
INTRODUCTION
Peripheral T-cell lymphoma (PTCL) is an aggressive subset of Non-Hodgkin’s lymphoma (NHL) with poor clinical outcomes, affecting ~7,000 individuals in the United States annually [1, 2]. The World Health Organization recognizes >25 subtypes of PTCL; each further characterized by heterogeneity including oncogenic mutations [1, 3, 4]. For the majority of these PTCL subtypes, patient mortality is not only high, but relapse is common and 5-year overall survival is 10-30% [2, 5, 6]. PTCL is currently treated with chemotherapy regimens designed for aggressive NHLs, but the rate and duration of response remain inferior to those of the B-cell lymphomas for which these treatments were designed [3, 7]. Recent clinical trials for treatment of PTCL include many novel agents, in addition to adaptations of traditional chemotherapy regimens. Novel therapeutic approaches under study include histone deacetylase (HDAC) inhibitors, immunoconjugates, antifolates, monoclonal antibodies, immunomodulatory agents, nucleoside analogues, proteasome inhibitors, and kinase inhibitors. Progress has been complicated by low numbers of PTCL patients and randomized prospective phase III data is currently limited, making it difficult to inform clinical management [5]. As such, there has been an increasing interest in identifying and developing additional therapeutic targets and treatment approaches to improve PTCL patient outcomes.
Research has been dedicated to the identification of potential oncogenic mutations and novel therapeutic targets in PTCL [8–19]. Several of these studies have revealed a recurrence of activating, oncogenic mutations in the JAK/STAT signaling pathway, specifically in the signal transducer and activator of transcription (STAT) STAT5B and STAT3 transcription factors, in multiple subtypes of PTCL [11, 15, 20–28]. The JAK/STAT signaling pathway plays an important role in T-cell activation during the immune response [29]. In physiologic JAK/STAT signaling through the common gamma chain, cytokine binding to the receptor complex triggers intracellular domain associated JAK1/3 molecules to autophosphorylate and activate the receptor, stimulating the recruitment and phosphorylation of STATs [30]. Activated STAT molecules dimerize and enter the nucleus to regulate the transcription of genes involved in cell proliferation and survival. Constitutive pathway activation, through mutation or overexpression of JAK1, JAK3, STAT3, STAT5, and the common gamma chain, have been reported in PTCL and shown to be oncogenic [11, 15, 31–34]. Of interest to our research, activating STAT5 mutations have been observed in multiple PTCL subtypes at frequencies of 21-36% in T-cell prolymphocytic leukemia (T-PLL), 33% in hepatosplenic T-cell lymphoma (HSTL), 63% in epitheliotropic intestinal T-cell lymphoma (EITL), 3% in Sézary syndrome (SS), and 2% in T-cell large granular lymphocytic leukemia (T-LGL) [11, 15, 20, 22–24]. The activating STAT5B mutation, N642H, which is observed repeatedly in PTCL, leads to a more aggressive clinical course in T-LGL and a significantly reduced probability of event free survival and an increase in cumulative incidence of relapse in T-cell acute lymphoblastic leukemia (T-ALL) [11, 35]. Activation of STAT3, both by mutation and overexpression, has also been observed in multiple PTCL subtypes [8, 10]. This is most prevalent in T-LGL, at a frequency of 40%, and also leads to poor patient outcomes [8, 10]. These studies demonstrate the substantial frequency of oncogenic STAT mutations in PTCL, particularly mutations in STAT5B.
Due to this prevalence of STAT5 mutations and their negative prognostic indications, inhibition of STAT molecules may be an appealing therapeutic approach in PTCL. JAK inhibitors have demonstrated clinical efficacy in hematologic malignancies driven by JAK activation, which has also been commonly reported in PTCL [36, 37]. However, the efficacy of JAK inhibition may be limited in cases with constitutively active STAT5, as STAT5 is downstream of these inhibitors’ site of action. Thus, there is substantial interest in pursuing the development of downstream STAT5 inhibitors for the treatment of STAT5-driven PTCL. A high throughput screen identified the drug pimozide to specifically inhibit STAT5 in chronic myelogenous leukemia and acute myeloid leukemia with FLT3 mutations [38, 39]. Pimozide is FDA approved as a neuroleptic, although its exact mechanism of action remains unknown. We sought to gain a more complete understanding of the therapeutic potential of STAT5 inhibition in PTCL in order to help advance the development of STAT5 inhibitors for ultimate clinical application.
Our study furthers this goal by assessing the effect of STAT5 inhibition in PTCL. We evaluate the utility of STAT5 inhibition in PTCL cell lines as well as primary T-PLL patient samples and shed light on the mechanism by which pimozide induces PTCL cell death. This research aims to inform the development of STAT5 inhibitors as a novel therapeutic approach in this malignancy.
RESULTS
STAT5 is constitutively active in PTCL
To study the inhibition of STAT5 in PTCL, we identified patient derived cell line models. Analysis of six malignant T-cell lines cultured in the absence of cytokine stimulation revealed three lines, HuT102, HuT78, and Kit225, with high constitutive STAT5 activation (Figure 1). HuT102 and HuT78 were derived from PTCL patients with mycosis fungoides and SS subtypes, respectively, and are cytokine independent [40]. IL-2-dependent Kit225 cells were derived from a T-PLL patient, the same PTCL subtype as the patient samples included in this study [41]. We thus focused our analysis on Kit225 cells, as well as the HuT102 line based on its comparable level of STAT5 expression. .
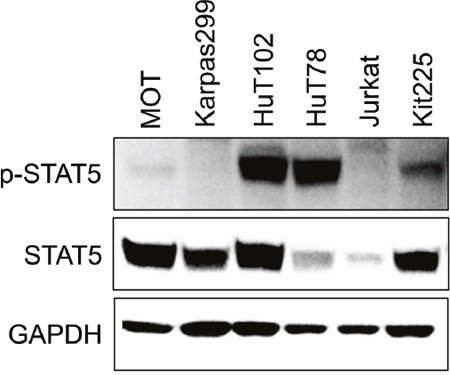
Figure 1: STAT5 is constitutively active in multiple PTCL cell lines. Western blots show STAT5 and phospho-STAT5 expression in six T-cell malignancy derived cell lines in absence of cytokine stimulation.
Pimozide inhibits STAT5 in PTCL
To determine the ability of pimozide to inhibit STAT5 activation in PTCL, we cultured HuT102 and Kit225 cells in a range of drug concentrations for 48h [38, 39]. Immunoblots for p-STAT5 protein expression revealed a concentration dependent decrease in STAT5 phosphorylation (Figure 2A). We quantified the number of viable PTCL cells after culture with pimozide using resazurin dye assay and demonstrated a drug dose-dependent decrease in PTCL cell viability, which corresponded to the observed reduction in STAT5 activation for both cell lines (Figure 2A-2B). We calculated an IC50 for pimozide of 15μM in Kit225 and 11μM in HuT102 cells.
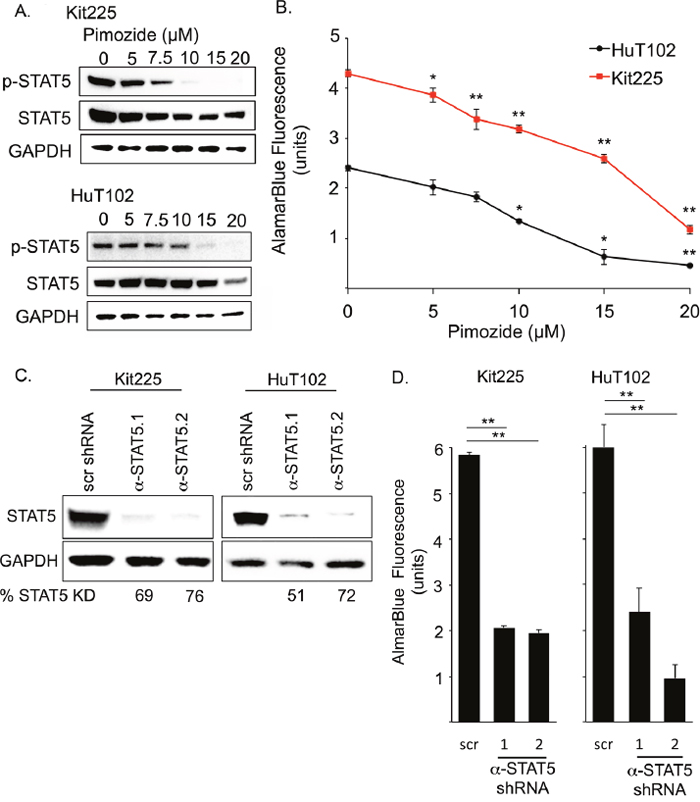
Figure 2: Disruption of STAT5 activity reduces PTCL cell viability. (A) Western blots show phospho-STAT5 (Y694) (90kDa), STAT5 (90kDa), and GAPDH (37kDa) protein expression in PTCL cell lines. (B) Resazurin dye assay (AlamarBlue©) quantifies change in viable PTCL cell number after 48h with increasing pimozide concentrations. (C) Western blots show STAT5 expression 6 days after transduction with anti-STAT5 targeted shRNA (α-STAT5.1 and 2) in PTCL cell lines compared to non-targeted scramble (scr) control. (D) Graph of AlamarBlue© assay quantifies PTCL cell viability. Significant p-values *=P<0.05, **=P<0.01.
