
INTRODUCTION
Apoptosis is a tightly controlled and evolutionarily conserved process of programmed cell death that is crucial for tissue homeostasis [1]. Evasion of apoptosis is critical for tumor development as well as maintenance and represents an almost universal hallmark of cancer which, importantly, also underpins resistance to diverse anti-cancer treatments [2]. BCL-2 family proteins are crucial regulators of the mitochondrial apoptotic pathway and are characterized by the presence of up to four conserved BCL-2 Homology (BH) domains. BCL-2, the first identified member of this family was originally discovered as part of the t(14;18) chromosomal translocation which occurs in human B cell lymphomas [3, 4]. BCL-2 family proteins are divided into 3 functionally distinct groups: (i) the pro-survival proteins (BCL-2, BCL-XL, BCL-W, MCL-1 and BFL-1/BCL2A1/A1), (ii) the multi-BH domain cell death effectors (BAX, BAK, BOK) and (iii) the BH3-only apoptosis initiators (e.g. BIM, BAD, NOXA) [5]. Dysregulation of the BCL-2 family altering the balance between pro-survival and pro-death members provides a common mechanism by which cancer cells acquire a survival advantage [6]. Dynamic interactions between members of BCL-2 family subgroups, involving binding of the BH3 domain of the pro-apoptotic members to a groove at the surface of the pro-survival proteins, control commitment to apoptosis [7]. Following activation by cellular stress, BH3-only proteins initiate apoptosis by inhibiting the pro-survival BCL-2 proteins [8] and potentially by directly activating BAX and BAK [9]. The subsequent activation and oligomerization of the death effectors BAX and BAK result in mitochondrial outer membrane permeabilization and apoptosis.
The pro-survival BCL-2 members exert their function by sequestering pro-apoptotic members through binding to their BH3 domains. Inhibiting these intracellular protein-protein interactions is therefore an attractive strategy to target the aberrant survival of cancer cells caused by BCL-2 family dysregulation [10]. ABT-737, which was discovered using structure-based drug design, targets BCL-2, BCL-XL and BCL-W and was the first example of an inhibitor of the pro-survival BCL-2 members demonstrating druggability of such targets [11]. Encouraging clinical activity against lymphoid malignancies thought to be BCL-2 dependent were observed in early trials with Navitoclax (ABT-263), the orally bioavailable analog of ABT-737 [12, 13, 14]. However, as BCL-XL has a crucial survival function in circulating platelets [15], Navitoclax induced a rapid thrombocytopenia due to its inhibition of BCL-XL, which limited its clinical utility. It was therefore hypothesized that a BCL-2 selective inhibitor should exhibit limited thrombocytopenia while maintaining antitumor activity in BCL-2 dependent lymphoid malignancies. Subsequent rational drug discovery research efforts led to the development of Venclexta™ (ABT-199), the first orally active BCL-2 selective (BCL-XL-sparing) inhibitor [16] that has been recently approved in high risk patients with relapsed or refractory Chronic Lymphocytic Leukemia (CLL) [17]. Here we describe S55746 (also known as BCL201), a novel, orally active BCL-2 specific inhibitor that has a partially overlapping but distinct BCL-2 hydrophobic groove binding mode compared to ABT-199. S55746, currently in phase I clinical trials in heamatological malignancies (Trials registration ID: NCT02920697, NCT02920541 and NCT02603445) displays all the hallmarks of a BCL-2 specific BH3-mimetic and exhibits robust antitumor activity in BCL-2 dependent lymphoid tumor xenograft models while sparing platelets.
RESULTS
S55746 is a selective inhibitor of BCL-2
Starting from a moderately active literature compound [18, 19] and using a structure-based drug design approach, S55746 was developped as described in Le Diguarher et al. 2013 [20] (Figure 1A and Supplementary Materials and Methods). Fluorescence polarization (FP) data, using Fluorescent-PUMA as a binder, demonstrates that S55746 is a potent inhibitor of BCL-2 (Ki = 1.3 nM). The selectivity of S55746 for BCL-2 versus BCL-XL ranges from ~70 to 400 folds depending on the assay used (Table 1). No significant binding to MCL-1 and BFL-1 was observed (Table 1). S55746 occupies the region typically referred to as S1/2/3 in contrast to the ABT-199 analog [16], which occupies a greater portion of the protein surface area including S2/3/4/5. S55746 adopts essentially the same binding mode as described in Porter et al. 2009 [18]. S55746 forms a single hydrogen bond to the backbone carbonyl of residue A149 (Figure 1B) buried deep into S2. The size-independent enthalpic efficiency (0.83) for S55746 binding to BCL-2 is suggestive of optimal polar and Van der Waal’s interactions, indicative of highly specific binding (Figure 1B) [21].
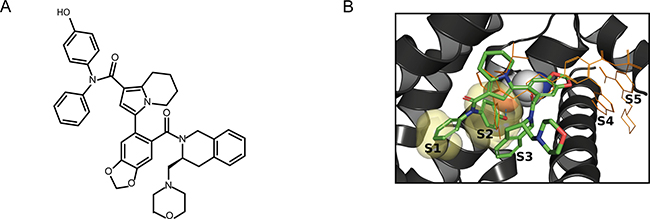
Figure 1: Chemical structure and binding mode of S55746. (A) Chemical structure of S55746. (B) Superposition of S55746 (green sticks) and ABT-199 analogue from PDB 4MAN (orange lines) X-ray crystal structures. Ala149 is highlighted (space-filling spheres) and the hydrogen bond from S55746 phenol to backbone carbonyl of Ala149 is shown (cyan dashed line). The key S1 and S2 binding pockets are shown as translucent yellow surfaces.By comparison with the ABT-199 analogue it can be seen that S55746 forms a compact structure that only accesses pockets S1/S2/S3 and not S4/S5. Image was prepared using Pymol.
Table 1: Associated affinity and selectivity data of ABT-263, S55746 and ABT-199
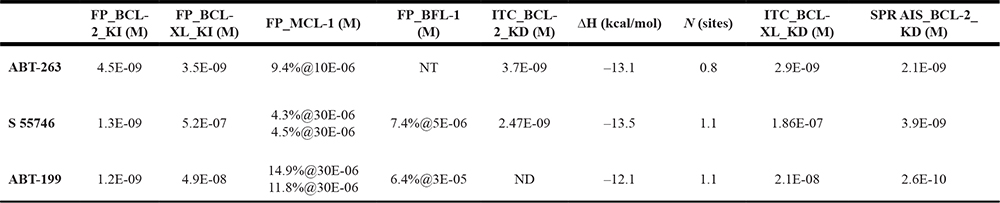
The KI were determined by Fluorescence polarisation and KD by direct binding SPR. X%@10E-05 means X% of inhibition at the maximal tested concentration (M). FP: Fluorescence Polarization; SPR AIS: Surface Plasmon Resonance Affinity In Solution; ITC: Isothermal Titration Calorimetry; ΔH : enthalpy delta. NT, not tested. ND, not determinable (too potent). Of note, bottom limit for the FP assay was 1.2 nM.
