
INTRODUCTION
Epithelial-mesenchymal transition (EMT) is intently related to the metastasis of breast cancer. Tumor cells that have undergone EMT could acquire the ability to disseminate from primary epithelial tumors [1, 2]. EMT cells are responsible for degrading the surrounding matrix to enable invasion and intravasation of both EMT and non-EMT cells [3]. It has been known that the inflammatory factors that exist in the tumor milieu could induce EMT of tumor cells [4]. Although several inflammatory cytokines can induce EMT, many cells undergo EMT only in the presence of multiple cytokines [5–9], suggesting that different cytokines may act synergistically to induce EMT. Therefore, exploring the mechanism underlying the synergistic effect of different cytokines will lead to better understanding the initiation of EMT.
TGF-β1 is a multi-functional cytokine that is abundantly expressed in many epithelial tumors to promote tumor growth [10]. It is well accepted that TGF-β1 is the best characterized EMT inducer [11]. However, TGF-β1 alone could not induce EMT in many breast cancer epithelial cells, presumably because it does not effectively activate the Smad pathway in these cells [12, 13]. In addition, TNF-α is another widely studied cytokine that induces EMT in many epithelial cells as well [14–16]. TNF-α is secreted mainly by tumor-associated macrophages that can also promote tumor growth [17]. Previous studies showed that TNF-α could accelerate TGF-β1-induced EMT in tumor cells, although TNF-α alone can also induce EMT in these cells [6, 14]. However, it is unclear whether TNF-α is able to induce EMT in TGF-β1-insensitive breast cancer cells, and whether TNF-α and TGF-β1 have synergistical effect to induce EMT in these cells.
The efficient activation of signaling pathways is the important driving force of cytokine-induced EMT. Smad2/3 is the canonical pathway of TGF-β signaling and plays a critical role in the TGF-β1-induced EMT and tumor development [18, 19]. In parallel, TGF-β1 also activates non-Smad pathways such as those depending on MAPK and NF-κB [20]. In fact, MAPK and NF-κB signaling pathways were all reported to participate in the TGF-β1-induced EMT [21, 22]. In addition, NF-κB is a canonical pathway of TNF-α signaling, which is also required for the TNF-α-induced EMT and invasiveness of tumor cells [15, 23]. Interestingly, TGF-β activated kinase 1 (TAK1), one of MAPKKKs, plays a critical role both in non-Smad pathway of TGF-β1 and canonical pathway of TNF-α. The activated TAK1 can activate MAPKs and NF-κB through MAPKKK/MAPKK/MAPK and IKK/IκBα pathways, respectively [24]. Concurrently, TAK1 was reported to be involved in the TGF-β1-induced and TNF-α-accentuated EMT by modulating multiple signaling pathways in mesothelial cells and bronchial epithelial cells [8, 25], suggesting that TAK1 might be a core factor for the effect of TGF-β1 and TNF-α. In this study, we investigated whether TNF-α, alone or together with TGF-β1, could induce EMT of breast cancer epithelial cells, and the role of TAK1 in the induction process. Our data showed that TNF-α alone could induce EMT of breast cancer epithelial cells and enhance the responsiveness of TGF-β1-insensitive cells by promoting TβRs expression. In turn, TGF-β1 cooperates with TNF-α to enhance the activation of multiple signaling pathways by enhancing TAK1 activation, thus promoting the EMT and invasiveness of tumor cells.
RESULTS
TGF-β1 cooperates with TNF-α to induce EMT and invasion in non-invasive breast cancer epithelial cells
To investigate whether TGF-β1 and TNF-α could induce EMT of non-invasive breast cancer epithelial cells, we cultured non-invasive MCF-7 and T-47D cells in presence of TGF-β1 and TNF-α for different days, and detected the protein level of epithelial marker E-cadherin and the mesenchymal marker vimentin. The results showed that the expression of E-cadherin was gradually up-regulated, while vimentin was gradually down-regulated both in MCF-7 and T-47D cells during prolonged stimulation (Figure 1A). Coincidentally, the invasive ability of TGF-β1/TNF-α-co-stimulated cells was gradually increased (Figure 1B). Next, we treated MCF-7 and T-47D cells with TGF-β1 and TNF-α, alone or in combination, for 6 days. TGF-β1 alone did not influence the morphology and the expression levels of E-cadherin and vimentin in these cells (Figure 1C). However, tumor cells treated with TNF-α alone displayed a fibroblast-like morphology, accompanied with decreased-expression of E-cadherin and increased-expression of vimentin (Figure 1C). Intriguingly, although TGF-β1 alone had no effect, co-application of TGF-β1 and TNF-α induced more pronounced fibroblast-like morphological changes as well as changes in the E-cadherin and vimentin expression than TNF-α applied alone (Figure 1C). Furthermore, co-stimulation also increased the invasive ability of these cells, while TGF-β1 alone had no effect (Figure 1E). Consistently, co-application also increased the proportion of highly polymerized F-actin (Figure 1D) and the expression and secretion of MMP-9 (Figure 1F), which could facilitate the invasive ability of tumor cells. These results indicated that TGF-β1 could synergistically enhance the effects of TNF-α on EMT induction and invasion in non-invasive breast cancer epithelial cells.
