
INTRODUCTION
Diffuse large B-cell lymphoma (DLBCL) is the most common lymphoma that accounts for 30%-50% of lymphoma cases and is characterized by both biological and clinical heterogeneity. Rituximab-added CHOP chemotherapy (cyclophosphamide, doxorubicin, vincristine, and prednisolone) has improved the long-term outcomes of DLBCL with low clinical risk factors [6-year overall survival (OS): 90.1%; 6-year progression-free survival (PFS): 80.2%] [1]. However, in refractory cases, complete response or complete response unconfirmed (CR/CRu) rate in salvage therapy was only 38%, and the 3-year event-free survival (EFS) was 31% [2].
Gene expression profiling was first introduced in 2000 as a tool for the stratification of DLBCL [3]. DLBCL was classified into two subgroups, which were then designated as germinal center B cell (GCB)-like and activated B cell (ABC)-like subgroups, which were used to define prognostic categories, with ABC-like subgroups showing poorer prognoses [3]. Since then, considerable efforts have directed the stratification of DLBCL, based on mutation profiling via next-generation sequencing, [4–8] although somatic mutations detected in these studies matched only in 10-20% reflecting the genetic diversity of DLBCL [9]. Several somatic mutations have been reported as prognostic factors for DLBCL after treatment with R-CHOP or similar regimens. In a previous study, targeted capture sequencing of selected 34 genes in 215 DLBCL patients revealed that TNFAIP3 and GNA13 mutations were significantly associated with poorer prognosis in ABC-like DLBCL patients subjected to R-CHOP treatment [10]. Whole-exome sequencing (WES) of 14 relapsed/refractory large B-cell lymphoma patients (nine DLBCL and five primary mediastinal large B-cell lymphoma) identified several frequently altered genes in the cohort; however, non-relapsed/refractory cases were not sequenced for comparison [11]. Morin et al. performed WES of 38 relapsed/refractory DLBCL and detected TP53, FOXO1, KMT2C, CCND3, NFKBIZ, and STAT6 as top candidate genes in which mutations were related to treatment resistance [12]. In Korea, six refractory DLBCL patients and seven responsive DLBCL patients were analyzed via WES and transcriptome sequencing [13]. Missense mutations in TP53 were observed exclusively in refractory patients (3/6), and TP53 copy number deletions were also detected in the same three patients [13]. A Chinese group reported the results of targeted capture sequencing of 27 genes in 196 DLBCL patients. Mutations or copy number deletions of CD58 and TP53 were found to be poor prognostic factors in their cohort [14].
Herein, we report alterations in TP53 (a combination of point mutation and gene deletion) and OSBPL10 (point mutation) as prognostic indicators for DLBCL. These indicators were identified via WES of 35 samples from DLBCL patients with extremely poor or excellent prognosis upon treatment with R-CHOP or similar regimens. Results were validated in an additional 85 cases as independent prognostic factors from the International Prognostic Index (IPI) for OS and PFS.
RESULTS
Whole-exome sequencing in the discovery cohort
Clinical features and pathological characteristics of the discovery cohort are summarized in Table 1 and Supplementary Table 1. Significant differences in two IPI items (LDH and extranodal lesion) were found between groups with poor prognosis (Dp) and those with good prognosis (Dg) in the discovery cohort (Table 1). All double expressor cases (MYC >60% and BCL2 score 3+ [15]) were found in the poor prognosis group (Dp) (Table 1). WES was performed on 35 matched tumor-normal DNA (nine and 26 patients with poor and good prognoses, respectively). The average estimated tumor content was 56.47% (30.98 - 89.16%) (Supplementary Table 2). In both prognostic groups, CT/GA transversions were the most frequent variants, followed by AG/TC transversions; other mutations were relatively infrequent (Supplementary Figure 1A, 1B, and 1C). Mutations as triplets, XCG XTG/CGX CAX, were frequently observed (Supplementary Figure 1D). Somatic mutations filtered through pipeline are shown in Figure 1A and Supplementary Table 3.
