
INTRODUCTION
Ovarian cancer is the fifth-leading cause of cancer related deaths in women, with an estimated 22,280 new cases and 14,240 deaths in the United States in 2016 [1]. Thereinto, approximately 70% is high-grade serous carcinomas [2]. Up to now, despite the effective treatments including radical resection, systemic chemotherapy, and targeted drugs for patients, the average 5-year survival is still only at 46% [1]. Ovarian cancer is a multifactorial disease caused by the interaction of genetic and epigenetic factors. DNA methylation, as the most prominent epigenetic alteration, could occur at CpG island in the promoter region, 5’ or 3’ untranslated regions, and even in gene body of tumor suppressor genes (TSGs). Hypermethylation in the proximal promoter region often contributes to the transcriptional down-regulation but methylation in exons is associated with active transcription [3, 4]. Recently, mounting evidences demonstrated that DNA methylation was involved in ovarian cancer [5–7]. Therefore, identifying the role of TSG methylation in patients with ovarian cancer is of value.
P16INK4a (also known as CDKN2A), a classical TSG, is located on chromosome 9p21 and plays an important role in cell cycle regulation by decelerating cells progression from G1 to S phase [8, 9]. It has become clear that the expression of P16 is reduced by DNA methylation [10–12]. And P16INK4a inactivation upregulates retinoblastoma (RB) protein by stimulating the cyclin dependent kinases (CDKs) and RB pathway, which leads to dysfunction of cell proliferation and apoptosis, thereby further facilitating carcinogenesis [13]. Indeed, several types of cancer, including ovarian cancer, exhibit a methylation phenotype of P16INK4a[14–16].
To date, even though abundant studies have been conducted to explore the role of P16INK4a methylation in ovarian cancer, the results are still inconclusive. Several studies reported that P16INK4a methylation was associated with an increasing trend in ovarian cancer [17–20]. While, other studies suggested that P16INK4a methylation was not related to the occurring of ovarian cancer [21–27]. Interestingly, even the conclusions in two published meta-analyses were inconsistent. Xiao et al. reported that aberrant methylation of P16INK4a promoter was significantly associated with ovarian carcinogenesis [28], while Jiang et al. suggested no association between P16INK4a promoter methylation and epithelial ovarian cancer [29].
Considering these conflicting conclusions on the role of methylated P16INK4a in ovarian cancer, we performed an adaptive synthesized analysis to quantitatively evaluate the occurrence frequency, clinicopathological features and potential prognostic significance of P16INK4a methylation in ovarian cancer. Moreover, we searched The Cancer Genome Atlas (TCGA) database, collecting hundreds of ovarian cancer samples with whole genome DNA methylation datasets to validate our meta-analysis.
RESULTS
Identification of relevant studies
The procedure of study selection is outlined in Figure 1. We identified 233 articles in the initial literature search. A total of 153 references remained after removing duplicates. After reading titles and abstracts, 84 records were identified for further full-text assessment, which further excluded 60 more articles. Finally, 24 studies from 1997 to 2015 were included in this meta-analysis [14, 17, 19-27, 30-42].
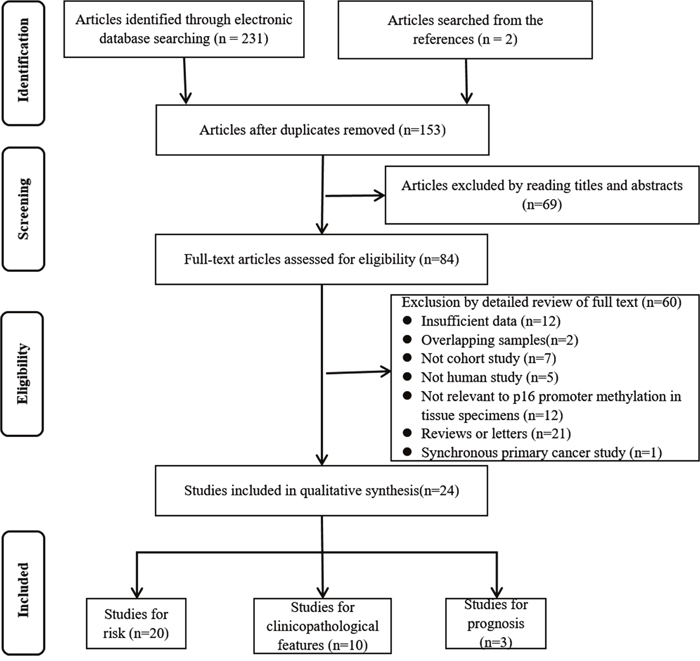
Figure 1: Flow diagram of study selection.
Baseline characteristics of included studies
Out of the 24 studies, 11 studies were conducted in Asia, 7 in Europe, 4 in America, 1 in Africa and 1 in Oceania. The detection methods of methylation in 20 studies were methylation-specific PCR (MSP) and real-time quantitative MSP (qMSP), while methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) was used in 2 studies, MethyLight was used in 1 study, and Southern analysis was used in 1 study. Among the 24 articles, 20 studies [14, 17, 19-27, 30-33, 35, 38-40, 42] addressed the risk of P16INK4a methylation in ovarian cancer, 10 studies [17, 22, 25, 26, 31, 34, 36, 37, 40, 41] covered clinicopathological features, and 3 studies [17, 35, 36] discussed prognosis. To explore the relationship between P16INK4a methylation and ovarian cancer risk, three groups, i.e., normal tissues, benign tissues and low malignant potential or borderline tumor (LMP) tissues, were compared. The Newcastle Ottawa Scale (NOS) scores of all case-control studies were greater than or equal to 5. The basic characteristics of all included studies are summarized in Tables 1 and 2.