To explore whether this reduction in PTCL cell viability was due to STAT5 inhibition, we assessed the effect of STAT5 knockdown on PTCL cell viability. Two anti-STAT5 shRNA constructs reduced total STAT5 protein expression by >50% in all experiments (Figure 2C). Both PTCL cell lines demonstrated an approximately 2/3 reduction in viable cell number after STAT5 knockdown, as compared to each cell line transduced with control shRNA (Figure 2D). These experiments show that reduction of STAT5 expression reduces PTCL cell viability.
Pimozide induces apoptosis in PTCL cells
We demonstrated that PTCL cell viability is diminished in response to inhibition of STAT5 phosphorylation by pharmacologic and genetic means. To determine the mechanism by which pimozide reduces viable PTCL cell number, we analyzed PTCL cell death for evidence of apoptosis over a range of pimozide concentrations. PTCL cells were cultured with pimozide for 48h and assessed by flow cytometry. Evidence of early apoptotic cell death includes positive stain by AnnexinV, while remaining 7-AAD negative. Double positive cells are indicative of late-stage apoptosis or another cell death mechanism [42]. Our results demonstrated pimozide induced a concentration dependent increase in the proportion of PTCL cells undergoing apoptosis (Figure 3A). Apoptotic Kit225 cells increased from 2.5% to >30% with 20μM of pimozide compared to control. In Hut102 cells, apoptosis increased ~5 fold, from <5% to >25% of cells. Cell death due to apoptosis for both cell lines was furthermore confirmed by immunoblot for caspase 3 cleavage (Figure 3B).
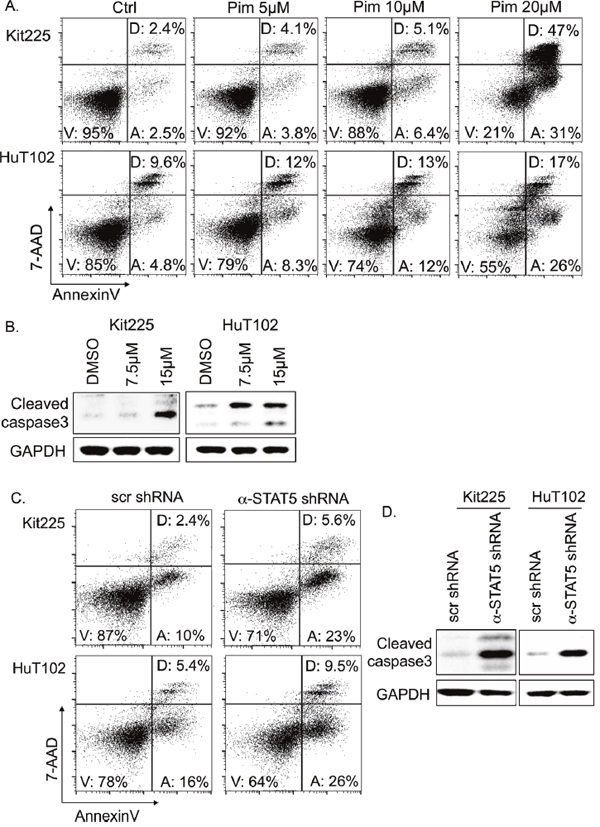
Figure 3: Apoptosis in PTCL cells following STAT5 knockdown and inhibition. (A) Flow cytometry identifies percentage of early apoptotic (“A”), viable (“V”), and dead/late apoptotic cells (“D”) by APC-AnnexinV/7-AAD following 48h with indicated pimozide concentrations. At least 40,000 events were collected. (B) Western blots show protein expression of cleaved caspase 3 (17 and 19kDa) versus GAPDH control (37 kDa). (C) Flow cytometry indicates proportion of apoptotic cells 6 days after anti-STAT5 shRNA knockdown verses control (scr). (D) Western blots show protein expression of cleaved caspase 3 versus GAPDH.
To investigate if induction of apoptosis is related to pimozide’s STAT5 inhibition, we also assessed PTCL cell survival after STAT5 knockdown. In Kit225 cells expressing <30% of endogenous STAT5 levels, apoptotic cells were increased from 10% to 23% verses control (Figure 3C). Apoptotic HuT102 cells increased from 16% to 26% after STAT5 knockdown. Apoptosis was confirmed as the mechanism of cell death in both cell lines using immunoblot for cleaved caspase 3 activation (Figure 3D).
STAT5 knockdown does not decrease BCL-2 family member expression in PTCL cell lines
To evaluate the mechanism by which STAT5 knockdown and inhibition with pimozide induced apoptosis in PTCL, we next studied molecules influencing apoptosis that are subject to transcriptional regulation by STAT5 in human T-cells [43–47]. Apoptotic regulators regulated by STAT5 in human CD4+ T-cells included six candidate molecules: MCL-1, BCL-2, BCL-xL, TRAF1, TP53BP2, and caspase 8 (Casp8), [47, 48]. The mRNA expression of these candidate molecules was assessed in PTCL cell lines after STAT5 knockdown and no significant differences in expression were observed compared to control conditions (Figure 4A). Similarly, protein levels of anti-apoptotic BCL-2 family members BCL-xL, BCL-2, and MCL-1 were not significantly altered, despite indication that STAT5 regulates apoptosis via their transcriptional downregulation in other models (Figure 4B) [45, 47, 49–51].
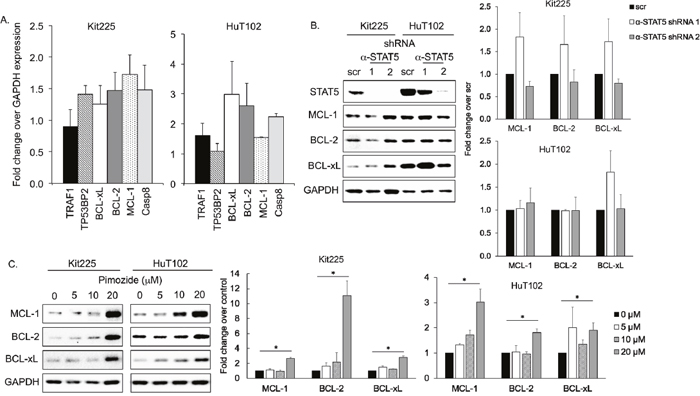
Figure 4: Expression of anti-apoptotic BCL-2 family after STAT5 knockdown/inhibition. (A) mRNA expression of six apoptotic mediators regulated by STAT5 relative to GAPDH control by RT qPCR in PTCL cell lines after STAT5 knockdown. (B) Western blots show protein expression of three anti-apoptotic BCL-2 family members after STAT5 knockdown with shRNA (α-STAT5.1 and 2) versus control (scr). Quantification bar graphs are shown to the right. Variations are not statistically significant. (C) MCL-1 (40 kDa), BCL-2 (28 kDa), BCL-xL (30 kDa), and GAPDH protein expression after 48h in indicated concentration of pimozide. Quantification bar graphs are shown to the right, *=P<0.05.
We also assessed BCL-2, BCL-xL, and MCL-1 protein expression in HuT102 and Kit225 after culture with pimozide. Our data demonstrated no significant change in expression by immunoblot when cultured with 5-10μM of pimozide for 48h (Figure 4C). Protein expression of these anti-apoptotic molecules were increased following 20μM pimozide, which may be compensatory or due to general organelle decomposition. Our results do not support a role for BCL-2 family members contributing to apoptosis following STAT5 inhibition in our PTCL models.
