfected with siRNA against RhoA (D, upper panel), Rac1 (E, upper panel) or Cdc42 (F, upper panel). Quantification of the protein expression of FoxM1 in HepG2 cells transfected with these siRNAs (lower panels). The values of the protein expression were normalized to the control. Data are expressed as mean ± SEM, **p < 0.01.
FoxM1 is associated with the mevalonate pathway in HCC patients
To confirm the clinical significance regarding the mevalonate pathway-dependent regulation of FoxM1 in human HCC, we finally examined the expressions of FoxM1 and the mevalonate pathway-related genes in tumor tissues of HCC patients, whose clinicopathological features are summarized in Table 1. The quantitative real-time RT-PCR analysis showed that the expression of FoxM1 had a significant positive correlation with that of HMGCR, a major enzyme of the mevalonate pathway, in these tissues (Figure 6A, r = 0.29, p < 0.05). This analysis also revealed a positive correlation between the expression of FoxM1 and that of sterol regulatory element-binding protein 2 (SREBP2), a main transcriptional factor which regulates HMGCR expression (Figure 6B, r = 0.35, p < 0.01). The overall survival of FoxM1- and HMGCR-high group was found to be significantly lower than that of FoxM1- and/or HMGCR-low group (Figure 6C). Likewise, the overall survival of FoxM1- and SREBP2-high group was found to be significantly lower than that of FoxM1- and/or SREBP2-low group (Figure 6D). As shown in Supplementary Tables 1 and 2, FoxM1- and HMGCR-high group had a statistically significant lower rate of liver cirrhosis of liver histology than FoxM1- and/or HMGCR-low group (p = 0.0337) and FoxM1- and SREBP2-high group had a statistically significant higher PIVKA-II level than FoxM1- and/or SREBP2-low group (p = 0.0294). Furthermore, as shown in Supplementary Tables 3 and 4, multivariate analysis showed that FoxM1- and SREBP2-high expression in tumor tissues was identified as an independent prognostic factor of overall survival (hazard ratio: 3.24; 95% confidence interval: 1.18-8.89; p = 0.0238). Collectively, these results would suggest the clinical implications of the mevalonate-FoxM1 pathway in human HCC.
Table 1. Clinicopathological features of HCC patients
n = 64 |
|
|---|---|
Age (y.o.), mean (range) |
64.2 (36–84) |
Gender : Male/Female |
57/7 |
HBs-Ag : Negative/Positive |
49/15 |
HCV-Ab : Negative/Positive |
31/33 |
Child-Pugh score : A/B |
57/7 |
AFP (ng/ml), median (range) |
20.5 (3–390000) |
PIVKA-II (mAU/ml), median (range) |
308 (13–361200) |
Liver histology : NL/CH/LC |
7/40/17 |
Maximum tumor size (mm), mean (range) |
44.4 (7–320) |
NL, normal liver; CH, chronic hepatitis; LC: liver cirrhosis
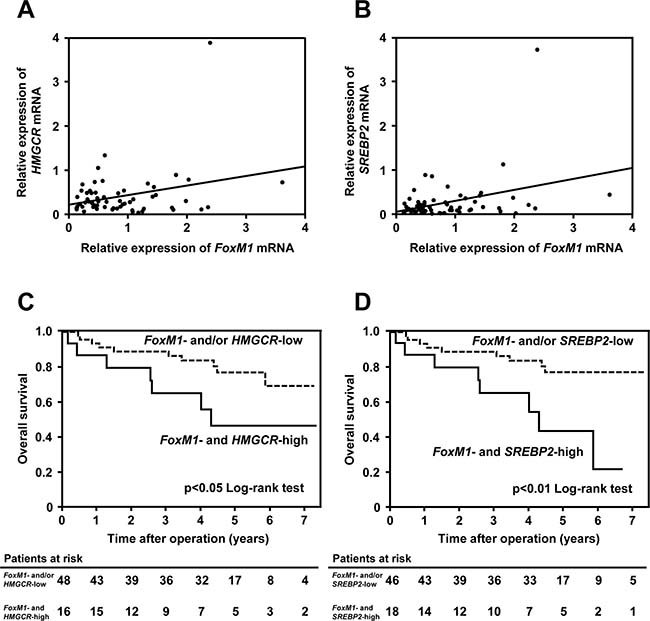
Figure 6: FoxM1 is associated with the mevalonate pathway in HCC patients. (A) Correlation between the gene expression of FoxM1 and HMGCR in tumor tissues of HCC patients (r = 0.29, p < 0.05). (B) Correlation between the gene expression of FoxM1 and SREBP2 in tumor tissues of HCC patients (r = 0.35, p < 0.01). (C and D) The relationship between the overall survival of FoxM1 and HMGCR (C)/SREBP2 (D) expression in human HCC. High group is defined as higher gene expression than median.