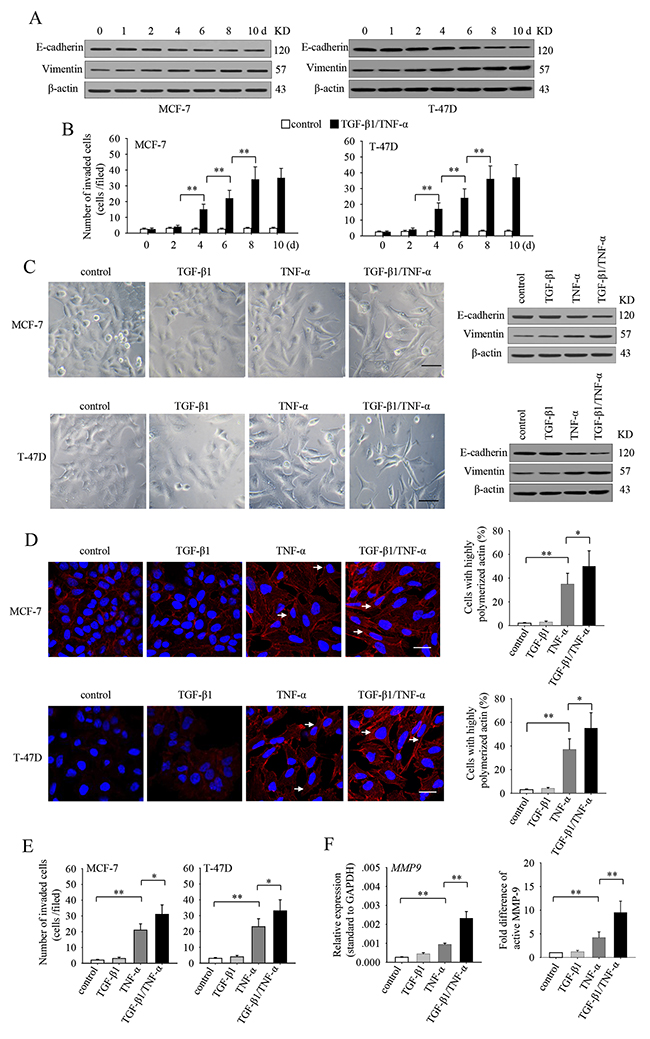
Figure 1: TGF-β1 cooperates with TNF-α to induce EMT and invasiveness in non-invasive breast cancer cells. (A-B) MCF-7 and T-47D cells were cultured in presence of TGF-β1 (10 ng/ml) and TNF-α (10 ng/ml) for the indicated time. The expression of E-cadherin, vimentin and β-actin was detected by Western blot (A) and used for Matrigel invasion assay (B). (C-F) MCF-7 and T-47D cells were cultured in presence of TGF-β1 and/or TNF-α for 6 days. The cells were then used for the following experiments. (C) The morphology of cells was photographed under an inverted phase contrast microscope (left). The scale bar represents 50 μm. The expression of E-cadherin and vimentin of cells were detected by Western blot (right). Data are the representatives of three independent experiments. (D) The cells were then cultured in presence of matrigel for 5 h. Cell F-actin was visualized by staining with rhodamine-phalloidin (red). Cell nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI, blue) (left). Arrows indicate representative cells with highly polymerized actin. The scale bar represents 25 μm. The percentage of cells with highly polymerized actin in total cells was calculated (right). (E) The cells were used for Matrigel invasion assay. (F) The cells were then cultured in presence of matrigel for 48 h. The expression of MMP-9 was detected by real-time RT-PCR (left). The MMP-9 in supernatants was detected by zymography assay, and the fold difference of active MMP-9 was calculated after densitometric analysis of the gel (right). P values, * P<0.05, **P<0.01.
TGF-β1 cooperates with TNF-α to augment the sustained activation of Smad3, MAPKs and NF-κB signaling pathways
The requirement for prolonged stimulation implied that the sustained activation of signaling pathways might be important for TGF-β1 and TNF-α to effectively induce an EMT and invasive phenotype of non-invasive breast cancer cells. We therefore analyzed the activation of signaling pathways that might be potentially involved in the effect of TGF-β1 and TNF-α. The results showed that the long-lasting co-stimulation with TGF-β1 and TNF-α gradually enhanced the sustained activation of Smad2, Smad3, p38 MAPK, ERK, JNK and NF-κB (Figure 2A and 2B). Next, we compared the effect of TGF-β1 and TNF-α, alone or in combination, in activating these signaling pathways after long-lasting stimulation. TGF-β1 could induce a wake sustained activation of Smad2 and Smad3, while TNF-α did not influence the phosphorylation of Smad2 and Smad3 (Figure 2C). TNF-α alone, but not TGF-β1 alone, efficiently induced the sustained activation of p38 MAPK, ERK, JNK and NF-κB (Figure 2D and 2E). Intriguingly, co-stimulation with TGF-β1 and TNF-α induced a significantly enhanced activation of all of these signaling pathways (Figure 2D and 2E). These results suggested that the prolonged co-stimulation with TGF-β1 and TNF-α is required not only for the sustained activation of Smad pathway but also for more efficiently activation of p38 MAPK, ERK, JNK and NF-κB pathways.
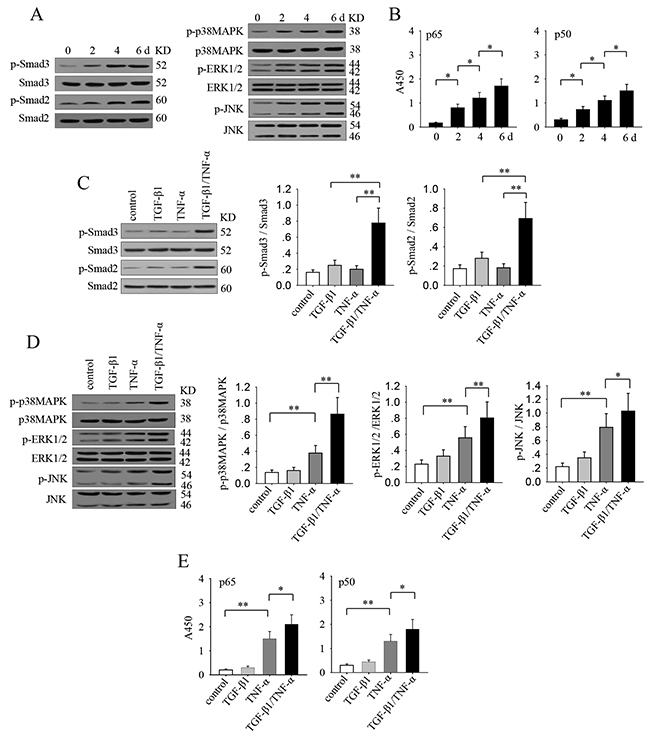
Figure 2: TGF-β1 cooperates with TNF-α to augment the sustained activation of Smad3, MAPKs and NF-kB signaling pathways. (A-B) MCF-7 cells were stimulated with TGF-β1 (10 ng/ml) and TNF-α (10 ng/ml) for the indicated time. (A) The phospho-Smad3, Smad3, phospho-Smad2, Smad2, phospho-p38 MAPK, p38 MAPK, phospho-ERK1/2, ERK1/2, phospho-JNK and JNK were detected by Western blot assay. (B) The activity of NF-κB was assayed as described in Methods. (C-E) MCF-7 cells were stimulated with TGF-β1 and/or TNF-α for 6 days. (C) The phospho-Smad3, Smad3, phospho-Smad2 and Smad2 were detected by Western blot assay (left). The ratio of phosphorylated protein to total protein of the indicated proteins were calculated after densitometric analysis of the blots (right). (D) The phospho-p38 MAPK, p38 MAPK, phospho-ERK1/2, ERK1/2, phospho-JNK and JNK were detected by Western blot assay (left). The ratio of phosphorylated protein to total protein of the indicated proteins were calculated after densitometric analysis of the blots (right). (E) The activity of NF-κB was assayed as described in Methods. Data are representative of three independent experiments, or pooled from three independent experiments. P values, * P<0.05, **P<0.01.
