
INTRODUCTION
Anabolic androgenic steroids (AAS) are one of the most commonly used drugs among athletes to improve physical performance. The use of AAS was prohibited by the International Olympic Committee in 1976 and more recently the World Anti-Doping Agency included these compounds in the list of prohibited substances [1]. Despite those bans, AAS abuse is continuing to increase particularly in the general population at fitness centers, mainly for aesthetic purposes [1–3]. The non-medical use of AAS among athletes and specific subsets of the general population (high school and college students) is considered a major and widespread public health issue [4–6]. It is becoming increasingly clear that the abuse of AAS is associated with serious adverse effects to the liver [7], cardiovascular [8, 9], central nervous [10–12], musculoskeletal [13–15], endocrine [11], fertility and reproductive [16–18] systems.
AAS are synthetic derivatives of testosterone and their pharmacodynamics is similar to all other steroid hormones. AASs are membrane-permeable and influence the nucleus of cells by direct action. When the exogenous hormone penetrates the membrane of the target cell, the first step is the link to an androgen receptor (AR) located in the cytoplasm of the cell. From there, the compound hormone-receptor diffuses into the nucleus, where it either alters gene expression [19], or activates processes that send signals to other parts of the cell [20]. AASs exert their actions by several different mechanisms: (i) they modulate androgen receptor expression as a consequence of intracellular metabolism; (ii) they affect directly the androgen receptor and thus subsequent interaction with co-activators and transcriptional activity; (iii) they interfere with glucocorticoid receptor expression eliciting an anticatabolic effect; and (iv) they act on the CNS resulting in behavioral changes, following the genomic and non-genomic pathways [21].
Thanks to drug designers, to date, more than 100 AAS compounds were synthesized. Analyzing their chemical structure, metabolic half-life, and physiological effects, three classes of AAS can be identified. The first class was obtained by the esterification of the 17β-hydroxyl group of testosterone and includes testosterone propionate and testosterone cypionate. The second class was composed by AASs esterified, connected with the long side chain moieties, with a substitution of a hydrogen for the methyl group at C19. The third class of AAS comprises those compounds that are alkylated at C17, such as 17α-methyltestosterone and stanozolol [22].
Many animal and “in vitro” studies have demonstrated that supraphysiologic doses of AAS enhance the expression of oxidative stress proteins as well as of inflammatory and proapoptotic mediators [23]. To the best of our knowledge, only a few studies have been focused on the specific effect of ASS on carcinogenicity.
A relationship between AAS use and cancer was previously suspected and recently proven. Clinical cases reported a causal connection between AAS use and both hepatocellular adenomas and adenocarcinomas. The physiopathological mechanism hidden behind this connection remains unclear. What is known is that the AAS induced biological effect is carried out by the androgen receptor (AR), an intracellular receptor located not only into reproductive organ cells but also into bone, muscle, brain, liver, kidney cells and adipocytes. AAS-AR binding initiates a cascade of events: receptor dimerization, nuclear translocation, binding to the specific promoter of genetic sequences and expression modifications [24].
FROM AAS GENOTOXICITY TO CARCINOGENICITY
Testosterone is mainly produced by the Leydig cells of testes in males, and ovaries and theca cells in females [25–26]. Smaller amounts are also synthesized by the adrenal gland in both sexes [27]. The production is regulated by a complex neuroendocrine mechanism which includes the pulsatile release of luteinizing hormone (LH) and subsequent cAMP activation of steroidogenic cascade and numerous steroidogenic stimuli and intratesticular factors that play a role in the intricate regulatory network of testosterone [28]. Cholesterol is the common substrate for all steroid hormones biosynthesis which is completed in the mitochondria. Steroidogenic acute regulatory protein (STAR) transfers cholesterol to the inner membrane of mitochondria. Through the mobilization and delivery from the outer to the inner mitochondrial membrane, cholesterol is converted to pregnenolone by the cytochrome P450 cholesterol side-chain cleavage enzyme (CYP11A1, also known as P450scc) [29]. Pregnenolone is further metabolized to progesterone by mitochondrial or microsomal 3b-hydroxysteroid dehydrogenases (HSD3B1). In Leydig cells, maturation of progesterone to androstenedione is catalyzed by the 17a-hydroxylase/C17–20lyase (CYP17A1 also known as P45017A1); further conversion of androstenedione to testosterone is dependent on the activity of 17b-hydroxysteroid dehydrogenase (17bHSD), steroid dehydrogenase specific for androgen production [27, 30] (Figure 1).
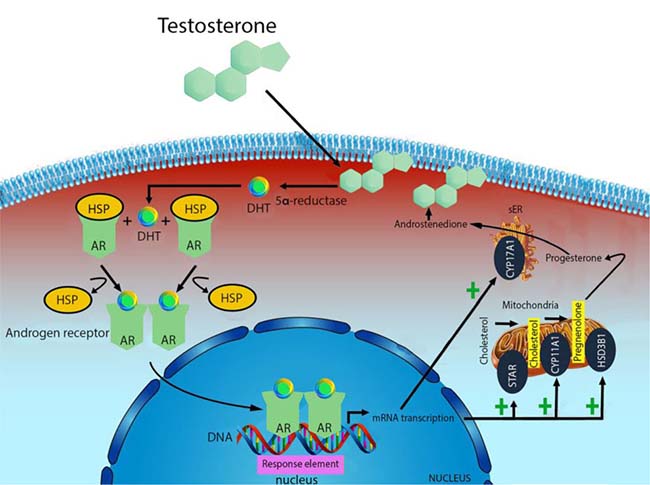
Figure 1: Mechanism of testosterone action. Free testosterone is transported into target tissue cell cytoplasm, where it can either bind the androgen receptor, or be reduced to 5α-di-hydro-testosterone (DHT) by the cytoplasmic enzyme 5-alpha reductase. The T-receptor complex undergoes to a structural change that allows its translocation into the cell nucleus, where it directly binds to specific nucleotide sequences of the chromosomal DNA. The produced mRNA promotes the biosynthesis of testosterone.
There are three main pathways in which testosterone exerts its effects. It can act directly on androgen receptor, or via dihydrotestosterone (DHT) produced by the action of 5-a-reductase, or via estrogen receptor by means of estradiol produced by CYP19 aromatase [31]. NR3A (also known GRIN3A) is an androgen nuclear receptor normally linked to heat shock proteins (HSP). When testosterone and DHT, bind to this receptor in the presence of bio-available androgens, it undergoes a conformational change, making free-form HSP, dimerization and moving through the nucleus. Here, this can activate DNA transcription of specific responsive genes through the DNA-binding domains to androgen response elements (ARE), by means of its zinc-finger motif. [32].
AAS mimics testosterone physiological effect, by inducing expression alterations on of DNA sequences. For example, Nandrolone is transported into the target tissue cell cytoplasm as testosterone. However, the mRNA produced interferes with the physiological biosynthesis of testosterone, reducing cholesterol conversion to pregnenolone through CYP11A1. Analogously, the conversion from progesterone to androstenedione through CYP17A1 is reduced resulting in diminishing endogenous testosterone production [33].