S55746 induces cell death selectively
The selectivity and potency of S55746 was firstly evaluated in the well-described BCL-2-dependent acute lymphoblastic leukemia (ALL) RS4;11 cell line, which express high levels of BCL-2 but low levels of BCL-XL [22] (Figure 2A, left panel). S55746 potently induces RS4;11 cell killing after 72 h of treatment with an IC50 of 71.6 nM (Figure 2A, right panel and Supplementary Figure 1A). Interestingly, S55746 exhibits a much weaker activity in H146 (IC50 1.7 μM), a BCL-XL-dependent cell line [23], which expresses a low level of BCL-2 and high level of BCL-XL (Figure 2A, left panel) whereas ABT-263, which targets BCL-2 and BCL-XL [14], induces equivalent cell killing in both RS4;11 and H146 (41.5 nM and 49.7 nM, respectively; Figure 2A, right panel and Supplementary Figure 1B). Of note, the BCL-2 selective inhibitor ABT-199 displays identical selectivity profile than S55746 in these two cell lines. Selective targeting of BCL-2 by S55746 was confirmed by co-immunoprecipitation experiments, which showed a concentration-dependent disruption of the BCL-2/BAX complex in RS4;11 cells following treatment with increasing concentrations of S55746 (Figure 2B). In BCL-2-overexpressing HeLa cells, S55746 affects BCL-2/BAX complex without any significant effect on the BCL-XL/BAX complex in BCL-XL-overexpressing HeLa cells (Figure 2C, left panel). In contrast, ABT-263 was shown to disrupt both BCL-2/BAX and BCL-XL/BAX complexes (Figure 2C, right panel).
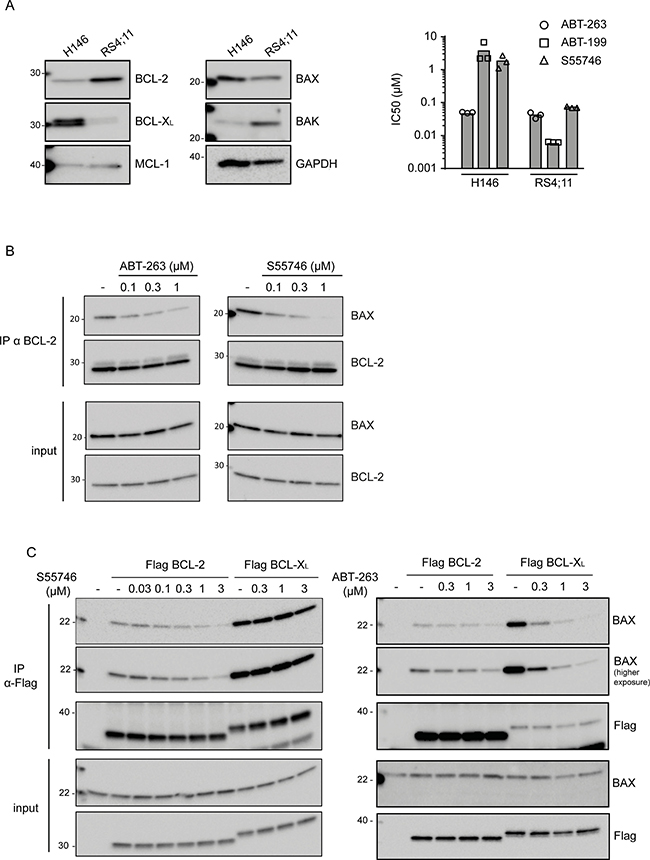
Figure 2: S55746 is a potent and selective inhibitor of BCL-2. (A) Expression of BCL-2, BCL-XL, MCL-1, BAX and BAK was assessed by immunoblotting in H146 and RS4;11 cell lines. GAPDH was used as a loading control (left panel). Viability of H146 and RS4;11 cell lines after incubation with increasing concentration of either ABT-263 (open circles), S55746 (open triangles) or ABT-199 (open squares) for 72 h. IC50 indicates concentration at which 50% of inhibition is reached. Mean and individual data of 3 independent experiments are shown (right panel). (B) Co-immunoprecipitation assay of RS4;11 cells treated with different concentrations of S55746 or ABT-263 for 2 h. ‘−’ indicates that cells were treated with DMSO only. Cells were lysed after treatment and immunoprecipitated (IP) with anti-BCL-2 antibody. The cell lysates (input) and immunoprecipitates were analyzed by immunoblotting with BAX or BCL-2 antibodies as indicated. (C) Co-immunoprecipitation assay of HeLa cells transfected with plasmids expressing Flag-tagged BCL-2 or Flag-tagged BCL-XL as indicated. Cells were treated with different concentrations of S55746 or ABT-263 for 2 h. ‘−’ indicates that cells were treated with DMSO only. Cells were lysed after treatment and immunoprecipitated (IP) with anti-Flag antibody. The cell lysates (input) and immunoprecipitates were analyzed by immunoblotting with BAX or Flag antibodies as indicated. Representative blots of three independent experiments are shown.
S55746 selectively induces apoptosis through BCL-2 inhibition in a BAX/BAK-dependent manner
S55746 rapidly induced apoptosis in a concentration dependent-manner, as monitored by externalization of phosphatidylserine (PS+ cells) in RS4;11 cells (Figure 3A). Cleavage of caspase-3 and Poly ADP-ribose polymerase (PARP) was also observed upon treatment by S55746 in a concentration dependent manner in the same cellular model (Supplementary Figure 1C and 1D, respectively). S55746-induced apoptosis in RS4;11 is mediated in part by the BAX effector protein since PARP cleavage (Figure 3B) is markedly inhibited in BAX-deficient RS4;11 cells, generated using a shRNA-based approach compared to control RS4;11 cells upon treatment with increasing concentration of S55746. The sensitivity of BAX-deficient RS4;11 cells was strongly reduced compared to control RS4;11 (≈ 50-fold, IC50 = 2.7 μM versus 0.057 μM, Figure 3C). The dependence of the killing activity of S55746 on BAX and BAK was further confirmed by CRISPR/Cas9 genome editing in the THP-1 AML cell line (Supplementary Figure 2).