The enhanced activation of multiple signaling pathways are modulated by TAK1 during prolonged co-stimulation
To explore the mechanisms underlying the gradually enhanced activation of multiple signaling pathways by TGF-β1 and TNF-α, we focused on TAK1, which is potentially involved in both TGF-β1 and TNF-α signaling [24, 26]. The results showed that co-stimulation with TGF-β1 and TNF-α gradually enhanced the sustained activation of TAK1 (Figure 3A). TGF-β1 alone was inefficient in inducing TAK1 activation, while TNF-α alone effectively activated TAK1 (Figure 3B). Intriguingly, co-application of TGF-β1 and TNF-α was more efficient in inducing the activation of TAK1 (Figure 3B). The similar dynamic change of TAK1 activation and the activation of Smad3, NF-κB and MAPKs implied that there may be linkage among them. To identify this, we treated MCF-7 cells with TGF-β1 and TNF-α in presence of (5Z)-7-oxozeaenol which specifically inhibits TAK1 activation (Figure 3C). The results showed that the sustained activation of Smad2, Smad3, MAPKs, and NF-κB were all significantly attenuated in the presence of (5Z)-7-oxozeaenol (Figure 3C and 3D). These results suggested that the TGF-β1/TNF-α-induced activation of Smad2/3, NF-κB and MAPKs were all modulated by TAK1. Then we wondered whether these TAK1-modulated pathways might influence the activation of TAK1. To ascertain this, we detected the phosphorylation level of TAK1 after stimulation with TGF-β1 and TNF-α in presence of SIS3 (Smad3 inhibitor), QNZ (NF-κB inhibitor), SB203580 (p38MAPK inhibitor), PD98059 (ERK pathway inhibitor), or SP600125 (JNK inhibitor) (Figure 3E). Among them, ERK inhibitor and p38 MAPK inhibitor were most effective in suppressing the sustained activation of TAK1. In addition, Smad3 inhibitor or NF-κB inhibitor slightly attenuated TAK1 activation. However, inhibiting JNK did not influence the activation of TAK1. These results suggested that the activation of ERK and p38 MAPK may have positive feed-back effect to promote the sustained activation of TAK1.
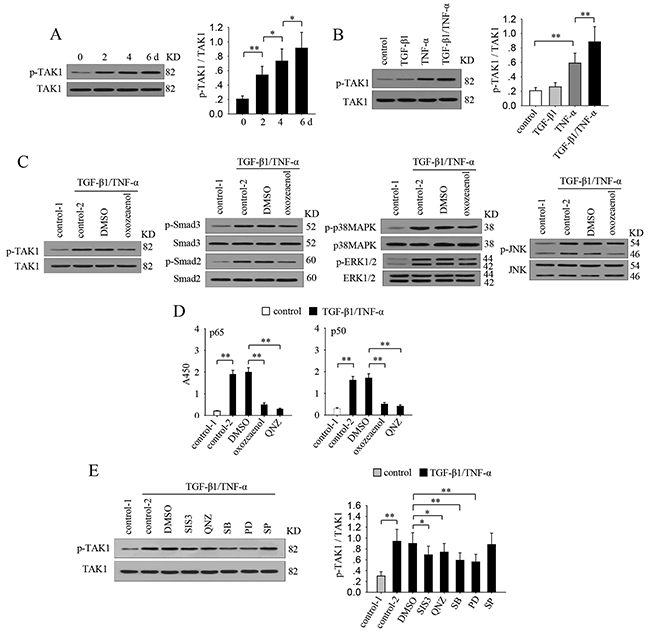
Figure 3: TAK1 modulates the enhanced activation of multiple signaling pathways. (A) MCF-7 cells were cultured in presence of TGF-β1 and TNF-α for the indicated time. Phospho-TAK1 and TAK1 were detected by Western blot (left). Relative phosphorylation of TAK1 was calculated after densitometry assay (right). (B) MCF-7 cells were cultured in presence of TGF-β1 and/or TNF-α for 6 days. Phospho-TAK1 and TAK1 were detected by Western blot (left). Relative phosphorylation of TAK1 was calculated after densitometry assay (right). (C) MCF-7 cells were cultured in presence of TGF-β1/TNF-α with or without (5Z)-7-oxozeaenol (TAK1 inhibitor, 600 nM) for 6 days. The Phospho-TAK1, TAK, Phospho-Smad3, Smad3, Phospho-p38 MAPK, p38 MAPK, phospho-ERK1/2, ERK1/2, phospho-JNK and JNK were detected by Western blot. (D) MCF-7 cells were cultured in presence of TGF-β1/TNF-α with or without (5Z)-7-oxozeaenol or QNZ (NF-κB inhibitor, 40 nM) for 6 days. The activity of NF-κB was assayed as described in Methods. (E) MCF-7 cells were untreated or treated for 6 days with TGF-β1/TNF-α in absence or presence of SIS3 (10 μM), QNZ (40 nM), SB203580 (SB, 10 μM), PD98059 (PD, 10 μM), SP600125 (SP, 10 μM). The phospho-TAK1 and TAK1 were detected by Western blot (left). The ratio of phospho-TAK1 and TAK1 was calculated after densitometry analysis of Western blots (right). Data are representative of three independent experiments, or pooled from three independent experiments. P values, * P<0.05, **P<0.01.