Table 1: Comparison of characteristics between the patients with and without TP53 or OSBPL10 aberrations
Discovery cohort |
Validation cohort |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Dp |
Dg |
P |
V |
TP53 M + D |
TP53 W, M or D |
P |
OSBPL10 M |
OSBPL10 W |
P |
||
N |
9 |
26 |
85 |
6 |
79 |
21 |
64 |
||||
age |
63.33 ± 3.77 |
60.58 ± 2.51 |
0.55 |
66.64 ± 1.37 |
71.50 ± 5.33 |
66.27 ± 1.42 |
0.38 |
65.24 ± 3.39 |
67.09 ± 1.46 |
0.62 |
|
sex |
male |
7 |
12 |
46 |
4 |
42 |
13 |
33 |
|||
female |
2 |
14 |
0.14 |
39 |
2 |
37 |
0.68 |
8 |
31 |
0.46 |
|
IPI |
low |
1 |
15 |
30 |
0 |
30 |
10 |
20 |
|||
low intermediate |
2 |
6 |
20 |
2 |
18 |
3 |
17 |
||||
high intermediate |
2 |
4 |
25 |
3 |
22 |
5 |
20 |
||||
high |
4 |
1 |
0.008 |
10 |
1 |
9 |
0.16 |
3 |
7 |
0.43 |
|
Clinical stage |
I/II |
2 |
16 |
47 |
3 |
44 |
11 |
36 |
|||
III/IV |
7 |
10 |
0.06 |
38 |
3 |
35 |
1 |
10 |
28 |
0.80 |
|
LDH |
normal |
1 |
19 |
43 |
1 |
42 |
11 |
32 |
|||
high |
8 |
7 |
0.002 |
42 |
5 |
37 |
0.11 |
10 |
32 |
1 |
|
ECOG-PS |
0, 1 |
7 |
26 |
72 |
2 |
70 |
20 |
52 |
|||
2, 3, 4 |
2 |
0 |
0.06 |
13 |
4 |
9 |
0.004 |
1 |
12 |
0.17 |
|
Extranodal lesion |
<2 |
4 |
22 |
63 |
4 |
59 |
16 |
47 |
|||
≥2 |
5 |
4 |
0.03 |
22 |
2 |
20 |
0.65 |
5 |
17 |
1 |
|
Hans algorithm |
GCB |
6 |
12 |
35 |
2 |
33 |
9 |
26 |
|||
Non-GCB |
3 |
14 |
0.44 |
42 |
4 |
38 |
0.68 |
12 |
30 |
0.80 |
|
Double expressor |
negative |
3 |
20 |
58 |
5 |
53 |
16 |
42 |
|||
positive |
2 |
0 |
0.03 |
8 |
0 |
8 |
1 |
0 |
8 |
0.18 |
|
CD5 (IHC) |
negative |
8 |
19 |
68 |
5 |
63 |
21 |
47 |
|||
positive |
1 |
6 |
0.64 |
9 |
1 |
8 |
0.54 |
0 |
9 |
0.10 |
|
MYC split FISH |
negative |
6 |
18 |
||||||||
positive |
1 |
3 |
1 |
||||||||
BCL2 split FISH |
negative |
5 |
19 |
||||||||
positive |
2 |
2 |
0.25 |
||||||||
BCL6 split FISH |
negative |
6 |
15 |
||||||||
positive |
1 |
6 |
0.64 |
||||||||
Double expressor: MYC >60% and BCL2 score 3+.
Dp: poor prognosis in the discovery cohort, Dg: good prognosis in the discovery cohort, V: Validation cohort, M: mutation, D: deletion, W: wild type, IHC: immunohistochemistry.
