
INTRODUCTION
Metastatic disease is the leading cause of death in patients with breast cancer and other malignancies. The high mortality reflects the limited treatment options available for progressed mammary carcinoma and highlights the need for new therapeutic approaches. An inflammatory microenvironment is an integral part of basically all tumors, even when they are not initiated by chronic inflammation. The reciprocal interactions between the inflammatory microenvironment and the tumor cells have a profound effect on tumor growth, metastasis and treatment resistance [1, 2]. Despite the progress that has been made in recent years, our understanding of how the microenvironment affects tumor progression and metastatic disease is incomplete and still requires further investigation.
Tumor cells have to overcome several obstacles and undergo a multi-step process called the metastatic cascade before giving rise to metastasis. First, they have to survive and thrive in the primary tumor and secrete factors that induce the pre-metastatic niche. Subsequently, they have to locally invade and enter the circulation to escape the primary tumor. Upon arrival at the metastatic site, the disseminated tumor cells have to egress from the blood stream, seed the tissue and expand to form metastases [3].
All steps of the metastatic cascade are strongly influenced by the immune system and the local microenvironment [1]. On the one hand, tumor cells have to constantly evade growth suppression and elimination by the anti-tumor immune response, especially cytotoxic T-cells and NK cells [4–7]. On the other hand, immune cells, in particular myeloid cells, are recruited and educated by tumor cells to actively facilitate their progression along the metastatic cascade. Macrophages comprise the most abundant myeloid cell type in human breast tumors [8] and their abundance in primary tumors is associated with poor prognosis [9, 10]. Pre-clinical studies have provided insights into the numerous ways macrophages can promote tumor progression and metastasis. They enhance tumor cell survival and proliferation by paracrine factors [11–13], trigger the angiogenic switch [14–17], promote local invasion [18, 19] and suppress CD8+ T-cells [9, 20]. Moreover, macrophages support tumor cell intravasation at the primary tumor [21] as well as extravasation at the distant site [22] and they have been shown to provide survival signals to tumor cells which colonize the distant site [23].
In parallel, intratumoral myeloid Ly6G+ cells drive tumor progression by stimulating tumor cell proliferation [24] and tumor neo-vascularization [25, 26], as well as inhibiting T-cell responses [27–29]. Furthermore, Ly6G+ cells are recruited to the metastatic site prior to colonization and establish the pre-metastatic niche, a microenvironment that is favorable for metastasis-initiating cells [30–33].
In human breast cancer specimens, NF-κB activation has been reported in both tumor cells as well as in the tumor stroma [34]. In addition, several cytokines which are known activators of NF-κB, including IL-1β, TNFα and IL-6, were reported to be upregulated in mammary tumors [35]. Moreover, previous studies of our lab using in vivo models of carcinogen-induced colon cancer [11, 12] demonstrated a tumor-promoting role of NF-κB signaling in myeloid cells during tumor promotion and progression. Given these reports and the high abundance of myeloid cells in mammary tumors [8, 9], we hypothesized that NF-κB signaling in myeloid cells might drive tumor progression in breast cancer. To test our hypothesis, we specifically deleted Ikkβ in myeloid cells in a well-established mouse model of metastatic breast cancer. The IKKβ subunit of the IKK complex is required for canonical NF-κB. Its activation leads to IκBα phosphorylation which upon ubiquitination is degraded by the proteasome. Subsequently, this triggers the release of NF-κB dimers that can now translocate to the nucleus to bind DNA and to induce transcription [36].
We show that IKKβ dependent NF-κB activation in myeloid cells is dispensable for primary tumor growth but required for establishing a lung microenvironment that supports the development of metastases.
RESULTS
To study the role of canonical NF-κB signaling in myeloid cells in breast cancer we crossed LysM-Cre/IkkβF/F (IkkβΔmye) mice [11] with mice that carry the polyoma middle T oncogene under the control of the MMTV promoter (MMTV PyMT) [37]. IkkβΔmye mice have a deletion of Ikkβ in myeloid cells preventing canonical NF-κB activation [11], whereas MMTV-PyMT mice develop spontaneous mammary carcinomas that metastasize with high incidence to the lung [37].
In the resulting PyMT IkkβΔmye mice primary tumor burden was not significantly altered compared to Ikkβ-proficient PyMT littermate controls and histological appearance of tumors from both genotypes was similar (Figure 1A). Consistently, proliferation and apoptosis of tumor cells determined by Ki-67 and cleaved caspase 3 immunohistochemistry, respectively, were comparable in both genotypes in the primary tumor at 15 weeks of age (Figure 1B). However, while all PyMT IkkβF/F animals had developed microscopically visible metastases at 12 weeks of age in the lung, 25% of PyMT IkkβΔmye mice were metastasis free (Figure 1C). At 15 weeks of age, the number of lung metastases in PyMT IkkβF/F control animals was more than four times higher compared to PyMT IkkβΔmye mice (Figure 1C). The size (Figure 1C) of established metastatic foci, nevertheless, was similar in PyMT IkkβF/F and PyMT IkkβΔmye animals, as was the number of Ki-67 and cleaved caspase 3 positive metastatic cells (Figure 1D). Thus, deletion of Ikkβ in myeloid cells does not affect primary tumor growth but potently suppresses formation of metastatic foci in the lung.
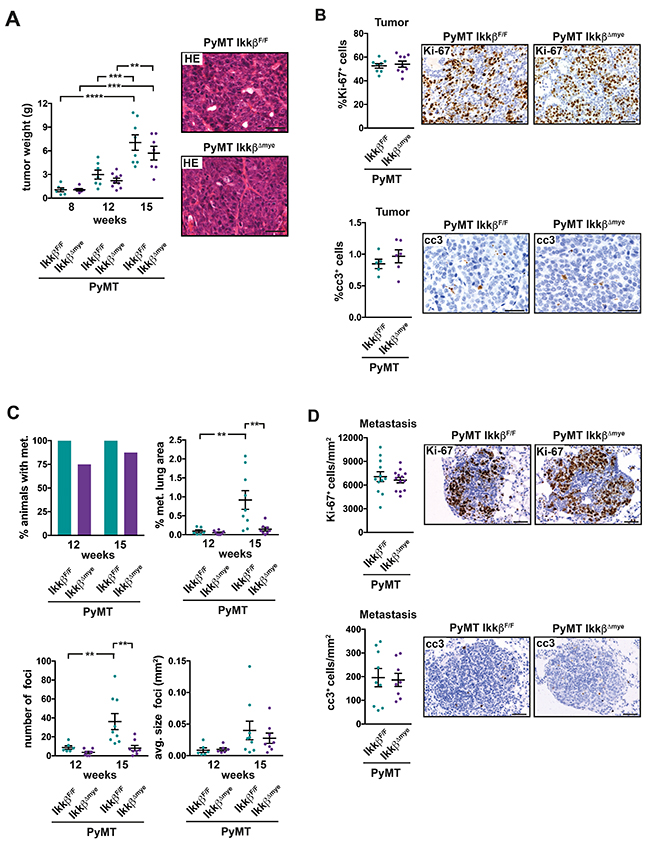
Figure 1: Deletion of Ikkβ in myeloid cells does not affect primary tumor growth but suppresses lung metastasis in the PyMT breast cancer model. (A) Combined weight of all mammary tumors per animal from PyMT IkkβF/F and PyMT IkkβΔmye mice at 8, 12 and 15 weeks of age (each n≥6) and representative H&E-stained primary tumor tissue at 15 weeks of age. (B) Percentage of Ki-67 positive (Ki-67+) cells and cleaved caspase 3 positive (cc3+) in tumors of PyMT IkkβF/F and PyMT IkkβΔmye mice at 15 weeks of age. Two tumors per animal were analyzed, depicted is the mean for each animal. Ki-67+ cells were quantified in a full section of the tumor (n≥8); cc3+ cells were quantified in 6 random 20x fields (n≥5). (C) Percentage of animals with lung metastasis, percentage of metastatic area, number and average size of metastatic foci in the lungs of PyMT IkkβF/F and PyMT IkkβΔmye mice at 12 and 15 weeks of age (each n≥6). (D) Ki-67+ cells and cc3+ cells per mm2 metastasis in metastatic foci from n≥5 PyMT IkkβF/F and PyMT IkkβΔmye mice at 15 weeks of age. Data are mean ± SEM. **p≤0,01 ***p≤0,001 ****p.≤0,0001. Scale bar is 0,05mm.