However, the genotoxicity mechanism is not well understood. Torres-Bugarín et al. [34], in a review analyzing androgen effects on cellular functions, concluded that a combination of genetic and epigenetic factors is the cause of toxicity, mutagenicity, genotoxicity, and carcinogenicity of sexual hormones. Epigenetic factors include three molecular mechanisms, controlling genetic transcription: DNA methylation, histone modifications and chromatin condensation [35]. DNA methylation inhibits the bind between transcriptional factors and their target sequences, both promoters, and introns, impeding the activation of transcriptional expression. In an animal model, 5α-DHT increased DNA methylation of srd5α2 (steroid 5α-reductases type 2) [36]. The latter is also regulated by chromatin condensation degree [37].
On taking account genetic factors, testosterone synthetic derivatives can be metabolized to 17β-estradiol in adipose, cerebral and testicular tissues. As previously described, the 17β-estradiol (E2) has an important role in estrogen-dependent breast cancer, and it is described as a potential mutagenic and carcinogenic mediator [38]. Furthermore, its metabolites are also considered inducers of cell proliferation.
During their catabolism, AAS reveal their oxidative role, increasing ROS, which are highly unstable and extremely reactive oxygen species, which easily lose hydrogen atoms. In this manner, they form covalent bonds with DNA bases or sequences, inducing a known genetic damage [39]. This mechanism was supposed to be connected to hepatocellular alterations because of its interference with bilirubin metabolism and vascular and cellular hyperplasia. For this kind of tumorigenesis action, different patterns of evolution were described, such as hepatic peliosis and focal-nodular hyperplasia/liver adenomas [40–41].
Moreover, in a study conducted by Seraj et al. [42] about testosterone derivative genotoxicity, its ability in generating DNA adducts was established. These processes, individually or in combination, can induce micronuclei formation among animals exposed to higher concentrations of AAS. Micronuclei are strictly related to several mutagenic stresses and are formed following chromosomal damage. These are chromatin particles derived from acentric chromosomal fragments, which are not incorporated into the daughter nucleus after mitosis. A variety of factors influences micronuclei formation, such as; age, sex, genetic constitution, physical and chemical agents. One of these agents has been proven high-dose nandrolone exposition, which determines a significant DNA damage in blood, liver, bone marrow, brain and testicle cells in experimental animals exposed [43]. On the other hand, AAS can elicit profound modifications in genetic sequences by means of alterations in telomerase activity [44]. Nourbakhsh et al. [45], tried to verify the implications of androgens in ovarian carcinogenesis. They demonstrated that both testosterone and androstenedione increased ovarian cancer cell viability via the expression, activity, and phosphorylation of telomerase, and by blocking phosphatidylinositol 3-kinase pathway inhibitors. Consequently, it was stated that the PI3K/Akt pathway triggered this upregulation and that the hTERT mRNA levels were significantly increased with exposure to testosterone and androstenedione, thanks to quantitative PCR analyses. hTERT is a gene capable to encode the telomerase catalytic subunit, thus, allowing sustained cell proliferation, which has already been revealed to be regulated in human prostate cancer cells in vivo by androgens. In addition, androgen use has been reported to improve telomerase expression and behavior. Tamoxifen and letrozole inhibit estradiol and androgens effects on telomerase activity which is not affected by flutamide (an androgen receptor antagonist) administration [46]. Alterations in telomerase activity are at the basis of stanozolol-induced DNA-damaging effects.
With regards to the theoretical genotoxic effects on DNA, it should be pointed out that AAS effects are linked to dosage and frequency of administration. Therefore abusers abide by strict and controlled administration regimens resorting to specific strategies. “Stacking”, one of the strategies employed, involves the use of multiple AAS in order to lower doses of each substance and their adverse effects [47]. A common occurrence is the simultaneous administration of AAS and growth hormone (GH). It is estimated that one in each four sportsman takes both these drugs [48–49].
AASs promote muscle fiber mass and hypertrophy by augmentation of satellite cell proliferation, myonuclei number and muscle protein synthesis [50]. GH, instead, directly regulates muscle protein expression and production, by binding its receptors but also indirectly by activation of the IGF-1 receptor, which can activate the PI3K/Akt pathway in order to endorse myocellular proliferation (Figure 2). As far as myogenesis is concerned, IGF-1 is a positive key signaling molecule inducing not also satellite cell multiplying but also myoblast differentiation and subsequent myoblast fusion into myotubes. Consequently, overexpression of IGF-1 causes significant hypertrophy and excess in proliferation [51]. Furthermore, IGF-1 seems to mediate the growth-promoting influences of anabolic steroids. Based on that, AAS and GH or IGF-1 are combinations with a high performance-enhancing potential. Testosterone stimulates the mitotic activity of satellite cells, while GH or IGF-1 mediates these effects on skeletal muscle cell growth and differentiation [52]. This combined effect on myocells, should be considered in the light that supra-physiological doses of GH are associated with increased incidences of colorectal, thyroid, breast, and prostate cancers. Moreover, high-level detection of plasmatic IGF-1 has been associated with cancer risk and cancer prognosis [53]. IGF-1 has been involved in tumor development and progression because cancer cells have an increased expression of IGF-1 receptors. In addition, Cappello et al. [54] have demonstrated that Heat shock proteins (Hsps) perform critical functions in maintaining protein homeostasis under physiological and stressful conditions. Hsp60 is a constitutive mitochondrial protein with specific functions related to mitochondrial protein folding, especially in response to oxidative stress [55–56]. Hsp90 was found to be overexpressed in multiple cancers, including prostate cancer. Hsp90 is able to activate over 200 proteins, including the AR, where Hsp90 prevents proteasome degradation and stabilizes AR conformation poised for ligand activation [57]. AR binds to Hsp90 in its inactive form. Upon binding of androgens, the receptor detaches from Hsp90 and becomes activated [58–60].
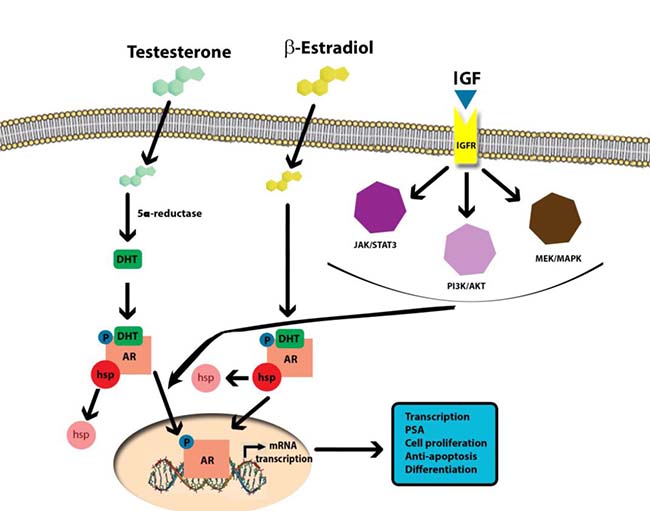
Figure 2: Molecular mechanisms of AAS-induced carcinogenicity.
Sirianni et al. [61] using a human breast cancer cell line, MCF-7, as an experimental model, demonstrated that stimulating aromatase expression and estrogen production through IGF-1 can promote cell proliferation. High doses of nandrolone (aromatizable) and stanozolol (non aromatizable), could potentially increase breast cancer risk because in cases of high bioavailability, these compounds can attach to the ER, inducing its nuclear translocation in vivo [62–64]. Rapidly increase in the concentration of IGF-1R, ERK1/2 and AKT phosphorylation, is demonstrated by the ICI block on AAS-dependent kinase activation. Considering the above data, estrogens induce cell proliferation in target cells via the increased expression of up-regulated CCND1, encoding cyclin D1 regulating cell cycle G1 phase, which determines breast cancer cell proliferation [65–67]. Nandrolone and stanozolol magnified cyclin D1 concentration, inducing cell proliferation.
