
INTRODUCTION
Chronic myeloid leukemia (CML) is a hematopoietic disorder in which myeloid cells proliferate excessively given to the abnormal activation of the chimaeric protein BCR-ABL1. The reciprocal translocation t(9;22)(q34;q11) gives rise to the fusion gene BCR-ABL1, which encodes a constitutively activated protein with tyrosine kinase activity [1]. Patients are treated with specific tyrosine-kinase inhibitors (TKI), such as Imatinib, Nilotinib and Dasatinib, and the disease is monitored by measuring the level of BCR-ABL1 transcripts in peripheral blood (PB) [2]. Despite most patients achieve a good response to TKI treatment [3], very low levels of disease can persist even after many years of successful treatment, making the decision of stopping TKI mostly restricted to clinical trials in which patients are closely monitored for rising levels of BCR-ABL1 transcripts in PB.
Although treatment duration, deep molecular response maintenance, and Sokal score have been reported as prognostic factors of molecular relapse after TKI withdrawal [4], an explanation for the heterogeneous response after treatment discontinuation is still lacking. Molecular relapse has been attributed to the persistence of leukemic stem cells (LSCs), which are independent of BCR-ABL1 kinase activity for survival in short term in vitro assays [5–7]. On the other hand, a recent report suggests that their elimination in vivo would not be a requisite for successful TKI discontinuation [8]. However, whether the functionality of residual LSCs can vary among patients in deep molecular response remains unknown. Their potential relevance is evidenced by the great efforts that are being taken in order to develop LSCs-targeting drugs [9–11]. Therefore, the quantification and characterization of LSCs in patients under prolonged TKI treatment is of great interest in the context of assessing residual disease. This task is specially challenging in such patients because of the low frequency of LSCs in the hematopoietic system, and their low level of BCR-ABL1 expression [12]. Therefore, new methods that allow greater sensitivity for LSCs detection are needed.
Breakpoints that lead to BCR-ABL1 chimaeric gene are dispersed over a ~3kbp region in BCR (intron 13 or intron 14), and a ~140kbp region in ABL1 (intron 1). Therefore, each patient harbors a specific genomic breakpoint, which requires personalized characterization in order to design patient-specific primers for PCR detection at the DNA level. Previous reports have shown that BCR-ABL1 can be detected at the DNA level in patients under sustained deep molecular response, even in those that attained a treatment-free remission (TFR) after TKI discontinuation [13]. However, it is unknown whether the persistent BCR-ABL1-positive cells constitute a primitive fraction, i.e. LSCs, with the potential capacity of causing disease, or whether they represent a population of cells unable to resume the leukemia.
In order to study the dynamics of primitive and progenitor fractions, we report the evaluation of such populations at the RNA level by functional in vitro assays, in CML patients at diagnosis and during TKI treatment. In addition, we show that it is possible to detect residual primitive cells by in vitro functional assays at the DNA level, which enables independence from BCR-ABL1 expression levels. This method is of potential interest for assessing residual disease in future TKI discontinuation trials.
RESULTS
Detection based on quantification of BCR-ABL1 mRNA in primitive and progenitor fractions
Long-term Culture Initiating Cell (LTC-IC) and short-term Colony Forming Units (CFU) assays are functional in vitro assays that enable to retrospectively quantify cells in the primitive and lineage-restricted progenitor fraction, respectively. Aiming to evaluate whether the dynamics of these populations can vary among patients at diagnosis and under TKI treatment, we performed both assays with PB or bone marrow (BM) samples from CML patients (Table 1). In order to distinguish leukemic from normal primitive or progenitor cells, we assessed the level of BCR-ABL1 transcripts by RT-qPCR in individual or pooled colonies plucked from methylcellulose (Figure 1). Whereas initially, detection was performed on individual colonies, we observed that the evaluation of pooled colonies increased the sensitivity of the assay, allowing to scan a higher number of colonies per patient, and increasing the yield of RNA for subsequent molecular analysis.