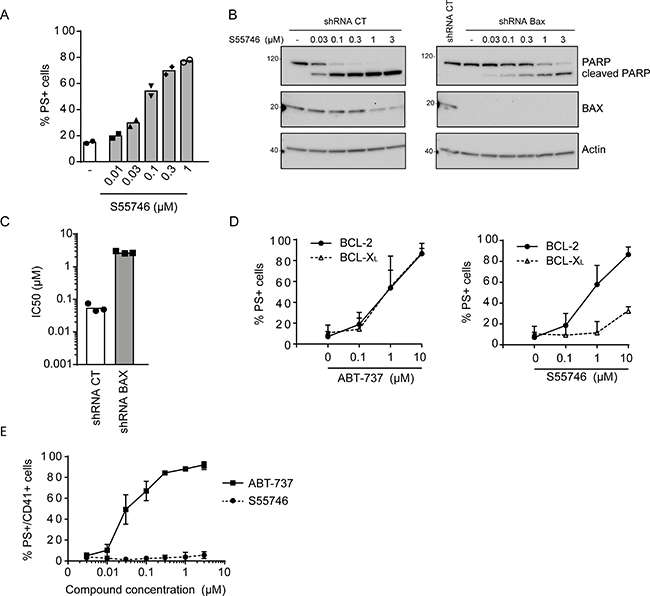
Figure 3: S55746 induces apoptosis in a BAX-dependent manner. (A) Apoptosis induction in RS4;11 cells treated with S55746 at the indicated concentration for 2 h. Cells were analyzed by flow cytometry for PI and annexin V-FITC labeling. Mean and individual points from two biological replicates are shown. ‘−’ indicates that cells were treated with DMSO only. (B) Expression of PARP and BAX assessed by immunoblotting in RS4;11 cells transduced with shRNA CT (control) or shRNA BAX upon treatment with increasing concentrations of S55746. Actin was used as a loading control. ‘−’ indicates that cells were treated with DMSO only. (C) Viability of RS4;11 cells transduced either with shRNA CT or shRNA BAX treated with increasing concentration of S55746 for 72 h. IC50 indicates concentration at which 50% of inhibition is reached. Mean and individual points from n = 3 biological replicates are shown. (D) FL5.12 cells either expressing BCL-2 (filled circles, thick line) or BCL-XL (open triangles, dashed line) were treated with increasing concentration of either ABT-737 (left panel) or S55746 (right panel) for 24 h upon IL-3 withdrawal. Cells were then analyzed by flow cytometry for PI and annexin V-FITC labeling. Mean ± s.d. of 4 biological replicates are shown. (E) Isolated platelets from 4 healthy donors were treated with increasing concentrations of ABT-737 (squares, thick line) or S55746 (circles, dashed line) for 3 h. CD41-PE positive cells were then gated and analyzed for annexin V-FITC labeling. PS+: Phosphatidylserine positive cells. EC50: half maximal effective concentration.
To further assess the selectivity of S55746 on apoptosis induction, S55746 was tested in the IL3-dependent murine pro-B lymphocytic FL5.12 cell line expressing either BCL-2 or BCL-XL as a required protein for cell survival in the absence of IL-3 [24]. Upon IL-3 withdrawal, increasing concentrations of ABT-737, an analogue inhibitor of ABT-263 that targets BCL-2 and BCL-XL, efficiently induced apoptosis, as illustrated by the increasing percentage of PS+ cells in either BCL-2 or BCL-XL-dependent FL5.12 cells (Figure 3D, left panel). However, S55746 was shown to strongly induce apoptosis in BCL-2-dependent FL5.12 cells with a minor effect in BCL-XL-dependent FL5.12 cells (Figure 3D, right panel). Of note is that isolated platelets from healthy volunteers, known to be strictly dependent on BCL-XL for survival [15], were totally insensitive to S55746 (EC50 > 3 μM) in contrast to ABT-737 which strongly induced apoptosis (EC50 = 0.03 μM; Figure 3E). Altogether, these data demonstrate that S55746 kills cancer cells through on-target activity, meaning activation of a BAX/BAK-dependent mitochondrial apoptotic pathway by direct inhibition of the BCL-2 pro-survival protein.
Activity of S55746 in hematological cell malignancies in vitro and ex vivo
Activity of S55746 was assessed in a panel of non-Hodgkin lymphoma cell lines including Diffuse Large B-Cell Lymphoma (DLBCL; Figure 4A), Mantle Cell Lymphoma (MCL; Figure 4B) and Burkitt Lymphoma (BL; Figure 4C). After 72 h of treatment, we found that six out of eleven DLBCL cell lines tested had an IC50 below 1 μM. Two MCL cell line out of five tested displayed an IC50 below 1 μM for S55746. In contrast, all Burkitt lymphoma cell lines tested had an IC50 above 10 μM for S55746. Cellular results for the BCL-2 selective inhibitor ABT-199 are shown for comparison (Figure 4).
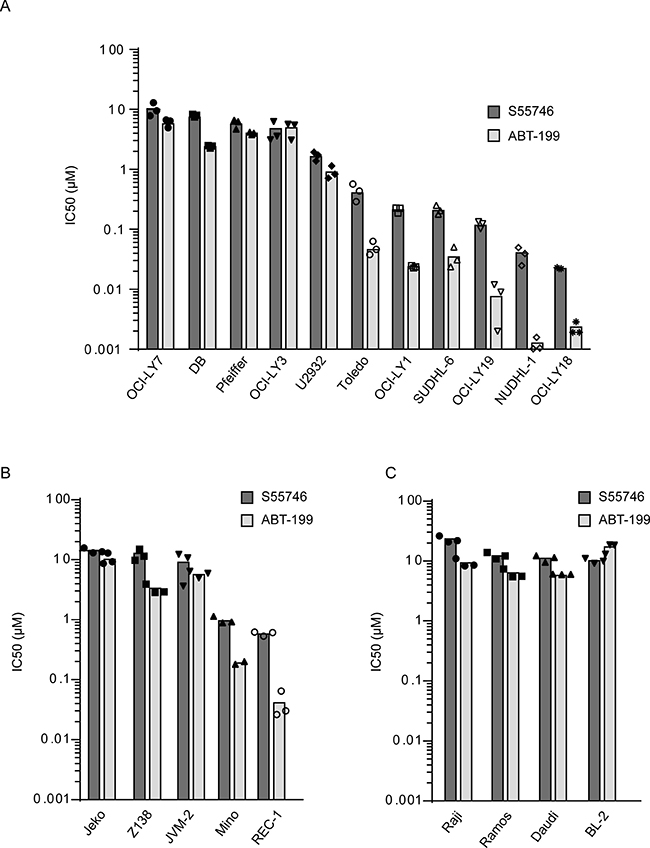
Figure 4: S55746 induces cell death in a panel of non-Hodgkin lymphoma cell lines. Viability of a panel of Diffuse Large B-Cell (DLBCL, A), Mantle Cell Lymphoma (MCL, B) and Burkitt lymphoma (BL, C) cell lines treated with increasing concentration of S55746 and ABT-199 for 72 h was determined using Cell Titer Glo. IC50 indicates concentration at which 50% of inhibition is reached. Mean and individual points from n = 3 biological replicates are shown. Origins of the cell lines are described in supplementary materials and methods.