Table 1: Characteristics of studies included for the association between P16INK4a methylation and ovarian cancer risk
First author |
Year |
Country |
Geographical location |
Sample size* |
Case number |
Age(year) |
Sample type |
Method |
Methylation site |
NOS score |
|||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
C(M/n) |
LMP(M/n) |
B(M/n) |
N(M/n) |
||||||||||
Moselhy |
2015 |
Saudi Arabia |
Asia |
Small |
14/18 |
- |
12/32 |
- |
52.3±12.1 |
FFPET |
MSP |
Promoter |
7 |
Bhagat |
2014 |
India |
Asia |
Large |
58/134 |
5/23 |
11/26 |
0/15 |
49.55±9.72 |
FFT |
qMSP |
Promoter |
7 |
Ozdemir |
2012 |
Turkey# |
Asia |
Large |
1/75 |
- |
- |
0/75 |
- |
Tissue |
MS-MLPA |
Promoter |
7 |
Ho |
2012 |
Taiwan |
Asia |
Small |
1/47 |
- |
6/29 |
- |
50(32-66) |
FFT |
MS-MLPA |
Promoter |
7 |
Cuľbová M |
2011 |
Slovakia |
Europe |
Small |
5/13 |
0/2 |
5/19 |
- |
54.8(34-74) |
FFT |
MSP |
Promoter |
6 |
Abou-zeid |
2011 |
Egypt$ |
Africa |
Large |
21/52 |
- |
9/43 |
4/40 |
60(49-74) |
FFT |
qMSP |
Promoter |
7 |
Gu |
2009 |
China |
Asia |
Large |
8/87 |
- |
13/42 |
- |
51(21-69) |
Tissue |
MethyLight |
Promoter |
7 |
Shen |
2008 |
China |
Asia |
Large |
13/63 |
- |
- |
0/30 |
52.8(33-76) |
FFT |
MSP |
Promoter |
6 |
Wu |
2007 |
Norway |
Europe |
Large |
0/52 |
0/2 |
0/2 |
- |
- |
FFT |
MSP |
Promoter |
6 |
Tam |
2007 |
Hong Kong |
Asia |
Large |
17/89 |
1/16 |
1/19 |
4/16 |
53.1±1.4 |
FFT |
MSP |
Promoter |
7 |
Wiley |
2006 |
Italy |
Europe |
Large |
89/215 |
4/19 |
- |
- |
57.7±11.4 |
FFT |
MSP |
Promoter |
7 |
Li |
2006 |
China |
Asia |
Small |
6/18 |
- |
- |
0/10 |
- |
Tissue |
MSP |
Promoter |
5 |
Makarla |
2005 |
USA |
America |
Small |
7/23 |
5/23 |
3/23 |
0/16 |
51.5(20-86) |
FFT |
MSP |
Promoter |
7 |
Liu |
2005 |
USA |
America |
Large |
13/52 |
- |
- |
15/40 |
61.5±9.4 |
FFT |
MSP |
Promoter |
5 |
Dhillon |
2004 |
India |
Asia |
Small |
10/25 |
- |
- |
1/75 |
- |
Tissue |
MSP |
Promoter |
7 |
Rathi |
2002 |
USA |
America |
Small |
5/49 |
- |
- |
0/16 |
56(40-79) |
FFT |
MSP |
Promoter |
7 |
Strathdee |
2001 |
UK |
Europe |
Large |
0/93 |
- |
- |
0/18 |
- |
FFT |
MSP |
Promoter |
6 |
Brown |
2001 |
UK |
Europe |
Small |
0/30 |
0/13 |
0/14 |
- |
- |
FFT |
MSP |
Promoter |
5 |
McCluskey |
1999 |
USA |
America |
Small |
21/37 |
11/15 |
14/20 |
- |
- |
FFT |
MSP |
Promoter |
6 |
Shih |
1997 |
Australia |
Oceania |
Small |
0/45 |
0/3 |
0/2 |
- |
- |
Tissue |
Southern analysis |
Promoter |
5 |
Abbreviations: C: cancer tissues; LMP: low malignant potential or borderline tumor tissues; B: benign tissues; N: normal tissues; M: methylated; n: number of patients; FFPET: formalin fixed and paraffin embedded tissues; FFT: fast frozen tissues; MSP: methylation-specific PCR; qMSP: real-time quantitative MSP; MS-MLPA: methylation-specific multiplex ligation-dependent probe amplification.
*We defined n<50 as small size and ≥50 as large size. n, the number of patients in case group.
# Turkey is a transcontinental Eurasian country and is usually assigned to Asia internationally.
$ Egypt is a transcontinental country spanning the northeast corner of Africa and southwest corner of Asia, usually assigned to Africa internationally.
Table 2: Characteristics of studies included for the association between P16INK4a methylation and clinicopathological features of ovarian cancer
First author |
Year |
Country |
Geographical location |
No. of patients |
Age (year) |
Stage |
Grade |
Histological subtype |
|||
|---|---|---|---|---|---|---|---|---|---|---|---|
I~II |
III~ IV (M/n) |
1~2 |
3 |
Serous |
Non-serous |
||||||
Bhagat |
2014 |
India |
Asia |
134 |
49.55±9.72 |
19/41 |
39/93 |
18/45 |
40/89 |
32/76 |
26/58 |
Cuľbová M |
2011 |
Slovakia |
Europe |
13 |
54.8(34-74) |
- |
- |
- |
- |
2/6 |
3/7 |
Shen |
2008 |
China |
Asia |
63 |
52.8(33-76) |
4/22 |
9/41 |
6/36 |
7/27 |
7/34 |
6/29 |
Yang |
2006 |
Hong Kong |
Asia |
49 |
48.8(26-79) |
4/24 |
5/25 |
6/22 |
3/25 |
3/17 |
6/32 |
Makarla |
2005 |
USA |
America |
23 |
51.5(20-86) |
- |
- |
- |
- |
2/9 |
5/14 |
Liu |
2005 |
USA |
America |
52 |
61.5±9.4 |
- |
- |
10/41 |
3/11 |
- |
- |
Katsaros |
2004 |
Italy |
Europe |
249 |
57(19-82) |
22/68 |
68/152 |
26/75 |
64/141 |
40/86 |
50/141 |
Hashiguchi |
2001 |
Japan |
Asia |
46 |
- |
4/21 |
2/20 |
7/33 |
0/13 |
2/14 |
5/32 |
McCluskey |
1999 |
USA |
America |
29 |
- |
- |
- |
- |
- |
1/14 |
1/15 |
Milde-langosch |
1998 |
Germany |
Europe |
44 |
- |
- |
- |
- |
- |
3/11 |
13/33 |
Abbreviations: M: methylated; n: number of patients.
Quantitative data synthesis
Association between P16INK4a methylation and ovarian cancer risk
A total of 1,217 ovarian cancers, 116 LMP cancers, 271 benign patients and 351 normal controls were quantitatively synthesized in this analysis. Results indicated that the frequency of P16INK4a methylation in cancer tissues was significantly elevated than that in normal tissues (odds ratio (OR) = 5.01, 95% confidence interval (CI) = 1.55-16.14) and LMP tissues (OR = 1.88, 95% CI = 1.10-3.19), but similar to benign tissues (OR = 1.18, 95% CI = 0.52-2.65) (Figure 2). Further analyses showed that the frequencies of P16INK4a methylation in benign tissues and LMP tissues were not higher than that in normal tissues (OR = 2.28, 95% CI = 0.37-14.09; OR = 2.28, 95% CI = 0.15-34.73, respectively) (Figure 3).