TESTICULAR CANCER
Testicular cancer represents 1% of male neoplasms and 5% of urological tumors, with 3–10 new cases occurring per 100,000 males/per year in Western society [68–69]. In the last decades, the incidence of this cancer is constantly increasing, especially in industrialized countries [70–72]. Leydig cell tumors are usually benign, but approximately 10% are malignant. The malignant variants occur only in adults.
Table 1 subdivides testicular cancers according to a pathological classification, in germ cell tumors, sex cord/gonadal stromal tumors and miscellaneous non-specific tumors [73].
Table 1: Subdivision of testicular cancers under pathological classification
Germ cell tumors |
Sex cord/gonadal stromal tumors |
Miscellaneous non-specific tumors |
|---|---|---|
• Intratubular germ cell neoplasia (IGCNU), unclassified type • Seminoma (including cases with syncytiotrophoblastic cells) • Spermatocytic seminoma (mention if there is a sarcomatous component) • Embryonal carcinoma • Yolk sac tumour • Choriocarcinoma • Teratoma (mature, immature, with malignant component) • Tumours with more than one histological type (specify percentage of individual components). |
• Leydig cell tumour • Malignant Leydig cell tumour • Sertoli cell tumour - lipid-rich variant - sclerosing - large cell calcifying • Malignant Sertoli cell tumour • Granulosa cell tumour - adult type - juvenile type • Thecoma/fibroma group of tumours • Other sex cord/gonadal stromal tumours - incompletely differentiated - mixed • Tumours containing germ cell and sex cord/gonadal stromal (gonadoblastoma) |
• Ovarian epithelial tumours • Tumours of the collecting ducts and rete testis • Tumours (benign and malignant) of non-specific stroma. |
Leydig cell tumors represent the most common kind of the sex/gonadal stromal category [74–75]. Their insurgency is located between the third and sixth decades of adult life: when it occurs before, it is frequently related to hormones therapy/abuse.
Adults with androgen-secreting tumors are generally asymptomatic. Adult clinical manifestations of estrogen-secreting tumors include loss of libido, erectile dysfunction, infertility, gynecomastia, feminine hair distribution, and gonadogenital atrophy [76]. Taking into account the etiology, Leydig cell tumors are associated with cryptorchidism, testicular atrophy, infertility, germline mutations in fumarate hydratase, hereditary leiomyomatosis and renal cell carcinoma [77]. These symptoms are commonly found in AAS abusers. In fact, the testicular atrophy represents one of the most frequent side-effects related to AAS abusers, so a relationship between Leydig cell cancer and AAS must be taken into consideration.
AASS AND LEYDIG CELL CANCER
To investigate the side effects of AASs abuse on testicular cells, several animal studies were performed. Dohle et al. [78] showed that exogenous administration of synthetic testosterone caused a negative alteration on the hypothalamic-pituitary axis, inhibiting the secretion of both Follicle-stimulating hormone (FSH) and luteinizing hormone (LH). This mechanism could lead to a decreased serum androgens concentration and cause hypogonadotropic hypogonadism with subsequent testicular atrophy. Androgen action is mediated by binding to androgen receptors (AR) both in testis and in other tissues [79]. Inside Sertoli cells, the receptor activation will represent the start impulse of spermatogenesis [78]. In rats’ testis, ARs are expressed in the somatic Leydig cells, in peritubular myoid cells, and in Sertoli cells as well as in rete testis, the epithelial cells of the epididymis, and prostate [80]. AASs effects are strictly dependent on AR presence and distribution, suggesting an AASs influence on the male reproductive system.
AASs abuse induces testicular damage by triggering oxidative stress via inflammatory cytokines, matrix metalloproteinases, cell adhesion molecules, apoptotic markers, and DNA damage [81–83]. These mechanisms interfere with testis development, morphology, function, and sperm features. In this context, Noorafshan et al. [84] showed that weight and volume of testis decreased in animals that received high doses of AASs in comparison to the control animals. Other studies, performed on animal models confirmed that AASs administration in animal model induced testicular weight reduction [85]. Furthermore, it is well described that in animal model AASs assumption can cause morphological changes, such as reduction of number and size of Leydig cells, cytoplasmic vacuolization, on seminiferous tubule length and lipid droplet deposition [81, 83–84]. Moreover, several sperm alterations were observed in animals undergoing AASs administration. Treatment with relatively high doses of AASs leads to a decrease sperm count, and sperm motility [84, 86–89]. Both high and low doses of AASs significantly lowered the sperm motility compared to the control group. These findings suggest relevant sperm alterations due to AAS abuse. Finally, it is well known that anabolic agents may induce cell proliferation. Several studies reported the relationship between AASs assumption with infertility and carcinogenesis progression [90–91].
Barone et al. [92], demonstrated blood–testis barrier (BTB) alterations. Tight junctions (TJs), basal ectoplasmic specializations (ES) and desmosome–gap junctions (D-GJs) compose the BTB in order to distinguish the basal and adluminal compartments in the seminiferous tubules. This study showed an increase in gene coding of TJ-integral membrane protein adaptor, TJP1 and an abnormal distribution with immunofluorescence staining was revealed in cell cytoplasm of the basal compartment and in some cells at the adluminal compartment. TJP1 anomalous localization has been linked to epithelial carcinoma in situ [93–96]. Furthermore, an altered induction of MUC1 was suspected after nandrolone administration. This protein is a component of the mucosal glycocalyx associated with the testicular germ cell line and impaired spermatogenesis [97–98]. Usually intracytoplasmic, MUC1 expression was detected by Barone et al. [92] in the nuclei of many seminiferous tubule spermatids in different mice treated with different nandrolone doses. MUC1 nuclear translocation from the cytoplasm has been associated with transcription control and cell proliferation, mimicking an oncoprotein [91, 99–100].
Using rat Leydig tumor cell line, R2C cells, as an experimental model, Sirianni et al. [101], demonstrated, that high concentration of androgens promotes Leydig cell aromatase metabolism, determining the presence of local estrogen quantities. The resulting aromatase over-expression in this tumor cells leads to considerable higher-than-normal E2 levels, which can either initiate or cause progression of Leydig cell tumor.
This mechanism is mediated differently by different AAS classes, as shown by the comparison of nandrolone, an aromatizable androgen, with stanozolol, a non-aromatizable one. Stanozolol is not susceptible to aromatase metabolism, resulting in a high concentration of testosterone, which provokes aromatase over-expression, transcription and production. Nandrolone is easily and rapidly converted to estradiol, because of constitutively aromatase activation in Leydig R2C cells. This causes an immediate increase in estrogen quantities with less androgen concentration, promoting aromatase gene, CYP19, transcription. In an experimental animal model also conducted on R2C cells, Pomara et al. [102] reported that testosterone levels increase when lower nandrolone concentration are administrated to Leydig cells. The increment stopped after treatment with higher androgenic concentrations. Different dose-dependent effects are caused by the nandrolone-induced modification in genetic expression in testosterone synthesis molecules, in particular, steroidogenic acute regulatory protein (StAR) and CYP17A1. The exact mechanism by which nandrolone exerts its effect is currently not known yet, but it could possibly be through miRNA regulation, post-translational modification or protein degradation.