Immune cells shape the local microenvironment during tumorigenesis and are important modulators of the metastatic cascade [1, 38]. To determine whether deletion of Ikkβ in myeloid cells affects the microenvironment in the primary tumor, we characterized tumor infiltrating immune cell populations by flow cytometry (Figure 2A, Supplementary Figure 1). Additionally, we determined the expression of several genes related to inflammation, epithelial to mesenchymal transition (EMT) and metastasis by RT-qPCR (Figure 2B). Myeloid cells, specifically CD11b+ F4/80+ Gr1- tumor-associated macrophages (TAMs) were by far the most abundant immune cell population in tumors of both PyMT IkkβΔmye and PyMT IkkβF/F control animals (Figure 2A). Yet, in tumors of PyMT IkkβΔmye animals, the proportion of viable F4/80+ Gr1- macrophages was significantly reduced. The proportion of F4/80- Gr1+ granulocytes, CD3+ T-cells and B220+ B-cells, on the other hand, was comparable to PyMT IkkβF/F controls (Figure 2A). Interestingly, we could not detect any differences in mRNA expression of genes involved in inflammation and metastasis in tumors from PyMT IkkβΔmye and PyMT IkkβF/F controls (Figure 2B).
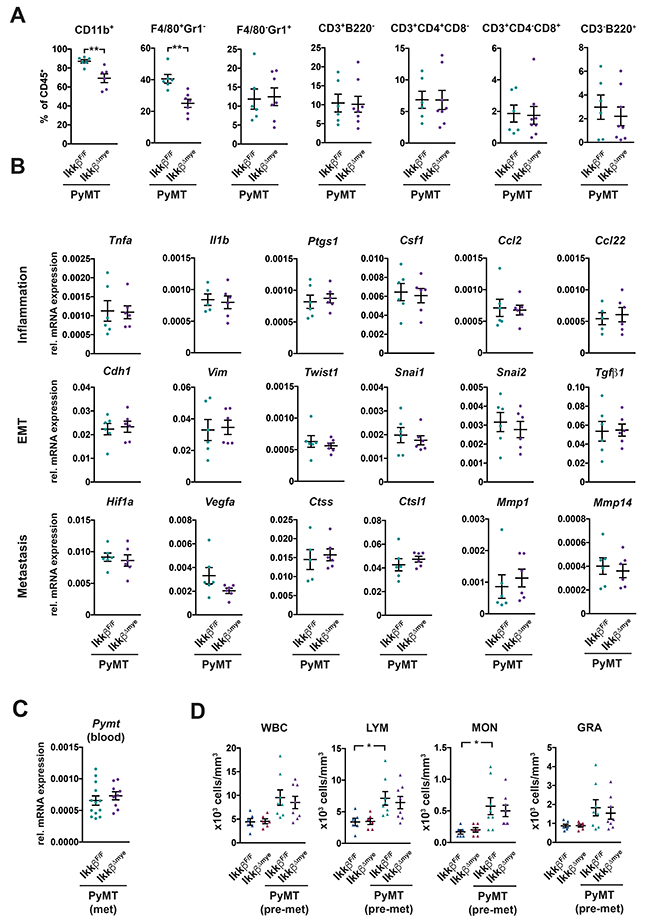
Figure 2: Deletion of Ikkβ in myeloid cells has only minor effects on the primary tumor microenvironment and does not affect dissemination. (A) Flow cytometric analysis of immune cell populations in tumors of PyMT IkkβF/F and PyMT IkkβΔmye mice at 12 weeks of age. Two or more tumors per animal were analyzed, depicted is the mean for each animal (n≥6). (B) Quantitative PCR of the indicated genes from tumor tissue of PyMT IkkβF/F and PyMT IkkβΔmye mice at 12 weeks of age. Two tumors per animal were analyzed, depicted is the mean for each animal (n≥5). (C) Pymt oncogene expression in the blood of PyMT IkkβF/F and PyMT IkkβΔmye mice (n≥9) at 15 weeks of age. (D) Immune cells in the peripheral blood of tumor free IkkβF/F and IkkβΔmye mice as well as tumor-bearing PyMT IkkβF/F and PyMT IkkβΔmye mice (n≥6) at 8 weeks of age. Depicted are total white blood cells (WBC), lymphocytes (LYM), monocytes (MON) and granulocytes (GRA). Data are mean ± SEM. **p≤0,01
To determine whether dissemination of tumor cells from the primary site was altered in PyMT IkkβΔmye mice, we quantified circulating tumor cells by measuring the mRNA expression of the Pymt oncogene in the blood. We found no difference in Pymt expression in the blood between PyMT IkkβΔmye and PyMT IkkβF/F control mice (Figure 2C). Mobilization of immune cells in the peripheral blood in response to the primary tumor was unaffected by myeloid-specific loss of Ikkβ as well (Figure 2D).
Collectively, these findings suggest that loss of Ikkβ in myeloid cells has only minor influence on primary tumor growth and the primary tumor microenvironment and is dispensable for the early events of the metastatic cascade. Thus, we hypothesized that metastasis in PyMT IkkβΔmye might be impaired at the end of the metastatic cascade at the distant site. Therefore, we tried to identify alterations in the lungs of PyMT IkkβΔmye mice that might account for the reduction in metastatic foci.
Since myeloid cells at the distant site have been suggested as crucial modulators of metastasis [6, 22, 30, 32, 39], we determined the numbers of granulocytes and macrophages in the lung parenchyma. Immunohistochemical analysis of MPO+ cells revealed a strong and gradual increase of granulocytes in the lung parenchyma of tumor bearing mice compared to tumor free animals in both genotypes (Figure 3A). However, lungs from PyMT IkkβΔmyemice in the metastatic stage displayed significantly reduced numbers of MPO+ granulocytes compared to PyMT IkkβF/F controls (Figure 3A). In contrast, the number of CD68+ macrophages in the lung parenchyma of metastatic animals was significantly increased upon myeloid Ikkβ deletion (Figure 3B). Interestingly, although containing lower numbers of CD68+ cells compared to metastatic PyMT IkkβΔmye mice, lungs from tumor free IkkβΔmye animals showed a clear trend towards increased presence of CD68+ cells as well.