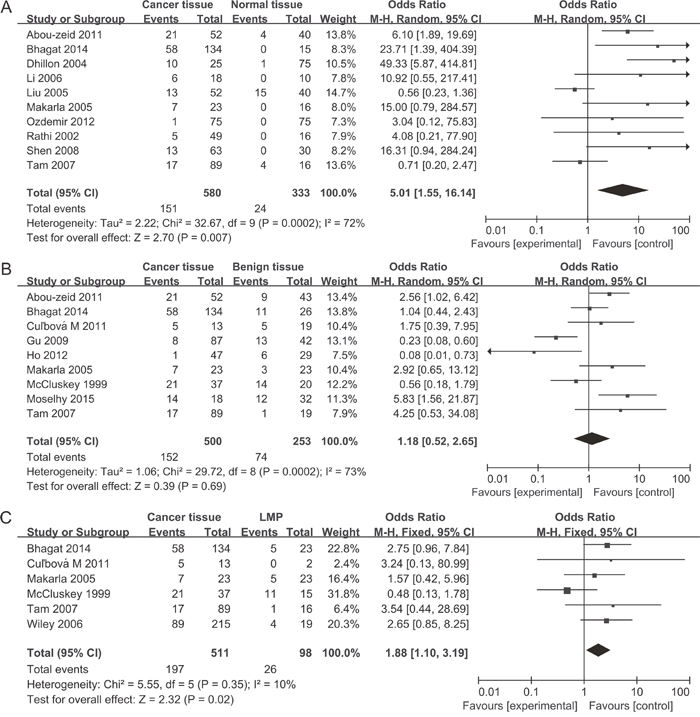
Figure 2: Forest plots for the association between P16INK4a methylation and ovarian cancer risk. (A) Cancer tissues vs. normal tissues; (B) Cancer tissues vs. benign tissues; (C) Cancer tissues vs. LMP tissues.
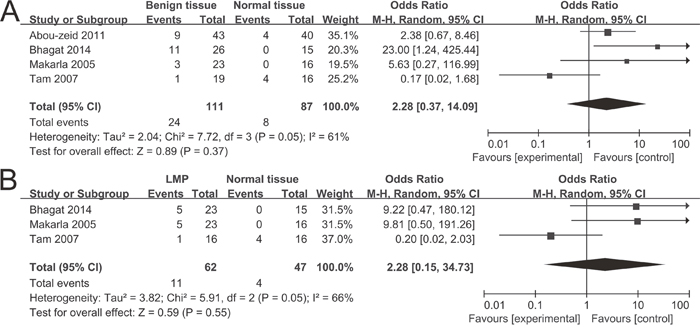
Figure 3: Forest plots for the association between P16INK4a methylation and ovarian diseases. (A) Benign tissues vs. normal tissues; (B) LMP tissues vs. normal tissues.
With large heterogeneity, meta-regression and subgroup analyses were conducted by the publication year, geographical location, method and case sample size in the comparison of cancer tissues vs. normal tissues. Meta-regression found that case sample size was significantly correlated with the inter-study heterogeneity (P = 0.041) while other covariates was not (Table 3). Furthermore, as shown in Table 3, subgroup analyses revealed that the OR was 5.69 (95% CI = 0.42-76.14) for the publication year ≤2005 and 4.71 (95% CI = 1.30-17.07) for >2005 under the random effects model. For geographical location, the OR was 7.85 (95% CI = 1.33-46.32) in Asia, 2.31 (95% CI = 0.24-22.01) in America and 6.10 (95% CI = 1.89-19.69) in Africa under random effects model. For test method, the OR for MSP was 4.49 (95% CI = 0.97-20.64) under random effects model and 8.11 (95% CI = 2.93-22.40) for other methods under fixed effects model. In addition, the OR was 15.75 (95% CI = 4.05-61.34) for sample size <50 in fixed effects model and 2.21 (95% CI = 1.33, 3.67) for that ≥ 50 in random effects model.
Table 3: Meta-regression and subgroup analyses of P16INK4a methylation in comparison of cancer tissues vs. normal tissues
Stratified analysis |
No. of studies |
Pooled OR (95% CI) |
Meta-regression |
Heterogeneity |
||
|---|---|---|---|---|---|---|
Random |
Fixed |
P-value |
I2 (%) |
P-value |
||
Publication year |
0.376 |
|||||
≤2005 |
4 |
5.69 (0.42, 76.14) |
2.17 (1.17, 4.05) |
84% |
0.0003 |
|
>2005 |
6 |
4.71 (1.30, 17.07) |
4.65 (2.32, 9.30) |
56% |
0.05 |
|
Geographical location |
0.161 |
|||||
Asia |
6 |
7.85 (1.33, 46.32) |
5.87 (2.70, 12.78) |
70% |
0.005 |
|
America |
3 |
2.31 (0.24, 22.01) |
1.15 (0.56, 2.37) |
68% |
0.05 |
|
Africa |
1 |
6.10 (1.89, 19.69) |
6.10 (1.89, 19.69) |
- |
- |
|
Method |
0.651 |
|||||
MSP |
7 |
4.49 (0.97, 20.64) |
2.33 (1.38, 3.92) |
77% |
0.0003 |
|
others |
3 |
6.79 (2.43, 18.94) |
8.11 (2.93, 22.40) |
0% |
0.57 |
|
Case sample size |
0.041 |
|||||
<50 |
4 |
17.21 (4.54, 65.28) |
15.75 (4.05, 61.34) |
0% |
0.58 |
|
≥50 |
6 |
2.21 (1.33, 3.67) |
2.74 (0.71, 10.53) |
75% |
0.001 |
|
Association between P16INK4a methylation and clinicopathological features in patients with ovarian cancer
10 studies comprising 680 samples were enrolled to assess whether or not the abnormal P16INK4a methylation was associated with ovarian cancer clinicopathological characteristics. As displayed in Figure 4, no statistically significant correlation was found between P16INK4a methylation and age of patients (≥60 vs. <60: OR = 1.39, 95% CI = 0.66-2.92), clinical stage (III~IV vs. I~II: OR = 1.21, 95% CI = 0.81-1.82), grade (3 vs. 1~2: OR = 1.20, 95% CI = 0.82-1.1.75) as well as histological subtype (serous vs. non-serous: OR = 1.09, 95% CI = 0.76-1.55).
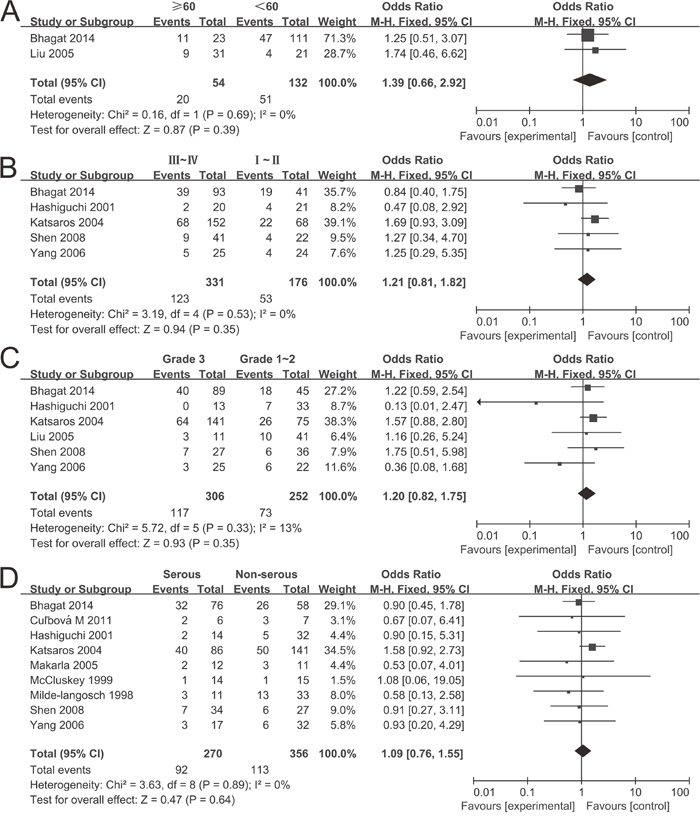
Figure 4: Forest plots for the association between P16INK4a methylation and clinicopathological features in ovarian cancer. (A) Age; (B) Clinical stage; (C) Tumor grade; (D) Histological subtype.