DISCUSSION
Evidence has accumulated regarding the crucial role of FoxM1 in cancer development and progression [18, 23, 30], suggesting the possible application of FoxM1 as a therapeutic target against cancer. In this study, for the first time, we have shown that FoxM1 acts as a downstream target for the mevalonate pathway of cholesterol biosynthesis in human HCC. Our in vitro findings showed that statins, well-known HMGCR inhibitors, reduced FoxM1 expression, indicating that statins could be novel inhibitors to reduce FoxM1 in HCC. We also demonstrated that protein geranylgeranylation was responsible for the regulation of FoxM1 expression in the mevalonate pathway. Using clinical samples, we observed a significant positive correlation between the gene expression of FoxM1 and that of HMGCR, a rate limiting enzyme for mevalonate pathway, or SREBP2, a master regulator of the mevalonate pathway [14], in HCC tissues. Furthermore, the expression of FoxM1 along with that of HMGCR or SREBP2 was found to define the prognosis of HCC patients. Therefore, we propose that the regulation of FoxM1 via the mevalonate pathway may open new avenues for the development of molecular targeted therapies against HCC.
FoxM1 is a well-defined transcription factor which is crucial for cell proliferation and cell cycle progression [31, 32]. Although FoxM1 is rarely expressed in quiescent or differentiated cells, its expression is highly elevated in proliferating cells or a variety of cancers [16, 17, 33]. In cancer cells, FoxM1 is shown to act downstream of growth signals, such as Ras-mitogen-activated protein kinase pathway or phosphatidylinositol 3-kinase-AKT pathway [34, 35]. Likewise, loss of tumor suppressor, such as p53 or p19Arf, is shown to control FoxM1 expression in cancer cells [20, 21, 36], suggesting that FoxM1 is involved in multiple hallmarks of cancer [37]. However, the links between FoxM1 and cancer metabolism has been yet fully investigated. Recently, it has been shown that FoxM1 is involved in glucose metabolism of pancreatic or ovarian cancers [24, 25]. Nevertheless, to date, it has remained unclear whether FoxM1 is involved in the mevalonate pathway of cholesterol biosynthesis, which is known to play a crucial role in lipid metabolism in cancer. In this study, we showed that the expression of FoxM1 is correlated with that of HMGCR or SREBP2 in tumor tissues of HCC patients. Our findings have raised the possibility that FoxM1 would link the mevalonate pathway of cholesterol biosynthesis to the oncogenic signals in HCC.
The mevalonate pathway is a major metabolic route to convert acetyl-CoA to cholesterol that is essential for cell proliferation [13, 14]. Dysregulation of the mevalonate pathway is known to promote oncogenesis in several cancers [38]. So far, several lines of evidence have shown that HMGCR has a key role in the oncogenic potential of the mevalonate pathway [14, 38]. In this study, inhibition of HMGCR by statins resulted in not only increased cell death but also the reduced expression of FoxM1 in human hepatoma cells. In addition, forced expression of FoxM1 restored statins-mediated cell death of human hepatoma cells, indicating that cell death in the mevalonate pathway might be regulated through FoxM1. This mevalonate pathway-mediated regulation of FoxM1 was confirmed by the expression of its known target genes, such as CCNB1 or BIRC5. The data also suggest that the additional treatment of MV did not seem to rescue the expression of these target genes completely. One possible explanation for this discrepancy is that other factors other than FoxM1, such as p53, might be involved in the regulation of CCNB1 or BIRC5 in the mevalonate pathway [39, 40]. Collectively, our data suggest that FoxM1 is a downstream target of the mevalonate pathway in HCC cells.