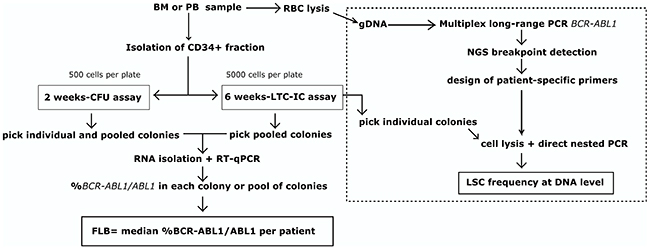
Figure 1: Workflow. Detection of primitive and progenitor fractions at RNA level by in vitro functional assays was performed on patient samples at diagnosis and during TKI treatment. Evaluation of the primitive fraction at DNA level was performed as proof of concept on one patient (dotted line). BM: bone marrow, PB: peripheral blood, RBC: red blood cell lysis, gDNA: genomic DNA, FLB: functional leukemic burden, NGS: next-generation sequencing, LSC: leukemic stem cell
Table 1: Clinical features of patients included in the study
At diagnosis |
MR1.0-MR2.0 |
MR3.0-MR5.0 |
|
|---|---|---|---|
(N=10) |
(N=13) |
(N=12) |
|
Age -yr |
|||
Median |
57 |
61 |
66 |
Range |
18-79 |
36-74 |
28-74 |
Female/Male sex, % |
30/70 |
15/85 |
50/50 |
Sokal score, no. (%) |
|||
Low |
4 (44.4) * |
5 (38.5) |
5 (45.5) ** |
Intermediate |
3 (33.4) * |
4 (30.8) |
5 (45.5) ** |
High |
2 (22.2) * |
4 (30.8) |
1 (9) ** |
TKI, no. (%) |
|||
Imatinib |
n/a |
13 (100) |
8 (66.7) |
Nilotinib 1st line |
n/a |
0 (0) |
3 (25) |
Nilotinib 2nd line |
n/a |
0 (0) |
1 (8.3) |
Time of TKI treatment, months |
|||
Median |
n/a |
6 |
20.4 |
Range |
n/a |
3-9 |
6-85 |
At diagnosis, before TKI treatment; MR- Molecular response in PB; n/a- not applicable, *1 missing value, % calculated over 9 cases; ** 1 missing value, % calculated over 11 cases
Given that the assessment of pooled colonies did not allow us to distinguish between the concurrently contribution of expression (i.e. BCR-ABL1 mRNA levels) and frequency (i.e. number of leukemic cells), we considered both factors as a single one, defined as primitive or progenitor “functional leukemic burden” (FLB). For each patient, the value of FLB was calculated as the median of %BCR-ABL1/ABL1 measured in each colony or pool of colonies (Figure 1). This definition enabled us to distinguish such effects from the previously reported “leukemic stem cell burden”, defined as the frequency of Philadelphia chromosome-positive cells (assessed by FISH) in phenotypically-defined primitive fractions sorted by flow cytometry [14, 15].
Results from patients at diagnosis before any TKI treatment showed variable levels of FLB both in the primitive and progenitor fractions, being higher in the latter (mean±SD: 6.3%±13.2% vs. 52.4%±21.5% respectively, Mann–Whitney U test, p<0.05, Figure 2A). As soon as patients underwent TKI treatment, the FLB was reduced both in the primitive and progenitor fractions (Figure 2B-2D). If we consider the proportion of BCR-ABL1+ single or pooled colonies according to the molecular response in PB, the primitive fraction showed a slower reduction than progenitor cells during treatment (Figure 3).
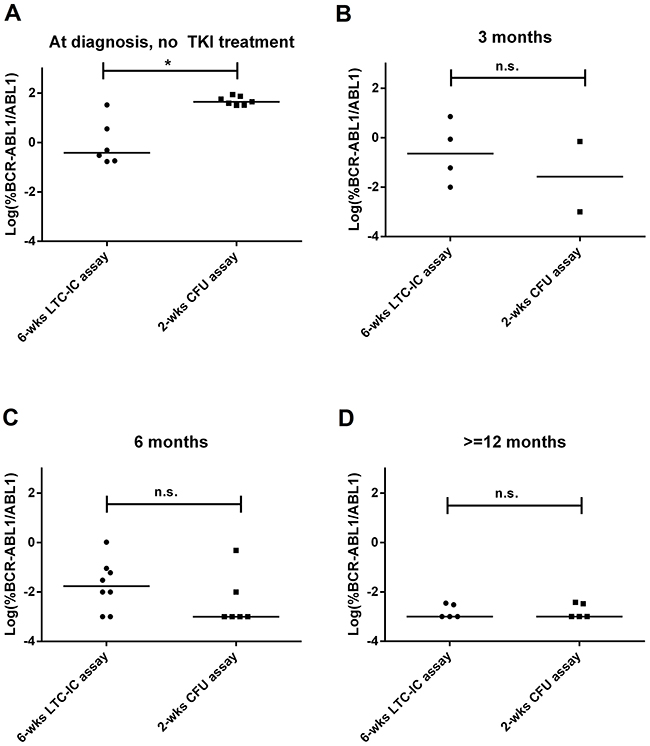
Figure 2: FLB in primitive and progenitor fractions at diagnosis and during TKI treatment. Each dot is the median value of individual or pooled colonies from each patient: (A) before TKI treatment, (B) at 3 months, (C) at 6 months, and (D) at 12 months of TKI treatment. In order to improve visualization, data was log-transformed (zero values were assigned a value of 0.001%, according to the theoretical limit of BCR-ABL1/ABL1 detection by RT-qPCR). Statistical analysis was performed before log-transformation. * = p<0.05, Mann–Whitney U test.