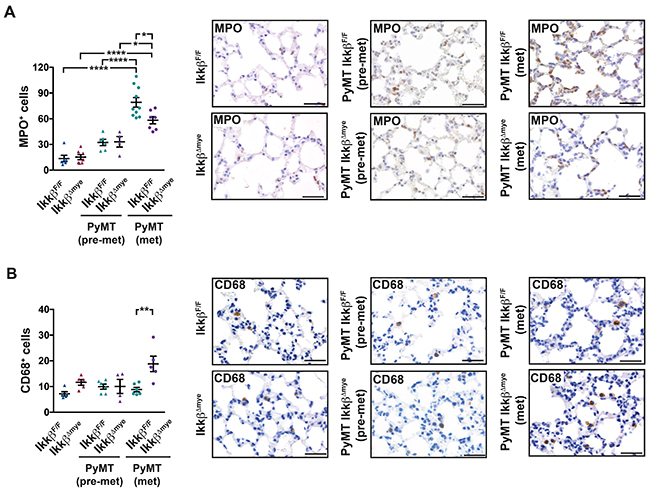
Figure 3: Lungs of PyMT IkkβΔmye mice have altered immune cell infiltration. Number of (A) MPO+ cells or (B) CD68+ cells per field in lungs of IkkβF/F and IkkβΔmye mice (tumor free; n=5) or PyMT IkkβF/F and PyMT IkkβΔmye mice at 8 weeks (pre-metastatic; n≥4) or 15 weeks of age (metastatic; n≥5), respectively. 9 random 40x fields per animal were analyzed. Data are mean ± SEM. *p≤0,05; **p≤0,01; ****p≤0,0001. Scale bar is 0,05mm.
To further characterize the alterations in the lung microenvironment upon myeloid-specific loss of Ikkβ, we analyzed the expression of genes related to inflammation and the pre-metastatic niche in lungs from tumor free mice, pre-metastatic mice (8 weeks) and metastatic mice (15 weeks). Deletion of Ikkβ in myeloid cells resulted in marked changes in expression of genes encoding numerous cytokines and chemokines in the lung (Figure 4A). Some of those genes were differentially expressed only in animals at the metastatic stage while others were also altered in tumor free animals. The subset of genes upregulated in lungs of both tumor free and metastatic Ikkβ-deficient animals comprised Tnfa, Ccl5, Ccl17, Ccl22, Cxcl1 and Csf2, although not all changes reached significance. However, only metastatic PyMT IkkβΔmye animals showed significant upregulation of GrzmB, S100a8, S100a9 and a clear trend towards increased expression of Tgfb1 and Ifng compared to PyMT IkkβF/F controls. Interestingly, expression of most genes was attenuated at 8 weeks of age in pre-metastatic lungs in both Ikkβ-proficient and deficient mice and only Tgfb1 was higher expressed in both genotypes compared to tumor free and metastatic animals.
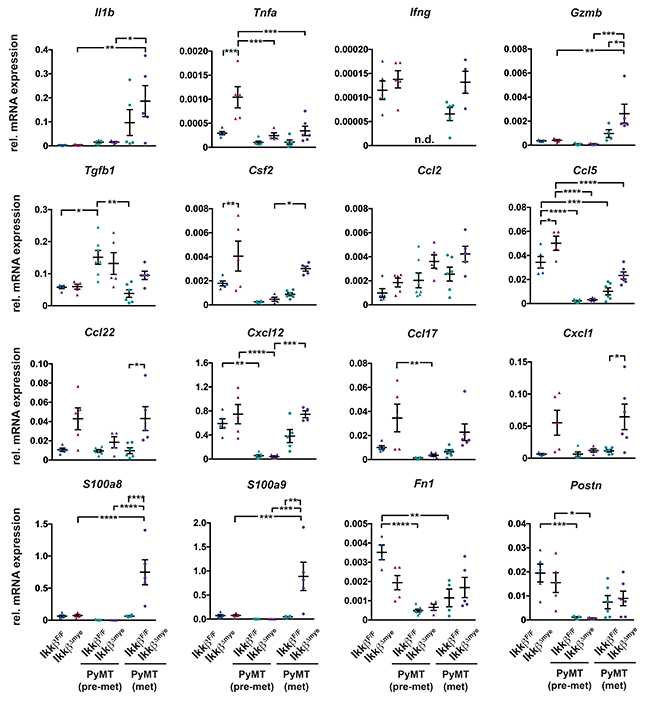
Figure 4: Deletion of Ikkβ in myeloid cells causes a pro-inflammatory lung microenvironment. Quantitative PCR of the indicated genes from lung tissue of IkkβF/F and IkkβΔmye mice (tumor free; n=5) or PyMT IkkβF/F and PyMT IkkβΔmye mice at 8 weeks (pre-metastatic; n≥4) or 15 weeks of age (metastatic; ≥4), respectively. Data are mean ± SEM *p≤0,05; **p≤0,01; ***p≤0,001; ****p≤0,0001
Some of the inflammatory mediators that were found elevated in the lungs of tumor-free IkkβΔmye animals (Figure 4A) also showed a trend towards slightly increased expression in the mammary glands of tumor-free IkkβΔmye animals and Il1b expression was significantly higher (Supplementary Figure 2).
The elevated mRNA expression of Ifng and GrzmB in metastatic lungs of PyMT IkkβΔmye animals suggested an increased presence of cytotoxic lymphocytes in that might confer increased protection from metastasis. To determine whether this is was the case, mononuclear cells from the lung were analyzed for IFN-γ and Granzyme B expression by FACS (Figure 5, Supplementary Figure 3). In lungs of PyMT IkkβΔmye mice we found significant more IFN-γ producing CD4+ and CD8+ T-cells compared to PyMT IkkβF/F controls (Figure 5). Moreover, PyMT IkkβΔmye mice showed a trend towards increased presence of Granzyme B expressing CD4+ T-cells in their lungs. Granzyme B expressing CD8+ cells on the other hand were only very rarely observed in mice of either genotype. The numbers of IFN-γ and Granzyme B expressing Nkp46+ Nk-cells were comparable in PyMT IkkβF/F and PyMT IkkβΔmye mice (Figure 5).
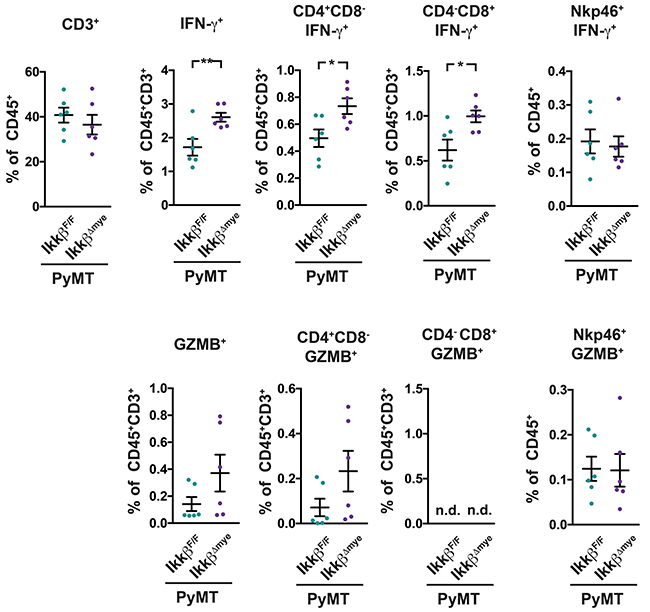
Figure 5: PyMT IkkβΔmye mice have increased levels of activated T-cells in their lungs. IFN-γ and Granzyme B expression in mononuclear cells from lungs of PyMT IkkβF/F and PyMT IkkβΔmye mice was determined at 11 weeks of age (n=6) by FACS. Data are mean ± SEM *p≤0,05; **p≤0,01
To test whether the pro-inflammatory lung microenvironment in PyMT IkkβΔmye mice has indeed tumor suppressive properties, we co-cultured PyMT tumor-derived TS1 cells in serum-reduced medium with lung tissue from either PyMT IkkβΔmye mice or PyMT IkkβF/F controls in a transwell system. The transwell system allows exchange of soluble factors but prevents physical contact (Figure 6A). When seeded at low density to mimic micrometastatic conditions, TS1 tumor cells co-cultured with lung tissue from PyMT IkkβΔmye mice showed a significant 25% reduction in growth compared to cells co-cultured with lung tissue from PyMT IkkβF/F mice (Figure 6B). Yet, when seeded at a higher density but still below confluence, no suppressive effect on the tumor cells was observed (Figure 6B). Sparse TS1 cells after co-culture were further characterized by FACS in terms of cell cycle progression and viability by staining with Hoechst 33342 and propidium iodide (PI). TS1 cells showed comparable cell cycle distribution after co-culture with lung tissue from PyMT IkkβF/F or PyMT IkkβΔmye mice. However, TS1 cells co-cultured with lung tissue from PyMT IkkβΔmye mice showed a significant increase in the fraction of PI+ dead cells (Figure 6C). Thus, loss of Ikkβ in myeloid cells leads to a tumor suppressive pro-inflammatory microenvironment in the lung.