Prognostic value of P16INK4a methylation in patients with ovarian cancer
Only two studies [35, 36] containing 464 patients evaluated the P16INK4a methylation on progression-free survival (PFS), and three studies [17, 35, 36] containing 600 patients on overall survival (OS). The combined results revealed P16INK4a methylation was significantly associated with a poor PFS by univariate Cox proportional hazards regression model (hazard ratio (HR) = 1.68, 95% CI = 1.26-2.24) (Figure 5A). After considering potential confounders by adjusting for age at diagnosis or surgery, disease stage, histological grade and residual tumor size, the pooled HR was 1.55 (1.15-2.08) (Figure 5B). Survival analysis also showed that P16INK4a methylation reduced OS in univariate and multivariate Cox regression models (HR = 1.28, 95% CI = 0.97-1.68; HR = 1.16, 95% CI = 0.87-1.55, respectively) (Figure 5C and 5D), but the differences were not statistically significant.
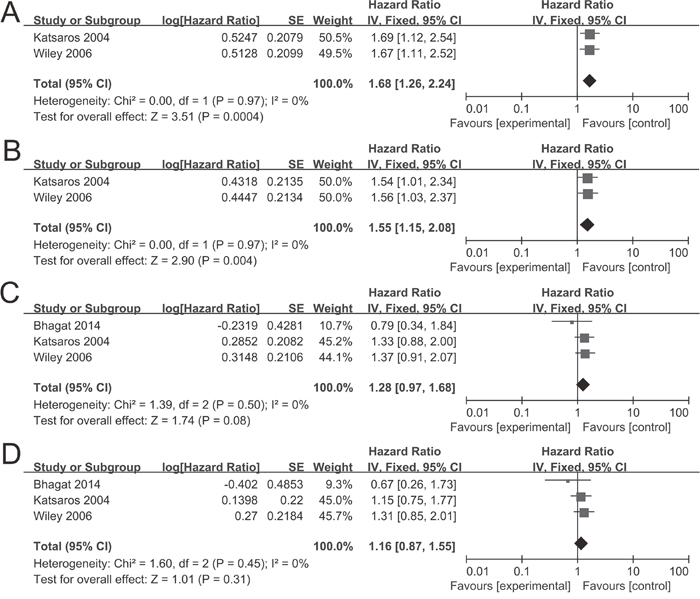
Figure 5: Forest plots for the evaluation of P16INK4a methylation on survival analysis in ovarian cancer. (A) PFS in univariate Cox regression model; (B) PFS in multivariate Cox regression model; (C) OS in univariate Cox regression model; (D) OS in multivariate Cox regression model.
Sensitivity analysis and publication bias
As presented in Figure 6 (6A, 6B, and 6C), no single study significantly affected the pooled ORs in the sensitivity analysis, indicating our analysis was relatively stable and credible. Funnel plots and Begg’s test were used to evaluate the publication bias. The funnel plots were largely symmetric suggesting there were no publication biases in the meta-analysis of P16INK4a methylation and ovarian cancer risk, which was confirmed by the Begg’s test (Figure 6D, 6E, and 6F).
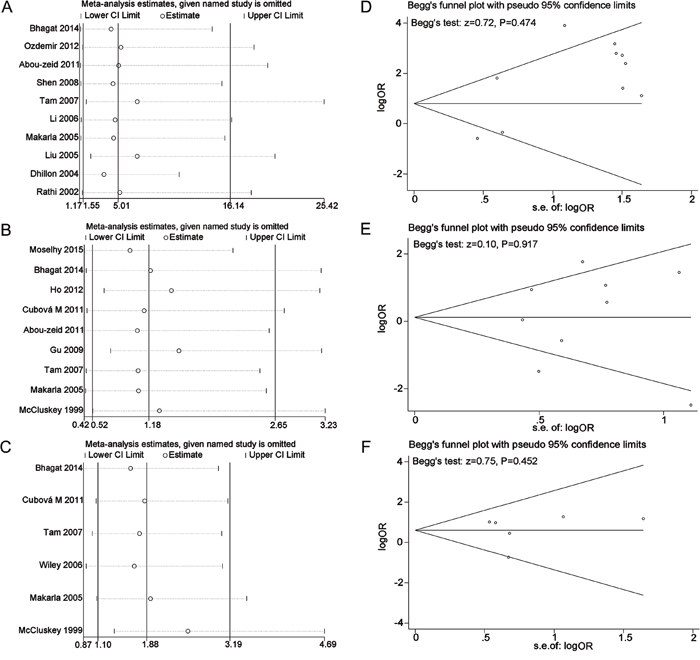
Figure 6: Sensitivity analyses and funnel plots for the publication bias of P16INK4a methylation during the carcinogenesis of ovarian cancer. (A) Sensitivity analysis for the comparison of cancer tissues vs. normal tissues; (B) Sensitivity analysis for the comparison of cancer tissues vs. benign tissues; (C) Sensitivity analysis for the comparison of cancer tissues vs. LMP tissues. (D) Funnel plot for the publication bias of cancer tissues vs. normal tissues; (E) Funnel plot for the publication bias of cancer tissues vs. benign tissues; (F) Funnel plot for the publication bias of cancer tissues vs. LMP tissues.
Methylation level of P16INK4a measured by TCGA program
To further explore the methylation level of P16INK4a in ovarian tumor tissues, we extracted DNA methylation data of P16INK4a CpG sites measured with Illumina HumanMethylation27 BeadChip from TCGA program. As shown in Table 4, the beta value of 582 ovarian tumor tissues and 12 normal ovarian tissues were extracted for analysis. Obviously, the methylation levels of 7 out of 9 CpG sites were significantly increased in the ovarian tumor tissues compared with the normal tissues (cg03079681, cg07752420, cg09099744, cg10895543, cg11653709, cg12840719, and cg26673943). Among these regions, methylation level of probe cg26673943 region (located at the promoter region of P16INK4a) was negatively associated with P16INK4a expression in ovarian cancer patients (adjusted P value < 0.000001). However, methylation levels of the rest 6 probes, which located at non-promoter region tended to positively associate with P16INK4a gene expression. Additionally, we found that methylation level of probe cg13479669 region was lower in tumor tissues compared with normal tissues, and negatively associated with P16INK4a gene expression in tumor tissues. These results suggest that hypermethylation of P16INK4a might be correlated with ovarian carcinogenesis and development. Nevertheless, it seems that the methylation at promoter region or non-promoter region has contrary effects on P16INK4a gene expression.