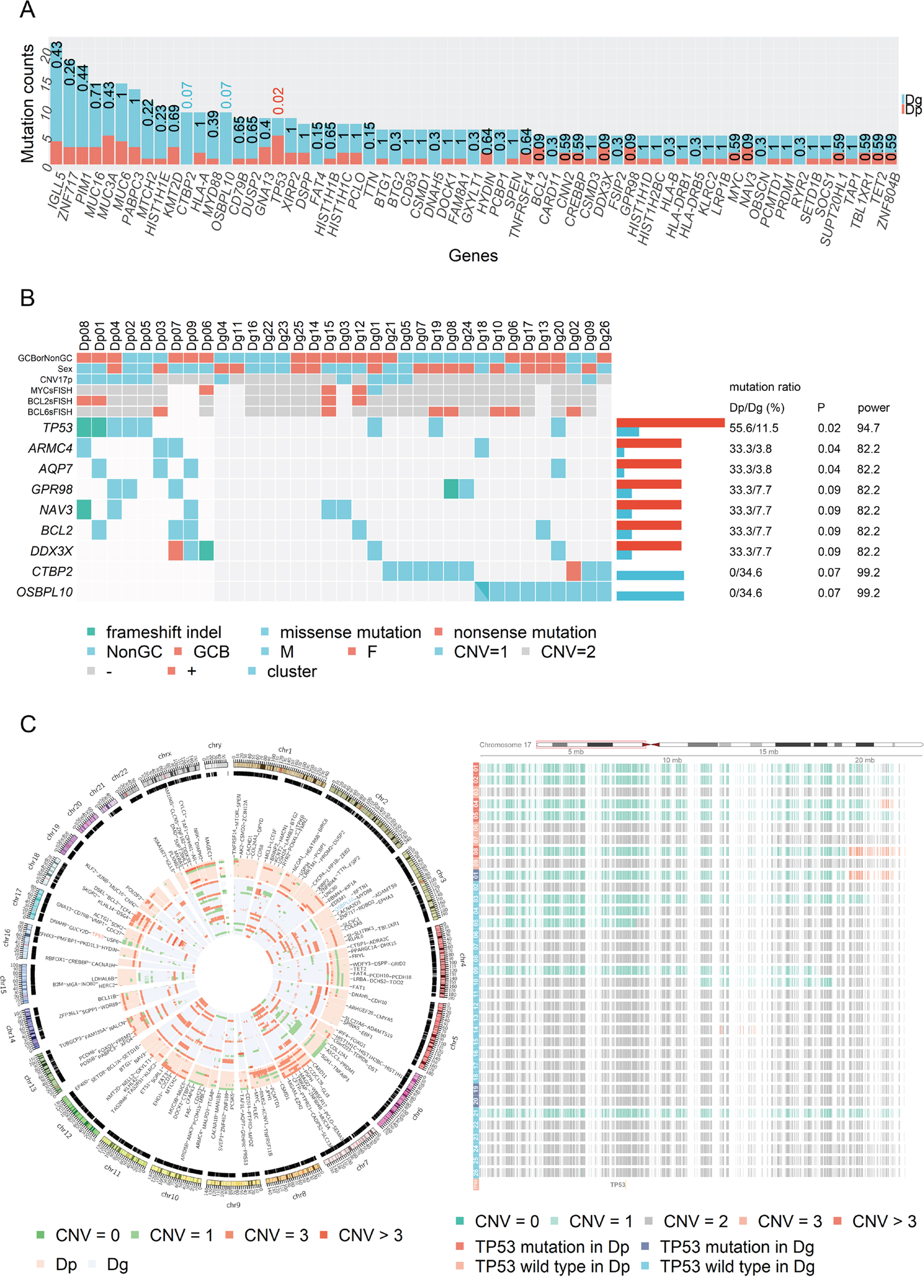
Figure 1: Mutational landscape and copy number variation in the discovery cohort. (A) The numbers of cases with mutations stratified based on prognostic group in the discovery cohort are presented. Numbers above each bar represent P-values for Fisher’s exact test. All detected mutations before manual inspection are listed. (B) Genes that showed different mutations between the positive and poor prognostic groups are shown in each case of the cohort. Statistical power was calculated based on the method reported by Lawrence et al. [44] (C) The CIRCOS plot of copy number variation in the discovery cohort. The figure on the right is an enlarged view of 17p. Six of eight (75%) TP53 mutations and six of 11 (55%) 17p deletions were found to coexist in the discovery cohort.