We next sought to determine if increased apoptosis was due to a collapse of metabolic mitochondrial function by assessing the bioenergetic profile of PTCL cell lines through ATP production in the context of STAT5 inhibition. ATP bioluminescent assay demonstrated that, in HuT102 and Kit225 cell lines, ATP production decreases in a pimozide dependent manner (Supplementary Figure 1). ATP production was also significantly reduced in both cell lines following STAT5 knockdown (Supplementary Figure 2). These data suggest that inhibition of STAT5 interferes with metabolic function of PTCL cell lines. Based on these results, pimozide likely induces apoptosis in PTCL cells by means of interference with cellular metabolism outside of the context of BCL-2 family survival protein expression.
Pimozide induces apoptosis via the TRAIL/DR4 dependent extrinsic apoptotic pathway
We subsequently explored whether the mechanism of apoptosis involves the intrinsic/mitochondrial pathway or extrinsic/death receptor pathway. First, we assessed changes in mitochondrial membrane potential (MMP) in apoptotic PTCL cells by JC-1 dye [52]. Accounting for slight decrease in MMP with advanced stages of apoptosis, our results indicate no significant change in MMP in pre-apoptotic PTCL cells cultured with pimozide versus control (Figure 5A). These results do not support the intrinsic pathway of apoptosis as a mechanism of cell death in PTCL cells treated with pimozide.
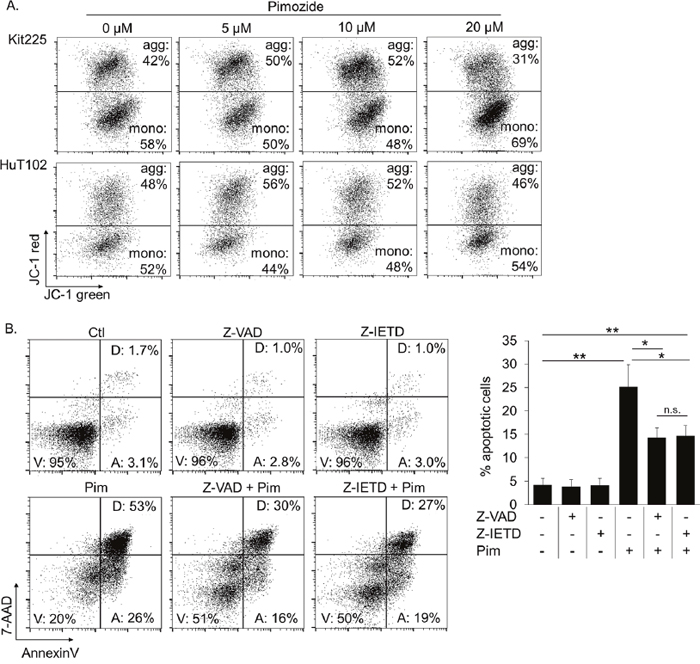
Figure 5: Pimozide induced apoptosis is caspase 8 dependent. (A) AnnenxinV negative pre-gated FACS plots show proportion of mitochondria with viable (JC-1 hi, aggregates) versus compromised (JC-1 red low, monomers) membrane integrity following 24h pimozide. (B) Proportion of PTCL cells undergoing apoptosis following 24h pimozide 20μM +/- pretreatment with caspase 8 inhibitor, Z-IETD, or pan-caspase inhibitor, Z-VAD. Gates “V,” “A,” and “D” contain viable, early apoptotic, and late apoptotic/dead cells, respectively. Significance indicated *=P<0.005, **=P<0.0005.
We next investigated the extrinsic pathway as a potential mechanism for pimozide induced apoptosis. The extrinsic apoptotic pathway signals through death receptors to activate caspases, bypassing the mitochondria [53–55]. Caspase 8 activation is exclusive to the extrinsic pathway. We compared the abilities of a caspase 8 inhibitor, Z-IETD-FMK, and a pan-caspase inhibitor, Z-VAD-FMK, which prevents apoptosis through both extrinsic and intrinsic pathways, to prevent apoptosis in pimozide treated PTCL cells. Significantly fewer pimozide treated cells underwent apoptosis after pretreatment with either of the caspase inhibitors, indicating that apoptosis is occurring by means of caspase 8 activity in this context (Figure 5B). Thus, our data suggest that pimozide induces cell death in PTCL cells by means of the extrinsic apoptotic pathway.
The extrinsic pathway is initiated by ligand (FasL, TNF-α, TRAIL) binding to corresponding death receptors (FAS, TNFαR, DR4/DR5) on the cell surface [55]. To investigate this pathway’s involvement further, we assessed several cell surface death receptors after culture with pimozide for changes in expression. We observed that expression of the TRAIL death receptor DR4 (TRAIL-R1) was upregulated in a pimozide concentration-dependent manner (Figure 6A). FAS expression was also upregulated on the surface of Kit225 cells. Combination of pimozide and TRAIL enhanced Kit225 cell death in vitro (Figure 6B). Addition of a TRAIL neutralizing antibody restored cells to near baseline levels of apoptosis, supporting that this cell death is TRAIL dependent (Figure 6C). These results suggest that TRAIL/DR4 signaling may be involved in the mechanism of pimozide induced apoptosis in PTCL cells.
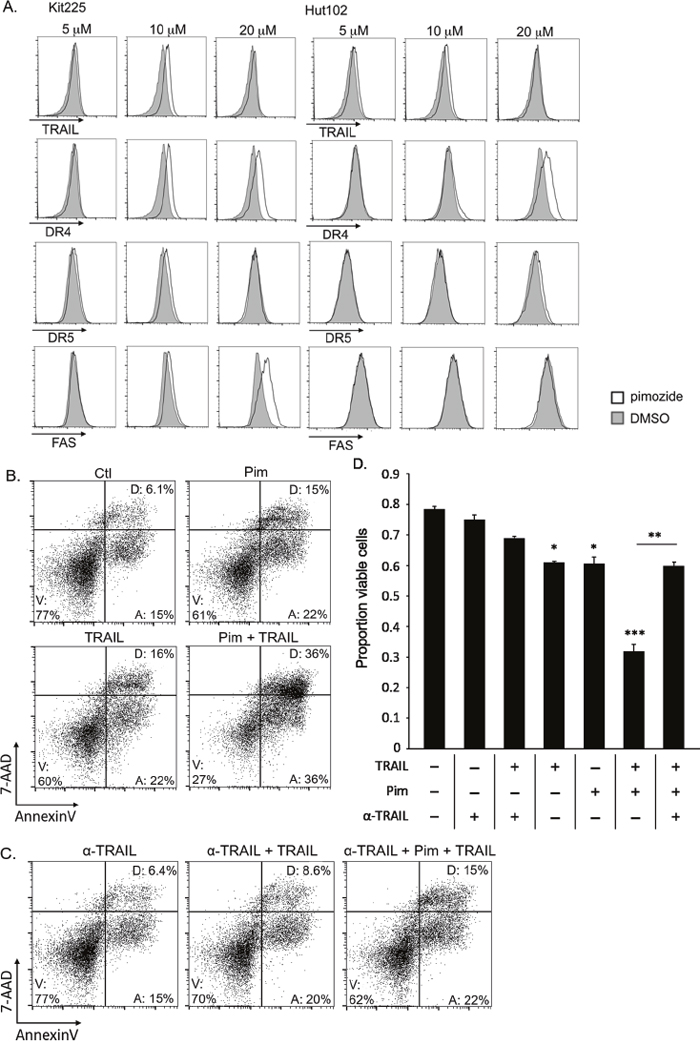
Figure 6: Pimozide enhances TRAIL/DR4 dependent apoptosis in PTCL. (A) Histograms show difference in TRAIL, DR4, DR5, and FAS surface expression on AnnexinV negative Kit225 and HuT102 cells after 48h pimozide (white) versus control (gray). (B) FACS plots show viable Kit225 cells with combination of 15μM pimozide and 10 ng/mL TRAIL after 24h. (C) FACS plots show viable cells from same experiment shown above with addition of TRAIL neutralizing antibody (α-TRAIL). (D) Bar graph quantifies viable (AnnexinV, 7-AAD negative) PTCL cells from 3 independent experiments shown in parts B and C. The 4th, 5th, and 6th bars are significant compared to the first three control bars at P value indicated, *=P<0.05, **=P<0.01, ***=P<0.005.
Pimozide inhibits STAT5 and induces apoptosis in primary patient PTCL
To assess our findings in patient primary malignant PTCL cells, we investigated the effect of pimozide on T-PLL patient samples ex vivo. Patient samples maintained under dimethyl sulfoxide (DMSO) vehicle conditions had substantial STAT5 phosphorylation, supporting the prevalence of STAT5 activation in T-PLL (Figure 7A). In comparison, pimozide reduced p-STAT5 expression by ≥70% in all samples. These results support that pimozide inhibits STAT5 in primary PTCL, consistent with the findings from established cell lines. Pimozide also reduced the number of viable primary PTCL cells, resulting in a 74-94% decrease in viability verses control, due to increased apoptotic cell death (Figure 7B-7C). Percent of apoptotic primary PTCL cells increased in all samples by an average of ~20% versus control. Our data, therefore, support pimozide induced STAT5 inhibition in primary patient PTCL.