TAK1 activation is enhanced by gradually up-regulated TGF-β receptors during long-lasting co-stimulation
To further investigate the mechanisms underlying the gradual activation of TAK1, we analyzed whether the expression of TGF-β1 receptors and TNF-α receptors were influenced by TGF-β1 and TNF-α. The results showed that the mRNA expressions of TβRI and TβRII were gradually increased during co-stimulation with TGF-β1 and TNF-α (Figure 4A and 4B). However, the expression of TNFRI and TNFRII were not significantly changed after co-stimulation (data not shown). TGF-β1 alone could not influence the expression of its receptors. Intriguingly, TNF-α alone promoted the expression of TβRI and TβRII, and co-application of TGF-β1 further up-regulated the expression of these receptors (Figure 4A-4C). We then analyzed whether signaling pathways were involved in modulating the expression of TGF-β receptors. To do this, we detected the mRNA expressions of TβRI and TβRII after stimulation with TGF-β1 and TNF-α in presence of SIS3, QNZ, SB203580, PD98059, or SP600125. The results showed that the up-regulation of TβRI and TβRII was suppressed when inhibiting p38 MAPK or ERK pathway (Figure 4D). Considering that inhibiting these pathways also decreased TAK1 activation, we then investigated whether TβRI or TβRII were involved in the enhanced activation of TAK1 during prolonged co-stimulation. To do it, we silenced TβRI or TβRII by transducting the shRNA lentiviral particles (Figure 4E). Intriguingly, silencing TβRI or TβRII not only attenuated the activation of TAK1 but also decreased the sustained activation levels of Smad2, Smad3, MAPKs and NF-κB (Figure 4F-4H). These results suggested that the up-regulated TβRs contribute to the enhanced activation of TAK1, which is required for the subsequent activation of down-stream signaling pathways.
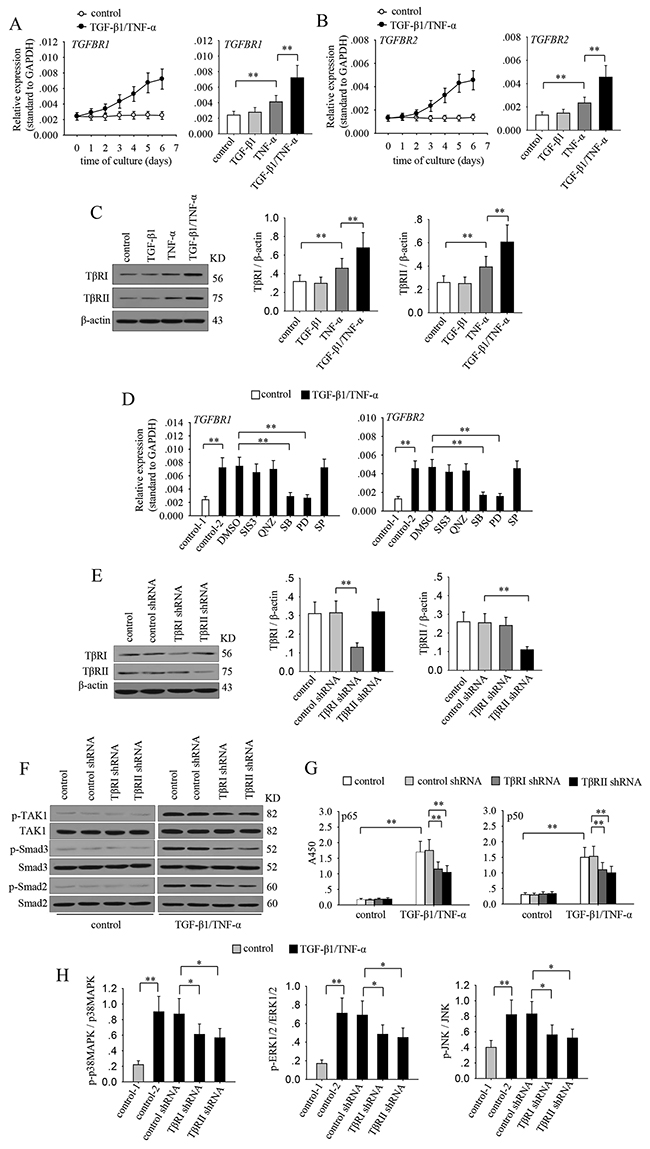
Figure 4: The up-regulation of TGF-β receptors contributes to the gradually enhanced activation of TAK1 during long-lasting co-stimulation. (A-C) MCF-7 cells were cultured in absence or presence of TGF-β1/TNF-α (left) for the indicated time. Or the cells were cultured for 6 days in presence of TGF-β1 and or TNF-α. The expression of TGF-βRI (A), and TGF-βRII (B) was detected by real-time RT-PCR. (C) The expression of TβRI and TβRII was detected by Western blot after 6-d culture (left). Relative expression of TβRI and TβRII were calculated after densitometry assay as standardized by β-actin (right). (D) MCF-7 cells were unstimulated or stimulated with TGF-β1/TNF-α in absence or presence of SIS3 (10 μM), QNZ (40 nM), SB203580 (SB, 10 μM), PD98059 (PD, 10 μM) and SP600125 (SP, 10 μM) for 6 days. The expression of TGF-βRI (left) and TGF-βRII (right) was detected by real-time RT-PCR. (E-H) MCF-7 cells were transducted with control, TβRI or TβRII shRNA lentivirus. And then the cells were selected for stable expression using puromycin. (E) The expression of TβRI and TβRII was detected by Western blot (left). Relative expression of TβRI and TβRII was calculated after densitometry assay as standardized by β-actin (right). (F) The phospho-TAK1, TAK1, phospho-Smad2, Smad2, phospho-Smad3 and Smad3 were detected by Western blot. (G) The activity of NF-κB was assayed as described in Methods. (H) The ratio of phosphorylated protein to total protein of p38 MAPK, ERK1/2 and JNK was calculated after densitometric analysis of the blots. Data are representative of three independent experiments, or pooled from three independent experiments. P values, * P<0.05, **P<0.01.
The sustained activation of signaling pathways and up-regulated SLUG are required for the co-stimulation-induced EMT and invasion of non-invasive breast cancer cells
To ascertain the molecular basis underlying the TGF-β1/TNF-α-mediated induction of EMT and invasion process, we next analyzed whether the activated signaling pathways are required for the effects of co-stimulation. To do this, we cultured MCF-7 cells with TGF-β1 and TNF-α in presence of 7Z-oxozeaenol, SIS3, QNZ, SB203580, PD98059, or SP600125. The results showed that inhibiting TAK1 nearly abolished the effects of TGF-β1/TNF-α-induced down-regulation of E-cadherin, up-regulation of vimentin and invasiveness in MCF-7 cells (Figure 5A and 5B). The ERK1/2, p38 MAPK, Smad3 and NF-κB pathways were all involved in the effects of TGF-β1 and TNF-α (Figure 5C and 5D). Among them, ERK1/2 and p38 MAPK pathway were more efficient than Smad3 and NF-κB pathway. However, JNK pathway had no effect (Figure 5C and 5D). These results suggested that the sustained activation of multiple signaling pathways is indeed required for TGF-β1/TNF-α-induced EMT and invasion of breast cancer cells.