Table 4: Methylation of P16INK4a CpG sites on Illumina HumanMethylation 27 BeadChip from TCGA datasets
Probe (Illumina Human Methylation 27) |
CpG island location (chromosome: DNA range) |
Normal tissue Beta value (mean, n = 12) |
Tumor tissue Beta value (mean, n = 582) |
Adjusted P value# |
Pearson Correlation Coefficient |
Adjusted P value* |
|---|---|---|---|---|---|---|
cg00718440 |
9: 21983444-21986348 |
0.016 |
0.016 |
0.960249 |
0.194104 |
0.001719 |
cg03079681 |
9: 21983444-21986348 |
0.015 |
0.026 |
< 0.000001 |
0.012972 |
1.0 |
cg07752420 |
9: 21958106-21958899 |
0.149 |
0.653 |
< 0.000001 |
0.569887 |
< 0.000001 |
cg09099744 |
9: 21958106-21958899 |
0.099 |
0.642 |
< 0.000001 |
0.630768 |
< 0.000001 |
cg10895543 |
9: 21958106-21958899 |
0.120 |
0.651 |
< 0.000001 |
0.624147 |
< 0.000001 |
cg11653709 |
9: 21958106-21958899 |
0.144 |
0.610 |
< 0.000001 |
0.555400 |
< 0.000001 |
cg12840719 |
9: 21958106-21958899 |
0.092 |
0.594 |
< 0.000001 |
0.627484 |
< 0.000001 |
cg13479669 |
9: 21983444-21986348 |
0.045 |
0.027 |
0.004226 |
-0.150891 |
0.0333435 |
cg26673943 |
9: 21983444-21986348 |
0.047 |
0.056 |
0.042428 |
-0.269361 |
< 0.000001 |
# P value of t test of the difference between normal tissue Beta value and tumor tissue Beta value;
* P value of Pearson’s correlation between the tumor tissues’ Beta value and CDKN2A expression (n = 368).
DISCUSSION
Ovarian cancer is one of the leading causes of cancer-related death in women [43]. Identification of early disease indicators for diagnosis and prognosis is of clinical value. P16INK4a, which resembles classic TSGs such as P53, is an important negative regulator of cell growth and proliferation [13]. It has been synthetically evaluated for aberrant P16INK4a methylation in numerous cancers [44–47], including ovarian cancer [28, 29]. Considering the conflicting conclusions in two meta-analyses, and the lack of comprehensive assessment on the role of methylated P16INK4a in ovarian cancer, we performed an adaptive synthesized analysis to investigate the relationships between P16INK4a methylation and ovarian cancer risk, as well as clinicopathological features and prognostic value in ovarian cancer. Meanwhile, we searched TCGA data to validate our meta-analysis.
Our meta-analysis demonstrated that P16INK4a methylation in cancer tissues was significantly higher than that in normal tissues (P < 0.05), but not much increased than that in benign tissues. Compared with normal tissues, the frequency of P16INK4a methylation was 2.28-fold higher in both benign tissues and LMP tissues (P > 0.05), but the differences were not statistically significant. Although not establishing a strong correlation between P16INK4a methylation and cancer progression, the above results do suggest a possibility that epigenetic alteration of P16INK4a methylation might play a certain role in ovarian carcinogenesis and might be useful in distinguishing malignant tumor from healthy ovarian tissues. Considering the evident heterogeneity, we conducted subgroup analyses based on probable covariates in the comparison of cancer tissues vs. normal tissues. For geographical location, P16INK4a methylation is a risk factor in Asia and Africa, but not in America. The divergence may be underscored in a large part to a combination of differences in allele frequencies and complex epistasis or gene-environment interactions [48]. The similar findings appeared in the subgroup analyses of different methods and publication year. Kurdyukov et al. [49] suggested that it was essential to choose an appropriate method in a suitable region to answer a particular biological question in studies of DNA methylation. Additionally, the 95% CI was large in the group of small sample size while relatively small in the group of large sample size, implying the conclusion may not be reliable unless studies should be conducted using a sufficient number of samples. Previous studies also demonstrated that the methylation status in blood samples or fluids might be different from that in tissues [50, 51]. Thus, our results should be interpreted with caution because sample types were limited to tissues in studies included in this meta-analysis.
Previous studies indicated that P16INK4a methylation was associated with poorly differentiated tumors and was different in histological subtype in ovarian cancer [19, 36]. However, we could not establish any significant correlations between P16INK4a methylation and clinicopathological features, including age, clinical stage, tumor differentiation or histological subtype in this study. Therefore, it might not be essential to predict the invasion and metastasis of ovarian cancer.
Katsaros et al. [36] and Wiley et al. [35] reported association of P16INK4a methylation with PFS and OS in ovarian cancer, while Bhagat et al. [17] found no significant value in predicting prognosis. In the present study, we discovered that P16INK4a methylation represented a risk factor for PFS. For OS, patients with P16INK4a methylation also had a slightly elevated risk, though the differences are not statistically significant. This trend was also observed in other types of cancer [44, 47]. However, its statistical confirmation requires large studies. The data from TCGA also indicated that methylation level of probe cg26673943 region (located at the promoter region of P16INK4a) in the ovarian tumor tissues was higher than normal ovarian tissues. Increased methylation of CpG island at the promoter region was negatively associated with P16INK4a gene expression, while methylation of CpG islands at non-promoter regions were positively associated with P16INK4a gene expression.
Compared with previous meta-analyses [28, 29], our meta-analysis had several improvements. Firstly, the development of ovarian cancer is a multistep procedure involving normal tissues, benign disease, low malignant potential or borderline tumor and malignant tumor [17]. We compared malignant ovarian cancer with LMP tumors, benign disease, and normal samples to give more rigorously to the analysis. Secondly, with 1,217 malignant ovarian cancer patients, 116 LMP, 271 benign patients and 351 normal samples, the sample size in our study is much larger than that of all previous meta-analyses. Finally, we included the clinicopathological features and prognostic significance of P16INK4a methylation in ovarian cancer for more comprehensive understanding of the underlying pathogenesis of ovarian cancer. These strengths make our study a useful effort in seeking better understanding of the P16INK4a methylation in ovarian cancer.