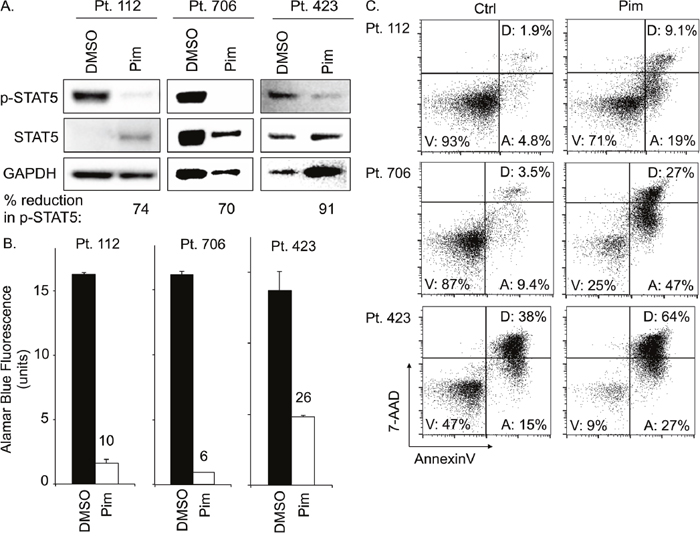
Figure 7: Pimozide inhibits STAT5 activation and induces apoptosis in primary PTCL patient samples. (A) Protein expression of phospho-STAT5, STAT5, and GAPDH for three different ex vivo PTCL patient samples (T-PLL subtype) after 24h pimozide 20μM versus control (Ctrl). (B) AlamarBlue© assay quantifies viable cells from PTCL patient samples after 48h pimozide versus control. (C) FACS plots show proportion of apoptotic patient PTCL cells (“A”) after 48h ex vivo culture with 20μM pimozide versus control.
DISCUSSION
We explore STAT5 as a therapeutic target in PTCL. Activating STAT5 mutations have been observed in multiple PTCL subtypes and are associated with a more aggressive clinical course [11, 15, 20, 22-25, 35]. In hematologic malignancies with activating JAK mutations, JAK inhibitors have proved clinically useful, however, they target upstream of STAT5 and may be ineffective in PTCL driven by activating STAT5 mutations [15, 36, 37]. Thus, STAT5 inhibition is a promising approach. We show that p-STAT5 is important in propagation of PTCL, as studied in two cell lines and in three patient samples. When inhibited by pharmacologic or genetic means, PTCL cell viability is reduced through induction of TRAIL mediated apoptosis. These results demonstrate that pimozide inhibits STAT5 and support the utility of STAT5 inhibition as a therapeutic strategy in PTCL.
We provide initial evidence of a mechanism by which STAT5 inhibition with pimozide induces apoptosis. Previous research demonstrates that pimozide decreases viability of two T-cell lines and two T-PLL patient cases in vitro [15], and the work presented here extends those findings to include a mechanism for evidence of cell death. We show that pimozide reduces PTCL cell viability in two additional cell lines and three T-PLL patient samples and this induction of apoptosis is caspase 8 and TRAIL dependent, associated with upregulation of the cell surface expression of TRAIL death receptor, DR4. These results support that pimozide induces apoptosis in PTCL cells via the extrinsic, TRAIL/DR4 dependent, apoptotic pathway. A study by Kanai, et. al. utilized chromatin immunoprecipitation with sequencing (ChIP-seq) with qPCR validation to identify STAT5A and STAT5B targeted genes in human CD4+ T-cells following 3 days in culture with IL-2 [47]. Their data show that TRAIL, also known as TNFSF10, is dominantly regulated by STAT5B. STAT5B was found to bind directly to the regulatory sequence “TTCCAAGAA” in the TRAIL promoter. These findings, in conjunction with our own, support that TRAIL induced cell death may be regulated by STAT5 and suggest a mechanism for apoptosis induced by STAT5 inhibition. In context, our results provide insight into targeting PTCL cells and improve our understanding of an incompletely characterized pharmaceutical for STAT5 inhibition.
It is noteworthy that BCL-2, BCL-xL, and MCL-1 do not appear to play a role in the induction of apoptosis following STAT5 knockdown or inhibition in our analysis. Prior research by others suggests that STAT5 knockdown triggers apoptosis through anti-apoptotic BCL-2 signaling via the intrinsic pathway in various hematologic malignancies and non-malignant T-cells [45, 47, 49–51]. However, in our study, BCL-2, BCL-xL, and MCL-1 expression were not decreased after STAT5 knockdown. MMP was also not affected by STAT5 inhibition, suggesting that pimozide does not induce apoptosis via the intrinsic, BCL-2 family dependent, pathway. This finding may represent a mechanism unique to pimozide and/or PTCL.
Targeting the JAK/STAT and apoptotic pathways in combination presents a promising therapeutic approach in PTCL. Combination therapies with BCL-2/BCL-xL inhibitors, targeting the intrinsic pathway, and JAK inhibitor ruxolitinib have demonstrated synergy in pre-clinical models of adult T-cell leukemia lymphoma and T-ALL [25, 56, 57]. Our research suggests that targeting the extrinsic pathway may offer a mechanistic advantage and implicates a role for combination of STAT5 inhibition with death receptor signaling agonists as a future therapeutic approach in PTCL, although effective TRAIL agonists are still in development [58].
It is important to note, that while inhibition of STAT5 is promising for PTCL therapy, alternative STAT5 inhibitors, aside from pimozide, may have greater clinical potential. Pimozide is FDA approved and has been used for decades as a neuroleptic with manageable side effects. Clinical dosing, however, is recommended not to exceed 10 mg/day and it is unknown if pimozide has any effect as a STAT5 inhibitor in vivo. Novel applications of pimozide are currently being studied in just over a dozen clinical trials for psychotic and neurologic disorders including schizophrenia, ALS, and tic disorders (clinicaltrials.gov). Furthermore, pimozide has not been shown to be a direct STAT5 inhibitor [38, 39]. Other STAT5 inhibitors have been developed specifically for direct, targeted STAT5 inhibition. Promising among these is a compound referred to as 13a, which was developed using in silico modeling and demonstrated to specifically inhibit STAT5 with an IC50 of ~3.5μM in a FLT3-ICD driven cell line [59]. Application of this and other novel specific targeted STAT5 inhibitors in primary patient PTCL samples will be an important future direction of this research before clinical utility can be assessed.
Although substantial pre-clinical research remains to be conducted with STAT5 inhibitors prior to their application in clinical trials, clinical trials with many other agents are ongoing for PTCL. As a result of recent trials, two HDAC inhibitors, romidepsin and belinostat, have been approved by the FDA for use in relapsed/refractory PTCL [3, 60, 61]. Immunoconjugates, such as brentuximab vedotin, have been studied in clinical trials in PTCL patients with promising results [62–65]. Several monoclonal antibodies are also in development. Alemtuzumab (anti-CD52), mogamulizumab (anti-CCR4), and zanolimumab (anti-CD4) have all been shown to induce a complete remission in a fraction of patients with relapse/refractory PTCL [3, 66, 67]. Studies of other agents such as lenalidomide, nucleoside analogs, proteasome inhibitors, the mTOR inhibitor everolimus, and the JAK inhibitor ruxolitinib are also currently in clinical trials for PTCL [1-3, 68-70]. These agents may demonstrate clinical utility alone, or in combination with current therapeutic options.
In addition to activating mutations in STAT5, JAK mutations have also been identified in PTCL. Constitutively activating mutations in JAK kinases, JAK1 and JAK3, have been observed in approximately one third to one half of cases of the T-PLL subtype of PTCL [15, 24, 34, 71, 72]. Furthermore, T-PLL patients with activating JAK3 mutations have experienced inferior clinical outcomes, including significantly decreased overall survival [15]. This observation supports application of JAK-targeted tyrosine kinase inhibitors in PTCL. JAK inhibitors ruxolitinib and tofacitinib have been shown to limit the proliferation of several different malignant T-cell lines as well as primary patient cells expressing activated JAK mutations ex vivo [25, 32, 73–76]. It is important to note, however, that JAK inhibitor efficacy is dependent on the mechanism of JAK/STAT pathway activation. In PTCL cases where STATs are directly activated without involving upstream signaling, JAK inhibitors have proven ineffective [15, 21, 77, 78]. Combination of JAK and STAT5 inhibition may increase efficacy and minimize development of drug resistance. Our future research will explore some of these applications.