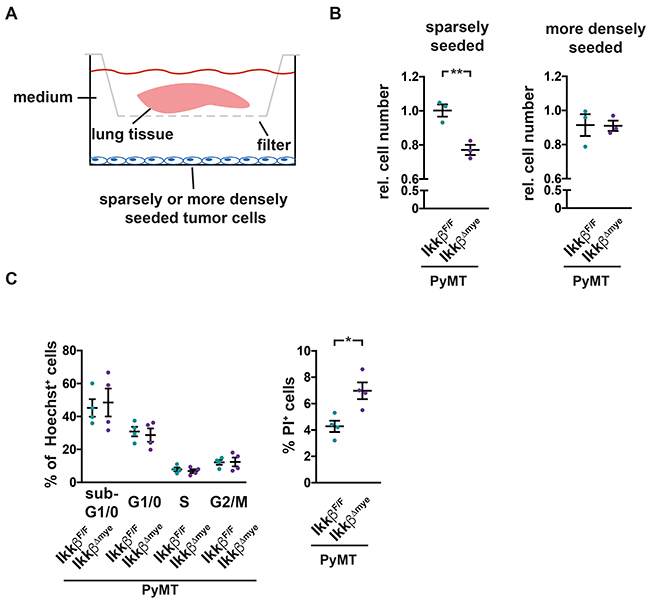
Figure 6: Soluble factors in lungs of PyMT IkkβΔmye mice suppress tumor cell survival. (A) TS1 tumor cells, seeded sparsely (15.000 cells/well) or more densely (50.000 cells/well) were co-cultured in serum-reduced medium with lung tissue from 12 weeks old, early metastatic PyMT IkkβF/F or PyMT IkkβΔmye mice in a transwell system for 16h (pore diameter 0,4μM). Lung tissue was removed and cell numbers of co-cultured cells were analyzed 24h later. (B) Depicted are relative cell numbers of TS1 cells after co-culture from three independent experiments. Each condition was measured with four to five replicates per experiment. (C) Cell cycle stage distribution and viability of sparsely seeded TS1 cells after co-culture were determined by Hoechst 33342 and Propidium iodide (PI) staining in FACS. Depicted are two independent experiments with two replicates each. Data are mean ± SEM *p≤0,05; **p≤0,01.
DISCUSSION
Our data reveals canonical NF-κB signaling in myeloid cells as an important mediator of breast to lung metastasis by regulating the microenvironment at the distant site. While deletion of Ikkβ in myeloid cells in the PyMT breast cancer model does not have a major impact on primary tumor growth and the primary tumor microenvironment it significantly reduces lung metastasis. Expression of metastasis-related genes in the primary tumor and comparable numbers of circulating tumor cells in PyMT IkkβF/F and PyMT IkkβΔmye indicate that the observed reduction in lung metastasis is not a consequence of altered dissemination from the primary tumor. Concomitantly, lungs of PyMT IkkβΔmye animals exhibit a pro-inflammatory signature that comprises numerous chemokines and cytokines that most likely shape a tumor suppressive environment. While the individual role of these inflammatory mediators is not clear, their integrated effects are tumor-suppressive, as demonstrated in co-culture assays. Some of the factors upregulated in lungs of PyMT IkkβΔmye mice have known cytotoxic properties. For instance, IFN-γ and Granzyme B have been described as potent effector molecules of the anti-cancer defense that can inhibit metastasis [5, 40–42]. TGFβ1 and TNFα are pleiotropic cytokines, which can have both tumor promoting and tumor suppressive effects depending on the context [43–45]. It is possible that in the lungs of PyMT IkkβΔmye mice they directly or indirectly contribute to a tumor hostile environment.
Strikingly, we observed a reduced number of metastatic foci but no difference in their size, proliferation or apoptosis. Thus, once a metastatic focus is successfully established and has progressed to a certain size, it is unaffected by the altered lung microenvironment in PyMT IkkβΔmye mice. This might reflect particular vulnerability of single metastasis-initiating cells compared to tumor cells in the bulk of an established metastasis. Our findings in an in vitro co-culture system, where factors secreted from lungs of pre-metastatic PyMT IkkβΔmye mice have suppressive properties only on sparse tumor cells but not on more confluent cells support this notion.
Ikkβ-deficiency in myeloid cells leads to the upregulation of pro-inflammatory markers in the lung of tumor free and metastatic mice. Yet, while partly overlapping, there are marked differences in the pro-inflammatory signature in the lungs of tumor free IkkβΔmye and metastatic PyMT IkkβΔmye mice. These differences might be the result of primary tumor-induced systemic effects on the lung microenvironment during the later stages of tumorigenesis. Alternatively, but not mutually exclusive, the particular microenvironment in metastatic PyMT IkkβΔmye mice might develop due to activation of immune cells in the lung when encountering metastasized tumor cells.
The changes in cytokine expression in lungs of PyMT IkkβΔmye mice are accompanied by alterations in immune cell populations. We observed a significant increase in IFN-γ producing CD4+ and CD8+ T-cells in the lung of PyMT mice IkkβΔmye, which may be responsible for the death of metastasis initiating cells. Also, the number of CD68+ macrophages was elevated in the lungs of PyMT IkkβΔmye mice. These cells might reflect bone marrow-derived macrophages (BMDMs) recruited in response to chemotactic cues [22]. A certain macrophage subset has been reported to promote extravasation of tumor cells during metastatic seeding [22, 46]. Yet, it has become clear in recent years that the functional polarization of immune cell populations determine their tumor-promoting or suppressive properties [47, 48]. Intriguingly, targeting NF-κB signaling in macrophages can polarize them towards a more cytotoxic phenotype [49].
Before onset of metastasis, tumor cells in the primary tumor secrete systemically acting soluble factors that prime distant tissues for metastasis. The metastatic niche provides a microenvironment that facilitates seeding and outgrowth of metastasis-initiating cells. Earlier studies have emphasized the importance of granulocytes for making the distant site more susceptible to metastatic seeding [30, 31, 33, 39]. Granulocytes have been reported to promote outgrowth of metastasis-initiating cells through leukotriene secretion [30], enhanced retention of circulating tumor cells by neutrophil extracellular traps [31, 50]. Moreover, granulocytic myeloid cells can suppress anti-tumor immunity and thereby contribute to metastasis [39, 51–53]. In line with these reports, we observed gradual recruitment of granulocytic cells to the lung with progressing tumorigenesis. Interestingly, this recruitment was significantly impaired in PyMT IkkβΔmye mice at the metastatic stage. Thus, impaired granulocyte recruitment to lungs of PyMT IkkβΔmye mice might contribute to reduced lung metastasis by affecting the metastatic niche and T-cell activation in the lung.