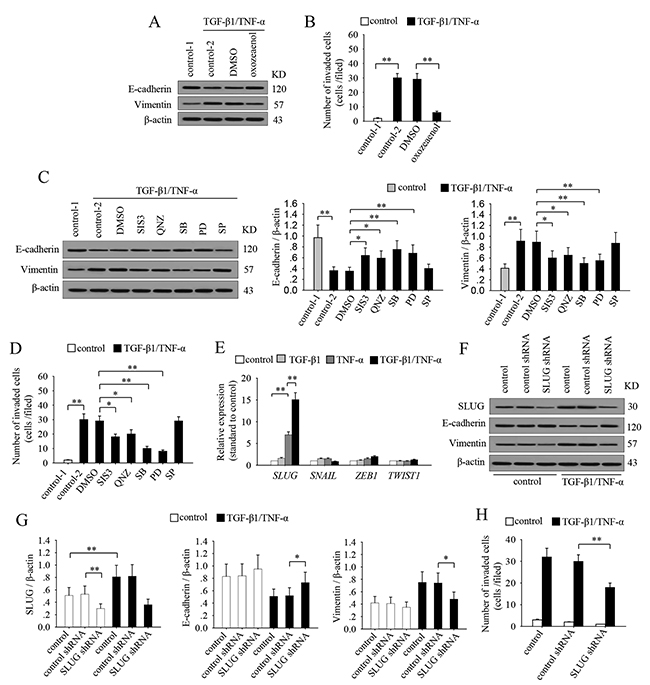
Figure 5: The sustained activation of signaling pathways and up-regulation of SLUG are required for inducing EMT and invasiveness in breast cancer cells. (A-B) MCF-7 cells were stimulated or unstimulated with TGF-β1 and TNF-α for 6 days in absence or presence of (5Z)-7-oxozeaenol. (A) The expression of E-cadherin and vimentin was detected by Western blot. (B) The cells were used for matrigel invasion assay. (C-D) MCF-7 cells were stimulated or unstimulated with TGF-β1 and TNF-α for 6 days in absence or presence of SIS3, QNZ, SB203580, PD98059 and SP600125 for 6 days. (C) The expression of E-cadherin and vimentin was detected by Western blot (left). Relative expression of E-cadherin and vimentin were calculated after densitometry assay as standardized by β-actin (right). (D) The cells were used for matrigel invasion assay. (E) MCF-7 cells were cultured in presence of TGF-β1 and/or TNF-α for 6 days. The expression of SLUG, SNAIL, ZEB1 and TWIST1 was detected by real-time RT-PCR. (F-H) MCF-7 cells were transducted with control or SLUG shRNA lentivirus, and then selected for stable expression using puromycin. (F) The expression of SLUG, E-cadherin, vimentin and β-actin was detected by Western blot. (G) Relative expression of SLUG, E-cadherin and vimentin were calculated after densitometry assay as standardized by β-actin. (H) The cells were used for matrigel invasion assay. Data are representative of three independent experiments, or pooled from three independent experiments. P values, * P<0.05, **P<0.01.
Considering that EMT-related transcriptional factors directly contribute to the EMT process, we then detected the mRNA expression of SNAIL, SLUG, ZEB1 and TWIST1 factors that intently related to EMT after co-stimulation with TGF-β1 and TNF-α. Among them, the expression of SLUG was increased most significantly, while the expression of ZEB1 was slightly up-regulated, but the expression of TWIST1 and SNAIL was not significantly changed (Figure 5E). Consistently, both Smad and ERK pathway are reported to be involved in up-regulating SLUG [11, 27]. TGF-β1 and TNF-α could induce the sustained activation of these pathways, thus promoting the expression of SLUG. Moreover, SLUG could promote the expression of TβRII [28], which forms a positive feed-back effect to further promote signaling pathways. Next, we knocked down SLUG in MCF-7 cells through transducting SLUG shRNA lentiviral particles (Figure 5F and 5G). Intriguingly, the effect of TGF-β1/TNF-α on EMT and invasion of MCF-7 cells was abolished after silencing SLUG (Figure 5F-5H), suggesting that SLUG is required for the effects of co-stimulation.
DISCUSSION
Both TGF-β1 and TNF-α exist in tumor microenvironment due to the inflammatory responses [29]. They could induce EMT and enhance metastatic potential of tumor cells. In this study, we found that TGF-β1 alone could not induce EMT in non-invasive breast cancer cells, MCF-7 and T-47D, in agreement with previous reports [12]. However, we identified that TNF-α alone could induce EMT and invasion in MCF-7 and T-47D cells, which has not been reported in TGF-β1-insensitive cells. Importantly, co-application of TGF-β1 and TNF-α synergistically induce EMT and invasion in these cells as well as in TGF-β1-sensitive cells [6, 7].
Both Smad and non-Smad pathways are involved in the TGF-β1-induced EMT and metastatic potential of tumor cells [13, 30]. Our data and previous report showed that the expression of TGF-β receptors is very low in MCF-7 and T-47D cells [13]. In line with this, TGF-β1 could not induce the sustained activation of not only Smad but also non-Smad pathways in MCF-7 and T-47D cells, which may explain why TGF-β1 could not induce EMT in non-invasive breast cancer cells. However, TNF-α could promote the expression of both TGF-βRI and TGF-βRII by activating ERK1/2 and p38 MAPK signaling pathways. The up-regulation of TβRs increased the response of the cells to TGF-β1. In turn, the co-application with TGF-β1 could further enhance these signaling pathways. The enhanced activation of these pathways forms a positive feed-back regulation to further promote the effect of TGF-β1 by up-regulating TGF-β receptors. In this context, TGF-β1/TNF-α-induced EMT and invasiveness of breast cancer cells need prolonged co-stimulation, since the TGF-β receptors were gradually increased.