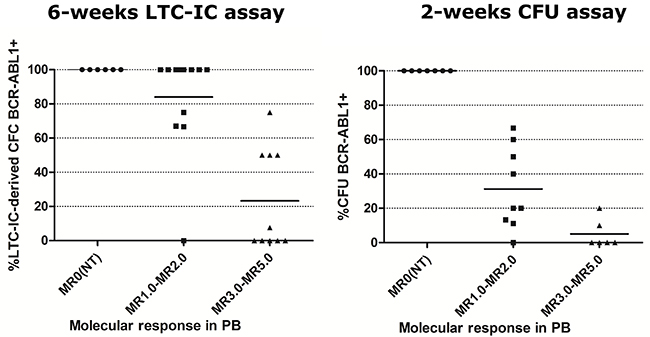
Figure 3: Percentage of BCR-ABL1+ single or pooled colonies in patients at diagnosis and under TKI treatment, according to their molecular response in PB. Individual or pooled colonies were evaluated by RT-qPCR from each patient. Horizontal lines show the mean value for each group. NT= at diagnosis, before TKI treatment.
Early reduction of BCR-ABL1 transcript burden in PB, such as BCR-ABL1/ABL1IS ≤ 10% at 3 months, or <1% at 6 months, is associated with optimal response to first-line TKI treatment [16]. Although most of these patients can sustain an optimal response over time (No-Resistant, NR), a minority of them can develop secondary resistance (SR) to treatment. Patients that never responded to TKI treatment were considered as primary resistant cases (PR, see Materials and Methods for further details). Given that additional prognostic markers could be of clinical relevance in order to better predict resistance to TKI, we compared the primitive and progenitor FLB from both groups at different time points upon TKI treatment.
Despite the fact that at diagnosis we did not observe significant differences neither in the progenitor nor in the primitive fraction between patients in the NR and PR/SR groups (Figure 4), at 6 months of TKI treatment patients in the PR/SR group showed significantly higher levels of primitive FLB than those in the NR group (p<0.05, Mann–Whitney U test, Figure 5).
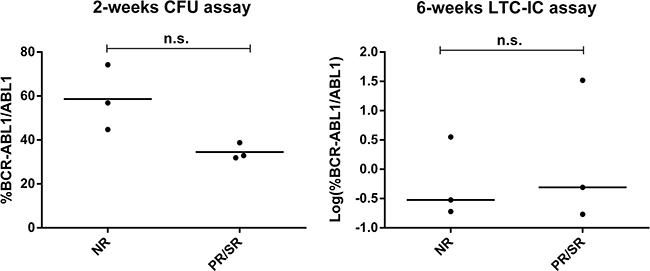
Figure 4: Evaluation of FLB at diagnosis. %BCR-ABL1/ABL1 was measured by RT-qPCR in individual and pooled colonies from patients at diagnosis, before any TKI treatment, in the progenitor (left) and primitive (right) fractions. Each dot is the median value of individual or pooled colonies from each patient. In order to improve visualization, data from 6-wks-LTC-IC assay was Log-transformed. Statistical analysis was performed before log-transformation, n.s: p>0.05, Mann–Whitney U test.
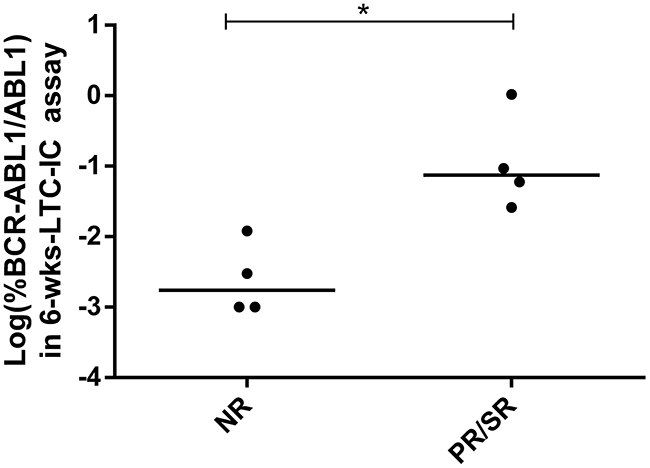
Figure 5: Primitive FLB in patients at 6 months of TKI treatment. %BCR-ABL1/ABL1 was measured by RT-qPCR in colonies from 6-wks-LTC-IC assays. Each dot is the median value of pooled colonies from each patient. In order to improve visualization, data was log-transformed (zero values were assigned a value of 0.001%, according to the theoretical limit of BCR-ABL1/ABL1 detection by RT-qPCR). Statistical analysis was performed before log-transformation. N=4 per group, * = p<0.05, Mann–Whitney U test.