Chronic Lymphocytic Leukemia (CLL) cells are highly dependent on BCL-2 expression for survival [25]. Efficacy of S55746 was evaluated in primary CLL cells freshly isolated from 7 patients (Supplementary Table 1). S55746 caused rapid induction of apoptosis, reflected by the presence of phosphatidylserine positive cells. All primary CLL cells tested had an EC50 in a low nanomolar range (from 4.4 to 47.2 nM following 4 hours of treatment; Figure 5A, top panel and Supplementary Table 1). In two representative CLL patient samples, S55746 induced characteristic ultrastructural changes of apoptosis such as chromatin condensation (data not shown) in addition to extensive processing of caspase-9 and caspase-3 as well as cleavage of PARP (Figure 5A, lower panel). All these data are consistent with activation of the intrinsic apoptotic pathway in CLL cells by S55746. In contrast to the data obtained on cell lines (Figure 4B), S55746 potently induced apoptosis in primary MCL cells with EC50 ranging from 2.5 to 110 nM following 24h of S55746 treatment (Figure 5B and Supplementary Table 2). Of note, ABT-199 has also been shown to be more potent on primary MCL cells versus MCL cell lines ; this potency shift is likely due to higher expression of BCL- XL in cell lines compare to primary cells [26]. Altogether, these data demonstrate that S55746 is a potent BCL-2 inhibitor that induces apoptotic cell death in cells that are dependent on BCL-2 for survival.
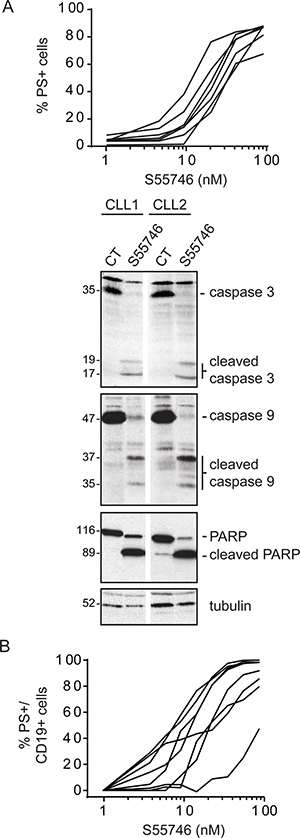
Figure 5: S55746 induces cell death in primary CLL and MCL cells. (A) Primary Chronic Lymphocytic Leukemia (CLL) cells freshly isolated from 7 patients were treated for 4 h with increasing concentrations of S55746. Cells were then analyzed by flow cytometry for PI and annexin V-FITC labeling. Individual curves of cells treated with S55746 are shown (top panel). CLL cells isolated from 2 patients were exposed to S55746 (3 nM) for 4 h. Total and cleaved caspase-3, caspase-9 and PARP were detected by immunoblotting using specific antibodies. Tubulin was used as a loading control (lower panel). (B) Primary Mantle Cell Lymphoma (MCL) cells freshly isolated from 8 patients were cultured with increasing concentrations of S55746 for 24 h. Cells were then analyzed by flow cytometry for annexin V-FITC and CD19-APC positive labeling. Individual curves of cells treated with S55746 are demonstrated. PS+: Phosphatidylserine positive cells.
S55746 is an effective agent that induces tumor regression in vivo
In order to confirm apoptosis induction by S55746 in vivo, caspase-3 activity was assessed on RS4;11 tumor xenografts 16 hours following single oral gavage treatment. Caspase-3 activity after S55746 treatment at 25 and 100 mg/kg was about 11 and 28 times higher than in vehicle-treated animals, respectively. ABT-263 induced caspase-3 activation about 20 times higher than in vehicle-treated group (Figure 6A). Consistent with the in vitro data (Figure 2 and Figure 3), S55746 treatment did not induce platelet loss in vivo at 25 and 100 mg/kg, while a strong decrease of platelet counts was observed upon treatment with ABT-263 at 100 mg/kg (Figure 6B).
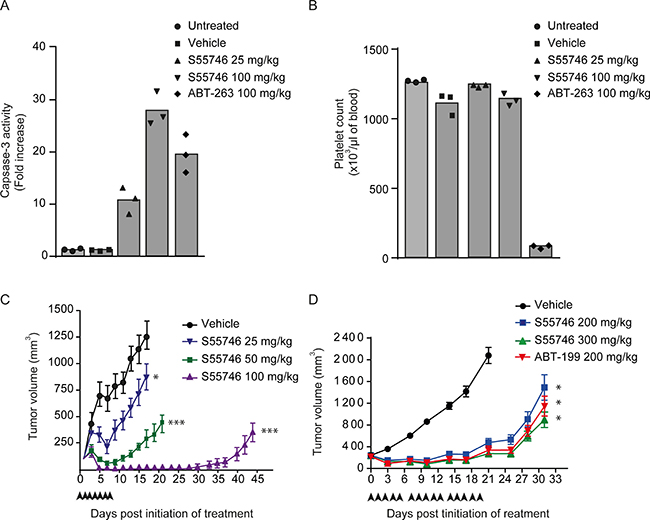
Figure 6: S55746 inhibits xenograft growth in RS4;11 and Toledo models. (A and B) Female SCID mice were inoculated with 1 × 107 RS4;11 cells and randomized 26 days after grafting. After randomization, mice were either not treated, or dosed once orally with vehicle, S55746 (25 mg/kg and 100 mg/kg) and ABT-263 (100 mg/kg). (A) Tumor samples were collected 16 h post dosing and analyzed for caspase-3 activity. Mean and individual data of caspase-3 activity fold increase over untreated group of 3 animals per treatment group are shown. (B) Platelet cells were counted 16 h post dosing. Mean and individual data of platelets counts of 3 animals per treatment group are shown. (C) Female SCID mice were inoculated with 1 × 106 RS4;11 cells and randomized 27 days after grafting. After randomization, mice were either not treated (black circles) or treated orally with S55746 every day for 7 consecutive days at 25 mg/kg (blue reverse triangles), 50 mg/kg (green squares) or 100 mg/kg (purple triangles). Mean tumor volumes with error bars depicting s.e.m. of 8 animals per treatment group are shown. One way ANOVA on day 17, followed by Dunnett post hoc comparison was performed and always compared to vehicle, (* p < 0.05; ***p < 0.001). (D) Female SCID mice were inoculated with 3 × 106 Toledo cells and randomized 24 days after grafting. After randomization, mice were either not treated (black circles) or treated orally five times a week with ABT-199 at 200 mg/kg (inverted red triangles) and S55746 at 200 mg/kg (blue squares) or 300 mg/kg (green triangles) for 3 weeks. Mean tumor volumes with error bars depicting s.e.m. of 6–7 animals per treatment group are shown. One way ANOVA on day 21, followed by Dunnett post hoc comparison was performed and always compared to vehicle, (* p < 0.05).