The accumulation of intermediate metabolites in the mevalonate pathway induces post-translational modification of signaling proteins that is required for cell proliferation, survival, and migration [13, 14]. This modification is referred to as prenylation, including protein geranylgeranylation and protein farnesylation [13, 14]. In this study, we showed that protein geranylgeranylation rather than protein farnesylation is required for the regulation of FoxM1 expression in the mevalonate pathway. We further identified Rho-family of small GTPases, RhoA, Rac1 and Cdc42, as geranylgeranylated proteins that regulate FoxM1 expression. Furthermore, as shown in Supplementary Figure 3, we found that the knockdown of FoxM1, HMGCR or Cdc42 caused a statistically significant increase in cell death, whereas the knockdown of RhoA or Rac1 did not. Our results showing the induction of cell death in HMGCR- or FoxM1-depleted HepG2 cells would be consistent with the results of statins treatment, suggesting the possibility that cell death is indeed by a similar mechanism in both cases. Although RhoA, Rac1 and Cdc42, were associated with the regulation of FoxM1 expression, the induction of cell death was observed in Cdc42-depleted HepG2 cells but not in RhoA- or Rac1-depleted HepG2 cells. One possible explanation for this discrepancy might be that the role of these Rho family proteins in the downstream of the mevalonate-FoxM1 pathway might differ in cell death of human hepatoma cells. At this stage, the detailed molecular mechanisms by which RhoA, Rac1 and Cdc42 coordinate FoxM1 expression in HCC cells has remained unclear. Further investigation will be needed to elucidate this issue.
The liver X receptors (LXRs), such as LXRα or LXRβ, are known to regulate the uptake, transport, and efflux of cholesterol in the liver and macrophages [41]. There have been several evidences showing the involvement of LXRs in a variety of cancers including HCC [42]. A previous report showed that the activation of LXRα resulted in the downregulation of FoxM1 and the suppression of proliferation in human hepatoma cells [43]. As shown in Supplementary Figure 4, the administration of pitavastatin significantly increased the gene expression of LXRα in HepG2 cells and re-exposure to mevalonate restored the increase of LXRα gene expression induced by pitavastatin. These data indicate that the up-regulation of LXRα by statin might be one possible mechanism underlying the inhibition of FoxM1 expression by statin. However, at this stage, the relation between the mevalonate pathway and the LXRα-mediated pathway in the regulation of FoxM1 has remained unclear. Further investigation will be needed to understand detailed mechanisms.
So far, several therapeutic strategies have been proposed to target FoxM1 in cancer. In mouse models, a cell-penetrating ARF peptide that reduces FoxM1 transcriptional activity has been shown to be effective to treat HCC [20, 21]. A high-throughput screening identified a thiazole antibiotics Syomycin A as a potent FoxM1 inhibitor [44] and thiostrepton, a similar type of thiazole antibiotics, has been shown to inhibit FoxM1 transcriptional activity by direct binding to FoxM1 in several human cancer cells including HCC cells [45]. Likewise, another screening also identified a small molecule FDI-6 as a potent FoxM1 inhibitor in breast cancer cells [46]. Despite these findings, no drugs targeting FoxM1 are available in clinical use for cancer therapy so far. In this study, we identified statins, FDA-approved cholesterol-lowering drugs, as novel FoxM1 inhibitors against HCC. In this study, we used various pharmaceutical inhibitors for the mevalonate pathway and showed that zoledronic acid or GGTI-298 had a potential to inhibit FoxM1 expression same as statins. Previous reports showed the possible toxic effects of these pharmaceutical inhibitors which we used in our study; pitavastatin may exhibit myotoxicity including myopathy, myalgia, myositis, or rhabdomyolysis [47]; zoledronic acid may induce hypocalcaemia, secondary hyperparathyroidism, or renal toxicity [48]. Because other inhibitors, such as GGTI-298 [49], FTI-277 [50], or YM-53601 [51], are still in preclinical stages, toxic effects in clinical use remain unclear. Collectively, it is worth noting that drugs already available in the clinical practice have an inhibitory effect on FoxM1 expression in HCC.
In conclusion, in this study, we demonstrated that oncogenic FoxM1 transcription factor functions downstream of the mevalonate pathway in HCC. Our findings also provided new insights on the interplay between the mevalonate pathway and oncogenic signals. Thus, the inhibition of FoxM1 by targeting the mevalonate pathway would be a novel therapeutic option for the treatment of HCC.