Two previous studies analyzed the role of NF-κB signaling in myeloid cells in experimental lung colonization assays. Myeloid-specific deletion of Ikkβ strongly increased experimental lung colonization using BrafV600E/Pten-/- or B16F0 melanoma cells presumably due to impaired anti-tumor function of macrophages [54]. Moreover, enforced NF-κB activation in myeloid cells through inducible overexpression of Ikkβ under the control of the Csf1r promoter inhibited experimental colonization of lungs with breast cancer cells [55]. However, in contrast to tumor cell injection into the tail vein the spontaneous PyMT model should be considered as a more adequate model of lung colonization as it also considers tumor evolution. Unlike tail vein injection, it takes into account potential systemic effects that a spontaneously developing primary tumor might have on metastasizing tumor cells and especially at the pre-metastatic niche. For instance, myeloid cells, which are normally recruited to the lung before onset of metastasis are not present in tail vein injection experiments and their potential effects are therefore neglected. The influence of other metastasis-regulating factors of the pre-metastatic niche is likewise lacking. Furthermore, different myeloid cell populations are affected when driving Cre recombinase expression using the Csf1r promoter compared to the Lysozyme M promoter. Unlike the Lysozyme M promoter, the Csf1r promoter is not active in mature granulocytes [11, 56–58].
In a colitis-associated colon cancer model, we previously found that deletion of Ikkβ in myeloid cells reduces the incidence as well as the size of adenomas due to decreased expression of pro-inflammatory cytokines which promote tumor cell proliferation [11]. On the other hand, in a sporadic colon carcinoma model, deletion of Ikkβ in myeloid cells reduced prevented lymph node metastasis but did not affect primary tumor growth [12]. In PyMT IkkβΔmye mice, primary tumor burden was unaltered but the formation of metastatic foci at the distant site was impaired. In contrast, in melanoma and lung cancer models NF-κB signaling in myeloid cells seems to confer a tumor-suppressive role [54]. Thus, myeloid-specific loss of Ikkβ can have diverse effects on tumorigenesis, underscoring the complex, context-dependent role of NF-κB signaling in different cancer models and during different stages of tumorigenesis.
Therapeutic targeting of immune cells and other stromal cells in the tumor microenvironment might be advantageous over interfering with tumor cell intrinsic mechanism since they are genetically more stable and therefore less likely to develop resistance [59, 60]. Due to the pivotal role of NF-κB in the inflammatory tumor microenvironment, IKKβ as a central component of the signaling pathway is in principle a promising drug target for cancer therapy. However, due to the multifaceted role of IKKβ in myeloid cells in tumorigenesis, a clear definition is required in which context patients can profit from targeting NF-κB. Our findings suggest that it might be beneficial to target canonical NF-κB in myeloid cells in metastatic breast cancer.
MATERIALS AND METHODS
Ethics statement
The investigation has been conducted in accordance to national and international guidelines and has been approved by the authors’ institutional review board.
Animals
MMTV PyMT [37] (The Jackson Laboratory) and LysM-Cre/IkkβF/F (IkkβΔmye) mice [11] have been described previously. The two strains, both on FvB background, were crossed to generate PyMT IkkβΔmye mice. Genotyping of mice was performed with the following primers:
Cre recombinase fwd |
ACCTGAAGATGTTCGCGATTATCT |
Cre recombinase rev |
ACCGTCAGTACGTGAGATATCTT |
IkkβΔ (“lox”) fwd |
CACAGTGCCCACATTATTTAGATA |
IkkβΔ (“lox”) rev |
GTCTTCAACCTCCCAAGCCTT |
PyMT (control) fwd |
CAAATGTTGCTTGTCTGGTG |
PyMT (control) rev |
GTCAGTCGAGTGCACAGTTT |
PyMT (transgene) fwd |
GGAAAGTCACTAGGAGCAGGG |
PyMT (transgene) rev |
GGAAGCAAGTACTTCACAAGGG |
Determining primary tumor burden
To determine tumor weight, mice were sacrificed and the complete mammary tumor tissue was isolated and weighted.
Histological analysis
For histological analysis, tissue was fixed over night at 4°C in 4% paraformaldehyde in PBS. After fixation, tissue was dehydrated, embedded in paraffin and sectioned. Tissue sections were deparaffinized, rehydrated and stained with Hämatoxylin/Eosin or processed for immunohistochemistry. To determine metastatic burden, number and size of metastatic foci was quantified in Hämatoxylin/Eosin (H&E)-stained 100μm serial sections of whole lungs with Aperio Image Scope Software. The metastatic index of an animal was calculated as percentage of collective metastatic area in all sections to total lung area. Total lung area was determined by addition of the areas of individual lung lobes at their biggest section. Metastatic incidence was calculated as percentage of animals with metastasis to total animals. Immune cell aggregates in the lung were quantified in the same way as metastases.
Immunohistochemistry
For immunohistochemistry, heat-mediated antigen unmasking was performed and incubation with the primary antibody was done as indicated in the following: Anti-Ki-67 1:1500, 30min RT, 12202S (Cell signaling); Anti-cleaved caspase 3 (Asp175) 1:200, 3h RT, 39579S (Cell signaling); Anti-CD3 1:2, 30min RT, IS503 (Dako); Anti-CD68 1:100, 30min RT, ab12521 (abcam); Anti-MPO 1:50, over night 4°C, ab139748 (abcam). Biotin-conjugated secondary antibodies (Vectorlabs) were used for primary antibody detection according to the DAB protocol (Vectorlabs). In tumors, Ki-67 positive cells were quantified automatically with Aperio Image Scope software version 12.3.2.5030 (Leica) in the whole section while cleaved caspase 3 positive cells were counted manually in six random 20x-fields. In lung metastases, Ki-67 and cleaved caspase 3 positive cells were counted manually and were depicted as positive cells per mm2 with respect to the area of the corresponding metastases.
CD3-, CD68- and MPO-staining in lung parenchyma were quantified in nine random 40x-fields from three different lung lobes. Positive cells were manually counted in ImageJ 1.49c. Normalization to a reference image with a given amount of lung tissue was performed since amount of lung tissue in a 40x-field could differ.