Detection based on amplification of BCR-ABL1 DNA in the primitive fraction
Regarding the possibility of assessing residual disease in patients showing deep molecular response, we were not able to detect BCR-ABL1 transcripts in most patients with the LTC-IC assay (Table 2). Given that detection is limited by the level of expression of BCR-ABL1 mRNA by individual colonies, we designed patient-specific primers in order to evaluate BCR-ABL1 rearrangement at the DNA level. A sample of PB at diagnosis was used for genomic DNA (gDNA) extraction and sequencing of the rearrangement breakpoint by long-range multiplex-PCR followed by next-generation sequencing (NGS) (Supplementary Figure 1). The patient showed declining levels of BCR-ABL1 transcripts in PB under Imatinib treatment (400mg daily), attaining a MR3.0 at 18 months. A PB sample at 18 months of TKI treatment was used for CD34+ cells isolation, followed by a 6-weeks-LTC-IC assay (Figure 1). We assessed the presence of BCR-ABL1-positive individual colonies by an in-house method of direct nested-PCR, without a gDNA extraction step (see Materials and methods). Quality of the starting material was evaluated by detecting an independent gene at the DNA level, and colonies from a 2-wks-CFU assay at 6 months of treatment were used as positive controls. Nested PCR showed improved sensitivity compared to single PCR (data not shown), and it was able to detect a low proportion of BCR-ABL1+ individual colonies in the primitive fraction (5 out of 20) (Figure 6).
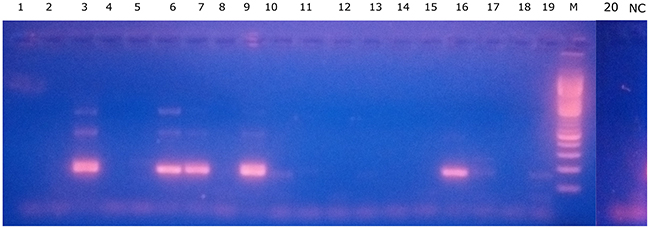
Figure 6: Patient-specific BCR-ABL1 gDNA detection in the primitive fraction. 2.5% agarose gel electrophoresis result from nested PCR using patient-specific primers. Estimated size of 2nd-round specific PCR product: 166bp. Lanes 1-20: individual CFU-Cs, NC= negative control (surrounding methylcellulose with no CFU-Cs), M: 100-bp molecular weight marker. Lanes 3, 6, 7, 9 and 16 show BCR-ABL1+ 2nd-round-PCR product.
Table 2: Percentage of patients with at least one pool of colonies positive for BCR-ABL1 mRNA
Molecular response in PB |
6-weeks LTC-IC assay |
|---|---|
MR0 (No treatment) |
100% (6/6) |
MR1.0-MR3.0 |
88% (15/17) |
MR4.0-MR5.0 |
20% (1/5) |
DISCUSSION
The assessment of individual LTC-IC-derived cells is particularly challenging: first, LSCs can be highly quiescent and, therefore, might not generate any detectable progeny in these assays [5]. Second, this cell population, being in the apex of the hematopoietic system, is of very low frequency, emphasizing the methodological difficulty in detecting these cells in BM or PB samples. In designing the present study, we first sought a method that would allow the functional isolation from a CD34+ cell population of a viable fraction of stem-like leukemic cells. For this, we used the most rigorous in vitro LSC detection technique (6-weeks LTC-IC) to evaluate the effects of TKI therapies on the most primitive leukemic cell compartment. As stated above, we considered the FLB to reflect a population of primitive or progenitor cells able to self-renew, give rise to daughter cells during in vitro functional assays, and produce variable levels of BCR-ABL1 mRNA. We had to adopt this definition because the evaluation of pooled colonies did not allow us to distinguish the effects of variable frequency of BCR-ABL1-positive colonies, from the effects of variable levels of BCR-ABL1 expression among individual colonies. Despite this limitation, dynamics of the primitive fraction during TKI treatment measured by FLB was comparable to previous reports based on phenotypically defined LSCs [14, 15], complementing such studies. As far as we are concerned, this is the first time that the primitive fraction is measured by LTC-IC assays in patients during their first year of TKI treatment.
Previous reports suggested that BCR-ABL1 overexpression in primitive cells (lin-CD34+CD38-) and mature (CD34+CD38+) progenitors may contribute to the failure of TKIs to prevent disease progression [17]. However, disease behavior is very variable for individual patients, with some progressing within a few months, and others remaining in stable CP for many years. This heterogeneity may relate to mutations later acquired in the BCR-ABL1 gene, subtype of stem cell in which BCR-ABL1 is expressed, differences in transcript expression patterns between patients, and differential interactions with the BM microenvironment and the immune system [18–23]. Here we show that, at six months of treatment, the primitive FLB in CP CML patients could be a prognostic marker correlating with resistance to TKIs. Although a larger cohort of patients should be studied in order to confirm our observations, these results reinforce previously reported data about the impact of LSC burden on TKI therapy outcome [14, 15], adding new evidence based on functional in vitro assays.