Nandrolone is an androgen receptor agonist. On binding to the AR receptor, it may induce the release of the AR receptor from Hsp90 and its translocation to the nucleus [103]. In our previous experiments, higher nandrolone concentrations induced a more pronounced increase in Hsp90 levels of expression and phosphorylation. This result is an indirect demonstration that nandrolone binds to AR-receptor and induces its activation. AR-receptor may act as a transcription factor binding to HREs or rapidly activating the MAP kinase pathway; and activates the CREB transcription factor via phosphorylation of ERK1/2 or through the direct binding of CREB in the cytoplasm [104]. The expression of STAR, HSD3B1, CYP11A1, CYP17A1 as well as of other enzymes of the testosterone biosynthesis pathway is activated by cAMP steroidogenesis. The latter is activated by the MAP kinase and CREB pathway, which is triggered by the non-conventional activation of the AR-receptor. In our experiments, nandrolone induced over-expression of STAR and HSD3B1, but downregulated CYP17A1 and CYP11A1. The down-regulation of CYP17A1 and CYP11A1 can be explained as an effect of nandrolone binding to the AR while the overexpression of STAR and HSD3B1 is consistent with the progesterone-like activity of nandrolone, overall leading to a decrease of testosterone synthesis [105].
Thus, Chimento et al. [106], suspected that pathophysiology of AAS-induced cancer induction and proliferation depends on two mechanisms. Firstly, the so-called estrogen-dependence that is related to estrogen production and not related to their androgenic effect. Secondly, the IGF-1R implication: this complex process was apparently demonstrated through the observation that ICI, an ER antagonist, reduces AAS-dependent cell proliferation. The same effect is generated by administration of IGF-1R pathway inhibitors (including IGF1R, PKC and PI3K inhibitors) which provoke a decrease of aromatase expression.
This consideration is not very innovative, considering that previous scientific literature extensively reported the estrogenic actions of androgens and the IGF-1R activation secondary to the binding of AAS to a membrane ER [107–108]. In human primary prostatic stromal cell cultures, DHT and T have already reported influencing IGF-1 protein expression. This was not the case E2 [109].
Whatever the underlying mechanism is (either a non-genomic AAS molecular process involving IGF-1 dependent signaling pathways or AAS-activated IGF-1R signaling through a membrane AR) the result is an activation of the IGF-1-mediated cascade pathway.
In turn, IGF-1 signaling activation of PI3K/ AKT and PLC/PKC pathways, results in an increase in aromatase expression and estrogen production inducing cell proliferation in Leydig cell cancer [101].
DISCUSSION
AASs are a known cause various functional disorders, which may affect liver, cardiovascular, reproductive, musculoskeletal, endocrine, renal, immunologic or neuropsychiatric systems. Their effect is related to dosage, frequency, and patterns of use [110–111]. Beyond these deleterious macro-effects, testosterone-derived androgens may act directly on cellular functions, with either genetic or epigenetic factors determining toxic, mutagen, genotoxic and carcinogenic results. AAS can also influence cancer cell proliferation via genomic and non-genomic mechanisms, such as the so-called estrogen-dependence mediated by ER, aromatase expression and IGF-1 production, which can even amplify each other. Therefore, IGF-1 can be considered as a well-known cancer inducer and promoter affecting each stage of tumor development, from cellular proliferation to the metastatic phase.
In the study conducted by Chimento et al. [106], aromatizable and non-aromatizable androgens promoted testicular tumor development in rat Leydig cells via the pathophysiology described above. This suggests that further and analogous consideration to human Leydig cell cancer should be considered. However, these are not the only mechanisms suspected to be involved in cancer development after AAS use. Barone et al. [92], analyzed alterations that occur at the blood–testis barrier (BTB). They reported modifications induced on Leydig cells different from those induced by IGF-1. This study showed an increase in gene coding of TJ-integral membrane protein adaptor, TJP1 and an anomalous localization linked to epithelial carcinoma in situ [93–96]. Furthermore, after nandrolone administration, MUC-1 nuclear translocation has been associated with transcription control and cell proliferation, mimicking an oncoprotein [91,100].
Other mechanisms hypothesized involved DNA-damage at different levels include: micronuclei formation [34, 42], DNA methylation [36], ROS-mediated DNA covalent binding [39], and alterations in telomerases [44–45].
CONCLUSIONS
The negative role of AAS in supraphysiological dosage impairs the expression of enzymes involved in testosterone biosynthesis. The side effects on the natural synthesis of testosterone play a potential role on the hormonal changes/regulation and could be suspected to be at the base of certain carcinogenic mechanisms. Furthermore, easily accessible and commonly diffused AAS, such as nandrolone and stanozolol, have the potential to induce and cause progression of particular kinds of cancer, such as Leydig cell tumor through multiple processes pathways. Their deleterious effect is further augmented by the fact, that such tumors have a high incidence in young people, the cohort of people abusing AAS. Further studies are necessary to investigate the potential link between AAS abuse and cancer.
CONFLICTS OF INTEREST
The authors declare that there are no conflict of interests, financial or otherwise, regarding the publication of this paper.
FUNDING
The authors received no specific funding for this work.
REFERENCES
1. Hartgens F, Kuipers H. Effects of androgenic-anabolic steroids in athletes. Sports Medicine. 2004; 34:513–554. https://doi.org/10.2165/00007256-200434080-00003.
2. Baker JS, Graham MR, Davies B. Steroid and prescription medicine abuse in the health and fitness community: A regional study. European Journal of Internal Medicine. 2006; 17:479–484. https://doi.org/10.1016/j.ejim.2006.04.010.
3. Melnik BC. Androgen abuse in the community. Current Opinion in Endocrinology, Diabetes and Obesity. 2009; 16:218–223. https://doi.org/10.1097/MED.0b013e32832afdfe.
4. Basaria S. Androgen abuse in athletes: Detection and consequences. Journal of Clinical Endocrinology and Metabolism. 2010; 95:1533–1543. https://doi.org/10.1210/jc.2009-1579.
5. Nikolopoulos DD, Spiliopoulou C, Theocharis SE. Doping and musculoskeletal system: Short-term and long-lasting effects of doping agents. Fundamental and Clinical Pharmacology. 2011; 25:535–563. https://doi.org/10.1111/j.1472-8206.2010.00881.x.
6. Kadi F. Cellular and molecular mechanisms responsible for the action of testosterone on human skeletal muscle. A basis for illegal performance enhancement. British Journal of Pharmacology. 2008; 154:522–528. https://doi.org/10.1038/bjp.2008.118.
7. Wakabayashi T, Onda H, Tada T, Iijima M, Itoh Y. High Incidence of Peliosis Hepatis in Autopsy Cases of Aplastic Anemia With Special Reference to Anabolic Steroid Therapy. Pathology International. 1984; 34:1079–1086. https://doi.org/10.1111/j.1440-1827.1984.tb07637.x.
8. Fineschi V, Neri M, Di Padua M, Fiore C, Riezzo I, Turillazzi E. Morphology, TNFα expression and apoptosis in the hearts of patients who died of abdominal compartment syndrome: An immunohistochemical study. International Journal of Cardiology. 2007; 116:236–241. https://doi.org/10.1016/j.ijcard.2006.02.020.