In conclusion, this study determines that targeting STAT5 is an effective therapeutic approach in PTCL and has the potential to be further developed for clinical application. We show that inhibition of STAT5 induces apoptosis in primary PTCL cells and are the first to demonstrate that STAT5 inhibition with pimozide induces apoptosis via the TRAIL/DR4-dependent extrinsic pathway. Our novel understanding of this mechanism may provide opportunities for new clinical and research applications. Our data suggest that PTCL patients with activating STAT5 mutations could benefit from STAT5 inhibitors either as mono-therapy or in combination with additional therapeutic strategies to provide more efficacious, less toxic treatment.
MATERIALS AND METHODS
Primary PTCL specimens
Peripheral blood specimens were collected from PTCL patients at the University of Maryland Greenebaum Comprehensive Cancer Center with the approval of the University of Maryland, Baltimore Institutional Review Board (UMB IRB). Written consent was obtained from all patients using a UMB IRB approved consent procedure. Peripheral blood mononuclear cells were isolated from each specimen by Ficoll gradient centrifugation according to the manufacturer’s protocol (GE Healthcare). Cells were cultured for up to 48h with IL-2 stimulation.
Flow cytometry
We performed flow cytometry on PTCL cell lines, HuT102 and Kit225, as well as primary PTCL samples. Cells were stained with AnnexinV-APC (eBioscience, Santa Clara, CA) and 7-AAD (7-amino actinomycin D) viability solution (BioLegend, San Diego, CA) to assay for apoptosis. MMP was assayed by staining with JC-1 dye (eBioscience) and AnnexinV. Cell surface expression of death receptors was determined by flow cytometry with separate stains using PE-conjugated anti-human CD253 (TRAIL; clone: RIK-2), PE-conjugated anti-human CD261 (DR4, TRAIL-R1; clone: DJR1), PE-conjugated anti-human CD262 (DR5, TRAIL-R2; clone: DJR2-4), FITC-conjugated anti-human CD95 (FAS; clone: DX2) (BioLegend).
Cell culture and conditions
Malignant human T-cell lines studied include Jurkat, Karpas299, HuT78, MoT, and HuT102, supplied by ATCC, and Kit225 provided by Dr. Thomas Waldmann at the National Institutes of Health. All lines were cultured in RPMI1640 medium with 10% FBS and maintained at a density below 1x106 cells/mL. Kit225, an IL-2 dependent cell line, was cultured with 100U/mL of IL-2. Primary patient PTCL cells were cultured in RPMI with 10% FBS and 100U/mL of IL-2 for ex vivo experiments. PTCL cells were treated with pimozide (Sigma Aldrich, St. Louis, MO) at a range of concentrations from 5-20μM for 24-48h with DMSO vehicle not exceeding 0.01% in culture. Kit225 cells were cultured with recombinant human TRAIL at 1-10 ng/mL +/- pimozide for 24h (Biolegend). TRAIL neutralization was performed with human anti-TRAIL antibody at 10μg/ml for 24h (clone # 75411, R&D Systems).
Anti-STAT5 shRNA knockdown
Four different anti-STAT5A and four different anti-STAT5B human lentiviral shRNA sequences in puromycin selective pLKO.1 plasmids were obtained from The RNAi Consortium (GE Dharmacon; clone IDs TRCN0000019304-8, TRCN0000019354-8). A control scramble shRNA, not targeted to the human genome, also in pLKO.1 plasmid was provided by Dr. Tami Kingsbury at the University of Maryland, Baltimore. Anti-STAT5 lentivirus for transduction was generated by calcium phosphate transfection of HEK293T cells as previously described using 7.5μg of shRNA plasmid, 5μg of pSP2, and 2.5μg of pMD2G viral helper plasmids [79]. PTCL cell lines were transduced with each anti-STAT5 shRNA plasmid using RetroNectin® according to the manufacturer’s Supernatant Infection Method protocol (Takara, Japan). Clones TRCN0000019354 and TRCN0000019357, referred to as anti-STAT5 shRNA 1 and 2, respectively in this study, consistently produced the most efficient knockdown of total STAT5 expression and were selected for subsequent experiments.
Western blot analysis
Cells were lysed with NP-40 lysis buffer and protein concentration was determined by Bradford assay. Samples were subjected to gel electrophoresis and transferred to PVDF membrane by dry transfer (iBlot; Invitrogen). Membranes were blocked for 1h with 5% BSA. Primary antibodies for p-STAT5 (Y694), GAPDH (14C10), BCL-xL, BCL-2 (D55G8), MCL-1 (D5V5L) and cleaved caspase 3 (D175) (Cell Signaling Technology) and STAT5 (sc-835, Santa Cruz Biotechnology) were used. Primary antibodies were diluted 1:1000 in 1% BSA and incubated overnight at 4°C. Anti-rabbit IgG HRP secondary antibody (Cell Signaling Technology) diluted 1:2000 was incubated for 1h at room temperature. Blots were developed using ECL Prime Western Blotting Detection Reagent (GE Healthcare) and imaged/analyzed using Image Lab software (BioRad).
Cell viability assays
The number of viable cells was quantified with trypan blue viability dye by automated cell counter (Countess, Invitrogen). Viability was also assessed by resazurin dye (AlamarBlue©) assay according to the manufacturer’s protocol (Invitrogen), imaged by spectrophotometer (Perkin Elmer Victor X3 workstation).
Apoptosis assays
Proportions of PTCL cells undergoing apoptosis were assessed by flow cytometry following staining with AnnexinV (eBioscience) and 7-AAD (BioLegend) according to the manufacturer’s protocols. Apoptosis was also determined by Western blot for caspase 3 cleavage. We assessed involvement of the extrinsic pathway using caspase inhibitors. Cells were pretreated with 50μM of a caspase 8 inhibitor, Z-IETD-FMK, or a pan-caspase inhibitor, Z-VAD-FMK, for 30min, then cultured with 5-20μM pimozide or DMSO for 24h (BD Biosciences). Involvement of the intrinsic pathway was assessed using JC-1 MMP dye according to the manufacturer’s protocol (eBioscience).
ATP production assay
Bioenergetic profile of PTCL cell lines cultured with varying concentrations of pimozide (5-20μM) and following anti-STAT5 shRNA knockdown vs. control was assessed using a ATP bioluminescent somatic cell assay kit according to the manufacturer’s protocol (SigmaAldrich). Imaging was performed by spectrophotometer (Perkin Elmer Victor X3 workstation).
RNA isolation and quantitative real-time RT-PCR
RNA was isolated using RNAqueous kit according to the manufacturer’s protocol (Life Technologies). Reverse transcription was performed according to the First-Strand cDNA Synthesis protocol (Thermo Fischer Scientific, Halethorpe, MD). Real-time quantitative PCR (qPCR) was performed using Taqman probes for human TRAF1, TP53BP2, Casp8, BCL-2, BCL-xL, and MCL-1 (Thermo Fischer Scientific) using the QuantStudio 6 Flex (Applied Biosystems Life Technologies) according to the manufacturer’s protocol.
Statistics
All data are representative of at least 3 independent experiments. Results were expressed as mean ± standard error of the mean. ANOVA and two-tailed student T-test were used to determine significance. P-values less than 0.05 were considered significant.
Abbreviations
ATP: Adenosine triphosphate; Casp8: Caspase 8; Ctrl: Control; DMSO: Dimethyl sulfoxide; EITL: Epitheliotropic intestinal T-cell lymphoma; HSTL: Hepatosplenic T-cell lymphoma; MMP: Mitochondrial membrane potential; NHL: Non-Hodgkin’s lymphoma; PTCL: Peripheral T-cell lymphoma; Scr: Scramble; SS: Sézary syndrome; STAT: Signal transducer and activator of transcription; T-ALL: T-cell acute lymphoblastic leukemia; T-LGL: T-cell large granular lymphocytic leukemia; T-PLL: T-cell prolymphocytic leukemia; TRAIL: TNF-related apoptosis-inducing ligand; UMB IRB: University of Maryland, Baltimore Institutional Review Board.
Author contributions
H.M.S. designed the research, performed experiments, analyzed and interpreted results, made the figures, and wrote the manuscript. A.F., K.S., and C.I.C. analyzed and interpreted results. A.B. designed the research, analyzed and interpreted results, and wrote the manuscript.