Gene expression analysis in tumors, lungs, mammary glands and blood
For mRNA expression analysis tumor, lung, mammary gland or blood tissue were homogenized in RLT lysis buffer (Qiagen) with one 20s pulse at 5000rpm of the precellys 24 homogenizator (bertin instruments). Lysates were centrifuged at 4°C and 13.000 rpm for 5min. RNA was isolated from lysates with RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. cDNA synthesis was performed with 0,5 to 1μg of RNA and SuperScript II Reverse Transcriptase (Invitrogen) according to the manufacturer’s protocol. cDNA level were subsequently determined by quantitative real time PCR with a SYBR-Green MasterMix (Roche) on a StepOnePlus Real Time PCR system (Applied Biosystems). Expression levels were normalized based on the expression of housekeeping gene cyclophilin. The following primers were used:
Il1b fwd |
TGTCTTTCCCGTGGACCTTC |
Il1b rev |
CCCATGAGTCACAGAGGATGG |
Ptgs2 fwd |
AGACAGATCATAAGCGAGGACC |
Ptgs2 rev |
CCATCCTTGAAAAGGCGCAGT |
Twist1 fwd |
GTCCCACTAGCAGCGGAG |
Twist1 rev |
TGTCCATTTTCTCCTTCTCTGGAA |
Cdh1 fwd |
AACGCTCCTGTCTTCAACCC |
Cdh1 rev |
GGTCACTTTGAGTGTGGCGA |
Hif1a fwd |
ACACAGAAATGGCCCAGTGA |
Hif1a rev |
GGCTTGTTAGGGTGCACTTC |
Ifng fwd |
TTACTGCCACGGCACAGTCA |
Ifng rev |
AGTTCCTCCAGATATCCAAGAAGAGA |
Vim fwd |
ACGAGTACCGGAGACAGGTG |
Vim rev |
AGTTAGCAGCTTCAAGGGCAA |
Ctsl fwd |
TTCAGGAACCGCTGATGCTT |
Ctsl rev |
CTGTCCTTCTAGGCAACCCG |
Ccl17 fwd |
AATGTAGGCCGAGAGTGCTG |
Ccl17 rev |
GCCCTGGACAGTCAGAAACA |
SnaI1 fwd |
GTCTGCACGACCTGTGGAAA |
SnaI1rev |
GGTCAGCAAAAGCACGGTTG |
Ctss fwd |
GGACTGGAGAGAGAAGGGCT |
Ctss rev |
AGCTTCCCCGTTTTCAGCTT |
Tgfb1 fwd |
TGTGGAGCAACATGTGGAACT |
Tgfb1 rev |
TGCCGTACAACTCCAGTGAC |
Snai2 fwd |
GCACATTCGAACCCACACATT |
Snai2 rev |
TGCAGTGAGGGCAAGAGAAAG |
Cxcl1 fwd |
ACTGCACCCAAACCGAAGTC |
Cxcl1 rev |
TGGGGACACCTTTTAGCATCTT |
Tnfa fwd |
CCCACGTCGTAGCAAACCA |
Tnfa rev |
GTACAACCCATCGGCTGGC |
Mmp14 fwd |
CCAAGGCAGCAACTTCAGC |
Mmp14 rev |
GTGAGCGTTGTGTGTGGGTA |
Csf1 fwd |
GCCAAGGAGGTGTCAGAACA |
Csf1 rev |
AGGCAATCTGGCATGAAGTCT |
Postn fwd |
CCCGCAGTGATGCCTATTGA |
Postn rev |
CAGCTTCGAGACATCGGAGT |
Gzmb fwd |
CCCAAAGACCAAACGTGCTT |
Gzmb rev |
CGTGGAGGTGAACCATCCTTA |
Cccl5 fwd |
TGCCCACGTCAAGGAGTATTT |
Cccl5 rev |
TTCTCTGGGTTGGCACACA |
S1008a fwd |
CAAGGAAATCACCATGCCCT |
S1008a rev |
ATATTCTGCACAAACTGAGGACAC |
S1009a fwd |
CCTGACACCCTGAGCAAGAA |
S1009a rev |
GTCCTGGTTTGTGTCCAGGT |
Tnc fwd |
AGCTTCCTGCTTAAGTCCCTG |
Tnc rev |
TAGGTGGCACACAACTGCTC |
Fn1 fwd |
AAGAGGCAGGCTCAGCAAAT |
Fn1 rev |
GTCCGTTCCCACTGCTGATT |
Csf2 fwd |
TCAAAGAAGCCCTGAACCTCC |
Csf2 rev |
CGAATATCTTCAGGCGGGTCT |
Ccl2 fwd |
CAGCCAGATGCAGTTAACGC |
Ccl2 rev |
AGCCTACTCATTGGGATCATCTTG |
Ccl22 fwd |
TCATGGCTACCCTGCGTGTC |
Ccl22 rev |
CCTTCACTAAACGTGATGGCAGAG |
Vegfa fwd |
ACCCACGACAGAAGGAGAGC |
Vegfa rev |
GGTCTCAATCGGACGGCA |
Mmp1 fwd |
ACGCTGCTTATGAAGCTAGTATGA |
Mmp1 rev |
TCTCTGGGGAATCCTCTCAGT |
Cxcl12 fwd |
CGGTTCTTCGAGAGCCACAT |
Cxcl12 rev |
TTGTTGTTCTTCAGCCGTGC |
Flow cytometric analysis of immune cell populations
To characterize myeloid cell populations in tumors, tumor tissue was isolated and minced with a scalpel. Tissue pieces were enzymatically digested for 40min at 37°C and moderate shaking with 2mg/ml Collagenase (Sigma-Aldrich) and 100μg/ml DNase I (Roche) in 10ml DMEM with 2% fetal calve serum (Gibco). After digestion, 40ml PBS (Gibco) was added and the suspension was filtered through 0.7μm nylon strainers (Thermo Fisher Scientific). The suspension was centrifuged for 5min at 4°C and 500x g and the supernatant was discarded. The cell pellet was resuspended in red blood cell lysis buffer (Sigma-Aldrich) and incubated for 10min at room temperature. 40ml of PBS were added and cells were centrifuged again. Cells were resuspended in staining buffer containing 1:1000 efluor780 viability dye (Thermo Fisher Scientific), 1:200 CD16/CD32 Fc-blocking antibody (eBioscience) and 1:200 fluorochrome-conjugated antibodies.
Antigen |
Antibody |
Fluorochrome |
|---|---|---|
CD45 |
eBioscience, 30-F11 |
PerCP-Cyan5.5, efluor450 |
CD11b |
BD bioscience, M1/70 |
BV500, PE-Cy7 |
Gr1 |
eBioscience, RB6-8C5 |
APC |
F4/80 |
eBioscience, BM8 |
PE |
CD3 |
eBioscience, 17A2 |
APC, BV605 |
CD8b |
BD bioscience, H35-17.2 |
PE, APC |
CD4 |
eBioscience, RM4-5 |
efluor450, BV510 |
B220 |
eBioscience, RA3-6B2 |
FITC |
Ifn-y |
eBioscience, XMG1.1 |
PerCP Cy-5.5. |
GrzmB |
eBioscience, NGZB |
FITC |
NKp46 |
BD bioscience, 29A1.4 |
A700 |
After 20min of incubation with the primary antibody at room temperature in the dark, cells were washed twice with PBS and fixed in IC fixation buffer (eBioscience). Cells were washed again in PBS and analyzed by FACS. Staining of lymphoid cell populations in tumor and lung tissue was performed in the same way, however, mononuclear cells were purified prior to staining with density centrifugation in a three-layered Percoll (GE Healthcare) gradient (30%, 40% and 75%). The cell solution was carefully applied on top of the gradient and centrifuged for 20min at 220x g at 4°C with disabled rotor break. The interphase containing the mononuclear cells was isolated, washed with PBS and pelleted. For cytokine staining, purified mononuclear cells were stimulated for 4h at 37°C and 5%CO2 in RPMI (Gibco) with 10% fetal calf serum (Gibco), PMA (20ng/ml) (Sigma-Aldrich), Ionomycin (1ug/ml) (Sigma-Aldrich) and GolgiPlug (1:1000) (BD bioscience). Cells were pelleted and resuspended in staining buffer with antibodies against surface antigens for 20min at RT, washed with PBS (Gibco) containing 2% fetal calf serum (Gibco) and fixed in IC fixation buffer (eBioscience). Cells were washed with 1x permeabilization buffer (eBioscience) and intracellular staining was performed over night at 4°C. Cells were then washed again and analyzed by FACS.