TAK1 was first identified as a member of TGF-β signaling family that mediates the activation of JNK and p38 MAPK [31]. Additionally, TAK1 is also involved in TGF-β1- or TNF-α-induced the activation of NF-κB through TAK1/IKK/IκB signaling pathway [15, 32]. Our data in this study showed that TNF-α alone was more efficient in inducing TAK1 activation than TGF-β1 alone, and that co-stimulation with TNF-α and TGF-β1 could further enhance the activation of TAK1. Furthermore, the prolonged co-stimulation gradually enhanced the sustained activation of TAK1 by gradually up-regulating TβRs. Following the enhanced activation of TAK1, the activation of Smad and non-Smad pathways was also sustained and gradually enhanced during the long-lasting co-stimulation. Inhibiting these pathways, especially TAK1, significantly blocked TNF-α/TGF-β1-induced EMT and invasiveness of breast cancer cells. These results identified TAK1 as the cross-road that mediates TGF-β1/TNF-α-induced EMT and invasiveness in breast cancer epithelial cells. Consistently, the critical function of TAK1 in the EMT process was also reported in bronchiolitis epithelial and mesothelial cells [8, 25].
Although the critical role of TAK1 in inducing EMT has been widely accepted, the mechanisms underlying the modulatory effect of TAK1 in signaling pathways have not been fully understood. It has been identified that TAK1 can modulate the activation of NF-κB and MAPK [31–33], but the impact of TAK1 on Smad3 activation remains debatable. JNK/c-Jun, but not Smad3, was found to be the downstream and effective signal of TAK1 in a EMT model driven by TGF-β1 and accentuated by TNF-α [8]. However, in this study, for the TGF-β1-insensitive cells, TAK1 can modulate the activation of Smad2/3, NF-κB and MAPKs which are induced by TGF-β1/TNF-α. Our results also showed that these signaling pathways, but not JNK pathway, are critical for TGF-β1/TNF-α-induced EMT. It implies that the modulatory effect of TAK1 may be diverse in different experimental models. In agreement with our results, a previous study showed that inhibiting TAK1 can suppress multiple signaling pathways, including the phosphorylation and transcriptional activation of Smad3 in mesothelial cells [33]. On the other hand, TAK1 can indirectly enhance the transcriptional activity of Smad3 by promoting the degradation of Smad3 inhibitory molecule SnoN [34].
EMT-related transcriptional factors were reported directly contribute to the EMT of breast cancer cells [1]. Our data showed that the expression of SLUG was exclusively up-regulated after co-stimulation with TGF-β1 and TNF-α. Both Smad and ERK pathways were reported to induce the expression of SLUG [11, 27]. Therefore, TGF-β1 and TNF-α could promote SLUG expression by activating both Smad and ERK pathways. On the other hand, SLUG could promote the expression of TGF-βRII [28]. In line with this, SLUG also forms a positive feed-back effect to enhance the activation of signaling pathways induced by TGF-β1 and TNF-α. Importantly, silencing SLUG nearly abolished TGF-β1/TNF-α-induced EMT and invasiveness. Functionally, SLUG could not only suppress the expression of E-cadherin directly, but also activate another EMT transcriptional factor ZEB1 to lead to EMT indirectly [27]. Therefore, the up-regulation of SLUG contributes to the TGF-β1/TNF-α-induced EMT and invasiveness of breast cancer cells.
In summary, in this study we demonstrated that TGF-β1 cooperates with TNF-α to promote the up-regulation of TβRs, thus inducing the sustained activation of TAK1 and the following activation of Smad2/3, NF-κB and MAPKs signaling pathways. All of these pathways, except for JNK, were required for TGF-β1/TNF-α-induced EMT and invasiveness of breast cancer cells. These findings highlight the importance of sustained activation of signaling pathways in the EMT and invasiveness of tumor cells. So inhibiting these signaling pathways, especially TAK1, may effectively suppress the EMT of tumor cells, thereby inhibiting the metastatic potential of tumor cells.
MATERIALS AND METHODS
Cells and reagents
Human breast cancer cell lines MCF-7 and T-47D were purchased from china center for type culture collection (CCTCC, Wuhan, china), and cultured in Dulbcco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Human TNF-α and TGF-β1 were purchased from PeproTech (Rocky Hill, NJ). Matrigel was purchased from BD Biosciences (Bedford, MA, USA). SB203580, PD98059, SP600125, SIS3, (5Z)-7-oxozeaenol, 6-amino-4-(4-phenoxyphenyl-ethylamino) quinazoline (QNZ) were purchased from Merck4 Bioscience (Calbiochem, Germany). All inhibitors were dissolved in DMSO as a stock solution and diluted with culture medium to the desired concentration without toxicity to cells.
Real-time RT-PCR
Total RNA was extracted from cells with TRIzol reagent (Invitrogen, Carlsbad, CA). The relative quantity of mRNA was determined by real-time RT-PCR assays as described previously [35], Briefly, 100 ng of total RNA was used for reverse transcription using Superscript II RNase H reverse transcriptase (Invitrogen, Carlsbad, CA) in a volume of 25 μL. Then, 2 μL of cDNA was amplified with SYBR Green Universal PCR Mastermix (Bio-Rad, Rich-mond, CA) in duplicate. For sample analysis, the threshold was set based on the exponential phase of products, and CT value for samples was determined. The resulting data were analyzed with the comparative CT method for relative gene expression quantification against house keeping gene GAPDH. The primer sequences were as follows: MMP9, sense 5′-CAGTCCACCCTTGTGCTCTTCC-3′, antisense 5′-CTGCCACCCGAGTGTAACCAT-3′; GAPDH, sense 5′-TCATTGACCTCAACTACATGGTTT-3′, antisense 5′-GAAGATGGTG ATGGGATTTC-3′; TGF-βRI, sense 5′-TGAACAGAAGTTAAGGCCAAATATC-3′, anti sense 5′-CAGGCAAAGCTGTAGAATTACATTT-3′; TGF-βRII, sense 5′-CGGTTAATAAC GACATGATAGTCAC-3′, antisense 5′-TCATGGCAAACTGTCTCTAGTGTTA-3′; SNAIL, sense 5′-ACCTTCCAGCAGCCCTAC-3′, antisense 5′-CCTTTCCCACTGTCCTCAT-3′; SLUG, sense 5′-AGGAATCTGGCTGCTGTG-3′, antisense 5′-GGAGAAAATGCCTTTGG AC-3′; TWIST1, sense 5′-GAGTCCGCAGTCTTACGAG-3′, antisense 5′-TGAGGGTCTGA ATCTTGCT-3′; ZEB1, sense 5′-ACACCTTTGCATACAGAACCC-3′ antisense 5′-ACACC CAGACTGCGTCACAT-3′.