ACKNOWLEDGMENTS
We would like to acknowledge the input of Dr. Richard Siegel of the National Institutes of Health for his assistance with experimental design.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This study was supported by funding from The Gabriel’s Angel Foundation for Cancer Research, The Alliance for Cancer Gene Therapy, The American Cancer Society, The Marlene and Stewart Greenebaum Comprehensive Cancer Center, and the NIH (NHLBI, grant: HL093027).
REFERENCES
1. Intlekofer AM, Younes A. From empiric to mechanism-based therapy for peripheral T cell lymphoma. Int J Hematol. 2014; 99:249-62. https://doi.org/10.1007/s12185-014-1521-2.
2. Coiffier B, Federico M, Caballero D, Dearden C, Morschhauser F, Jager U, Trumper L, Zucca E, Gomes da Silva M, Pettengell R, Weidmann E, d’Amore F, Tilly H, et al. Therapeutic options in relapsed or refractory peripheral T-cell lymphoma. Cancer Treat Rev. 2014; 40:1080-8. https://doi.org/10.1016/j.ctrv.2014.08.001.
3. Zhang Y, Xu W, Liu H, Li J. Therapeutic options in peripheral T cell lymphoma. J Hematol Oncol. 2016; 9:37. https://doi.org/10.1186/s13045-016-0267-0.
4. Thoma G, Druckes P, Zerwes HG. Selective inhibitors of the Janus kinase Jak3--Are they effective? Bioorg Med Chem Lett. 2014; 24:4617-21. https://doi.org/10.1016/j.bmcl.2014.08.046.
5. Foss FM, Zinzani PL, Vose JM, Gascoyne RD, Rosen ST, Tobinai K. Peripheral T-cell lymphoma. Blood. 2011; 117:6756-67. https://doi.org/10.1182/blood-2010-05-231548.
6. Savage KJ. Update: peripheral T-cell lymphomas. Curr Hematol Malig Rep. 2011; 6:222-30. https://doi.org/10.1007/s11899-011-0100-3.
7. Skarbnik AP, Burki M, Pro B. Peripheral T-cell lymphomas: a review of current approaches and hopes for the future. Front Oncol. 2013; 3:138. https://doi.org/10.3389/fonc.2013.00138.
8. Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmaki H, Andersson EI, Lagstrom S, Clemente MJ, Olson T, Jalkanen SE, Majumder MM, Almusa H, Edgren H, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012; 366:1905-13. https://doi.org/10.1056/NEJMoa1114885.
9. Vasmatzis G, Johnson SH, Knudson RA, Ketterling RP, Braggio E, Fonseca R, Viswanatha DS, Law ME, Kip NS, Ozsan N, Grebe SK, Frederick LA, Eckloff BW, et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood. 2012; 120:2280-9. https://doi.org/10.1182/blood-2012-03-419937.
10. Jerez A, Clemente MJ, Makishima H, Koskela H, Leblanc F, Peng Ng K, Olson T, Przychodzen B, Afable M, Gomez-Segui I, Guinta K, Durkin L, Hsi ED, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 2012; 120:3048-57. https://doi.org/10.1182/blood-2012-06-435297.
11. Rajala HL, Eldfors S, Kuusanmaki H, van Adrichem AJ, Olson T, Lagstrom S, Andersson EI, Jerez A, Clemente MJ, Yan Y, Zhang D, Awwad A, Ellonen P, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013; 121:4541-50. https://doi.org/10.1182/blood-2012-12-474577.
12. Huang Y, de Reynies A, de Leval L, Ghazi B, Martin-Garcia N, Travert M, Bosq J, Briere J, Petit B, Thomas E, Coppo P, Marafioti T, Emile JF, et al. Gene expression profiling identifies emerging oncogenic pathways operating in extranodal NK/T-cell lymphoma, nasal type. Blood. 2010; 115:1226-37. https://doi.org/10.1182/blood-2009-05-221275.
13. Shimizu D, Taki T, Utsunomiya A, Nakagawa H, Nomura K, Matsumoto Y, Nishida K, Horiike S, Taniwaki M. Detection of NOTCH1 mutations in adult T-cell leukemia/lymphoma and peripheral T-cell lymphoma. Int J Hematol. 2007; 85:212-8. https://doi.org/10.1532/ijh97.06165.
14. Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, Carpenter Z, Abate F, Allegretta M, Haydu JE, Jiang X, Lossos IS, Nicolas C, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014; 46:166-70. https://doi.org/10.1038/ng.2873.
15. Kiel MJ, Velusamy T, Rolland D, Sahasrabuddhe AA, Chung F, Bailey NG, Schrader A, Li B, Li JZ, Ozel AB, Betz BL, Miranda RN, Medeiros LJ, et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. 2014; 124:1460-72. https://doi.org/10.1182/blood-2014-03-559542.
16. Manso R, Sanchez-Beato M, Monsalvo S, Gomez S, Cereceda L, Llamas P, Rojo F, Mollejo M, Menarguez J, Alves J, Garcia-Cosio M, Piris MA, Rodriguez-Pinilla SM. The RHOA G17V gene mutation occurs frequently in peripheral T-cell lymphoma and is associated with a characteristic molecular signature. Blood. 2014; 123:2893-4. https://doi.org/10.1182/blood-2014-02-555946.
17. Manso R, Rodriguez-Pinilla SM, Gonzalez-Rincon J, Gomez S, Monsalvo S, Llamas P, Rojo F, Perez-Callejo D, Cereceda L, Limeres MA, Maeso C, Ferrando L, Perez-Seoane C, et al. Recurrent presence of the PLCG1 S345F mutation in nodal peripheral T-cell lymphomas. Haematologica. 2015; 100:e25-7. https://doi.org/10.3324/haematol.2014.113696.
18. Kleppe M, Tousseyn T, Geissinger E, Kalender Atak Z, Aerts S, Rosenwald A, Wlodarska I, Cools J. Mutation analysis of the tyrosine phosphatase PTPN2 in Hodgkin’s lymphoma and T-cell non-Hodgkin’s lymphoma. Haematologica. 2011; 96:1723-7. https://doi.org/10.3324/haematol.2011.041921.
19. Nakamoto-Matsubara R, Sakata-Yanagimoto M, Enami T, Yoshida K, Yanagimoto S, Shiozawa Y, Nanmoku T, Satomi K, Muto H, Obara N, Kato T, Kurita N, Yokoyama Y, et al. Detection of the G17V RHOA mutation in angioimmunoblastic T-cell lymphoma and related lymphomas using quantitative allele-specific PCR. PLoS One. 2014; 9:e109714. https://doi.org/10.1371/journal.pone.0109714.
20. Nicolae A, Xi L, Pittaluga S, Abdullaev Z, Pack SD, Chen J, Waldmann TA, Jaffe ES, Raffeld M. Frequent STAT5B mutations in gammadelta hepatosplenic T-cell lymphomas. Leukemia. 2014; 28:2244–48. https://doi.org/10.1038/leu.2014.200.
21. Kucuk C, Jiang B, Hu X, Zhang W, Chan JK, Xiao W, Lack N, Alkan C, Williams JC, Avery KN, Kavak P, Scuto A, Sen E, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat Commun. 2015; 6:6025. https://doi.org/10.1038/ncomms7025.
22. Kiel MJ, Sahasrabuddhe AA, Rolland DC, Velusamy T, Chung F, Schaller M, Bailey NG, Betz BL, Miranda RN, Porcu P, Byrd JC, Medeiros LJ, Kunkel SL, et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in Sezary syndrome. Nat Commun. 2015; 6:8470. https://doi.org/10.1038/ncomms9470.
23. Nairismagi ML, Tan J, Lim JQ, Nagarajan S, Ng CC, Rajasegaran V, Huang D, Lim WK, Laurensia Y, Wijaya GC, Li ZM, Cutcutache I, Pang WL, et al. JAK-STAT and G-protein-coupled receptor signaling pathways are frequently altered in epitheliotropic intestinal T-cell lymphoma. Leukemia. 2016; 30:1311-9. https://doi.org/10.1038/leu.2016.13.
24. Lopez C, Bergmann AK, Paul U, Murga Penas EM, Nagel I, Betts MJ, Johansson P, Ritgen M, Baumann T, Aymerich M, Jayne S, Russell RB, Campo E, et al. Genes encoding members of the JAK-STAT pathway or epigenetic regulators are recurrently mutated in T-cell prolymphocytic leukaemia. Br J Haematol. 2016; 173:265-73. https://doi.org/10.1111/bjh.13952.