Several potential limitations in our current study should be also noted. Firstly, the heterogeneity was still large after subgroup analyses in the assessment of the association between P16INK4a methylation and ovarian cancer risk, which may affect the statistical power. Secondly, as a retrospective study, a potential unidentified confounding information and selection bias may exist in our meta-analysis. We could not eliminate the possibility of publication bias, where positive results are likely published than negative results. Thirdly, the total sample size was still relatively small for reliably assessing the prognostic value of P16INK4a methylation in ovarian cancer. Fourthly, none of the studies included in our meta-analysis defined the region considered as promoter or provided specific methylation sites. Therefore, we are unable to establish whether or not they focused on the same sequence of P16INK4a gene. However, the impact of methylation on transcriptional potential depends on the density of the methylated CpG islands and their location relative to the transcription start site. This highlights the importance of a uniform and full-scale reporting of study designs and outcomes. Additionally, previous researches showed that the occurrence of P16INK4a methylation may depend on the histological subtype [34, 41, 52]. However, we are unable to extract the sufficient data to analyze the association between P16INK4a methylation and HGSC because no detailed information of P16INK4a methylation in HGSC was provided in the eligible articles.
Although with certain limitations, our study is a comprehensive meta-analysis focusing on the correlation of aberrant P16INK4a methylation with the initiation, development, and prognosis of ovarian cancer to provide new insight into the pathogenesis of ovarian cancer.
In conclusion, our meta-analysis suggests that aberrant methylation of P16INK4a may be essential to the initiation of ovarian cancer and in distinguishing malignant from healthy ovarian tissues. Besides, P16INK4a methylation is a potential predictive factor for poor prognosis in ovarian cancer. This study indicates the need for multicenter large-scale studies to confirm the role of P16INK4a methylation in ovarian cancer.
MATERIALS AND METHODS
Search strategy and selection criteria
PubMed, EMBASE, Web of Science and China National Knowledge Infrastructure (CNKI) were searched up to October 12, 2016 by the following keywords and search items: (P16 OR P16INK4a OR CDKN2A) AND (methylation OR hypermethylation OR demethylation) AND (ovarian OR ovary) AND (cancer OR carcinoma OR neoplasm). The search was limited to human studies, without language restriction. Moreover, a manual search of the relevant references was implemented to identify the potentially additional articles.
The following criteria were used for screening eligible studies: (1) case-control studies evaluating the association between P16INK4a promoter methylation and ovarian cancer risk, or case only studies evaluating the association of P16INK4a promoter methylation with clinicopathological features or prognosis in ovarian cancer; (2) articles providing sufficient information for calculating an odds ratio (OR) and corresponding 95% confidence interval (95% CI), or study offering hazard ratio (HR) and 95% CI directly; (3) sample types limited to tissues; (4) studies with full text articles. It’s worth noting that when multiple reports were published from a same study population, only the most recent or complete information was included in this meta-analysis. Meanwhile, studies with NOS scores greater than or equal to 5 were enrolled.
Data extraction and quality assessment
With a preformed unified form, data were extracted independently by two investigators and disagreements were resolved by discussion till consensus were achieved. The following information was extracted from studies: the first author’s name, publication year, country, geographical location, sample size, age of patients in the case group, the frequencies of methylation in the case and control groups, methods for detecting methylation, methylation site, disease stage, tumor grade, histological subtype and effects on survival outcomes.
The quality of eligible case-control studies was assessed according to the NOS criteria [53]. The NOS criteria is based on three aspects: (1) subject selection: 0~4; (2) comparability of subject: 0~2; (3) clinical outcome: 0~3.
Statistical analysis
Our principal analysis was conducted using Review Manager 5.2 (Cochrane Collaboration, Oxford, UK). ORs with corresponding 95% CIs were calculated to estimate the association between P16INK4a promoter methylation and ovarian cancer risk or clinicopathological features. Meanwhile, HRs and 95% CIs were used to assess the prognosis of P16INK4a promoter methylation on ovarian cancer. Inter-study heterogeneity was estimated with the Cochran’s Q statistic and I2 tests. P<0.05 or I2 >50% indicated substantial heterogeneity, then the random effects model was applied. Otherwise, the fixed effects model was selected. We also explored sources of heterogeneity using meta-regression and subgroup analyses by publication year, geographical location, method and case sample size. Additionally, the Stata 12.0 (Stata Corporation, TX, USA) was performed to evaluate the sensitivity analysis and publication bias of our studies. Publication bias was evaluated by funnel plots and Begg’s test, P < 0.05 was considered statistically significant. It’s worth mentioning that, for some trials containing no events in both case and control arm, as no information supplied about likely magnitude of the effect, we excluded such trials when synthesizing data [54].
TCGA datasets extraction and analysis
We collected DNA methylation datasets of 582 ovarian cancer cases and 12 ovarian normal tissues from The Cancer Genome Atlas (TCGA, “TCGA-OV” project) program [https://cancergenome.nih.gov/]. The methylation measurement was performed using Illumina HumanMethylation27 BeadChip. Beta value of each CpG site was extracted to assess the methylation level of CDKN2A gene. Beta value was calculated based on the intensities of the methylated (M) and unmethylated (U) bead types [55]: beta value = M/ (M+U). The difference of DNA methylation level of CpG sites between ovarian tumor tissues and normal ovarian tissues in the TCGA database were analyzed by t-test on the means. Plus, P16INK4a gene expression value (fragments per kilobase of transcript per million mapped reads, FPKM) in ovarian tumor tissues (TCGA, “TCGA-OV” project) was also extracted. Pearson’s product-moment correlation between P16INK4a gene expression levels and methylation of its CpG islands were evaluated. Data analysis were performed using R software (R i386 3.4.0). P values were adjusted via Bonferroni correction.
Author contributions
P.X and F.W conceived and designed the study; P.X and X.G identified related studies as well as assessed the quality of included studies; P.X and Z.Z performed the statistical analysis and drafted the manuscript; Q.D, S.T, J.W and P.W revised the manuscript; M.Y revised and finalized the manuscript. All authors reviewed and approved the manuscript prior to submission.
ACKNOWLEDGMENTS AND FUNDING
This study was supported by National Natural Science Funds (No. 81472033 and No. 30901308), the National Science Foundation of Hubei Province (No. 2013CFB233 and No. 2013CFB235), the Scientific and technological project of Wuhan City (No. 2014060101010045), Hubei Province health and family planning scientific research project (WJ2015Q021) and Training Program of the science and technology innovation from Zhongnan Hospital of Wuhan University(cxpy20160054). We thank the Beijing Circle & Dot Technology Co., Ltd for assisting in analyzing the TCGA database.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7-30. https://doi.org/10.3322/caac.21332.
2. Prat J. New insights into ovarian cancer pathology. Ann Oncol. 2012; 23:x111–17. https://doi.org/10.1093/annonc/mds300.
3. Kazanets A, Shorstova T, Hilmi K, Marques M, Witcher M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim Biophys Acta. 2016; 1865:275-88. https://doi.org/10.1016/j.bbcan.2016.04.001.
4. Maunakea AK, Chepelev I, Cui K, Zhao K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013; 23:1256-69. https://doi.org/10.1038/cr.2013.110.