The fact that longer treatment is associated with lower relapse rates upon TKI interruption suggests that the risk of relapse might be related to the achievement of certain depth of MR below the detection limit of conventional RT-qPCR [24]. In multiple TKI-discontinuation trials, around 60% of patients relapse, whereas 40% can maintain TFR [4]. It has been shown that patients that achieve TFR can harbor BCR-ABL1-positive clones assessed at the DNA level [13], or at the RNA level [8]. However, it is not known whether such persistent cells are potentially clonogenic, i.e. LSCs that are able to self-renew and give rise to a full leukemic clone, or whether they are terminally differentiated cells that could survive TKI treatment by some unknown mechanism. A third possibility is that the immune system in TFR patients allows the presence of LSCs that are not able to develop their leukemic potential [25]. For this reason, strategies that increase PCR sensitivity for detection of residual LSCs may be relevant to better select patients with the highest likelihoods of remaining in remission without treatment. Although detectable levels of BCR-ABL1 mRNA in individual colonies from patients with undetectable deep molecular response have been reported, such levels were very low [26], suggesting that RNA-based methods will always be limited in patients under TKI treatment for long periods of time. In this work, we describe a strategy that, combining direct-PCR assessment for BCR-ABL1 DNA breakpoint with LTC-IC assay for separation of the primitive hematopoietic fraction, enables us to measure residual leukemic disease at the LSC level. Therefore, this method could be particularly useful in situations where the concurrently low burden of leukemic primitive cells and very low BCR-ABL1 mRNA expression levels, limit the sensitive detection of residual disease. With the purpose to validate this strategy, we will apply it in a currently recruiting TKI discontinuation trial in Argentina; we believe that in the future, this study may result of great interest in order to understand patients´ heterogeneous capacity of successfully maintaining TFR.
Overall, our results suggest that assessment of BCR-ABL1 mRNA levels in the primitive fraction during TKI treatment could be of clinical utility in order to improve and complement molecular monitoring in PB. Finally, we propose that the residual leukemic burden should be further assessed by gDNA PCR at the LSC level.
MATERIALS AND METHODS
Patient samples
The project was approved by the Institutional Review Board, at Instituto Alexander Fleming, Buenos Aires, Argentina. After giving written informed consent, BM or PB samples were obtained from newly diagnosed, untreated CML patients in CP, or from patients at different time points during TKI treatment. Mononuclear cells were isolated by density-gradient centrifugation (Ficoll-Paque PLUS, GE Healthcare Life Sciences) for 30 minutes at 480xg, followed by one wash in PBS, a red cell lysis step (EDTA 0.13mM, KHCO3 1 mM, NH4Cl 170mM, pH 7.3), and a low-speed centrifugation step (12-15 minutes at 200xg) for removal of the platelet-rich fraction. Up to 2x108 mononuclear cells were used for CD34+ cells isolation. Patients were classified according to their response to TKI treatment as follows: patients that were not able to achieve early response milestones and that would be considered as cases of warning or treatment failure according to the European Leukemia Net [16], were classified as cases of PR to treatment; patients that initially responded to treatment, and met early response (3 and 6 months) milestones, but later developed intolerance, rising levels of BCR-ABL1 transcripts, or secondary malignancies, were considered as SR patients; patients that sustained the requirements to be considered as optimal responders up to 1 year of TKI treatment, and did not develop SR events were considered as NR patients.
CD34+ cells isolation
In order to enrich samples in the progenitor and primitive fraction, we performed a positive selection using CD34 MicroBeads (Miltenyi Biotech), according to the manufacturer’s instructions. The CD34+ fraction was immediately used for functional assays, or cryopreserved for later use in freezing medium (DMEM 50%/HSA 40%/DMSO 10%). When available, 10,000 cells were used for purity assessment by flow cytometry.
CFU assay
500 CD34+ cells were plated in duplicate in p35 culture dishes containing 1mL of enriched methylcellulose (Methocult H4435 Medium, Stem Cell Technologies, Vancouver, Canada), and incubated at 37°C in a humid chamber. After 14-18 days, hematopoietic colonies were counted under the microscope, and individual or pooled colonies (made of 4 to 6 colonies) were plucked from methylcellulose and put into 500μl of RPMI medium (GIBCO, Thermo Fisher Scientific). Up to 10 pools of colonies were evaluated per patient, depending on the yield of the assay. All types of colonies were used for RNA isolation (CFU-GM, CFU-M, CFU-G, BFU-E, CFU-GEMM), and no differences in BCR-ABL1 mRNA expression were found among different types (data not shown). After centrifugation, pellets were resuspended in 100μl of lysis solution (RNAqueous-Micro Kit, Ambion, Thermo Fisher Scientific), and kept at -20°C until RNA extraction was performed.