MATERIALS AND METHODS
Reagents
Pitavastatin was a gift from Kowa Pharmaceutical Co., Ltd. (Tokyo, Japan). Simvastatin, fluvastatin sodium hydrate, mevalonolactone, GGTI-298 trifluoroacetate salt hydrate, FTI-277 trifluoroacetate salt, zoledronic acid monohydrate, farnesyl pyrophosphate ammonium salt, geranylgeranyl pyrophosphate ammonium salt, squalene, NSC23766 trihydrochloride, and ML141 were obtained from Sigma-Aldrich (Saint Louis, MO, USA). YM-53601 was obtained from Cayman Chemical Co. (Ann Arbor, MI, USA). C3 transferase was obtained from Cytoskeleton, Inc. (Denver, CO, USA).
Cell culture
HepG2, Huh7 and HLF cells were obtained from the Japanese Cancer Resources Bank (JCRB, Tokyo, Japan) between 2011 and 2015 and have been cryopreserved in liquid nitrogen until use. Cells were used within 10 passages after thawing and were cultured in DMEM (Sigma-Aldrich, Saint Louis, MO, USA) supplemented with 10% FCS and Antibiotic-Antimycotic (Thermo Fisher Scientific, Inc., Waltham, MA, USA) in an atmosphere of 5% CO2 in air at 37° C. Test for mycoplasmal contamination was performed using MycoAlert mycoplasma detection kit (Lonza, Walkersville, MD, USA). Cells were seeded on 6-well plate at 1.0 × 105 cells/well. After an overnight culture, cells were treated with reagents for 24 hours. Cells were then harvested to prepare protein extracts.
Western blot analysis
Protein extraction and Western blot analysis were performed as previously described [52]. The following primary antibodies were used: FoxM1 (D12D5), Cleaved PARP (Asp214) (D64E10), RhoA (67B9), Rac1/2/3, Cdc42 (11A11) (Cell Signaling Technology, Inc., Danvers, MA, USA), T7-tag monoclonal antibody (Merck KGaA, Darmstadt, Germany), GAPDH (Trevigen, Inc., Gaithersburg, MD, USA). Donkey anti-rabbit IgG-HRP (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and anti-mouse IgG-HRP (Promega Corp., Madison, WI, USA) were used as a secondary antibody.
Quantitative real-time RT-PCR
Cells were seeded on 6-well plate at 1.0 × 105 cells/well. After an overnight culture, cells were treated with reagents for 24 hours. Total RNA was extracted and purified using QIAshredder (Qiagen, Hilden, Germany) and RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions as previously described [52]. cDNA was reverse-transcribed from 1μg of total RNA using ReverTra Ace qPCR RT Master Mix (Toyobo Co., Ltd., Osaka, Japan). Quantitative real-time RT-PCR was performed with QuantiTect Primer Assays (FoxM1: QT00000140, HMGCR: QT00004081, CCNB1: QT00006615, BIRC5: QT01679664, SREBP2: QT00052052, RhoA: QT00044723, Rac1: QT00065856, Cdc42: QT00066528, LXRα (NR1H3): QT00065156, GAPDH: QT00079247, Qiagen, Hilden, Germany), using Light Cycler 1.5 (Roche Diagnostics, Basel, Switzerland) and Quant Studio 6 (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Relative quantitation of gene expression was analyzed with ΔΔCt method, using GAPDH as an internal control.
siRNA transfection
Stealth RNAi siRNA of FoxM1 (HSS177135), HMGCR (HSS104864), RhoA (VHS40471), Rac1 (VHS40447) and Cdc42 (VHS40393) were obtained from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). 20 nM of gene specific siRNA or Stealth RNAi siRNA Negative Control (Thermo Fisher Scientific, Inc., Waltham, MA, USA) was transfected with Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific, Inc., Waltham, MA, USA) after incubating in Opti-MEM I Reduced serum medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA) as previously described [52].