5. Dong A, Lu Y, Lu B. Genomic/Epigenomic Alterations in Ovarian Carcinoma: Translational Insight into Clinical Practice. J Cancer. 2016; 7:1441-51. https://doi.org/10.7150/jca.15556.
6. Koukoura O, Spandidos DA, Daponte A, Sifakis S. DNA methylation profiles in ovarian cancer: implication in diagnosis and therapy (Review). Mol Med Rep. 2014; 10:3-9. https://doi.org/10.3892/mmr.2014.2221.
7. Gloss BS, Samimi G. Epigenetic biomarkers in epithelial ovarian cancer. Cancer Lett. 2014; 342:257-63. https://doi.org/10.1016/j.canlet.2011.12.036.
8. Sharpless NE, DePinho RA. The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev. 1999; 9:22-30. doi:
9. Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993; 366:704-7. https://doi.org/10.1038/366704a0.
10. Qin Y, Liu JY, Li B, Sun ZL, Sun ZF. Association of low p16INK4a and p15INK4b mRNAs expression with their CpG islands methylation with human hepatocellular carcinogenesis. World J Gastroenterol. 2004; 10:1276-80.
11. Kim BN, Yamamoto H, Ikeda K, Damdinsuren B, Sugita Y, Ngan CY, Fujie Y, Ogawa M, Hata T, Ikeda M, Ohue M, Sekimoto M, Monden T, et al. Methylation and expression of p16INK4 tumor suppressor gene in primary colorectal cancer tissues. Int J Oncol. 2005; 26:1217-26.
12. Gao SJ, Zhang GF, Zhang RP. High CpG island methylation of p16 gene and loss of p16 protein expression associate with the development and progression of tetralogy of Fallot. J Genet. 2016; 95:831-7.
13. Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res. 2005; 576:22-38. https://doi.org/10.1016/j.mrfmmm.2004.08.021.
14. Moselhy SS, Kumosani TA, Kamal IH, Jalal JA, Jabaar HS, Dalol A. Hypermethylation of P15, P16, and E-cadherin genes in ovarian cancer. Toxicol Ind Health. 2015; 31:924-30. https://doi.org/10.1177/0748233713484657.
15. Di Vinci A, Perdelli L, Banelli B, Salvi S, Casciano I, Gelvi I, Allemanni G, Margallo E, Gatteschi B, Romani M. p16(INK4a) promoter methylation and protein expression in breast fibroadenoma and carcinoma. Int J Cancer. 2005; 114:414-21. https://doi.org/10.1002/ijc.20771.
16. Shima K, Nosho K, Baba Y, Cantor M, Meyerhardt JA, Giovannucci EL, Fuchs CS, Ogino S. Prognostic significance of CDKN2A (p16) promoter methylation and loss of expression in 902 colorectal cancers: Cohort study and literature review. Int J Cancer. 2011; 128:1080-94. https://doi.org/10.1002/ijc.25432.
17. Bhagat R, Kumar SS, Vaderhobli S, Premalata CS, Pallavi VR, Ramesh G, Krishnamoorthy L. Epigenetic alteration of p16 and retinoic acid receptor beta genes in the development of epithelial ovarian carcinoma. Tumour Biol. 2014; 35:9069-78. https://doi.org/10.1007/s13277-014-2136-1.
18. Bammidi LS, Neerukonda GN, Murthy S, Kanapuram RD. p16 gene alterations in human ovarian cancers: comparison between tissue and blood samples. Int J Gynecol Cancer. 2012; 22:553-60. https://doi.org/10.1097/IGC.0b013e31823fa90c.
19. Abou-Zeid AA, Azzam AZ, Kamel NA. Methylation status of the gene promoter of cyclin-dependent kinase inhibitor 2A (CDKN2A) in ovarian cancer. Scand J Clin Lab Invest. 2011; 71:542-7. https://doi.org/10.3109/00365513.2011.590224.
20. Dhillon VS, Aslam M, Husain SA. The contribution of genetic and epigenetic changes in granulosa cell tumors of ovarian origin. Clin Cancer Res. 2004; 10:5537-45. https://doi.org/10.1158/1078-0432.ccr-04-0228.
21. Ozdemir F, Altinisik J, Karateke A, Coksuer H, Buyru N. Methylation of tumor suppressor genes in ovarian cancer. Exp Ther Med. 2012; 4:1092-6. https://doi.org/10.3892/etm.2012.715.
22. Shen WJ, Dai DQ, Guo KJ, Li XM. Promoter hypermethylation of RASSF1A, BRCA1 and p16 gene in epithelial ovarian cancer and its clinical significance. China J Cancer Prev Treat. 2008; 15:530-3.
23. Tam KF, Liu VW, Liu SS, Tsang PC, Cheung AN, Yip AM, Ngan HY. Methylation profile in benign, borderline and malignant ovarian tumors. J Cancer Res Clin Oncol. 2007; 133:331-41. https://doi.org/10.1007/s00432-006-0178-5.
24. Li M, Huang ZJ, Dong WH, Li XY, Wang XY, He XH, Wang H, Wang ZH. [Disfigurement of p16INK4A gene expression in development of ovarian cancer and the mechanism]. [Article in Chinese]. Zhonghua Fu Chan Ke Za Zhi. 2006; 41:408-12.
25. Liu Z, Wang LE, Wang L, Lu KH, Mills GB, Bondy ML, Wei Q. Methylation and messenger RNA expression of p15INK4b but not p16INK4a are independent risk factors for ovarian cancer. Clin Cancer Res. 2005; 11:4968-76. https://doi.org/10.1158/1078-0432.ccr-04-2293.
26. Makarla PB, Saboorian MH, Ashfaq R, Toyooka KO, Toyooka S, Minna JD, Gazdar AF, Schorge JO. Promoter hypermethylation profile of ovarian epithelial neoplasms. Clin Cancer Res. 2005; 11:5365-9. https://doi.org/10.1158/1078-0432.ccr-04-2455.
27. Rathi A, Virmani AK, Schorge JO, Elias KJ, Maruyama R, Minna JD, Mok SC, Girard L, Fishman DA, Gazdar AF. Methylation Profiles of Sporadic Ovarian Tumors and nonmalignant Ovaries from High-Risk Women. Clin Cancer Res. 2002; 8:3324-31.
28. Xiao X, Cai F, Niu X, Shi H, Zhong Y. Association between P16INK4a Promoter Methylation and Ovarian Cancer: A Meta-Analysis of 12 Published Studies. PLoS One. 2016; 11:e0163257. https://doi.org/10.1371/journal.pone.0163257.
29. Jiang Y, Yan F, Liang L, Wan Y, Liu J, Cheng W. Meta-analysis demonstrates no association between p16ink4a promoter methylation and epithelial ovarian cancer. Arch Gynecol Obstet. 2017; 295:697–704. https://doi.org/10.1007/s00404-016-4264-x.