9. Albano GD, Sessa F, Messina A, Monda V, Bertozzi G, Maglietta F, Giugliano P, Vacchiano G, Gabriella M, Salerno M. AAS and organs damage: A focus on Nandrolone effects. Acta Medica Mediterranea. 2017; 6:939–946.
10. Bertozzi G, Sessa F, Albano GD, Sani G, Maglietta F, Roshan MHK, Volti GL, Bernardini R, Avola R, Pomara C, Salerno M. The Role of Anabolic Androgenic Steroids in Disruption of the Physiological Function in Discrete Areas of the Central Nervous System. Mol Neurobiol. 2017 Oct 2. [Epub ahead of print].
11. Shahidi NT. A review of the chemistry, biological action, and clinical applications of anabolic-androgenic steroids. Clinical Therapeutics. 2001; 23:1355–1390. https://doi.org/10.1016/S0149-2918(01)80114-4.
12. Petito A, Altamura M, Iuso S, Padalino FA, Sessa F, D’Andrea G, Margaglione M, Bellomo A. The relationship between personality traits, the 5HTT polymorphisms, and the occurrence of anxiety and depressive symptoms in elite athletes. PLoS One. 2016; 11:e0156601. https://doi.org/10.1371/journal.pone.0156601.
13. Al-Ismail K, Torreggiani WC, Munk PL, Nicolaou S. Gluteal mass in a bodybuilder: Radiological depiction of a complication of anabolic steroid use. European Radiology. 2002; 12:1366–1369. https://doi.org/10.1007/s00330-001-1169-1.
14. Turillazzi E, Perilli G, Di Paolo M, Neri M, Riezzo I, Fineschi V. Side effects of AAS abuse: An overview. Mini Rev Med Chem. 2011; 11:374–389. https://doi.org/10.2174/138955711795445925.
15. Maravelias C, Dona A, Stefanidou M, Spiliopoulou C. Adverse effects of anabolic steroids in athletes: A constant threat. Toxicology Letters. 2005; 158:167–175. https://doi.org/10.1016/j.toxlet.2005.06.005.
16. Mazzeo F, Motti ML, Messina G, Monda V, Ascione A, Tafuri D, Palmieri F, Messina A, Monda M. Use of nutritional supplements among south Italian students of physical training and sport university. Curr Top Toxicol. 2013; 9:21–6.
17. Deng Q, Zhang Z, Wu Y, Yu WY, Zhang J, Jiang ZM, Zhang Y, Liang H, Gui YT. Non-Genomic Action of Androgens is Mediated by Rapid Phosphorylation and Regulation of Androgen Receptor Trafficking. Cellular Physiology and Biochemistry. 2017; 43:223–236. https://doi.org/10.1159/000480343.
18. Drobnis EZ, Nangia AK. Exogenous androgens and male reproduction. Advances in Experimental Medicine and Biology. 2017; 1034:25–28. https://doi.org/10.1007/978-3-319-69535-8_4.
19. Sanchez AM, Flamini MI, Polak K, Palla G, Spina S, Mannella P, Genazzani AD, Simoncini T. Actin cytoskeleton remodelling by sex steroids in neurons. Journal of Neuroendocrinology. 2012; 24:195–201. https://doi.org/10.1111/j.1365-2826.2011.02258.x.
20. Hampl R, Kubátová J, Stárka L. Steroids and endocrine disruptors - History, recent state of art and open questions. J Steroid Biochem Mol Biol. 2016; 155:217–223. https://doi.org/10.1016/j.jsbmb.2014.04.013.
21. Kicman AT. Pharmacology of anabolic steroids. British Journal of Pharmacology. 2008; 154:502–521. https://doi.org/10.1038/bjp.2008.165.
22. Oberlander JG, Porter DM, Penatti CAA, Henderson LP. Anabolic androgenic steroid abuse: Multiple mechanisms of regulation of GABAergic synapses in neuroendocrine control regions of the rodent forebrain. Journal of Neuroendocrinology. 2012; 24:202–214. https://doi.org/10.1111/j.1365-2826.2011.02151.x.
23. Riezzo I, Di Paolo M, Neri M, Bello S, Cantatore S, D’Errico S, Dinucci D, Parente R, Pomara C, Rabozzi R, Turillazzi E, Fineschi V. Anabolic steroid - and exercise - induced cardio-depressant cytokines and myocardial β1 receptor expression in CD1 mice. Current Pharmaceutical Biotechnology. 2011; 12:275–284.
24. Gao W, Bohl CE, Dalton JT. Chemistry and Structural Biology of Androgen Receptor. Chemical Reviews. 2005; 105:3352–3370. https://doi.org/10.1021/cr020456u.
25. Magoffin DA. Ovarian theca cell. Int J Biochem Cell Biol. 2005; 37:1344–9.
26. Young JM, McNeilly AS. Theca: the forgotten cell of the ovarian follicle. Reproduction. 2010; 140:489–504. https://doi.org/10.1530/REP-10-0094.
27. Kostic TS, Stojkov NJ, Bjelic MM, Mihajlovic AI, Janjic MM, Andric SA. Pharmacological doses of testosterone upregulated androgen receptor and 3-beta-hydroxysteroid dehydrogenase/delta-5-delta-4 isomerase and impaired leydig cells steroidogenesis in adult rats. Toxicological Sciences. 2011; 121:397–407. https://doi.org/10.1093/toxsci/kfr063.
28. Janjic MM, Stojkov NJ, Andric SA, Kostic TS. Anabolic-androgenic steroids induce apoptosis and NOS2 (nitric-oxide synthase 2) in adult rat Leydig cells following in vivo exposure. Reproductive Toxicology. 2012; 34:686–693. https://doi.org/10.1016/j.reprotox.2012.10.003.
29. Stocco DM, Wang X, Jo Y, Manna PR. Multiple signaling pathways regulating steroidogenesis and steroidogenic acute regulatory protein expression: More complicated than we thought. Molecular Endocrinology. 2005; 19:2647–2659. https://doi.org/10.1210/me.2004-0532.
30. Miller WL, Auchus RJ. The Molecular Biology, Biochemistry, and Physiology of Human Steroidogenesis and Its Disorders. Endocrine Reviews. 2011; 32:81–151. https://doi.org/10.1210/er.2010-0013.
31. Handelsman DJ. Androgen Physiology, Pharmacology and Abuse. [Updated 2016 Dec 12]. In: De Groot LJ, Chrousos G, Dungan K, et al, editors. Endotext. South Dartmouth (MA): MDText.com, Inc.; 2000. https://www.ncbi.nlm.nih.gov/books/NBK279000/.
32. Hodgson MC, Astapova I, Cheng S, Lee LJ, Verhoeven MC, Choi E, Balk SP, Hollenberg AN. The Androgen Receptor Recruits Nuclear Receptor CoRepressor (N-CoR) in the Presence of Mifepristone via its N and C Termini Revealing a Novel Molecular Mechanism for Androgen Receptor Antagonists. J Biol Chem. 2005; 280:6511–6519. https://doi.org/10.1074/jbc.M408972200.
33. Svechnikov K, Izzo G, Landreh L, Weisser J, Sder O. Endocrine disruptors and leydig cell function. Journal of Biomedicine and Biotechnology, 2010; 2010: 684504.