Immunofluorescent staining
Cells were seeded on poly-L-lysine coated culture cover glass (13 mm, Matsunami Glass Industry, Ltd., Osaka, Japan) placed in 24-well plate at 3.0 × 104 cells/well. After an overnight culture, cells were treated with reagents for 24 hours. Cells were fixed with 4% paraformaldehyde for 20 minutes at room temperature. After permeabilized with 0.5% Triton-X for 20 minutes at room temperature, the cells were treated with 3% bovine serum albumin (Sigma-Aldrich, Saint Louis, MO, USA) for 30 minutes at room temperature. After this blocking procedure, cells were treated with a primary antibody for overnight at 4° C, and then a secondary antibody for 1 hour at room temperature. We used the following antibodies: FoxM1 (D12D5, Cell Signaling Technology, Inc., Danvers, MA, USA), Goat anti-rabbit IgG (H+L) Secondary Antibody, Alexa Flour 488 conjugate (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Cover glass was mounted with DAPI Fluoromount-G (Southern Biotechnology Associates, Inc., Birmingham, AL, USA) on slide glass.
Assessment of caspase3/7 activity
Cells were seeded on 96-well plate at 3.5 × 103 cells/well. After an overnight culture, cells were treated with reagents for 24 or 48 hours. The caspase 3/7 activity of supernatant was assessed with Caspase-Glo 3/7 Assay (Promega Corp., Madison, WI, USA), according to the manufacturer’s instructions.
WST assay
Cells were seeded on 96-well plate at 3.5 × 103 cells/well. After an overnight culture, cells were treated with reagents for 48 hours. The WST assay was assessed with Cell Count Reagent SF (Nacalai Tesque, Inc., Kyoto, Japan), according to the manufacturer’s instructions.
Plasmid transfection
T7-tagged FoxM1 plasmid (T7-FoxM1) was provided by Dr. Pradip Raychaudhuri [34]. After seeding cells and overnight culture, the plasmid was transfected with FuGENE HD Transfection Reagent (Promega Corp., Madison, WI, USA), according to the manufacturer’s instructions.
Patients
We examined 64 patients undergoing curative hepatic resection for HCC at the Department of Surgery, Osaka University Hospital from May 2001 to November 2011. None of the patients had undergone transcatheter arterial embolization or transcatheter arterial chemoembolization before surgery. This study was performed according to the ethical guidelines of the Declaration of Helsinki and was approval to use the resected samples from the Institutional Review Board (IRB) Committees at Osaka University (IRB No. 13556 and 15267), and informed consent was obtained from all patients. In this study, the “high” group is defined as the patients with higher gene expression than median; the “low” group is defined as those with lower gene expression than median.
Statistical analysis
Statistical analysis was performed with JMP Pro 12.2.0 (SAS Institute, Inc., Cary, NC, USA) using Student’s t-test. Data was represented as mean ± standard error of the mean (SEM) from three biological replicates. We used Pearson’s correlation coefficients to determine the correlation between the genes expression in human HCC. Overall survival was calculated using Kaplan–Meier method and compared by Log-rank test. Patients’ characteristics were compared using Mann–Whitney U test or Pearson’s chi-square test. The prognostic factors were identified using the Cox proportional hazards model; the factors chosen using simple Cox regression were further examined using multiple Cox regression. Statistical significance was defined as p < 0.05.
Abbreviations
FoxM1: Forkhead Box M1 transcription factor; FPP: farnesyl pyrophosphate; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GGPP: geranylgeranyl pyrophosphate; HCC: hepatocellular carcinoma; HMGCR: 3-hydroxy-3-methylglutaryl CoA reductase; RT-PCR: reverse-transcription polymerase chain reaction; siRNA: small-interfering RNA; SREBP2: sterol regulatory element-binding protein 2.
Author contributions
Conception and design: Y. Yoshida; Development of methodology: S. Ogura, Y. Yoshida, T. Kurahashi, M. Egawa; Acquisition of data: S. Ogura, Y. Yoshida, T. Kurahashi; Analysis and interpretation of data: S. Ogura, Y. Yoshida, T. Kurahashi, M. Egawa, K. Furuta, S. Kiso, Y. Kamada, H. Hikita, T. Tatsumi; Writing, review and/or revision of the manuscript: S. Ogura, Y. Yoshida, T. Takehara; Administrative, technical, or material support: H. Ogita, H. Eguchi, Y. Doki, M. Mori; Study supervision: T. Takehara.
ACKNOWLEDGMENTS
We thank members of our department for helpful discussions.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported in part by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS).
REFERENCES
1. Bruix J, Reig M, Sherman M. Evidence-Based Diagnosis, Staging, and Treatment of Patients With Hepatocellular Carcinoma. Gastroenterology. 2016; 150:835–53. https://doi.org/10.1053/j.gastro.2015.12.041.