Antitumor activity of S55746 was then evaluated in RS4;11 and Toledo models, two models that display different in vitro sensitivities towards S55746 (71.6 nM in RS4;11 (Figure 2A, right panel) vs 401 nM in Toledo (Figure 4A)). In RS4;11 bearing SCID mice, daily oral gavage treatment of S55746 for 7 consecutive days induced significant anti-tumor activity compared to untreated animals (p < 0.05 at 25 mg/kg and p < 0.001 at 50 and 100 mg/kg). This response increased in magnitude and in durability in a dose dependent manner (Figure 6C). Seventeen days after beginning of treatment at 25 mg/kg, 50 mg/kg and 100 mg/kg, tumor growth inhibition was 67.1, 16.3 and −93.8 T/C respectively, with complete regression observed in all animals treated at 100 mg/kg (Supplementary Figure 3A and Supplementary Table 3).
Efficacy of S55746 was also assessed in Toledo bearing mice treated by oral gavage five times a week for 3 weeks at 200 and 300 mg/kg and compared to ABT-199 at 200 mg/kg (Figure 6D). After 21 days of treatment, S55746 induced a significant tumor growth inhibition either at 200 or 300 mg/kg (13% and 2% T/C, respectively; p < 0.05; Supplementary Table 4). In this model, S55746 and ABT-199 showed similar anti-tumor efficacy (Figure 6D). Individual data at day 21 post-treatment are presented in Supplementary Figure 3B. Interestingly, body weight did not significantly differ in all treatment groups compared to the vehicle control group (Supplementary Figure 3C), even at doses up to 300 mg/kg of S55746. Altogether, these data indicate that S55746 is a highly efficacious and well-tolerated orally active BCL-2 inhibitor.
DISCUSSION
In the present study, we describe S55746, a novel bona fide BH3 mimetic compound which selectively inhibits BCL-2. Importantly, S55746 induces the critical hallmarks of the mitochondrial apoptosis pathway and kills BCL-2 dependent tumor cells in a BAX/BAK dependent manner. In vivo, S55746 is highly efficacious against BCL-2 dependent tumor models without causing platelet loss, in agreement with its strong selectivity for BCL-2 over BCL-XL, thus supporting its evaluation in clinical trials.
The role of BCL-2 in lymphoid malignancies is well recognized and BCL-2 overexpression found in such diseases is driven by multiple mechanisms. The t(14;18) chromosome translocation, bringing the BCL-2 gene under the control of the immunoglobulin heavy chain enhancer is found in all B-cell follicular lymphomas [3, 4] and in approximately 30% of DLBCL [27] while BCL-2 gene amplification is commonly found in MCL and DLBCL [28]. BCL-2 is also overexpressed in both acute [29, 30] and chronic [31] leukemias where loss of microRNAs regulating BCL-2 expression are common [32, 33]. The clinical activity of Venclexta™ in these BCL-2 overexpressing hematological malignancies and its recent approval for patients with relapsed or refractory CLL with 17p deletion provide very strong clinical proof-of-concept and seminal registration for drugs targeting the BCL-2 family.
Target mutations causing resistance have been demonstrated with various high-affinity targeted therapies [34]. In the context of experimentally induced ABT-199 resistance, BCL-2 mutations affecting its hydrophobic groove have been identified [35, 36]. The clinical relevance of these mutations remains unknown at this stage. Since ABT-199 and S55746 have different binding modes, one can theoretically speculate that mutation(s) in the BCL-2 protein could cause resistance to one compound and not the other. Importantly, the pro-survival proteins MCL-1 and BCL-XL have also emerged as resistance factors to the BCL-2 specific inhibitor ABT-199 [37]. The recent discovery of selective BCL-XL [38, 39] and MCL-1 specific [40, 41] inhibitors is therefore of particular interest as these new specific BH3 mimetic compounds provide very promising strategies to overcome BCL-2 specific resistance mechanisms. The discovery of S55746, a new BCL-2 specific inhibitor, currently in phase I trials, could offer other opportunities to investigate the clinical utility of BCL-2 inhibition in the clinic.
MATERIALS AND METHODS
Human cells
Peripheral blood samples from CLL patients were obtained with informed patient consent and local ethical committee approval (UHL-09723). Peripheral blood mononuclear cells were purified using Histopaque (Sigma Aldrich, Dorset, United Kingdom) and cultured in RPMI 1640 medium supplemented with 10% FCS and 2 mM L-glutamine (Life Technologies Inc, Paisley, United Kingdom) at 2 × 106 cells ml-1. Interphase fluorescence in situ hybridization (FISH) was performed for detection of 17p13 deletion. Mantle cell lymphoma (MCL) primary cells were obtained after informed consent from MCL patients treated at the department of clinical hematology from the University Hospital of Nantes, France. Peripheral MCL cells from blood or pleural effusion, were purified after Ficoll-Hypaque separation with immuno-magnetic anti-CD19 beads (Miltenyi, Paris, France). Primary cells were cultured in RPMI-1640 supplemented with 10% FCS and 2 mM glutamine. Human platelets were isolated from citrate blood donated by healthy volunteers. Washed platelets were cultured in Hepes-buffered saline (HBS, 10 mM Hepes, 150 mM NaCl, 5 mM KCl, 1 mM MgSO4, pH 7.4).
Chemical
ABT-737, Venclexta™ (ABT-199) and ABT-263 were obtained from Selleck-Chem. Synthesis of S55746 is described in the supplemental material and methods.
Fluorescence polarization (FP)
The assays were carried out in black-walled, flat bottomed, low binding, 384-well plates in buffer A (10 mM 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 150 mM NaCl, 0.05% Tween 20, pH 7.4 and 5% DMSO) in the presence of 10 nM Fluorescein-PUMA. Final concentrations of MCL-1, BFL-1, BCL-2 and BCL-XL proteins were 10, 10, 10 and 20 nM respectively. The assay plates were incubated for 2 hours at room temperature and the FP was measured on a Synergy 2 reader (Ex. 528 nm, Em. 640 nm, Cut-off 510 nm). The binding of increasing doses of compound was expressed as a percentage reduction in mP compared to the window established between ‘DMSO only’ and ‘total inhibition’ control (30 μM PUMA). The inhibitory concentrations that gave a 50% reduction in mP (IC50) were determined, from 11-point dose response curves, in XL-Fit using a 4-Parameter Logistic Model (Sigmoidal Dose-Response Model). The KI was subsequently calculated as previously described [42].