TP53, CTBP2, and OSBPL10 alterations were selected as candidate prognostic factors based on the following criteria, P < 0.1 and statistical power > 90 (Figure 1B). However, CTBP2 was discarded after manual inspection with Integrative Genomics Viewer (IGV) [16], because multiple mutations were detected from a single read of CTBP2 in both tumor and normal samples, probably due to mapping error (Supplementary Figure 2). Therefore, only TP53 and OSBPL10 mutations were further verified by Sanger sequencing (data not shown).
TP53 mutation sites were limited to the DNA-binding core domain (Figure 2A). Interestingly, in the poor prognostic group, five patients (Dp01, Dp02, Dp04, Dp05, and Dp08) harbored both TP53 mutations and 17p deletion, and the remaining four patients did not have either one. By contrast, in the good prognostic group, only one (Dg01) had the both aberrations, although TP53 mutations were detected in three patients and 17p deletion was detected in seven patients (Figure 1B, and 1C). Notably, the TP53 mutation and 17p deletion were found to be poor prognostic factors for OS (P = 0.00035) and PFS (P = 0.013) only when patients had both aberrations (Supplementary Figure 3A).
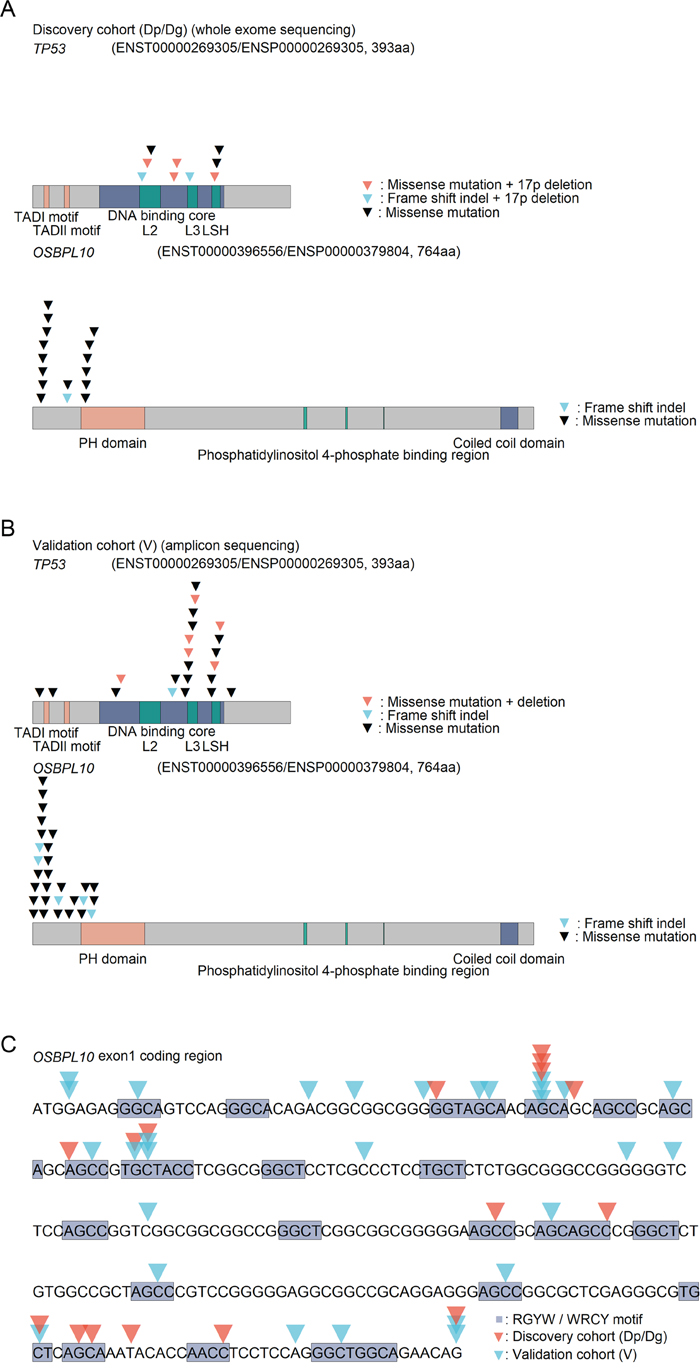
Figure 2: TP53 and OSBPL10 mutations in DLBCL. (A) Graphical view of TP53 and OSBPL10 mutations in the discovery cohort. (B) Graphical view of the TP53 and OSBPL10 mutations in the validation cohort. (C) Overview of detected mutations in the OSBPL10 exon 1 coding region.