2. Lencioni R. Loco-regional treatment of hepatocellular carcinoma. Hepatology. 2010; 52:762–73. https://doi.org/10.1002/hep.23725.
3. Yang JD, Roberts LR. Hepatocellular carcinoma: A global view. Nat Rev Gastroenterol Hepatol. 2010; 7:448–58. https://doi.org/10.1038/nrgastro.2010.100.
4. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, et al, and SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008; 359:378–90. https://doi.org/10.1056/NEJMoa0708857.
5. Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, Gerolami R, Masi G, Ross PJ, et al, and RESORCE Investigators. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017; 389:56–66. https://doi.org/10.1016/S0140-6736(16)32453-9.
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74. https://doi.org/10.1016/j.cell.2011.02.013.
7. Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013; 12:829–46. https://doi.org/10.1038/nrd4145.
8. Wada A, Fukui K, Sawai Y, Imanaka K, Kiso S, Tamura S, Shimomura I, Hayashi N. Pamidronate induced anti-proliferative, apoptotic, and anti-migratory effects in hepatocellular carcinoma. J Hepatol. 2006; 44:142–50. https://doi.org/10.1016/j.jhep.2005.09.022.
9. Honda Y, Aikata H, Honda F, Nakano N, Nakamura Y, Hatooka M, Morio K, Kobayashi T, Fukuhara T, Nagaoki Y, Kawaoka T, Hiramatsu A, Imamura M, et al. Clinical outcome and prognostic factors in hepatocellular carcinoma patients with bone metastases medicated with zoledronic acid. Hepatol Res. 2017; 47:1053–60. https://doi.org/10.1111/hepr.12844.
10. Chou R, Dana T, Blazina I, Daeges M, Jeanne TL. Statins for Prevention of Cardiovascular Disease in Adults: Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA. 2016; 316:2008–24. https://doi.org/10.1001/jama.2015.15629.
11. Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology. 2013; 144:323–32. https://doi.org/10.1053/j.gastro.2012.10.005.
12. Osmak M. Statins and cancer: current and future prospects. Cancer Lett. 2012; 324:1–12. https://doi.org/10.1016/j.canlet.2012.04.011.
13. Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nat Rev Cancer. 2005; 5:930–42. https://doi.org/10.1038/nrc1751.
14. Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer. 2016; 16:718–31. https://doi.org/10.1038/nrc.2016.76.
15. Lonardo A, Loria P. Potential for statins in the chemoprevention and management of hepatocellular carcinoma. J Gastroenterol Hepatol. 2012; 27:1654–64. https://doi.org/10.1111/j.1440-1746.2012.07232.x.
16. Okabe H, Satoh S, Kato T, Kitahara O, Yanagawa R, Yamaoka Y, Tsunoda T, Furukawa Y, Nakamura Y. Genome-wide analysis of gene expression in human hepatocellular carcinomas using cDNA microarray: identification of genes involved in viral carcinogenesis and tumor progression. Cancer Res. 2001; 61:2129–37.
17. Pilarsky C, Wenzig M, Specht T, Saeger HD, Grutzmann R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia. 2004; 6:744–50. https://doi.org/10.1593/neo.04277.
18. Yoshida Y, Wang IC, Yoder HM, Davidson NO, Costa RH. The forkhead box M1 transcription factor contributes to the development and growth of mouse colorectal cancer. Gastroenterology. 2007; 132:1420–31. https://doi.org/10.1053/j.gastro.2007.01.036.
19. Calvisi DF, Pinna F, Ladu S, Pellegrino R, Simile MM, Frau M, De Miglio MR, Tomasi ML, Sanna V, Muroni MR, Feo F, Pascale RM. Forkhead box M1B is a determinant of rat susceptibility to hepatocarcinogenesis and sustains ERK activity in human HCC. Gut. 2009; 58:679–87. https://doi.org/10.1136/gut.2008.152652.
20. Kalinichenko VV, Major ML, Wang X, Petrovic V, Kuechle J, Yoder HM, Dennewitz MB, Shin B, Datta A, Raychaudhuri P, Costa RH. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004; 18:830–50. https://doi.org/10.1101/gad.1200704.