Surface plasmon resonance (AIS)
The experiments were performed at 30° C in a running buffer HBS-EP pH 7.4 (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.05% P20) supplemented 1 mM TCEP, 2% glycerol and 1% DMSO. PUMA BH3 peptide was purchased from Biopeptide LLC and immobilized on a CM-5 sensor chip. Standard amine coupling protocol of Biacore GE Healthcare has been applied with EDC/NHS coupling followed by ethanolamine deactivation. A channel treated in the same manner but in the absence of peptide was used as the reference channel. Compounds were tested in double-diluted series of twelve concentrations with top concentrations ranging from 20 nM to 2000 nM. Samples were preincubated with 1–2 nM BCL-2 in a running buffer and injected over the generated surfaces at a flow rate 15 μl/min for 300 sec. The calibration curve was generated using the same procedure but without added compound. Affinity evaluations were performed using the Affinity In Solution Model of BIAevaluation 2.1 (BIAcore GE Healthcare Bio-SciencesCorp) software.
Isothermal titration calorimetry (ITC)
The protein was dialyzed prior to use in 2 L of 25 mM HEPES, 150 mM NaCl, 5% glycerol (v/v), 0.5% MTG (v/v) at pH 7.4 for 3 hours at room temperature. The protein was dialyzed using a 3.5 K MWCO Slide-A-Lyzer™ cassette. The protein solution was recovered, spin-filtered through a 0.22 μM frit. The filtered protein was quantified by UV absorbance at 280 nm using an extinction coefficient of 36440 M-1cm-1. 500 ml of dialysis buffer was retained (remainder used to rinse out SCHOT bottle) and degassed for a minimum of 30 mins under vacuum with constant stirring. Typically, 50 μM protein was titrated from the syringe into the cell containing 5 μM compound. The solutions were carefully matched to ensure same solvent and buffer conditions were present in both syringe and cell (this is to minimize heat of dilution effects). The final solution was the dialysis buffer plus 1% DMSO and 0.05% P20. The experiment was performed over 13 injections with stirring at 1000 rpm, gain setting low and at 25º C. Pure water was used in the reference cell. The first injection was 0.5 μl with a duration of 1 second with a gap of 150 seconds until the further 12 injections of 3.05 μl with a duration 6.1 seconds and a 240 second interval between injections. Data was analyzed using the vendor supplied software, PEAQ-ITC Analysis Software. The data model used was the single site model. The data shown is the average of 2 or 3 independent experiments. For compounds with affinities greater than 10 nM it is not possible to accurately determine the potency by ITC, however the enthalpy and stoichiometry are accurately determined. The data shows that each compound binds with a 1:1 stoichiometry and with high enthalpies on BCL-2 demonstrating a highly specific interaction.
Crystallography
Crystals of the BCL-2 complex with S55746 were grown using BCL-2 (His6-BCL-2 based on the 2W3I construct with additional mutation G118Q) at a concentration of 9.4 mg/ml [20 mM Tris buffer pH 7.5, 100 mM NaCl, 2 mM DTT, 1% glycerol] by the hanging drop vapour diffusion technique. 2 μl of protein solution was mixed with 2 μl of crystallization reservoir (0.1 M sodium acetate buffer pH 5.25, 20% Jeffamine 600, 10% PEG3350) in a hanging drop plate. Crystallization was initiated by the addition of micro-crystals of the BCL-2/S55748 complex (S55748 is a methylated version of S55746). The new crystals, which grew over 2–3 days, were crushed and used for a second micro-seeding of a fresh mixture BCL-2/S55746 solution and crystallization reservoir. The plate was incubated at 284 K and bi-prism crystals appeared overnight and grew to a maximum ~50 μm along the edge over 4 days. Suitable crystals for data collection were flash frozen in liquid nitrogen after cryoprotection using crystallization reservoir. The diffraction data was collected at the Proximal station, Soleil Synchrotron, at wavelength 0.97857 Å using Pilatus-6 M detector. The crystal diffracted to 1.39 Å, P212121 space group and cell dimensions a = 35.09, b = 46.9, c = 85.04, α,β,γ 90,90,90 and mosaicity of 0.2 degrees and one molecule per asymmetric unit. Several cycles of refinement were conducted with Refmac5 software from the ccp4 package, alternating with manual rebuilding using the COOT software package. The final model consisted of 144 residues, S55746 ligand and 152 molecules of water. S55746 topology restraints were defined using the ProDrug software package.
Cell sorting and flow cytometry analysis
CLL cells were exposed for 4 h with S55746 (1, 3, 10, 30, 100 nM) before analysis of apoptosis using staining with AnnexinV-FITC/propodium iodide and flow cytometry. EC50 values were calculated using GraphPadPrism (non-linear fit, sigmoidal dose response, variable slope). Washed human platelets were cultured at a final density of 2.5 × 107/ml and exposed for 3 h with S55746 (3, 10, 30, 100, 300, 1000, 3000 nM) before staining with CD41-PE (BD) and Annexin-FITC and flow cytometry. Mononuclear MCL cells were cultured for 24 h in presence or absence of S55746 (concentration ranging from 0.1 nM to 100 nM) in RPMI supplemented with 10% FCS and 2 mM glutamine. Cell death was assessed in the CD19+ primary MCL cells by annexinV-FITC staining combined with an analysis of altered cellular morphology (lower FCS). Fluorescence was analyzed on FACSCalibur. The EC50 value was determined by the S55746 concentrations resulting in 50% of annexinV positive cells.
RS4;11 cells were treated with the indicated compounds for 2h, centrifuged and washed with binding buffer (10 mM Hepes, 140 mM NaCl, 2.5 mM CaCl2). Cells were incubated with 200 μl of binding buffer containing AnnexinV–FITC (Invitrogen) and propidium iodide (PI, Sigma) during 15 min at 20° C in the dark. THP-1 cells were incubated with 200 μl of binding buffer containing AnnexinV–APC (BD Biosciences) and DAPI (Sigma). 400 μl of binding buffer was added and samples were kept at 4° C before cytometric analysis. For each sample, 104 cells were analyzed by flow cytometry on Epics XL/MCL flow cytometer (Beckman Coulter, France). Fluorescence was collected at 520 (FITC), 630 nm (PI), 660 nm (APC) and 470 (DAPI). Numbers of apoptotic cells (addition of primary apoptosis, secondary apoptosis and necrosis) was normalized to the total numbers of cells per tubes.
CellTiter-Glo luminescent cell viability assay
Cells were seeded into 96-well plates and treated at 8 points with 1:2 serial dilution of compounds. Cell viability was assessed and assayed for viability using CellTiter-Glo reagent (Promega) following the manufacturer’s instructions. Plates were read using Tecan luminescence plate reader. Results were normalized to the viability of cells without compounds (control wells). The IC50 values were calculated using nonlinear regression algorithms in XCell software.
In vivo xenografts
Experiments were performed in SCID/beige female mice from Charles River Laboratories, Massachusetts, USA. Animals, approximately 7-8 weeks of age at the start of the treatment were allowed to acclimate in animal facility with access to food and water ad libitum for 3 days prior to manipulation. Animals were handled in accordance with IACUC regulations and guidelines for experiments performed in the USA and with European and French regulation for the protection of vertebrate animals for experiments performed in France. Animal well-being and behavior, including grooming and ambulation were monitored at least once a day. General health of mice was monitored and mortality recorded daily. Any moribund animals were sacrificed.