34. Torres-Bugarín O, Covarrubias-Bugarín R, Zamora-Perez AL, Torres-Mendoza BMG, García-Ulloa M, Martínez-Sandoval FG. Anabolic androgenic steroids induce micronuclei in buccal mucosa cells of bodybuilders. British Journal of Sports Medicine. 2007; 41:592–596. https://doi.org/10.1136/bjsm.2006.032474.
35. Hashimoto H, Vertino PM, Cheng X. Molecular coupling of DNA methylation and histone methylation. Epigenomics. 2010; 2:657–69. https://doi.org/10.2217/epi.10.44.
36. Bissegger S, Langlois VS. Androgens modulate gene expression and specific DNA methylation pattern of steroid 5α-reductases in the frog Silurana tropicalis. Gen Comp Endocrinol. 2016; 234:123–32. https://doi.org/10.1016/j.ygcen.2016.03.021.
37. Schwarzenbach H. Impact of physical activity and doping on epigenetic gene regulation. Drug Test Anal. 2011; 3:682–7. https://doi.org/10.1002/dta.294.
38. Liehr JG. Is Estradiol a Genotoxic Mutagenic Carcinogen? Endocr Rev. 2000; 21:40–54. https://doi.org/10.1210/edrv.21.1.0386.
39. Souza LD, da Cruz LA, Cerqueira EM, Meireles J. Micronucleus as biomarkers of cancer risk in anabolic androgenic steroids users. Hum Exp Toxicol. 2017; 36:302–310. https://doi.org/10.1177/0960327116650005.
40. Socas L, Zumbado M, Pérez-Luzardo O, Ramos A, Pérez C, Hernández JR, Boada LD. Hepatocellular adenomas associated with anabolic androgenic steroid abuse in bodybuilders: a report of two cases and a review of the literature. Br J Sports Med. 2005; 39:e27. https://doi.org/10.1136/bjsm.2004.013599.
41. Giannitrapani L, Soresi M, La Spada E, Cervello M, D’Alessandro N, Montalto G. Sex hormones and risk of liver tumor. Ann N Y Acad Sci. 2006; 1089:228–36. https://doi.org/10.1196/annals.1386.044.
42. Meireles JR, Oliveira SV, Costa-Neto AO, Cerqueira EM. Genotoxic and cytotoxic effects of testosterone cypionate (deposteron(®)). Mutat Res. 2013; 753:72–5. https://doi.org/10.1016/j.mrgentox.2013.02.003.
43. do Carmo CA, Gonçalves ÁLM, Salvadori DMF, Maistro EL. Nandrolone androgenic hormone presents genotoxic effects in different cells of mice. J Appl Toxicol. 2012; 32:810–814. https://doi.org/10.1002/jat.1701.
44. Kara M, Ozcagli E, Fragkiadaki P, Kotil T, Stivaktakis PD, Spandidos DA, Alpertunga B. Determination of DNA damage and telomerase activity in stanozolol-treated rats. Experimental and Therapeutic Medicine. 2017; 13:614–618. https://doi.org/10.3892/etm.2016.3974.
45. Nourbakhsh M, Golestani A, Zahrai M, Modarressi MH, Malekpour Z, Karami-Tehrani F. Androgens stimulate telomerase expression, activity and phosphorylation in ovarian adenocarcinoma cells. Mol Cell Endocrinol. 2010; 330:10–16. https://doi.org/10.1016/j.mce.2010.07.021.
46. Calado RT, Yewdell WT, Wilkerson KL, Regal JA, Kajigaya S, Stratakis CA, Young NS. Sex hormones, acting on the TERT gene, increase telomerase activity in human primary hematopoietic cells. Blood. 2009; 114:2236–2243. https://doi.org/10.1182/blood-2008-09-178871.
47. Pope HG, Katz DL. Psychiatric and Medical Effects of Anabolic-Androgenic Steroid Use A Controlled Study of 160 Athletes. Arch Gen Psychiatry. 1994; 51:375–382. https://doi.org/10.1001/archpsyc.1994.03950050035004.
48. Baron DA, Martin DM, Abol Magd S. Doping in sports and its spread to at-risk populations: an international review. World Psychiatry. 2007; 6:118–123.
49. Jozkow P, Medras M. Growth hormone and IGF-1 as doping agents in competitive sport. Endokrynol Pol. 2009; 60:389.
50. Serra C, Bhasin S, Tangherlini F, Barton ER, Ganno M, Zhang A, Shansky J, Vandenburgh HH, Travison TG, Jasuja R, Morris C. The role of GH and IGF-I in mediating anabolic effects of testosterone on androgen-responsive muscle. Endocrinology. 2011; 152:193–206. https://doi.org/10.1210/en.2010-0802.
51. Coleman ME, DeMayo F, Yin KC, Lee HM, Geske R, Montgomery C, Schwartz RJ. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J. Biol. Chem. 1995; 270:12109–16.
52. Wu Y, Bauman WA, Blitzer RD, Cardozo C. Testosterone-induced hypertrophy of L6 myoblasts is dependent upon Erk and mTOR. Biochem Biophys Res Commun. 2010; 400:679–83. https://doi.org/10.1016/j.bbrc.2010.08.127.
53. Jenkins PJ, Mukherjee A, Shalet SM. Does growth hormone cause cancer? Clin. Endocrinol. 2006; 64:115–21.
54. Cappello F, De Macario EC, Gammazza AM, Bonaventura G, Carini F, Czarnecka AM, Farina F, Zummo G, Macario AJL. Hsp60 and human aging: Les liaisons dangereuses. Frontiers in Bioscience. 2013; 18:626–637. https://doi.org/10.2741/4126.
55. Sarangi U, Singh MK, Abhijnya KVV, Prasanna Anjaneya Reddy L, Siva Prasad A, Pitke VV, Paithankar K, Subbarao Sreedhar A. Hsp60 chaperonin acts as barrier to pharmacologically induced oxidative stress mediated apoptosis in tumor cells with differential stress response. Drug Target Insights. 2013; 7:35–51. https://doi.org/10.4137/DTI.S12513.
56. Cappello F, Gammazza AM, Piccionello AP, Campanella C, Pace A, De Macario EC, Macario AJL. Hsp60 chaperonopathies and chaperonotherapy: Targets and agents. Expert Opinion on Therapeutic Targets. 2014; 18:185–208. https://doi.org/10.1517/14728222.2014.856417.
57. Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010; 10:537–549. https://doi.org/10.1038/nrc2887.
58. Shafi AA, Cox MB, Weigel NL. Androgen receptor splice variants are resistant to inhibitors of Hsp90 and FKBP52, which alter androgen receptor activity and expression. Steroids. 2013; 78:548–54. https://doi.org/10.1016/j.steroids.2012.12.013.
59. Vanaja DK, Mitchell SH, Toft DO, Young CYF. Effect of geldanamycin on androgen receptor function and stability. Cell Stress and Chaperones. 2002; 7:55–64. https://doi.org/10.1379/1466-1268(2002)007<0055:EOGOAR>2.0.CO;2.
60. Salomone F, Li Volti G, Vitaglione P, Morisco F, Fogliano V, Zappalà A, Palmigiano A, Garozzo D, Caporaso N, D’Argenio G, Galvano F. Coffee enhances the expression of chaperones and antioxidant proteins in rats with nonalcoholic fatty liver disease. Translational Research. 2014; 163:593–602. https://doi.org/10.1016/j.trsl.2013.12.001.