25. Waldmann TA. JAK/STAT pathway directed therapy of T-cell leukemia/lymphoma: Inspired by functional and structural genomics. Mol Cell Endocrinol. 2017; 451:66– 70. https://doi.org/10.1016/j.mce.2017.02.019.
26. Hatzimichael E, Tsolas E, Briasoulis E. Profile of pacritinib and its potential in the treatment of hematologic disorders. J Blood Med. 2014; 5:143-52. https://doi.org/10.2147/jbm.s51253.
27. Ariyoshi K, Nosaka T, Yamada K, Onishi M, Oka Y, Miyajima A, Kitamura T. Constitutive activation of STAT5 by a point mutation in the SH2 domain. J Biol Chem. 2000; 275:24407-13. https://doi.org/10.1074/jbc.M909771199.
28. Simpson HM, Khan RZ, Song C, Sharma D, Sadashivaiah K, Furusawa A, Liu X, Nagaraj S, Sengamalay N, Sadzewicz L, Tallon LJ, Chen QC, Livak F, et al. Concurrent Mutations in ATM and Genes Associated with Common gamma Chain Signaling in Peripheral T Cell Lymphoma. PLoS One. 2015; 10:e0141906. https://doi.org/10.1371/journal.pone.0141906.
29. Lan RY, Selmi C, Gershwin ME. The regulatory, inflammatory, and T cell programming roles of interleukin-2 (IL-2). J Autoimmun. 2008; 31:7-12. https://doi.org/10.1016/j.jaut.2008.03.002.
30. Lee S, Margolin K. Cytokines in cancer immunotherapy. Cancers (Basel). 2011; 3:3856-93. https://doi.org/10.3390/cancers3043856.
31. Waldmann TA, Chen J. Disorders of the JAK/STAT Pathway in T Cell Lymphoma Pathogenesis: Implications for Immunotherapy. Annu Rev Immunol. 2017; 35:533–50. https://doi.org/10.1146/annurev-immunol-110416-120628.
32. Koo GC, Tan SY, Tang T, Poon SL, Allen GE, Tan L, Chong SC, Ong WS, Tay K, Tao M, Quek R, Loong S, Yeoh KW, et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2012; 2:591-7. https://doi.org/10.1158/2159-8290.cd-12-0028.
33. Degryse S, de Bock CE, Cox L, Demeyer S, Gielen O, Mentens N, Jacobs K, Geerdens E, Gianfelici V, Hulselmans G, Fiers M, Aerts S, Meijerink JP, et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood. 2014; 124:3092-100. https://doi.org/10.1182/blood-2014-04-566687.
34. Bergmann AK, Schneppenheim S, Seifert M, Betts MJ, Haake A, Lopez C, Maria Murga Penas E, Vater I, Jayne S, Dyer MJ, Schrappe M, Duhrsen U, Ammerpohl O, et al. Recurrent mutation of JAK3 in T-cell prolymphocytic leukemia. Genes Chromosomes Cancer. 2014; 53:309-16. https://doi.org/10.1002/gcc.22141.
35. Bandapalli OR, Schuessele S, Kunz JB, Rausch T, Stutz AM, Tal N, Geron I, Gershman N, Izraeli S, Eilers J, Vaezipour N, Kirschner-Schwabe R, Hof J, et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica. 2014; 99:e188-92. https://doi.org/10.3324/haematol.2014.104992.
36. Griesshammer M, Gisslinger H, Mesa R. Current and future treatment options for polycythemia vera. Ann Hematol. 2015; 94:901-10. https://doi.org/10.1007/s00277-015-2357-4.
37. Cazzola M, Kralovics R. JAK inhibitor in CALR-mutant myelofibrosis. N Engl J Med. 2014; 370:1169. https://doi.org/10.1056/NEJMc1400499.
38. Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, Gashin LB, Terrell S, Klitgaard JL, Santo L, Addorio MR, Ebert BL, Griffin JD, Frank DA. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2011; 117:3421-9. https://doi.org/10.1182/blood-2009-11-255232.
39. Nelson EA, Walker SR, Xiang M, Weisberg E, Bar-Natan M, Barrett R, Liu S, Kharbanda S, Christie AL, Nicolais M, Griffin JD, Stone RM, Kung AL, et al. The STAT5 Inhibitor Pimozide Displays Efficacy in Models of Acute Myelogenous Leukemia Driven by FLT3 Mutations. Genes Cancer. 2012; 3:503-11. https://doi.org/10.1177/1947601912466555.
40. Gazdar AF, Carney DN, Bunn PA, Russell EK, Jaffe ES, Schechter GP, Guccion JG. Mitogen requirements for the in vitro propagation of cutaneous T-cell lymphomas. Blood. 1980; 55:409-17.
41. Hori T, Uchiyama T, Tsudo M, Umadome H, Ohno H, Fukuhara S, Kita K, Uchino H. Establishment of an interleukin 2-dependent human T cell line from a patient with T cell chronic lymphocytic leukemia who is not infected with human T cell leukemia/lymphoma virus. Blood. 1987; 70:1069-72.
42. Atale N, Gupta S, Yadav UC, Rani V. Cell-death assessment by fluorescent and nonfluorescent cytosolic and nuclear staining techniques. J Microsc. 2014; 255:7-19. https://doi.org/10.1111/jmi.12133.
43. Baskiewicz-Masiuk M, Masiuk M, Machalinski B. The influence of STAT5 antisense oligonucleotides on the proliferation and apoptosis of selected human leukaemic cell lines. Cell Prolif. 2003; 36:265-78.
44. Qin JZ, Zhang CL, Kamarashev J, Dummer R, Burg G, Dobbeling U. Interleukin-7 and interleukin-15 regulate the expression of the bcl-2 and c-myb genes in cutaneous T-cell lymphoma cells. Blood. 2001; 98:2778-83.
45. Liu S, Walker SR, Nelson EA, Cerulli R, Xiang M, Toniolo PA, Qi J, Stone RM, Wadleigh M, Bradner JE, Frank DA. Targeting STAT5 in hematologic malignancies through inhibition of the bromodomain and extra-terminal (BET) bromodomain protein BRD2. Mol Cancer Ther. 2014; 13:1194-205. https://doi.org/10.1158/1535-7163.mct-13-0341.
46. Heltemes-Harris LM, Willette MJ, Vang KB, Farrar MA. The role of STAT5 in the development, function, and transformation of B and T lymphocytes. Ann N Y Acad Sci. 2011; 1217:18-31. https://doi.org/10.1111/j.1749-6632.2010.05907.x.
47. Kanai T, Seki S, Jenks JA, Kohli A, Kawli T, Martin DP, Snyder M, Bacchetta R, Nadeau KC. Identification of STAT5A and STAT5B target genes in human T cells. PLoS One. 2014; 9:e86790. https://doi.org/10.1371/journal.pone.0086790.
48. Perez J. Deathbase: Cell Death/Apoptosis Database. 2017.
49. Nam S, Scuto A, Yang F, Chen W, Park S, Yoo HS, Konig H, Bhatia R, Cheng X, Merz KH, Eisenbrand G, Jove R. Indirubin derivatives induce apoptosis of chronic myelogenous leukemia cells involving inhibition of Stat5 signaling. Mol Oncol. 2012; 6:276-83. https://doi.org/10.1016/j.molonc.2012.02.002.
50. Dumon S, Santos SC, Debierre-Grockiego F, Gouilleux-Gruart V, Cocault L, Boucheron C, Mollat P, Gisselbrecht S, Gouilleux F. IL-3 dependent regulation of Bcl-xL gene expression by STAT5 in a bone marrow derived cell line. Oncogene. 1999; 18:4191-9. https://doi.org/10.1038/sj.onc.1202796.
51. Lord JD, McIntosh BC, Greenberg PD, Nelson BH. The IL-2 receptor promotes lymphocyte proliferation and induction of the c-myc, bcl-2, and bcl-x genes through the trans-activation domain of Stat5. J Immunol. 2000; 164:2533-41.
52. Perelman A, Wachtel C, Cohen M, Haupt S, Shapiro H, Tzur A. JC-1: alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 2012; 3:e430. https://doi.org/10.1038/cddis.2012.171.
53. Fulda S. Targeting extrinsic apoptosis in cancer: Challenges and opportunities. Semin Cell Dev Biol. 2015; 39:20-5. https://doi.org/10.1016/j.semcdb.2015.01.006.
54. Ashkenazi A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev. 2008; 19:325-31. https://doi.org/10.1016/j.cytogfr.2008.04.001.
55. Ozoren N, El-Deiry WS. Cell surface Death Receptor signaling in normal and cancer cells. Semin Cancer Biol. 2003; 13:135-47.