21. Gusarova GA, Wang IC, Major ML, Kalinichenko VV, Ackerson T, Petrovic V, Costa RH. A cell-penetrating ARF peptide inhibitor of FoxM1 in mouse hepatocellular carcinoma treatment. J Clin Invest. 2007; 117:99–111. https://doi.org/10.1172/JCI27527.
22. Park HJ, Gusarova G, Wang Z, Carr JR, Li J, Kim KH, Qiu J, Park YD, Williamson PR, Hay N, Tyner AL, Lau LF, Costa RH, Raychaudhuri P. Deregulation of FoxM1b leads to tumour metastasis. EMBO Mol Med. 2011; 3:21–34. https://doi.org/10.1002/emmm.201000107.
23. Egawa M, Yoshida Y, Ogura S, Kurahashi T, Kizu T, Furuta K, Kamada Y, Chatani N, Hamano M, Kiso S, Hikita H, Tatsumi T, Eguchi H, et al. Increased expression of Forkhead box M1 transcription factor is associated with clinicopathological features and confers a poor prognosis in human hepatocellular carcinoma. Hepatol Res. 2017; 47:1196–205. https://doi.org/10.1111/hepr.12854.
24. Cui J, Shi M, Xie D, Wei D, Jia Z, Zheng S, Gao Y, Huang S, Xie K. FOXM1 promotes the warburg effect and pancreatic cancer progression via transactivation of LDHA expression. Clin Cancer Res. 2014; 20:2595–606. https://doi.org/10.1158/1078-0432.CCR-13-2407.
25. Wang Y, Yun Y, Wu B, Wen L, Wen M, Yang H, Zhao L, Liu W, Huang S, Wen N, Li Y. FOXM1 promotes reprogramming of glucose metabolism in epithelial ovarian cancer cells via activation of GLUT1 and HK2 transcription. Oncotarget. 2016; 7:47985–97. https://doi.org/10.18632/oncotarget.10103.
26. Major ML, Lepe R, Costa RH. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol Cell Biol. 2004; 24:2649–61. https://doi.org/10.1128/mcb.24.7.2649-2661.2004.
27. Leung TW, Lin SS, Tsang AC, Tong CS, Ching JC, Leung WY, Gimlich R, Wong GG, Yao KM. Over-expression of FoxM1 stimulates cyclin B1 expression. FEBS Lett. 2001; 507:59–66.
28. Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, Tan Y, Ackerson T, Costa RH. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol. 2005; 25:10875–94. https://doi.org/10.1128/MCB.25.24.10875-10894.2005.
29. Thurnher M, Nussbaumer O, Gruenbacher G. Novel aspects of mevalonate pathway inhibitors as antitumor agents. Clin Cancer Res. 2012; 18:3524–31. https://doi.org/10.1158/1078-0432.CCR-12-0489.
30. Raychaudhuri P, Park HJ. FoxM1: a master regulator of tumor metastasis. Cancer Res. 2011; 71:4329–33. https://doi.org/10.1158/0008-5472.CAN-11-0640.
31. Costa RH. FoxM1 dances with mitosis. Nat Cell Biol. 2005; 7:108–10. https://doi.org/10.1038/ncb0205-108.
32. Balli D, Ustiyan V, Zhang Y, Wang IC, Masino AJ, Ren X, Whitsett JA, Kalinichenko VV, Kalin TV. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J. 2013; 32:231–44. https://doi.org/10.1038/emboj.2012.336.
33. Ye H, Kelly TF, Samadani U, Lim L, Rubio S, Overdier DG, Roebuck KA, Costa RH. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol Cell Biol. 1997; 17:1626–41.
34. Park HJ, Carr JR, Wang Z, Nogueira V, Hay N, Tyner AL, Lau LF, Costa RH, Raychaudhuri P. FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO J. 2009; 28:2908–18. https://doi.org/10.1038/emboj.2009.239.
35. Ma RY, Tong TH, Cheung AM, Tsang AC, Leung WY, Yao KM. Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1c. J Cell Sci. 2005; 118:795–806. https://doi.org/10.1242/jcs.01657.
36. Barsotti AM, Prives C. Pro-proliferative FoxM1 is a target of p53-mediated repression. Oncogene. 2009; 28:4295–305. https://doi.org/10.1038/onc.2009.282.