LTC-IC assay
5,000 CD34+ cells were plated in collagen-coated p35 culture dishes containing 2mL of Long-term culture medium (Myelocult supplemented with freshly prepared hydrocortisone 10-6 M, both from Stem Cell Technologies), over a feeder layer composed of murine M2-10B4 and SI/SI cells producing hIL-3, hSCF and hGCSF (kindly provided by Stem Cell Technologies), previously irradiated at 80Gy. Transfected M2-10B4 and SI/SI cells were selected with G418 and Hygromicin B every 2 or 3 passages. One half of the medium was replaced every week. After 6 weeks, adherent and non-adherent cells were harvested, and 20,000 cells were used for short-term CFU assays, as described above.
RNA extraction and RT-qPCR
Total RNA was extracted from single or pooled colonies using RNAqueous-Microkit (Ambion, Thermo Fisher Scientific). RNA was eluted in 18μl of elution solution at 75°C; only samples with acceptable purity (A260/A280 ≥ 1.7) were used for retrotranscription using Superscript II (Invitrogen, Thermo Fisher Scientific). qPCR was performed using primers and probes for BCR-ABL1 and ABL1 from the European against cancer program [27]. Absolute quantification was performed using a calibration curve from a commercial kit (Molecular MD). Total RNA from PB samples was extracted using Trizol (Invitrogen, Thermo Fisher Scientific) after lysis of red blood cells. Results were harmonized to the International Scale by means of a conversion factor obtained by standardization with secondary cellular calibrators [28]. Some patients were monitored in other standardized centers, and so BCR-ABL1/ABL1 ratios were obtained directly from their data.
NGS detection of BCR-ABL1 breakpoint
High quality genomic DNA was extracted from PB or BM samples from newly diagnosed CML patients using DNAzol (Invitrogen, Thermo Fisher Scientific). Multiplex long-range PCR was performed using different sets of primers as previously described [13]. The PCR product was used for library generation in the Ion Torrent PGM platform (Thermo Fisher Scientific) according to manufacturer’s instructions. Briefly, PCR products were fragmented with Ion Shear Plus Reagents and purified with Agencourt AMPure XP (Beckman Coulter). Barcoded adapters were ligated according to manufacturer’s instructions with Ion Xpress Plus Fragment Library Kit, and the products were quantified by qPCR. Finally, samples were prepared for emulsion PCR, enriched with Ion PGM Template OT2 200 Kit, and sequenced using Ion 318TM Chip v2 with Ion PGM Sequencing 200 Kit v2.
Bioinformatics analysis of NGS data
In order to find the chromosomal translocation point, reads were aligned using the Subjunc aligner included in the Subread package (v1.5.0-p3) [29] and human genome reference UCSC version of the hg19. Sorting and indexing were performed with Picard (v1.119) (http://broadinstitute.github.io/picard). The fusion points were localized using CREST (Clipping Reveals Structure, v0.0.1) [30], which relays on soft-clipping signatures for identifying breakpoints. Finally, Blast2Seq algorithm was used against ABL1 and BCR reference sequences in order to identify each segment.
Detection of BCR-ABL1+ cells at DNA level using NGS-derived data
Sequence corresponding to patient-specific fusion breakpoint was used for PCR primer design (Primer3Plus). Short and long-term culture assays were performed as described above. Individual colonies were plucked from methylcellulose and subjected to direct nested PCR without a DNA extraction step. For this, cell lysis was performed by plucking individual colonies with a p20 tip set at 5μl, into 50 μL of the following mixture: GoTaqFlexi PCR buffer (Promega, USA) supplemented with NP-40 0.5%, Tween-20 0.5%, and proteinase K 0.91 mg/mL. Lysates were incubated for 1 hour at 60°C, followed by 15 minutes at 95°C. 10% of the final volume was used for first-round PCR. This PCR product was diluted ten-fold for second-round PCR.
Statistical analysis
Data was plotted using GraphPad Prism software or R project. Statistical analysis of the data was performed with Infostat Software (Córdoba, Argentina). When the assumptions of normality and homoscedasticity of the data were not met, variance was modeled previous to performing a parametric test (ANOVA), or analyzed by non-parametric tests (Mann–Whitney U test). A mixed-effects model was used in ANOVA (response to treatment as fixed-effect type, patient as a random-effect type).
Abbreviations
CML: chronic myeloid leukemia; TKI: tyrosine-kinase inhibitors; PB: peripheral blood; BM: bone marrow; RT-qPCR: quantitative reverse transcription polymerase chain reaction; LSC: leukemic stem cells; TFR: treatment-free remission; LTC-IC: Long-term Culture Initiating Cell; CFU: Colony Forming Units; FLB: functional leukemic burden; PR: primary resistance; SR: secondary resistance: NR: no resistance; gDNA: genomic DNA; NGS: next-generation sequencing; CP: chronic phase; MR: molecular response.