Relative quantification of circulating tumor cells
Blood from tumor-bearing animals was collected by cardiac puncture and transferred to an EDTA-containing S-monovette (Sarstedt) to prevent blood clotting. 5ml red blood cell lysis buffer was added to up to 500μl of blood. After 10min at room temperature, lysis buffer was diluted with 45ml of PBS and the cells were pelleted by centrifugation at 4°C and 500x g for 5min. The supernatant was aspirated and the cell pellet was resuspended in RLT lysis buffer (Qiagen) including 1% β-mercaptoethanol (Sigma-Aldrich). Samples were then stored at -80°C until RNA isolation. RNA isolation was performed as described above. On-column DNA digestion during isolation as well as additional off-column DNAse I digest was performed with 1U/μl RNAse-free DNAse I (Thermo Fischer Scientifc) for 15min at 37°C to remove residual genomic DNA. Digestion was stopped by heat inactivation at 65°C for 10min.
Peripheral blood count
Blood was collected from the facial vein and analyzed with a scil vet abc blood counter (scil).
Co-culture of tumor cells with lung tissue
PyMT tumor-derived TS1 tumor cells [61] were seeded on a 24 well plate at a density of 15.000 or 50.000 cells per well. On the next day, PyMT IkkβF/F or PyMT IkkβΔmye animals were sacrificed and their lungs were perfused with PBS via the right ventricle to flush out the blood. Lungs were isolated, lobes were separated and tissue was cut in equal pieces. Tumor cells were washed once with PBS and serum-reduced DMEM medium with Pen/Strep (Gibco), Glutamax (Gibco) and 0,1% fetal calve serum (Gibco) was added to each well. Transwells with a pore size of 0,4μM (Greiner Bio-One) were placed in the well and 40mg of lung tissue was placed in the upper compartment of the transwells in reduced DMEM. Cells and lung tissue were co-cultured over night for 15h, then, the lung tissue pieces were removed and cells were incubated for another 24h. Afterwards, cells were stained with trypan blue (Sigma-Aldrich) and counted with a hemocytometer (Laboroptik). To compare different experiments, the number of cells after co-culture with lung tissue from PyMT IkkβF/F or PyMT IkkβΔmye mice was normalized to the number of control cells seeded and grown in parallel at the corresponding density and time in complete DMEM with Pen/Strep (Gibco), Glutamax (Gibco) and 10% fetal calve serum (Gibco). For each of the three experiment depicted, four to five replicates per condition existed.
To determine cell cycle stage and viability of TS1 cells after co-culture with lung tissue, co-culture was performed as described above but the experiment was scaled up to yield an appropriate amount of cells for FACS analysis. 70.000 TS1 cells per well were plated in a 6 well plate and co-cultured with ≈90mg of lung tissue per well in 3,5ml serum-reduced DMEM medium. For each condition two replicates existed. After the incubation period, cells were trypsinized and their concentration was adjusted to a number of 0,5x106 cells/ml in DMEM with 2% FCS, 1:10 Penicilin/Streptomycin, Glutamax, 10mM Hepes and 5ug/ml Hoechst 33342 dye (Sigma-Aldrich). Cells were incubated for 30 min in a 2ml reaction tube in the water bath at 37°C and were mixed by inverting every 10min. Cells were then centrifuged for 5min at 500g and 4°C and resuspended in ice-cold Hanks buffered salt solution with 10mM HEPES and 2% FCS (HBSS+) with 2ug/ml propidium iodide (ebioscience). Cells were incubated on ice for 20min, washed once with HBSS+ buffered salt solution with 10mM HEPES and 2% FCS and analyzed by FACS.
Statistics
Statistics of data depicted in the figures was calculated with PRISM7 (Graphpad). When comparing two data sets, unpaired, two-tailed t-test was performed. When comparing more than two data sets, one-way ANOVA and Bonferroni’s multiple comparison were performed. For metastatic incidence no statistics was done since a single value is depicted in the graph (percentage). Differences in datasets are considered significant only when a p value smaller than p = 0,05 is indicated as in the following: *p < 0,05, **p < 0,01, ***p < 0,001, ****p < 0,0001.
Abbreviations
BMDMs, bone marrow-derived macrophages; EMT, epithelial to mesenchymal transition; GRA, granulocytes; LYM, lymphocytes; MON, monocytes; PI, propidium iodide; PyMT, MMTV polyoma middle T; TAMs, tumor-associated macrophages; TANs, Tumor-associated neutrophils; WBC, white blood cells.
Author contributions
T.N. and Ö.C. performed and analyzed experiments. F.R.G. designed the study, T.N. and F.R.G. wrote the manuscript.
ACKNOWLEDGMENTS
We thank Hana Kunkel, Kathleen Mohs, Christin Danneil, Eva Rudolf and Petra Dinse for expert technical assistance. We are grateful to Johanna Joyce for providing TS1 cells.
CONFLICTS OF INTEREST
The authors have no conflicting financial interest.
FUNDING
This work was supported in part by the LOEWE Center for Cell and Gene Therapy Frankfurt (CGT, III L 4-518/17.004) and institutional funds from the Georg-Speyer-Haus.
REFERENCES
1. Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015; 15:73–86. https://doi.org/10.1038/nri3789.
2. Martin M, Wei H, Lu T. Targeting microenvironment in cancer therapeutics. Oncotarget. 2016; 7:52575–83. https://doi.org/10.18632/oncotarget.9824.
3. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011; 147:275–92. https://doi.org/10.1016/j.cell.2011.09.024.
4. Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, Andrews D, Mikeska T, Mangan NE, Samarajiwa SA, de Weerd NA, Gould J, Argani P, et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med. 2012; 18:1224–31. https://doi.org/10.1038/nm.2830.
5. Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M, Tan TG, Zheng L, Ong LC, Jin Y, Kato M, Prévost-Blondel A, Chow P, et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest. 2010; 120:2030–9. https://doi.org/10.1172/JCI42002.
6. Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell. 2011; 20:300–14. https://doi.org/10.1016/j.ccr.2011.08.012.
7. Mlecnik B, Bindea G, Kirilovsky A, Angell HK, Obenauf AC, Tosolini M, Church SE, Maby P, Vasaturo A, Angelova M, Fredriksen T, Mauger S, Waldner M, et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci Transl Med. 2016; 8:327ra26. https://doi.org/10.1126/scitranslmed.aad6352.
8. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, Diehn M, West RB, Plevritis SK, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015; 21:938–45. https://doi.org/10.1038/nm.3909.
9. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, Rugo HS, Hwang ES, Jirström K, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011; 1:54–67. https://doi.org/10.1158/2159-8274.CD-10-0028.
10. Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996; 56:4625–9.
11. Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004; 118:285–96. https://doi.org/10.1016/j.cell.2004.07.013.
12. Schwitalla S, Ziegler PK, Horst D, Becker V, Kerle I, Begus-Nahrmann Y, Lechel A, Rudolph KL, Langer R, Slotta-Huspenina J, Bader FG, Prazeres da Costa O, Neurath MF, et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell. 2013; 23:93–106. https://doi.org/10.1016/j.ccr.2012.11.014.
13. Li H, Yang B, Huang J, Lin Y, Xiang T, Wan J, Chouaib S, Ren G. Cyclooxygenase-2 in tumor-associated macrophages promotes breast cancer cell survival by triggering a positive-feedback loop between macrophages and cancer cells. Oncotarget. 2015; 6:29637–50. https://doi.org/10.18632/oncotarget.4936.
14. Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001; 193:727–40. https://doi.org/10.1084/jem.193.6.727.
15. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006; 66:11238–46. https://doi.org/10.1158/0008-5472.CAN-06-1278.
16. Lin L, Chen YS, Yao YD, Chen JQ, Chen JN, Huang SY, Zeng YJ, Yao HR, Zeng SH, Fu YS, Song EW. CCL18 from tumor-associated macrophages promotes angiogenesis in breast cancer. Oncotarget. 2015; 6:34758–73. https://doi.org/10.18632/oncotarget.5325.