Western blot assay
Western blot assay was done as described previously [35]. In brief, harvested cells were lysed in lysis buffer contained protease cocktail inhibitors. The cell extract was separated by SDS-polyacrylamide gel electrophoresis, and transferred to nitrocellulose membranes. After blocking with TBST (Tris-buffered saline with 0.05% Tween-20) containing 5% nonfat milk, the membranes were incubated with antibodies against human p-TAK1 (Thr187), TAK1, p-Smad3 (Ser423/425), Smad3, p-Smad2 (Ser467), Smad2, p-p38MAPK (Thr180/Tyr182), p38MAPK, p-ERK1/2 (Thr202/Tyr204), ERK1/2, p-JNK (Thr183/Tyr185), JNK, E-cadherin, vimentin, TGF-βRI, TGF-βRII, SLUG and β-actin. All antibodies were purchased from Santa cruz biotechnology (Santa Cruz, CA) and cell signaling technology company (Beverly, MA). After incubation with the secondary antibody conjugated with horseradish peroxidase, membranes were extensively washed, and the immunoreactivity was visualized by enhanced chemiluminescence according to the manufacturer’s protocol (ECL kit; Santa Cruz Biotechnology, Santa Cruz, CA).
Matrigel invasion assay
Invasion assay was performed using Boyden chambers (Transwell, Corning, Inc, Corning, NY). The transwell filter inserts were coated with matrigel. The lower chambers were filled with DMEM medium containing 10% FBS. 1×105 tumor cells were placed in the upper compartment. After 24-h incubation at 37°C in a humidified incubator with 5% CO2, the non-invasive cells were removed. The cells attached to the lower surface of the membrane insert were fixed, stained, and counted under a microscope from five randomly chosen fields in each membrane. The average number of the cells per fields was calculated.
Cell transduction
Control shRNA lentiviral particles, TβRI lentiviral particles, TβRII shRNA lentiviral particles and SLUG shRNA lentiviral particles were all purchased from Santa Cruz Biotechnology. To down regulate TβRI or TβRII or SLUG in tumor cells, the cells were transduced with corresponding shRNA lentiviral particles according to the manufacturer’s protocol. After selection with puromycin, the cells were used for further experiments.
NF-κB activity assay
The nuclear extract was prepared with Nuclear Extraction Kit (Millipore, Billerica, MA). The activity of NF-κB in nuclear extract was determined by NF-κB Assay Kit (Millipore, Billerica, MA) according to the manufacturer’s protocol. In brief, cells were harvested and added cytoplasmic lysis buffer to separate the cytoplasmic protein and nucleus. The nuclear proteins from nucleus were extracted by using nuclear extraction buffer. The nuclear proteins, biotinylated double stranded oligonucleotide NF-kB capture probe and transcription factor assay buffer were mixed. The mixture was first incubated in a streptavidin-coated plate. After further incubation with primary antibody against NF-kB p50 or p65, and then incubation with a highly sensitive HRP-conjugated secondary antibody, the substrate TMB was added for detection. Last, the absorbance value was detected in a spectrophotometric plate reader with a standard OD450 filter.
Actin polymerization analysis
Tumor cells were incubated in matrigel-coated plate for 5 h. The cells were then fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and then stained with rhodamine-phalloidin (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol to visualize the cells with highly polymerized actin.
Statistics
Results were expressed as mean±SD and interpreted by one-way ANOVA. Differences were considered to be statistically significant when P <0.05.
ACKNOWLEDGMENTS AND FUNDING
We gratefully thank Prof. Xing-Zhen Chen (Department of Physiology, University of Alberta, Canada) and Dr. Feng-Hua Wu (Department of Physiology, Hubei University of Chinese Medcine, China) for modifying the manuscript and their valuable advice. This work was supported by National Natural Science Foundation of China (No.81472704, 81272314, 30830095), National Development Program (973) For Key Basic Research of China (No. 2009CB521806).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Foroni C, Broggini M, Generali D, Damia G. Epithelial-mesenchymal transition and breast cancer: role, molecular mechanisms and clinical impact. Cancer Treatment Review. 2012; 38:689-697.
2. Thiery JP. Epithelial-mesenchymal transitions in tumor progression. Nat Review Cancer. 2002; 2:442-454.
3. Tsuji T, Ibaragi S, Hu GF. Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Research. 2009; 69:7135-7139.
4. Soria G, Ofri-Shahak M, Haas I, Yaal-Hahoshen N, Leider-Trejo L, Leibovich-Rivkin T, Polina Weitzenfeld, Tsipi Meshel, Esther Shabtai, Mordechai Gutman, Adit Ben-Baruch. Inflammatory mediators in breast cancer: coordinated expression of TNFalpha & IL-1beta with CCL2 & CCL5 and effects on epithelial-to- mesenchymal transition. BMC cancer. 2011; 11:130.
5. Said NABM, Williams ED. Growth factors in induction of epithelial- mesenchymal transition and metastasis. Cells Tissues Organs. 2011; 193:85-97.
6. Yamauchi Y, Kohyama T, Takizawa H, Kamitani S, Desaki M, Takami K, Kawasaki S, Kato J, Nagase T. Tumor necrosis factor-alpha enhances both epithelial-mesenchymal transition and cell contraction induced in A549 human alveolar epithelial cells by transforming growth factor-β1. Experimental Lung Research. 2010; 36:12-24.
7. Kamitani S, Yamauchi Y, Kawasaki S, Takami K, Takizawa H, Nagase T, Kohyama T. Simultaneous stimulation with TGF-β1 and TNF-α induces epithelial mesenchymal transition in bronchial epithelial cells. International Archives of Allergy and Immunology. 2011; 155:119-128.
8. Gardner A, Fisher AJ, Richter C, Johnson GE, Moisey EJ, Brodlie M, Ward C, Krippner-Heidenreich A, Mann DA, Borthwick LA. The critical role of TAK1 in accentuated epithelial to mesenchymal transition in obliterative bronchiolitis after lung transplantation. The American Journal of Pathology. 2012; 180:2293-2308.