61. Sirianni R, Capparelli C, Chimento A, Panza S, Catalano S, Lanzino M, Pezzi V, Andò S. Nandrolone and stanozolol upregulate aromatase expression and further increase IGF-I-dependent effects on MCF-7 breast cancer cell proliferation. Mol Cell Endocrinol. 2012; 363:100–10. https://doi.org/10.1016/j.mce.2012.08.002.
62. Rochefort H, Garcia M. Androgen on the estrogen receptor I – Binding and in vivo nuclear translocation. Steroids. 1976; 28:549–560.
63. Turillazzi E, Vacchiano G, Luna-Maldonado A, Neri M, Pomara C, Rabozzi R, Riezzo I, Fineschi V. Tryptase, CD15 and IL-15 as reliable markers for the determination of soft and hard ligature marks vitality. Histology and Histopathology. 2010; 25:1539–1546.
64. Vyshka G, Vacchiano G. Severe flaccid paraparesis following spinal anaesthesia: A sine materia occurrence. BMJ Case Reports. 2014. https://doi.org/10.1136/bcr-2013-202071.
65. Arnold A, Papanikolaou A. Cyclin D1 in breast cancer pathogenesis. J Clin Oncol. 2005; 23:4215–4224.
66. Precenzano F, Ruberto M, Parisi L, Salerno M, Maltese A, D’Alessandro I, Grappa MF, Magliulo RM, Messina G, Roccella M. Borderline intellectual functioning and parental stress: An italian case-control study. Acta Medica Mediterranea. 2016; 32:1762–1765. https://doi.org/10.19193/0393-6384_2016_6_160.
67. Vacchiano G, Vyshka G. Balkans syndrome: A potential link between multiple sclerosis and hypervaccination. Journal of the Royal Army Medical Corps. 2012; 158:338–340. https://doi.org/10.1136/jramc-158-04-15.
68. La Vecchia C, Bosetti C, Lucchini F, Bertuccio P, Negri E, Boyle P, Levi F. Cancer mortality in Europe, 2000-2004, and an overview of trends since 1975. Annals of Oncology. 2009; 21:1323–1360. https://doi.org/10.1093/annonc/mdp530.
69. Curado MP, Edwards B, Shin HR, Storm H, Ferlay J, Heanue M, Boyle P. Cancer incidence in five continents, 2007; Vol IX. IARC Scientific Publication No. 160.
70. Nigam M, Aschebrook-Kilfoy B, Shikanov S, Eggener S. Increasing incidence of testicular cancer in the United States and Europe between 1992 and 2009. World Journal of Urology. 2015; 33:623–631. https://doi.org/10.1007/s00345-014-1361-y.
71. Znaor A, Lortet-Tieulent J, Jemal A, Bray F. International variations and trends in testicular cancer incidence and mortality. European Urology. 2014; 65:1095–1106. https://doi.org/10.1016/j.eururo.2013.11.004.
72. Shanmugalingam T, Soultati A, Chowdhury S, Rudman S, Van Hemelrijck M. Global incidence and outcome of testicular cancer. Clinical Epidemiology. 2013; 5:417–427. https://doi.org/10.2147/CLEP.S34430.
73. Albers P, Albrecht W, Algaba F, Bokemeyer C, Cohn-Cedermark G, Fizazi K, Horwich A, Laguna MP, Nicolai N, Oldenburg J. Guidelines on Testicular Cancer: 2015 Update. European Urology. 2015; 68:1054–1068. https://doi.org/10.1016/j.eururo.2015.07.044.
74. Kim I, Young RH, Scully RE. Leydig cell tumors of the testis. A clinicopathological analysis of 40 cases and review of the literature. Am J Surg Pathol. 1985; 9:177–192.
75. Forte F, Scarfini M, Spera E, De Carolis A, Sansalone S, Virgili G, Vespasiani G. Leiomyoma of the epididymis and testicular adnexa: Apropos of 3 cases. Arch Ital Urol Androl. 2004; 76:119–120.
76. García M, Dargallo T, Palacios M, Carames J, Gómez J, Sacristan F, Vela D. Leydig cell tumour: Enucleation as a therapeutic choice in a case with atypical symptoms [Tumor de celulas de leydig: Enucleación como tratamiento en un caso de presentación atípica]. Archivos Espanoles de Urologia. 2012; 65:897–899.
77. Carvajal-Carmona LG, Alam NA, Pollard PJ, Jones AM, Barclay E, Wortham N, Pignatelli M, Freeman A, Pomplun S, Ellis I, Poulsom R, El-Bahrawy MA, Berney DM, Tomlinson IPM. Adult leydig cell tumors of the testis caused by germline fumarate hydratase mutations. Journal of Clinical Endocrinology and Metabolism. 2006; 91:3071–3075. https://doi.org/10.1210/jc.2006-0183.
78. Dohle GR, Smit M, Weber RFA. Androgens and male fertility. World Journal of Urology. 2003; 21:341–345. https://doi.org/10.1007/s00345-003-0365-9.
79. Bremner WJ, Millar MR, Sharpe RM, Saunders PTK. Immunohistochemical localization of androgen receptors in the rat testis: Evidence for stage-dependent expression and regulation by androgens. Endocrinology. 1994; 135:1227–1234. https://doi.org/10.1210/endo.135.3.8070367.
80. Vornberger W, Prins G, Musto NA, Suarez-Quian CA. Androgen receptor distribution in rat testis: New implications for androgen regulation of spermatogenesis. Endocrinology. 1994; 134:2307–2316. https://doi.org/10.1210/endo.134.5.8156934.
81. Nagata S, Kurosawa M, Mima K, Nambo Y, Fujii Y, Watanabe G, Taya K. Effects of anabolic steroid (19-nortestosterone) on the secretion of testicular hormones in the stallion Journal of Reproduction and Fertility. 1999; 115:373–379.
82. Mae A. Amelioration of nandrolone decanoate-induced testicular and sperm toxicity in rats by taurine: Effects on steroidogenesis, redox and inflammatory cascades, and intrinsic apoptotic pathway. Toxicology and Applied Pharmacology. 2015; 282:285–296. https://doi.org/10.1016/j.taap.2014.12.007.
83. Tahtamouni LH, Mustafa NH, Hassan IM, Ahmad IM, Yasin SR, Abdalla MY. Nandrolone decanoate administration to male rats induces oxidative stress, seminiferous tubules abnormalities, and sperm DNA fragmentation. JJBS. 2010; 3:165–174.
84. Noorafshan A, Karbalay-Doust S, Ardekani FM. High doses of nandrolone decanoate reduce volume of testis and length of seminiferous tubules in rats. APMIS. 2005; 113:122–125. https://doi.org/10.1111/j.1600-0463.2005.apm1130205.x.
85. Mesbah SF, Shokri S, Karbalay-Doust S, Mirkhani H. The effect of nandrolone decanoate on the body, testis and epididymis weight and semen parameters in adult male rats. Iranian Journal of Medical Sciences. 2007; 32:93–99.
86. Takahashi M, Tatsugi Y, Kohno T. Endocrinological and pathological effects of anabolic-androgenic steroid in male rats. Endocrine Journal. 2004; 51:425–434. https://doi.org/10.1507/endocrj.51.425.
87. Karbalay-Doust S, Noorafshan A, Ardekani FM, Mirkhani H, Baker G. The reversibility of sperm quality after discontinuing nandrolone decanoate in adult male rats. Asian Journal of Andrology. 2007; 9:235–239. https://doi.org/10.1111/j.1745-7262.2007.00203.x.