17. Lewis JS, Landers RJ, Underwood JC, Harris AL, Lewis CE. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J Pathol. 2000; 192:150–8. https://doi.org/10.1002/1096-9896(2000)9999:9999<::AID-PATH687>3.0.CO;2-G.
18. Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, Berman T, Joyce JA. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010; 24:241–55. https://doi.org/10.1101/gad.1874010.
19. Goswami S, Sahai E, Wyckoff JB, Cammer M, Cox D, Pixley FJ, Stanley ER, Segall JE, Condeelis JS. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005; 65:5278–83. https://doi.org/10.1158/0008-5472.CAN-04-1853.
20. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO. The Cellular and Molecular Origin of Tumor-Associated Macrophages. Science. 2014; 344:921–5. https://doi.org/10.1126/science.1252510.
21. Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, Segall JE, Pollard JW, Condeelis J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007; 67:2649–56. https://doi.org/10.1158/0008-5472.CAN-06-1823.
22. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011; 475:222–5. https://doi.org/10.1038/nature10138.
23. Chen Q, Zhang XH, Massagué J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell. 2011; 20:538–49. https://doi.org/10.1016/j.ccr.2011.08.025.
24. Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, Stolz DB, Land SR, Marconcini LA, Kliment CR, Jenkins KM, Beaulieu KA, Mouded M, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. 2010; 16:219–23. https://doi.org/10.1038/nm.2084.
25. Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006; 103:12493–8. https://doi.org/10.1073/pnas.0601807103.
26. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, Matrisian LM, Carbone DP, Lin PC. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004; 6:409–21. https://doi.org/10.1016/j.ccr.2004.08.031.
27. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009; 16:183–94. https://doi.org/10.1016/j.ccr.2009.06.017.
28. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008; 111:4233–44. https://doi.org/10.1182/blood-2007-07-099226.
29. Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001; 166:678–89. https://doi.org/10.4049/jimmunol.166.1.678.
30. Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. 2015; 528:413–7. https://doi.org/10.1038/nature16140.
31. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, Bourdeau F, Kubes P, Ferri L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013 Jul 1. [Epub ahead of print]. https://doi.org/10.1172/JCI67484.
32. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, Psaila B, Kaplan RN, Bromberg JF, Kang Y, Bissell MJ, Cox TR, Giaccia AJ, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. 2017; 17:302–17. https://doi.org/10.1038/nrc.2017.6.
33. Liu Y, Gu Y, Han Y, Zhang Q, Jiang Z, Zhang X, Huang B, Xu X, Zheng J, Cao X. Tumor Exosomal RNAs Promote Lung Pre-metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell. 2016; 30:243–56. https://doi.org/10.1016/j.ccell.2016.06.021.
34. Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, Iglehart JD. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci U S A. 2004; 101:10137–42. https://doi.org/10.1073/pnas.0403621101.
35. Chavey C, Bibeau F, Gourgou-Bourgade S, Burlinchon S, Boissière F, Laune D, Roques S, Lazennec G. Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 2007; 9:R15. https://doi.org/10.1186/bcr1648.
36. Bollrath J, Greten FR. IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009; 10:1314–9. https://doi.org/10.1038/embor.2009.243.
37. Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992; 12:954–61. https://doi.org/10.1128/MCB.12.3.954.
38. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010; 140:883–99. https://doi.org/10.1016/j.cell.2010.01.025.
39. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJ, Ciampricotti M, Hawinkels LJ, Jonkers J, de Visser KE. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015; 522:345–8. https://doi.org/10.1038/nature14282.
40. Street SE, Cretney E, Smyth MJ. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood. 2001; 97:192–97. https://doi.org/10.1182/blood.V97.1.192.
41. Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, Schreiber RD. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci USA. 1998; 95:7556–61. https://doi.org/10.1073/pnas.95.13.7556.
42. Barth RJ Jr, Mulé JJ, Spiess PJ, Rosenberg SA. Interferon gamma and tumor necrosis factor have a role in tumor regressions mediated by murine CD8+ tumor-infiltrating lymphocytes. J Exp Med. 1991; 173:647–58. https://doi.org/10.1084/jem.173.3.647.
43. Pickup M, Novitskiy S, Moses HL. The roles of TGFβ in the tumour microenvironment. Nat Rev Cancer. 2013; 13:788–99. https://doi.org/10.1038/nrc3603.
44. Palen K, Weber J, Dwinell MB, Johnson BD, Ramchandran R, Gershan JA. E-cadherin re-expression shows in vivo evidence for mesenchymal to epithelial transition in clonal metastatic breast tumor cells. Oncotarget. 2016; 7:43363–75. https://doi.org/10.18632/oncotarget.9715.
45. Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009; 9:361–71. https://doi.org/10.1038/nrc2628.
46. Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, Lang RA, Pollard JW. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One. 2009; 4: e6562. https://doi.org/10.1371/journal.pone.0006562.
47. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014; 41:49–61. https://doi.org/10.1016/j.immuni.2014.06.010.
48. Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. 2016; 16:431–46. https://doi.org/10.1038/nrc.2016.52.
49. Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008; 205:1261–8. https://doi.org/10.1084/jem.20080108.
50. Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, Schott AF, Kinugasa-Katayama Y, Lee Y, Won NH, Nakasone ES, Hearn SA, Küttner V, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. 2016; 8:361ra138. https://doi.org/10.1126/scitranslmed.aag1711.
51. Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med. 2015; 66:97–110. https://doi.org/10.1146/annurev-med-051013-052304.
52. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006; 66:1123–31. https://doi.org/10.1158/0008-5472.CAN-05-1299.
53. Yan HH, Pickup M, Pang Y, Gorska AE, Li Z, Chytil A, Geng Y, Gray JW, Moses HL, Yang L. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. 2010; 70:6139–49. https://doi.org/10.1158/0008-5472.CAN-10-0706.
54. Yang J, Hawkins OE, Barham W, Gilchuk P, Boothby M, Ayers GD, Joyce S, Karin M, Yull FE, Richmond A. Myeloid IKKβ promotes antitumor immunity by modulating CCL11 and the innate immune response. Cancer Res. 2014; 74:7274–84. https://doi.org/10.1158/0008-5472.CAN-14-1091.
55. Connelly L, Barham W, Onishko HM, Chen L, Sherrill TP, Zabuawala T, Ostrowski MC, Blackwell TS, Yull FE. NF-kappaB activation within macrophages leads to an anti-tumor phenotype in a mammary tumor lung metastasis model. Breast Cancer Res. 2011; 13:R83. https://doi.org/10.1186/bcr2935.
56. Sasmono RT, Oceandy D, Pollard JW, Tong W, Pavli P, Wainwright BJ, Ostrowski MC, Himes SR, Hume DA. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003; 101:1155–63. https://doi.org/10.1182/blood-2002-02-0569.
57. Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, Sylvestre V, Stanley ER. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 2002; 99:111–20. https://doi.org/10.1182/blood.V99.1.111.
58. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999; 8:265–77. https://doi.org/10.1023/A:1008942828960.
59. Zardavas D, Irrthum A, Swanton C, Piccart M. Clinical management of breast cancer heterogeneity. Nat Rev Clin Oncol. 2015; 12:381–94. https://doi.org/10.1038/nrclinonc.2015.73.
60. Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M, Trivett M, Thompson ER, Ramakrishna M, Gorringe KL, Polyak K, Haviv I, Campbell IG. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet. 2008; 40:650–5. https://doi.org/10.1038/ng.117.
61. Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, Bell-McGuinn KM, Zabor EC, Brogi E, Joyce JA. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011; 25:2465–79. https://doi.org/10.1101/gad.180331.111.