RS4;11 Acute Lymphoblastic Leukemia cell lines was obtained from ATCC. Toledo (Diffuse Large B-Cell Lymphoma) human cell lines were obtained internally through Bioresources and the Novartis-Cancer Cell Line Encyclopedia [43]. Cells were free of Mycoplasma and murine viral contamination in the IMPACT VIII PCR assay panel (IDEXX RADIL, IDEXX laboratories INC, Westbrook, ME, USA). Cells used for subcutaneous implantation were cultured in RPMI plus 10% FBS at 37° C in a humidified atmosphere containing 5% carbon dioxide). For RS4;11 culture, the medium was also supplemented with 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 mM Hepes and 4.5 g/L glucose. Cells were resuspended in a 1:1 mixture of cold DPBS (Dulbecco’s Phosphate-Buffered Saline) and Matrigel™ (Becton-Dickinson #354234) at a concentration of 3 × 107 cells/ml for Toledo cells, and of 1 × 107 cells/ml for RS4;11 cells.
For each experiment, female SCID/beige mice were implanted subcutaneously (right axillary region) with 3 × 106 Toledo or RS4;11 (1 × 106 cells for efficacy studies and 1 × 107 cells for pharmacodynamics studies) suspended in 1:1 mixture of cold DPBS and Matrigel™ in a total volume of 100 μl. Body weights were recorded and tumors were measured with digital calipers twice to three times a week . Tumor volume was calculated using the formula: length × width2/2. Percent changes in body weights was calculated as (BWcurrent - BWinitial)/(BWinitial) × 100. Data is presented as percent body weight change from the day of treatment initiation.
When tumors reached approximately 200 mm3 for efficacy studies or 300 mm3 for pharmacodynamic studies, mice were randomized. S55746 was formulated in PEG300/EtOH/water (40/10/50). ABT-199 was formulated in PEG300/EtOH/Phosal (30/10/60). Mice were treated via oral gavage at 10 ml/kg with the doses and schedules described in the figure.
For pharmacodynamic studies, blood and tumors were removed 16 h post dosing and immediately snap-frozen. Total proteins were extracted from the tumors and caspase-3 activity was assessed in triplicate using CaspACE® Assay System (Promega). Platelet cell counts were determined 16 hours post dosing using Coulter AcT diff (Beckman).
For efficacy studies, percent treatment/control (T/C) values were calculated using the following formula: %T/C = 100 × ΔT/ΔC if ΔT > 0. Regression = 100 × ΔT/Tinitial if ΔT < 0 where: T = mean tumor burden of the drug-treated group on the final day of the study; ΔT = mean tumor burden of the drug-treated group on the final day of the study – mean tumor burden of the drug-treated group on initial day of dosing; Tinitial = mean tumor burden of the drug-treated group on initial day of dosing; C = mean tumor burden of the control group on the final day of the study; and ΔC = mean tumor burden of the control group on the final day of the study – mean tumor burden of the control group on initial day of dosing. If the tumor volume was less than 14 mm3 for more than 3 consecutive measurements, animals were considered in complete regression (CR).
Statistical analysis
All in vivo data were expressed as mean ± standard error of the mean (SEM). Delta tumor volume and body weight were used for statistical analysis. Between-group comparisons were carried out using the Kruskal-Wallis ANOVA followed by a post hoc Dunn’s test or Tukey’s test. For all statistical evaluations, the level of significance was set at p < 0.05. Significance compared to the vehicle control group is reported unless otherwise stated.
Author contributions
Conception and design : A Bruno, P Casara, I Chen, GM Cohen, A Claperon, J Davidson, O Geneste, C Graham, JM Henlin, T Le Diguarher, E Morris, J Murray, JB Starck, Y Wang. Development of methodology: J Davidson, G Guasconi, G Le Toumelin-Braizat, J Murray, S Qiu, F Rocchetti, A Studeny, Y Wang, N Whitehead. Acquisition of data : P Dokurno, AM Girard, F Gravé, G Guasconi, G Le Toumelin-Braizat, G Lysiak-Auvity, S Maiga, N Matassova, J Murray, C Pedder, S Qiu, F Rocchetti, E Schneider, A Studeny, M Vogler, Y Wang. Analysis and interpretation of data : M Amiot, A Bruno, P Casara, A Claperon, O Geneste, AM Girard, F Gravé, T Le Diguarher, G Lysiak-Auvity, S Maïga, E Morris, J Murray, E Schneider, JB Starck, M Vogler, Y Wang. Writing, review and/or revision of the manuscript: M Amiot, M Chanrion, A Claperon, F Colland, T Le Diguarher, O Geneste, L Kraus-Berthier, E Morris, J Murray, JB Starck, Y Wang. Administrative, technical, or material support: J Davidson, MJS Dyer, T Le Diguarher, JM Henlin, R Hubbard, Le Gouill, JB Starck, Y Wang. Study supervision: A Claperon, J Davidson, O Geneste, JA Hickman, R Hubbard, L Kraus-Berthier, E Morris, J Murray, Y Wang, M Wood.
ACKNOWLEDGMENTS
We thank Peter Kierstan for protein production support, Mandy Fallowfield, Jalanie D’Alessandro and Heather Simmonite for analytical support, Stéphanie Giraudet and Stéphane Léonce for flow cytometry and cell sorting analysis.
CONFLICTS OF INTEREST
P Casara and JA Hickman are former employees of Institut de Recherches Servier. M Chanrion, A Claperon, F Colland, O Geneste, AM Girard, F Gravé, G Guasconi, JM Henlin, G Le Toumelin-Braizat, G Lysiak-Auvity, F Rocchetti, E Schneider, JB Starck and A Studeny are full-time employees of Institut de Recherches Servier. T Le Diguarher is a full-time employee of Technology Servier. A Bruno and L Kraus-Berthier are full-time employees of Institut de Recherches Internationales Servier. E Morris, S Qiu and Y Wang are full-time employees of Novartis Institutes for BioMedical Research; E Morris and Y Wang are stock owner of Novartis. S LeGouill has served on advisory board for Servier. Amiot and Cohen’s laboratories have received research funds from Servier. I Chen, J Davidson, P Dokurno, C Graham, N Matassova, J Murray, C Pedder, N Whitehead, M Wood are full-time employees of Vernalis Ltd. R Hubbard is a part-time employee of Vernalis Ltd.
GRANT SUPPORT
None to declare.
REFERENCES
1. Green DR, Llambi F. Cell Death Signaling. Cold Spring Harb Perspect Biol. 2015; 7.
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74.
3. Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984; 226:1097–9.