A total of 16 mutations were identified in the OSBPL10 genes of nine patients. Interestingly, all identified mutations were confined to the exon 1 coding region (Figure 2A), and all patients harboring the mutations belonged to the good prognostic group. OSBPL10 mutations were found to be a highly reliable prognostic factor for improved PFS (P = 0.024) (Supplementary Figure 3B).
TP53 and OSBPL10 aberrations in the validation cohort
On the basis of the results obtained from WES of the discovery cohort, we further analyzed another 85 DLBCL cases (validation cohort). Clinical features and pathological characteristics are summarized in Table 1 and Supplementary Table 1. Mutations in the whole coding regions of TP53 and OSBPL10 exon 1 were examined via amplicon sequencing. The average read counts and mean coverage were 650,800 (284,998 - 1,529,726) and 30,258 (12,414 - 71,264), respectively. Twenty-two TP53 and 29 OSBPL10 mutations were detected in 18 (21%) and 21 (25%) of the 85 patients, respectively (Supplementary Table 4).
TP53 copy number loss (TP53 deletion) was identified in 13 out of the 85 patients via real-time quantitative genomic PCR analysis (Supplementary Table 5). Six patients (V51, V67, V76, V77, V80, and V83) harbored both TP53 mutation and deletion, and most mutations were confined to the DNA-binding core domain (Figure 2B). Although three patients had both TP53 and OSBPL10 mutations (V14, V25, and V31), none harbored all the three aberrations.
OSBPL10 in silico functional prediction
OSBPL10 mutations detected in our cohort were annotated based on protein functional prediction score (SIFT score [17] and Polyphen2 score [18]). Among the mutations that could be analyzed, 38.9% (7/18 cases) were classified as “deleterious” based on SIFT score and 30% (6/20 cases) were “possibly damaging” or “probably damaging” based on Polyphen2 score. On the other hand, 88.9% (16/18 cases) of TP53 mutations were determined to be “deleterious” based on SIFT score, and 94.1% (16/17 cases) were analyzed as “probably damaging” based on Polyphen2 score (Supplementary Table 4).
OSBPL10 as a target of somatic hypermutation
Interestingly, 30 out of the 45 OSBPL10 mutations (67%) were located in the RGYW/WRCY motif (Figure 2C), which is known as a region susceptible to somatic hyper mutation (SHM), a mechanism that causes highly frequent somatic mutations in normal and neoplastic B cells [19]. In the discovery cohort, the proportion of motif mutations to all somatic mutations was significantly higher in individuals harboring OSBPL10 mutations (32.6% vs. 26.2%; P = 0.01) (Figure 3A). The proportion of CT/GA mutations to all somatic mutations tended to be higher in individuals with OSBPL10 mutations (P = 0.08) (Figure 3B). Furthermore, OSBPL10 was identified as a SHM target based on the method reported by Khodabakhshi et al [20]. (Table 2, and Supplementary Table 6).