Author contributions
MB designed the study; MSR processed patient samples, performed LTC-IC and CFU assays, primer design, direct nested PCR experiments, collected clinical data and performed statistical analysis; MBS, LG and CM performed multiplex PCR and NGS of DNA breakpoints; DK and PY performed NGS bioinformatics analysis; MSR and MB analyzed the data and wrote the manuscript; SC, MRC, JF, CF, BM, CP, MAP, VV, JSA provided patient samples; JM, IL and MB supervised the entire study. All authors read and approved the manuscript in its final version.
ACKNOWLEDGMENTS
We thank Alicia Enrico, Jorge Milone, Paola Ochoa, Paula Rocca, and Adriana Vitriu for their contribution to this work.
CONFLICTS OF INTEREST
We state that this study was supported financially by a Novartis grant; Dr. Michele Bianchini has received speaker fees from Novartis; all other authors declare no competing financial interests.
FUNDING
We thank Novartis Oncology Argentina, Fundación Cáncer (FUCA), ANPCyT, CONICET, and Fundación Sales for their support to this investigation.
REFERENCES
1. Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008; 112:4808–17. https://doi.org/10.1182/blood-2008.
2. Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer. 2007; 7:345–56. https://doi.org/10.1038/nrc2126.
3. Holyoake TL, Vetrie D. The chronic myeloid leukemia stem cell: Stemming the tide of persistence. Blood. 2017; 129:1595–606. https://doi.org/10.1182/blood-2016-09-696013.
4. Saußele S, Richter J, Hochhaus A, Mahon FX. The concept of treatment-free remission in chronic myeloid leukemia. Leukemia. 2016; 30:1638–47. https://doi.org/10.1038/leu.2016.115.
5. Graham SM, Jørgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, Holyoake TL. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002; 99:319–25. https://doi.org/10.1182/blood.V99.1.319.
6. Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011; 121:396–409. https://doi.org/10.1172/JCI35721DS1.
7. Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, Nicolini FE, Müller-Tidow C, Bhatia R, Brunton VG, Koschmieder S, Holyoake TL. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012; 119:1501–10. https://doi.org/10.1182/blood-2010-12-326843.
8. Chomel JC, Bonnet ML, Sorel N, Sloma I, Bennaceur-Griscelli A, Rea D, Legros L, Marfaing-Koka A, Bourhis JH, Ame S, Guerci-Bresler A, Rousselot P, Turhan AG. Leukemic stem cell persistence in chronic myeloid leukemia patients in deep molecular response induced by tyrosine kinase inhibitors and the impact of therapy discontinuation. Oncotarget. 2016; 7:35293–301. https://doi.org/10.18632/oncotarget.9182.
9. Abraham SA, Hopcroft LE, Carrick E, Drotar ME, Dunn K, Williamson AJ, Korfi K, Baquero P, Park LE, Scott MT, Pellicano F, Pierce A, Copland M, et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature. 2016; 534:341–6. https://doi.org/10.1038/nature18288.
10. Nievergall E, Ramshaw HS, Yong ASM, Biondo M, Busfield SJ, Vairo G, Lopez AF, Hughes TP, White DL, Hiwase DK. Monoclonal antibody targeting of IL-3 receptor α with CSL362 effectively depletes CML progenitor and stem cells. Blood. 2014; 123:1218–28. https://doi.org/10.1182/blood-2012-12-475194.
11. Zhang B, Strauss AC, Chu S, Li M, Ho Y, Shiang KD, Snyder DS, Huettner CS, Shultz L, Holyoake T, Bhatia R. Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell. 2010; 17:427–42. https://doi.org/10.1016/j.ccr.2010.03.011.
12. Kumari A, Brendel C, Hochhaus A, Neubauer A, Burchert A. Low BCR-ABL expression levels in hematopoietic precursor cells enable persistence of chronic myeloid leukemia under imatinib. Blood. 2012; 119:530–9. https://doi.org/10.1182/blood-2010-08-303495.
13. Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Bartley PA, Slader C, Field C, Dang P, Filshie RJ, Mills AK, Grigg AP, Melo JV, Hughes TP. Patients with chronic myeloid leukemia who maintain a complete molecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR. Leukemia. 2010; 24:1719–24. https://doi.org/10.1038/leu.2010.185.
14. Mustjoki S, Richter J, Barbany G, Ehrencrona H, Fioretos T, Gedde-Dahl T, Gjertsen BT, Hovland R, Hernesniemi S, Josefsen D, Koskenvesa P, Dybedal I, Markevärn B, et al, and Nordic CML Study Group (NCMLSG). Impact of malignant stem cell burden on therapy outcome in newly diagnosed chronic myeloid leukemia patients. Leukemia. 2013; 27:1520–6. https://doi.org/10.1038/leu.2013.19.