88. Shokri S, Hemadi M, Bayat G, Bahmanzadeh M, Jafari-Anarkooli I, Mashkani B. Combination of running exercise and high dose of anabolic androgenic steroid, nandrolone decanoate, increases protamine deficiency and DNA damage in rat spermatozoa. Andrologia. 2014; 46:184–190.
89. Mohamed HM, Mohamed MAH. Effect of different doses of nandrolone decanoate on lipid peroxidation, DNA fragmentation, sperm abnormality and histopathology of testes of male Wister rats. Experimental and Toxicologic Pathology. 2015; 67:1–11.
90. Kumar P, Lindberg L, Thirkill TL, Ji JW, Martsching L, Douglas GC. The MUC1 extracellular domain subunit is found in nuclear speckles and associates with spliceosomes. PLoS One. 2012; 7:e42712.
91. Leng Y, Cao C, Ren J, Huang L, Chen D, Ito M, Kufe D. Nuclear import of the MUC1-C oncoprotein is mediated by nucleoporin Nup62. Journal of Biological Chemistry. 2007; 282:19321–19330. https://doi.org/10.1074/jbc.M703222200.
92. Barone R, Pitruzzella A, Marino Gammazza A, Rappa F, Salerno M, Barone F, Sangiorgi C, D’Amico D, Locorotondo N, Di Gaudio F, Cipolloni L, Di Felice V, Schiavone S, et al. Nandrolone decanoate interferes with testosterone biosynthesis altering blood-testis barrier components. J Cell Mol Med. 2017; 21:1636–1647. https://doi.org/10.1111/jcmm.13092.
93. Giallongo C, Tibullo D, La Cava P, Branca A, Parrinello N, Spina P, Stagno F, Conticello C, Chiarenza A, Vigneri P, Palumbo GA, Di Raimondo F. BRIT1/MCPH1 expression in chronic myeloid leukemia and its regulation of the G2/M checkpoint. Acta Haematologica. 2011; 126:205–210. https://doi.org/10.1159/000329911.
94. Fink C, Weigel R, Fink L, Wilhelm J, Kliesch S, Zeiler M, Bergmann M, Brehm R. Claudin-11 is over-expressed and dislocated from the blood-testis barrier in Sertoli cells associated with testicular intraepithelial neoplasia in men. Histochemistry and Cell Biology. 2009; 131:755–764. https://doi.org/10.1007/s00418-009-0576-2.
95. Bergmann M. Testicular biopsy, histology and TESE [Hodenbiopsie, histologie und TESE] Journal fur Urologie und Urogynakologie. 2007; 14:5–12.
96. Fink C, Weigel R, Hembes T, Lauke-Wettwer H, Kliesch S, Bergmann M, Brehm RH. Altered expression of ZO-1 and ZO-2 in sertoli cells and loss of blood-testis barrier integrity in testicular carcinoma in situ. Neoplasia. 2006; 8:1019–1027. https://doi.org/10.1593/neo.06559.
97. Seo JT, Lee JS, Jun JH, Yang MH. Expression of mucin genes in the human testis and its relationship to spermatogenesis. Yonsei Medical Journal. 2005; 46:667–672.
98. Franke FE, Kraus S, Eiermann C, Pauls K, Lalani EN, Bergmann M. MUC1 in normal and impaired spermatogenesis. Molecular Human Reproduction. 2001; 7:505–512.
99. Frati P, Busardò FP, Cipolloni L, Dominicis ED, Fineschi V. Anabolic Androgenic Steroid (AAS) Related Deaths: Autoptic, Histopathological and Toxicological Findings. Curr Neuropharmacol. 2015; 13:146–159. https://doi.org/10.2174/1570159X13666141210225414.
100. Bramanti V, Grasso S, Tibullo D, Giallongo C, Pappa R, Brundo MV, Tomassoni D, Viola M, Amenta F, Avola R. Neuroactive molecules and growth factors modulate cytoskeletal protein expression during astroglial cell proliferation and differentiation in culture. Journal of Neuroscience Research. 2016; 94:90–98. https://doi.org/10.1002/jnr.23678.
101. Sirianni R, Chimento A, Malivindi R, Mazzitelli I, Andò S, Pezzi V. Insulin-like growth factor-I, regulating aromatase expression through steroidogenic factor 1, supports estrogen-dependent tumor Leydig cell proliferation. Cancer Res. 2007; 67:8368–8377. https://doi.org/10.1158/0008-5472.CAN-06-4064.
102. Pomara C, Barone R, Marino Gammazza A, Sangiorgi C, Barone F, Pitruzzella A, Locorotondo N, Di Gaudio F, Salerno M, Maglietta F, Sarni AL, Di Felice V, Cappello F, et al. Effects of Nandrolone Stimulation on Testosterone Biosynthesis in Leydig Cells. Journal of Cellular Physiology. 2016; 231:1385–1391. https://doi.org/10.1002/jcp.25272.
103. Fang Y, Fliss AE, Robins DM, Caplan AJ. Hsp90 regulates androgen receptor hormone binding affinity in vivo. Journal of Biological Chemistry. 1996; 271:28697–28702. https://doi.org/10.1074/jbc.271.45.28697.
104. Fix C, Jordan C, Cano P, Walker WH. Testosterone activates mitogen-activated protein kinase and the cAMP response element binding protein transcription factor in Sertoli cells. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101:10919–10924. https://doi.org/10.1073/pnas.0404278101.
105. De Souza GL, Hallak J. Anabolic steroids and male infertility: A comprehensive review. BJU International. 2011; 108:1860–1865. https://doi.org/10.1111/j.1464-410X.2011.10131.x.
106. Chimento A, Sirianni R, Zolea F, De Luca A, Lanzino M, Catalano S, Andò S, Pezzi V. Nandrolone and stanozolol induce leydig cell tumor proliferation through an estrogen-dependent mechanism involving IGF-I system. Journal of Cellular Physiology. 2012; 227:2079–2088. https://doi.org/10.1002/jcp.22936.
107. Rochefort H, Garcia M. The estrogenic and antiestrogenic activities of androgens in female target tissues. Pharmacol Ther. 1983; 23:193–216.
108. Song RXD, Fan P, Yue W, Chen Y, Santen RJ. Role of receptor complexes in the extranuclear actions of estrogen receptor alpha in breast cancer. Endocr Relat Cancer. 2006; 13:S3–13.
109. Le H, Arnold JT, McFann KK, Blackman MR. DHT and testosterone, but not DHEA or E2, differentially modulate IGF-I, IGFBP-2, and IGFBP-3 in human prostatic stromal cells. Am J Physiol Endocrinol Metab. 2006; 290:E952-E960 https://doi.org/10.1152/ajpendo.00451.2005.
110. Pirompol P, Teekabut V, Weerachatyanukul W, Bupha-Intr T, Wattanapermpool J. Supra-physiological dose of testosterone induces pathological cardiac hypertrophy. Journal of Endocrinology. 2016; 229:13–23. https://doi.org/10.1530/JOE-15-0506.
111. Nieschlag E, Vorona E. Doping with anabolic androgenic steroids (AAS): Adverse effects on non-reproductive organs and functions. Reviews in Endocrine and Metabolic Disorders. 2015; 16:199–211. https://doi.org/10.1007/s11154-015-9320-5.