4. Tsujimoto Y, Yunis J, Onorato-Showe L, Erikson J, Nowell PC, Croce CM. Molecular cloning of the chromosomal breakpoint of B-cell lymphomas and leukemias with the t(11;14) chromosome translocation. Science. 1984; 224:1403–6.
5. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014; 15:49–63.
6. Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016; 16:99–109.
7. Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008; 9:47–59.
8. O’Neill KL, Huang K, Zhang J, Chen Y, Luo X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016; 30:973–88.
9. Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, Huang DC, Kluck RM, Adams JM, Colman PM. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013; 152:519–31.
10. Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov. 2017; 16:273–84.
11. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005; 435:677–81.
12. Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, Tulpule A, Dunleavy K, Xiong H, Chiu YL, Cui Y, Busman T, Elmore SW, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010; 11:1149–59.
13. Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DC, Xiong H, Cui Y, Busman TA, McKeegan EM, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012; 30:488–96.
14. Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008; 68:3421–8.
15. Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, Huang DC, Kile BT. Programmed anuclear cell death delimits platelet life span. Cell. 2007; 128:1173–86.
16. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013; 19:202–8.
17. Stilgenbauer S, Eichhorst B, Schetelig J, Coutre S, Seymour JF, Munir T, Puvvada SD, Wendtner CM, Roberts AW, Jurczak W, Mulligan SP, Bottcher S, Mobasher M, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016; 17:768–78.
18. Porter J, Payne A, de Candole B, Ford D, Hutchinson B, Trevitt G, Turner J, Edwards C, Watkins C, Whitcombe I, Davis J, Stubberfield C. Tetrahydroisoquinoline amide substituted phenyl pyrazoles as selective Bcl-2 inhibitors. Bioorg Med Chem Lett. 2009; 19:230–3.
19. Varadarajan S, Vogler M, Butterworth M, Dinsdale D, Walensky LD, Cohen GM. Evaluation and critical assessment of putative MCL-1 inhibitors. Cell Death Differ. 2013; 20:1475–84.
20. Le Diguarher T, Casara P, Starck JB, Henlin JM, Davidson JEP, Murray JB, Graham CJ, Chen IJ, Geneste O, Hickman J, Depil S, Le Tiran A, Nyerges M, et al. New indolizine compounds, a process for their preparation and pharmaceutical compositions containing them. 01/08/2013; WO2013110890A1.
21. Ferenczy GG, Keseru GM. Enthalpic efficiency of ligand binding. J Chem Inf Model. 2010; 50:1536–41.
22. Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood. 2008; 111:2300–9.
23. Shoemaker AR, Mitten MJ, Adickes J, Ackler S, Refici M, Ferguson D, Oleksijew A, O’Connor JM, Wang B, Frost DJ, Bauch J, Marsh K, Tahir SK, et al. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin Cancer Res. 2008; 14:3268–77.
24. Gottschalk AR, Boise LH, Oltvai ZN, Accavitti MA, Korsmeyer SJ, Quintans J, Thompson CB. The ability of Bcl-x(L) and Bcl-2 to prevent apoptosis can be differentially regulated. Cell Death Differ. 1996; 3:113–8.
25. Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007; 117:112–21.
26. Chiron D, Dousset C, Brosseau C, Touzeau C, Maïga S, Moreau P, Pellat-Deceunynck C, Le Gouill S, Amiot M. Biological rational for sequential targeting of Bruton tyrosine kinase and Bcl-2 to overcome CD40-induced ABT-199 resistance in mantle cell lymphoma. Oncotarget. 2015; 6:8750–59. https://doi.org/10.18632/oncotarget.3275.
27. Huang JZ, Sanger WG, Greiner TC, Staudt LM, Weisenburger DD, Pickering DL, Lynch JC, Armitage JO, Warnke RA, Alizadeh AA, Lossos IS, Levy R, Chan WC. The t(14;18) defines a unique subset of diffuse large B-cell lymphoma with a germinal center B-cell gene expression profile. Blood. 2002; 99:2285–90.
28. Iqbal J, Neppalli VT, Wright G, Dave BJ, Horsman DE, Rosenwald A, Lynch J, Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Campo E, Ott G, et al. BCL2 expression is a prognostic marker for the activated B-cell-like type of diffuse large B-cell lymphoma. J Clin Oncol. 2006; 24:961–8.
29. Coustan-Smith E, Kitanaka A, Pui CH, McNinch L, Evans WE, Raimondi SC, Behm FG, Arico M, Campana D. Clinical relevance of BCL-2 overexpression in childhood acute lymphoblastic leukemia. Blood. 1996; 87:1140–6.
30. Irish JM, Anensen N, Hovland R, Skavland J, Borresen-Dale AL, Bruserud O, Nolan GP, Gjertsen BT. Flt3 Y591 duplication and Bcl-2 overexpression are detected in acute myeloid leukemia cells with high levels of phosphorylated wild-type p53. Blood. 2007; 109:2589–96.
31. Marschitz I, Tinhofer I, Hittmair A, Egle A, Kos M, Greil R. Analysis of Bcl-2 protein expression in chronic lymphocytic leukemia. A comparison of three semiquantitation techniques. Am J Clin Pathol. 2000; 113:219–29.
32. Calin GA, Cimmino A, Fabbri M, Ferracin M, Wojcik SE, Shimizu M, Taccioli C, Zanesi N, Garzon R, Aqeilan RI, Alder H, Volinia S, Rassenti L, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008; 105:5166–71.
33. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005; 102:13944–9.
34. Garraway LA, Janne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012; 2:214–26.
35. Fresquet V, Rieger M, Carolis C, Garcia-Barchino MJ, Martinez-Climent JA. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood. 2014; 123:4111–9.
36. Tahir SK, Smith ML, Hessler P, Rapp LR, Idler KB, Park CH, Leverson JD, Lam LT. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer. 2017; 17:399.
37. Lin KH, Winter PS, Xie A, Roth C, Martz CA, Stein EM, Anderson GR, Tingley JP, Wood KC. Targeting MCL-1/BCL-XL Forestalls the Acquisition of Resistance to ABT-199 in Acute Myeloid Leukemia. Sci Rep. 2016; 6:27696.
38. Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, Baell JB, Colman PM, Deshayes K, Fairbrother WJ, Flygare JA, Gibbons P, Kersten WJ, et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat Chem Biol. 2013; 9:390–7.
39. Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, Belmont LD, Nimmer P, Xiao Y, Ma XM, Lowes KN, Kovar P, Chen J, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. 2015; 7:279ra40.
40. Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, Bruno A, Csekei M, Paczal A, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016; 538:477–82.
41. Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, Nimmer P, Jin S, Smith M, Xiao Y, Kovar P, Tanaka A, Bruncko M, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015; 6:e1590.
42. Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito NG, Stuckey JA, Wang S. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal Biochem. 2004; 332:261–73.
43. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483:603–7.