9. Uttamsingh S, Bao X, Nguyen KT, Bhanot M, Gong J, Chan JLK, Liu F, Chu TT, Wang LH. Synergistic effect between EGF and TGF-β1 in inducing oncogenic properties of intestinal epithelial cells. Oncogene. 2007; 27:2626-2634.
10. de Caestecker MP, Piek E, Roberts AB. Role of transforming growth factor-beta signaling in cancer. Journal of The National Cancer Institute. 2000; 92:1388-1402.
11. Xu J, Lamouille S, Derynck R. TGF-β-induced epithelial to mesenchymal transition. Cell Research. 2009; 19:156-172.
12. Brown KA, Aakre ME, Gorska AE, Price JO, Eltom SE, Pietenpol JA, Moses HL. Induction by transforming growth factor-β1 of epithelial to mesenchymal transition is a rare event in vitro. Breast Cancer Research. 2004; 6:R215.
13. Zhou YH, Liao SJ, Li D, Luo J, Wei JJ, Yan B, Sun R, Shu Y, Wang Q, Zhang GM, Feng ZH. TLR4 Ligand/H2O2 enhances TGF-b1 signaling to induce metastatic potential of non-invasive breast cancer cells by activating non-smad pathways. PLoS ONE. 2013; 8:e65906.
14. Bates RC, Mercurio AM. Tumor necrosis factor-alpha stimulates the epithelial-to- mesenchymal transition of human colonic organoids. Molecular Biology Cell. 2003; 14:1790-1800.
15. Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, Chao CH, Yamaquchi H, Yang NK, Ding Q, Wang Y, Lai YJ, LaBaff AM, Wu TJ, Lin BR, Yang MH, Hortobaqyi GN, Hung MC. Epithelial-mesenchymal transition induced by TNF-α requires NF-κB-mediated transcriptional up-regulation of Twist1. Cancer Research. 2012; 72:1290-1300.
16. Chuang MJ, Sun KH, Tang SJ, Deng MW, Wu YH, Sung JS, Cha TL, Sun GH. Tumor-derived tumor necrosis factor-alpha promotes progression and epithelial-mesenchymal transition in renal cell carcinoma cells. Cancer Science. 2008; 99:905-913.
17. Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer and Metastasis Review. 2006; 25:409-416.
18. Gupta J, Robbins J, Jilling T, Seth P. TGFbeta-dependent induction of interleukin-11 and interleukin-8 involves SMAD and p38 MAPK pathways in breast tumor models with varied bone metastases potential. Cancer Biology Therapy. 2011; 11:311-316.
19. Do TV, Kubba LA, Du H, Sturgis CD, Woodruff TK. Transforming growth factor-beta1, transforming growth factor-beta2, and transforming growth factor-beta3 enhance ovarian cancer metastatic potential by inducing a Smad3-dependent epithelial-to-mesenchymal transition. Molecular Cancer Research. 2008; 6:695-705.
20. Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Research. 2009; 19:128- 139.
21. Pechkovsky DV, Scaffidi AK, Hackett TL, Ballard J, Shaheen F, Thompson PJ, Thannickal VJ, Knight DA. Transforming growth factor beta1 induces alphavbeta3 integrin expression in human lung fibroblasts via a beta3 integrin-, c-Src-, and p38 MAPK-dependent pathway. Journal of Biology Chemistry. 2008; 283:12898-12908.
22. Huber MA, Azoitei N, Baumann B, Grünert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. Journal of Clinical Investigation. 2004; 114:569-581.
23. Maier HJ, Schmidt-Strassburger U, Huber MA, Wiedemann EM, Beug H, Wirth T. NF-kappaB promotes epithelial-mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Letter. 2010; 295:214-228.
24. Landstrom M. The TAK1-TRAF6 signalling pathway. The International Journal of Biochemistry & Cell Biology. 2010; 42:585-9.
25. Strippoli R, Benedicto I, Perez Lozano ML, Pellinen T, Sandoval P, Lopez-Cabrera M, del Pozo MA. Inhibition of transforming growth factor-activated kinase 1 (TAK1) blocks and reverses epithelial to mesenchymal transition of mesothelial cells. PLoS One. 2012; 7:e31492.
26. Sakurai H, Suzuki S, Kawasaki N, Nakano H, Okazaki T, Chino A, Doi T, Saiki I. Tumor necrosis factor-alpha-induced IKK phosphorylation of NF-kappaB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. Journal of Biology Chemistry. 2003; 278:36916-36923.
27. Joseph MJ, Dangi-Garimella S, Shields MA, Diamond ME, Sun L, Koblinski JE, Munshi HG. Slug is a downstream mediator of transforming growth factor-beta1-induced matrix metalloproteinase-9 expression and invasion of oral cancer cells. Journal of Cell Biochemistry. 2009; 108:726-736.
28. Dhasarathy A, Phadke D, Mav D, Shah RR, Wade PA. The transcription factors snail and slug activate the transforming growth factor-Beta signaling pathway in breast cancer. PLoS One. 2011; 6:e26514.
29. Martin M, Wei H, Lu T. Targeting microenvironment in cancer therapeutics. Oncotarget. 2016; 7:52575-52583.
30. Meulmeester E, ten Dijke P. The dynamic roles of TGF-β in cancer. Journal of Pathology. 2011; 223:206-219.
31. Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniquchi T, Nishida E, Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995; 270:2008-2011.
32. Neil JR, Schiemann WP. Altered TAB1:IkappaB kinase interaction promotes transforming growth factor beta-mediated nuclear factor-kappaB activation during breast cancer progression. Cancer Research. 2008; 68:1462-1470.
33. Sakurai H, Miyoshi H, Toriumi W, Sugita T. Functional interactions of transforming growth factor beta-activated kinase 1 with IkappaB kinases to stimulate NF-kappaB activation. Journal of Biology Chemistry. 1999; 274:10641-10648.
34. Kajino T, Omori E, Ishii S, Matsumoto K, Ninomiya-Tsuji J. TAK1 MAPK kinase kinase mediates transforming growth factor-beta signaling by targeting snoN oncoprotein for degradation. Journal of Biology Chemistry. 2007; 282:9475-9481.
35. Huang B, Lei Z, Zhang GM, Li D, Song C, Li B, Liu YY, Yuan Y, Unkeless J, Xiong HB, Feng ZH. SCF- mediated mast cell infiltration and activation exacerbate the inflammation and immunosuppression in tumor microenvironment. Blood. 2008; 112:1269-1279.