15. Thielen N, Richter J, Baldauf M, Barbany G, Fioretos T, Giles F, Gjertsen BT, Hochhaus A, Schuurhuis GJ, Sopper S, Stenke L, Thunberg S, Wolf D, et al. Leukemic stem cell quantification in newly diagnosed patients with chronic myeloid leukemia predicts response to nilotinib therapy. Clin Cancer Res. 2016; 22:4030–38.
16. Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, Cervantes F, Clark RE, Cortes JE, Guilhot F, Hjorth-Hansen H, Hughes TP, Kantarjian HM, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013; 122:872–84.
17. Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, Eaves C. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007; 21:926–35. https://doi.org/10.1038/sj.leu.2404609.
18. Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004; 103:4010–22. https://doi.org/10.1182/blood-2003-12-4111.
19. Yong AS, Szydlo RM, Goldman JM, Apperley JF, Melo JV. Molecular profiling of CD34+ cells identifies low expression of CD7, along with high expression of proteinase 3 or elastase, as predictors of longer survival in patients with CML. Blood. 2006; 107:205–12. https://doi.org/10.1182/blood-2005-05-2155.
20. Bolton-Gillespie E, Schemionek M, Klein HU, Flis S, Hoser G, Lange T, Nieborowska-Skorska M, Maier J, Kerstiens L, Koptyra M, Müller MC, Modi H, Stoklosa T, et al. Genomic instability may originate from imatinib-refractory chronic myeloid leukemia stem cells. Blood. 2013; 121:4175–83. https://doi.org/10.1182/blood-2012-11-466938.
21. Giustacchini A, Thongjuea S, Barkas N, Woll PS, Povinelli BJ, Booth CAG, Sopp P, Norfo R, Rodriguez-Meira A, Ashley N, Jamieson L, Vyas P, Anderson K, et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat Med. 2017; 23:692–702. https://doi.org/10.1038/nm.4336.
22. Landberg N, Hansen N, Askmyr M, Ågerstam H, Lassen C, Rissler M, Hjorth-Hansen H, Mustjoki S, Järås M, Richter J, Fioretos T. IL1RAP expression as a measure of leukemic stem cell burden at diagnosis of chronic myeloid leukemia predicts therapy outcome. Leukemia. 2016; 30:253–7. https://doi.org/10.1038/leu.2015.135.
23. Tarafdar A, Hopcroft LEM, Gallipoli P, Pellicano F, Cassels J, Hair A, Korfi K, Jørgensen HG, Vetrie D, Holyoake TL, Michie AM. CML cells actively evade host immune surveillance through cytokine-mediated downregulation of MHC-II expression. Blood. 2017; 129:199–208. https://doi.org/10.1182/blood-2016-09-742049.
24. Sobrinho-Simões M, Wilczek V, Score J, Cross NC, Apperley JF, Melo JV. In search of the original leukemic clone in chronic myeloid leukemia patients in complete molecular remission after stem cell transplantation or imatinib. Blood. 2010; 116:1329–35. https://doi.org/10.1182/blood-2009-11-255109.
25. Rea D, Henry G, Khaznadar Z, Etienne G, Guilhot F, Nicolini F, Guilhot J, Rousselot P, Huguet F, Legros L, Gardembas M, Dubruille V, Guerci-Bresler A, et al. Natural killer cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: the IMMUNOSTIM study. Haematologica. 2017; 102:1368–77. https://doi.org/10.3324/haematol.2017.165001.
26. Chomel J, Sorel N, Guilhot J, Guilhot F, Turhan AG, Dc W. BCR-ABL expression in leukemic progenitors and primitive stem cells of patients with chronic myeloid leukemia. Blood. 2012; 119:2964–5. https://doi.org/10.1182/blood-2011-12-396226.
27. Gabert J, Beillard E, van der Velden VH, Bi W, Grimwade D, Pallisgaard N, Barbany G, Cazzaniga G, Cayuela JM, Cavé H, Pane F, Aerts JL, De Micheli D, et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia - a Europe Against Cancer program. Leukemia. 2003; 17:2318–57.
28. Ruiz MS, Medina M, Tapia I, Mordoh J, Cross NCP, Larripa I, Bianchini M. Standardization of molecular monitoring for chronic myeloid leukemia in Latin America using locally produced secondary cellular calibrators. Leukemia. 2016; 30:2258–60. https://doi.org/10.1038/leu.2016.197.
29. Liao Y, Smyth GK, Shi W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013; 41. https://doi.org/10.1093/nar/gkt214.
30. Wang J, Mullighan CG, Easton J, Roberts S, Heatley SL, Ma J, Rusch MC, Chen K, Harris CC, Ding L, Holmfeldt L, Payne-Turner D, Fan X, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods. 2011; 8:652–54.