30. Ho CM, Huang CJ, Huang CY, Wu YY, Chang SF, Cheng WF. Promoter methylation status of HIN-1 associated with outcomes of ovarian clear cell adenocarcinoma. Mol Cancer. 2012; 11:53. https://doi.org/10.1186/1476-4598-11-53.
31. Cul’bová M, Lasabová Z, Stanclová A, Tilandyová P, Zúbor P, Fiolka R, Danko J, Visnovský J. [Methylation of selected tumor-supressor genes in benign and malignant ovarian tumors]. [Article in Slovak]. Ceska Gynekol. 2011; 76:274–79.
32. Gu XH, Lu Y, Ma D, Liu XS, Guo SW. [Model of aberrant DNA methylation patterns and its applications in epithelial ovarian cancer.]. [Article in Chinese]. Zhonghua Fu Chan Ke Za Zhi. 2009; 44:754–59.
33. Wu Q, Lothe RA, Ahlquist T, Silins I, Tropé CG, Micci F, Nesland JM, Suo Z, Lind GE. DNA methylation profiling of ovarian carcinomas and their in vitro models identifies HOXA9, HOXB5, SCGB3A1, and CRABP1 as novel targets. Mol Cancer. 2007; 6:45.
34. Yang HJ, Liu VW, Wang Y, Tsang PC, Ngan HY. Differential DNA methylation profiles in gynecological cancers and correlation with clinico-pathological data. BMC Cancer. 2006; 6:212. https://doi.org/10.1186/1471-2407-6-212.
35. Wiley A, Katsaros D, Chen H, Rigault de la Longrais IA, Beeghly A, Puopolo M, Singal R, Zhang Y, Amoako A, Zelterman D, Yu H. Aberrant promoter methylation of multiple genes in malignant ovarian tumors and in ovarian tumors with low malignant potential. Cancer. 2006; 107:299-308. https://doi.org/10.1002/cncr.21992.
36. Katsaros D, Cho W, Singal R, Fracchioli S, Rigault De La Longrais IA, Arisio R, Massobrio M, Smith M, Zheng W, Glass J, Yu H. Methylation of tumor suppressor gene p16 and prognosis of epithelial ovarian cancer. Gynecol Oncol. 2004; 94:685-92. https://doi.org/10.1016/j.ygyno.2004.06.018.
37. Hashiguchi Y, Tsuda H, Yamamoto K, Inoue T, Ishiko O, Ogita S. Combined analysis of p53 and RB pathways in epithelial ovarian cancer. Hum Pathol. 2001; 32:988-96. https://doi.org/10.1053/hupa.2001.27115.
38. Brown I, Milner BJ, Rooney PH, Haites NE. Inactivation of the p16INK4A gene by methylation is not a frequent event in sporadic ovarian carcinoma. Oncol Rep. 2001; 8:1359-62.
39. Strathdee G, Appleton K, Illand M, Millan DWM, Sargent J, Paul J, Brown R. Primary ovarian carcinomas display multiple methylator phenotypes involving known tumor suppressor genes. Am J Pathol. 2001; 158:1121-7.
40. McCluskey LL, Chen C, Delgadillo E, Felix JC, Muderspach LI, Dubeau L. Differences in p16 gene methylation and expression in benign and malignant ovarian tumors. Gynecol Oncol. 1999; 72:87-92. https://doi.org/10.1006/gyno.1998.5235.
41. Milde-Langosch K, Ocon E, Becker G, Loning T. p16/MTS1 inactivation in ovarian carcinomas: high frequency of reduced protein expression associated with hyper-methylation or mutation in endometrioid and mucinous tumors. Int J Cancer. 1998; 79:61-5.
42. Shih YC, Kerr J, Liu J, Hurst T, Khoo SK, Ward B, Wainwright B, Chenevix-Trench G. Rare mutations and no hypermethylation at the CDKN2A locus in epithelial ovarian tumours. Int J Cancer. 1997; 70:508-11.
43. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136:E359-86. https://doi.org/10.1002/ijc.29210.
44. Xing X, Cai W, Shi H, Wang Y, Li M, Jiao J, Chen M. The prognostic value of CDKN2A hypermethylation in colorectal cancer: a meta-analysis. Br J Cancer. 2013; 108:2542-8. https://doi.org/10.1038/bjc.2013.251.
45. Li J, Zhou C, Zhou H, Bao T, Gao T, Jiang X, Ye M. The association between methylated CDKN2A and cervical carcinogenesis, and its diagnostic value in cervical cancer: a meta-analysis. Ther Clin Risk Manag. 2016; 12:1249-60. https://doi.org/10.2147/tcrm.s108094.
46. Wang X, Zhu YB, Cui HP, Yu TT. Aberrant promoter methylation of p15 (INK(4)b) and p16 (INK(4)a) genes may contribute to the pathogenesis of multiple myeloma: a meta-analysis. Tumour Biol. 2014; 35:9035-43. https://doi.org/10.1007/s13277-014-2054-2.
47. Tang B, Li Y, Qi G, Yuan S, Wang Z, Yu S, Li B, He S. Clinicopathological Significance of CDKN2A Promoter Hypermethylation Frequency with Pancreatic Cancer. Sci Rep. 2015; 5:13563. https://doi.org/10.1038/srep13563.
48. Fraser HB, Lam LL, Neumann SM, Kobor MS. Population-specificity of human DNA methylation. Genome Biol. 2012; 13:R8. https://doi.org/10.1186/gb-2012-13-2-r8.
49. Kurdyukov S, Bullock M. DNA Methylation Analysis: Choosing the Right Method. Biology (Basel). 2016; 5:E3. https://doi.org/10.3390/biology5010003.
50. Chang H, Yi B, Li L, Zhang HY, Sun F, Dong SQ, Cao Y. Methylation of tumor associated genes in tissue and plasma samples from liver disease patients. Exp Mol Pathol. 2008; 85:96-100. https://doi.org/10.1016/j.yexmp.2008.07.001.
51. Zhu W, Qin W, Hewett JE, Sauter ER. Quantitative evaluation of DNA hypermethylation in malignant and benign breast tissue and fluids. Int J Cancer. 2010; 126:474-82. https://doi.org/10.1002/ijc.24728.
52. Niederacher D, Yan HY, An HX, Bender HG, Beckmann MW. CDKN2A gene inactivation in epithelial sporadic ovarian cancer. Br J Cancer. 1999; 80:1920-6. https://doi.org/10.1038/sj.bjc.6690621.
53. Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol. 2010; 25:603-5. https://doi.org/10.1007/s10654-010-9491-z.
54. Bradburn MJ, Deeks JJ, Berlin JA, Russell Localio A. Much ado about nothing: a comparison of the performance of meta-analytical methods with rare events. Stat Med. 2007; 26:53-77. https://doi.org/10.1002/sim.2528.
55. Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, Godwin AK, Gross J, Hartmann L, et al, and Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–15. https://doi.org/10.1038/nature10166.