37. Halasi M, Gartel AL. FOX(M1) news—it is cancer. Mol Cancer Ther. 2013; 12:245–54. https://doi.org/10.1158/1535-7163.MCT-12-0712.
38. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, Martirosyan A, Hakem A, Hakem R, Jurisica I, Penn LZ. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A. 2010; 107:15051–6. https://doi.org/10.1073/pnas.0910258107.
39. Innocente SA, Abrahamson JL, Cogswell JP, Lee JM. p53 regulates a G2 checkpoint through cyclin B1. Proc Natl Acad Sci U S A. 1999; 96:2147–52.
40. Jin Y, Wei Y, Xiong L, Yang Y, Wu JR. Differential regulation of survivin by p53 contributes to cell cycle dependent apoptosis. Cell Res. 2005; 15:361–70. https://doi.org/10.1038/sj.cr.7290303.
41. Hong C, Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov. 2014; 13:433–44. https://doi.org/10.1038/nrd4280.
42. Bovenga F, Sabba C, Moschetta A. Uncoupling nuclear receptor LXR and cholesterol metabolism in cancer. Cell Metab. 2015; 21:517–26. https://doi.org/10.1016/j.cmet.2015.03.002.
43. Hu C, Liu D, Zhang Y, Lou G, Huang G, Chen B, Shen X, Gao M, Gong W, Zhou P, Dai S, Zeng Y, He F. LXRalpha-mediated downregulation of FOXM1 suppresses the proliferation of hepatocellular carcinoma cells. Oncogene. 2014; 33:2888–97. https://doi.org/10.1038/onc.2013.250.
44. Radhakrishnan SK, Bhat UG, Hughes DE, Wang IC, Costa RH, Gartel AL. Identification of a chemical inhibitor of the oncogenic transcription factor forkhead box M1. Cancer Res. 2006; 66:9731–5. https://doi.org/10.1158/0008-5472.CAN-06-1576.
45. Hegde NS, Sanders DA, Rodriguez R, Balasubramanian S. The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nat Chem. 2011; 3:725–31. https://doi.org/10.1038/nchem.1114.
46. Gormally MV, Dexheimer TS, Marsico G, Sanders DA, Lowe C, Matak-Vinkovic D, Michael S, Jadhav A, Rai G, Maloney DJ, Simeonov A, Balasubramanian S. Suppression of the FOXM1 transcriptional programme via novel small molecule inhibition. Nat Commun. 2014; 5:5165. https://doi.org/10.1038/ncomms6165.
47. Tomaszewski M, Stepien KM, Tomaszewska J, Czuczwar SJ. Statin-induced myopathies. Pharmacol Rep. 2011; 63:859–66.
48. Papapetrou PD. Bisphosphonate-associated adverse events. Hormones (Athens). 2009; 8:96–110.
49. Chu UB, Duellman T, Weaver SJ, Tao Y, Yang J. Endothelial protective genes induced by statin are mimicked by ERK5 activation as triggered by a drug combination of FTI-277 and GGTI-298. Biochim Biophys Acta. 2015; 1850:1415–25. https://doi.org/10.1016/j.bbagen.2015.03.011.
50. Kim DM, Ryu SW, Choi C. Long-term treatment of farnesyltransferase inhibitor FTI-277 induces neurotoxicity of hippocampal neurons from rat embryo in a ROS-dependent manner. Biochem Biophys Res Commun. 2010; 403:91–6. https://doi.org/10.1016/j.bbrc.2010.10.123.
51. Ugawa T, Kakuta H, Moritani H, Matsuda K, Ishihara T, Yamaguchi M, Naganuma S, Iizumi Y, Shikama H. YM-53601, a novel squalene synthase inhibitor, reduces plasma cholesterol and triglyceride levels in several animal species. Br J Pharmacol. 2000; 131:63–70. https://doi.org/10.1038/sj.bjp.0703545.
52. Furuta K, Yoshida Y, Ogura S, Kurahashi T, Kizu T, Maeda S, Egawa M, Chatani N, Nishida K, Nakaoka Y, Kiso S, Kamada Y, Takehara T. Gab1 adaptor protein acts as a gatekeeper to balance hepatocyte death and proliferation during acetaminophen-induced liver injury in mice. Hepatology. 2016; 63:1340–55. https://doi.org/10.1002/hep.28410.