56. Zhang M, Mathews Griner LA, Ju W, Duveau DY, Guha R, Petrus MN, Wen B, Maeda M, Shinn P, Ferrer M, Conlon KD, Bamford RN, O’Shea JJ, et al. Selective targeting of JAK/STAT signaling is potentiated by Bcl-xL blockade in IL-2-dependent adult T-cell leukemia. Proc Natl Acad Sci U S A. 2015; 112:12480-5. https://doi.org/10.1073/pnas.1516208112.
57. Gavcovich TB, Walker KL, Bouchlaka MN, Capitini CM. Inhibition of the JAK/STAT and Bcl-2 Pathways Enhances Anti-Tumor Effects in an in vitro model of T-Cell Acute Lymphoblastic Leukemia (T-ALL). Blood. 2015; 126:2528.
58. Gieffers C, Kluge M, Merz C, Sykora J, Thiemann M, Schaal R, Fischer C, Branschadel M, Abhari BA, Hohenberger P, Fulda S, Fricke H, Hill O. APG350 induces superior clustering of TRAIL receptors and shows therapeutic antitumor efficacy independent of cross-linking via Fcgamma receptors. Mol Cancer Ther. 2013; 12:2735-47. https://doi.org/10.1158/1535-7163.mct-13-0323.
59. Cumaraswamy AA, Lewis AM, Geletu M, Todic A, Diaz DB, Cheng XR, Brown CE, Laister RC, Muench D, Kerman K, Grimes HL, Minden MD, Gunning PT. Nanomolar-Potency Small Molecule Inhibitor of STAT5 Protein. ACS Med Chem Lett. 2014; 5:1202-1206. doi:10.1021/ml500165r.
60. O’Connor OA, Horwitz S, Masszi T, Van Hoof A, Brown P, Doorduijn J, Hess G, Jurczak W, Knoblauch P, Chawla S, Bhat G, Choi MR, Walewski J, et al. Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol. 2015; 33:2492-9. https://doi.org/10.1200/jco.2014.59.2782.
61. Foss F, Horwitz S, Pro B, Prince HM, Sokol L, Balser B, Wolfson J, Coiffier B. Romidepsin for the treatment of relapsed/refractory peripheral T cell lymphoma: prolonged stable disease provides clinical benefits for patients in the pivotal trial. J Hematol Oncol. 2016; 9:22. https://doi.org/10.1186/s13045-016-0243-8.
62. Fanale MA, Horwitz SM, Forero-Torres A, Bartlett NL, Advani RH, Pro B, Chen RW, Davies A, Illidge T, Huebner D, Kennedy DA, Shustov AR. Brentuximab vedotin in the front-line treatment of patients with CD30+ peripheral T-cell lymphomas: results of a phase I study. J Clin Oncol. 2014; 32:3137-43. https://doi.org/10.1200/jco.2013.54.2456.
63. Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, Matous J, Ramchandren R, Fanale M, Connors JM, Yang Y, Sievers EL, Kennedy DA, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol. 2012; 30:2190-6. https://doi.org/10.1200/jco.2011.38.0402.
64. Fuentes AC, Szwed E, Spears CD, Thaper S, Dang LH, Dang NH. Denileukin Diftitox (Ontak) as Maintenance Therapy for Peripheral T-Cell Lymphomas: Three Cases with Sustained Remission. Case Rep Oncol Med. 2015; 2015:123756. https://doi.org/10.1155/2015/123756.
65. FDA. Current and Resolved Drug Shortages and Discontinuations Reported to FDA. 2014.
66. Weidmann E, Hess G, Chow KU, Krause SW, Subklewe M, Kruse J, Weisel KC, Soekler M, Kim SZ, Napieralski S, Rech J, Dreyling M, Jager E, et al. A phase II study of alemtuzumab, fludarabine, cyclophosphamide, and doxorubicin (Campath-FCD) in peripheral T-cell lymphomas. Leuk Lymphoma. 2010; 51:447-55. https://doi.org/10.3109/10428190903580402.
67. Ishida T, Joh T, Uike N, Yamamoto K, Utsunomiya A, Yoshida S, Saburi Y, Miyamoto T, Takemoto S, Suzushima H, Tsukasaki K, Nosaka K, Fujiwara H, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J Clin Oncol. 2012; 30:837-42. https://doi.org/10.1200/jco.2011.37.3472.
68. Dueck G, Chua N, Prasad A, Finch D, Stewart D, White D, van der Jagt R, Johnston J, Belch A, Reiman T. Interim report of a phase 2 clinical trial of lenalidomide for T-cell non-Hodgkin lymphoma. Cancer. 2010; 116:4541-8. https://doi.org/10.1002/cncr.25377.
69. Morschhauser F, Fitoussi O, Haioun C, Thieblemont C, Quach H, Delarue R, Glaisner S, Gabarre J, Bosly A, Lister J, Li J, Coiffier B. A phase 2, multicentre, single-arm, open-label study to evaluate the safety and efficacy of single-agent lenalidomide (Revlimid) in subjects with relapsed or refractory peripheral T-cell non-Hodgkin lymphoma: the EXPECT trial. Eur J Cancer. 2013; 49:2869-76. https://doi.org/10.1016/j.ejca.2013.04.029.
70. Toumishey E, Prasad A, Dueck G, Chua N, Finch D, Johnston J, van der Jagt R, Stewart D, White D, Belch A, Reiman T. Final report of a phase 2 clinical trial of lenalidomide monotherapy for patients with T-cell lymphoma. Cancer. 2015; 121:716-23. https://doi.org/10.1002/cncr.29103.
71. Bellanger D, Jacquemin V, Chopin M, Pierron G, Bernard OA, Ghysdael J, Stern MH. Recurrent JAK1 and JAK3 somatic mutations in T-cell prolymphocytic leukemia. Leukemia. 2014; 28:417-9. https://doi.org/10.1038/leu.2013.271.
72. Chen E, Staudt LM, Green AR. Janus kinase deregulation in leukemia and lymphoma. Immunity. 2012; 36:529-41. https://doi.org/10.1016/j.immuni.2012.03.017.
73. McGirt LY, Jia P, Baerenwald DA, Duszynski RJ, Dahlman KB, Zic JA, Zwerner JP, Hucks D, Dave U, Zhao Z, Eischen CM. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood. 2015; 126:508-19. https://doi.org/10.1182/blood-2014-11-611194.
74. Bouchekioua A, Scourzic L, de Wever O, Zhang Y, Cervera P, Aline-Fardin A, Mercher T, Gaulard P, Nyga R, Jeziorowska D, Douay L, Vainchenker W, Louache F, et al. JAK3 deregulation by activating mutations confers invasive growth advantage in extranodal nasal-type natural killer cell lymphoma. Leukemia. 2014; 28:338-48. https://doi.org/10.1038/leu.2013.157.
75. Coppo P, Gouilleux-Gruart V, Huang Y, Bouhlal H, Bouamar H, Bouchet S, Perrot C, Vieillard V, Dartigues P, Gaulard P, Agbalika F, Douay L, Lassoued K, et al. STAT3 transcription factor is constitutively activated and is oncogenic in nasal-type NK/T-cell lymphoma. Leukemia. 2009; 23:1667-78. https://doi.org/10.1038/leu.2009.91.
76. Ju W, Zhang M, Jiang JK, Thomas CJ, Oh U, Bryant BR, Chen J, Sato N, Tagaya Y, Morris JC, Janik JE, Jacobson S, Waldmann TA. CP-690,550, a therapeutic agent, inhibits cytokine-mediated Jak3 activation and proliferation of T cells from patients with ATL and HAM/TSP. Blood. 2011; 117:1938-46. https://doi.org/10.1182/blood-2010-09-305425.
77. Zamo A, Chiarle R, Piva R, Howes J, Fan Y, Chilosi M, Levy DE, Inghirami G. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene. 2002; 21:1038-47. https://doi.org/10.1038/sj.onc.1205152.
78. Roskoski R Jr. Anaplastic lymphoma kinase (ALK): structure, oncogenic activation, and pharmacological inhibition. Pharmacol Res. 2013; 68:68-94. https://doi.org/10.1016/j.phrs.2012.11.007.
79. Guo L, Wang L, Yang R, Feng R, Li Z, Zhou X, Dong Z, Ghartey-Kwansah G, Xu M, Nishi M, Zhang Q, Isaacs W, Ma J, et al. Optimizing conditions for calcium phosphate mediated transient transfection. Saudi J Biol Sci. 2017; 24:622-9. https://doi.org/10.1016/j.sjbs.2017.01.034.