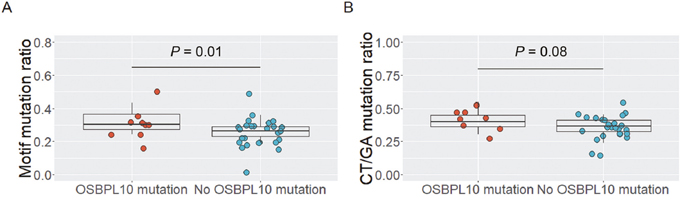
Figure 3: Analysis of OSBPL10 mutations and somatic hypermutation target motifs. (A) Proportion of RGYW/WRCY motif mutations to all somatic mutations. (B) Proportion of CT/GA mutations to all somatic mutations.
Table 2: OSBPL10 was identified as a SHM target
Gene |
Total SNVs |
Motif mutation |
Transition mutation |
C:G mutation |
SHM indicator |
|---|---|---|---|---|---|
PIM1* |
588 |
426 |
406 |
583 |
< 0.001 |
IGLL5 |
482 |
359 |
254 |
429 |
< 0.001 |
PABPC3 |
386 |
69 |
276 |
151 |
< 0.001 |
KCNJ18 |
214 |
0 |
102 |
214 |
< 0.001 |
CTBP2 |
209 |
31 |
93 |
111 |
0.0025 |
ZNF717 |
161 |
14 |
89 |
135 |
0.0018 |
MUC3A |
161 |
25 |
75 |
76 |
0.0068 |
ATAD3B |
120 |
0 |
120 |
120 |
< 0.001 |
MUC6 |
110 |
20 |
23 |
98 |
< 0.001 |
LDHAL6B |
108 |
27 |
84 |
30 |
0.0073 |
MTCH2 |
107 |
3 |
81 |
32 |
< 0.001 |
CD79B |
90 |
4 |
63 |
7 |
< 0.001 |
PABPC1 |
88 |
6 |
39 |
24 |
< 0.001 |
CDC27 |
76 |
30 |
66 |
0 |
< 0.001 |
HLA-DRB1 |
74 |
37 |
32 |
35 |
0.0408 |
MYD88 |
72 |
4 |
68 |
8 |
< 0.001 |
BTG1* |
71 |
53 |
54 |
67 |
0.0024 |
DUSP2* |
71 |
42 |
57 |
62 |
0.0273 |
ANKLE1 |
68 |
0 |
0 |
8 |
< 0.001 |
AK2 |
63 |
52 |
32 |
61 |
0.0013 |
SHANK3 |
61 |
21 |
61 |
17 |
0.0176 |
AQP7 |
58 |
1 |
32 |
57 |
0.0060 |
CNN2 |
43 |
31 |
39 |
24 |
0.0232 |
MPEG1 |
40 |
28 |
31 |
40 |
0.0307 |
HNRNPL |
39 |
0 |
36 |
0 |
< 0.001 |
OSBPL10 |
30 |
28 |
22 |
27 |
0.0175 |
KLRC2 |
29 |
25 |
4 |
25 |
0.0039 |
ARMC4 |
28 |
8 |
0 |
20 |
0.0109 |
FAM205A |
28 |
18 |
28 |
9 |
0.0387 |
*: previously reported gene.
Prognostic values of TP53 and OSBPL10 aberrations
In the validation cohort, as well as the discovery cohort, TP53 mutations and deletions were found to be poor prognostic factors for OS (P = 0.0016) and PFS (P = 0.023) only when they co-existed (Figure 4A). OSBPL10 mutation was validated as a highly reliable prognostic factor for better OS (P = 0.037) and PFS (P = 0.041) (Figure 4B). Significant differences were observed in OS and PFS when patients were stratified into three groups based on the presence of an OSBPL10 mutation (best prognosis) and coexistence of both TP53 mutation, deletion (poorest prognosis) (Figure 4C) and the others. Resulting values were designated as Genomic Prognostic Index (GPI).
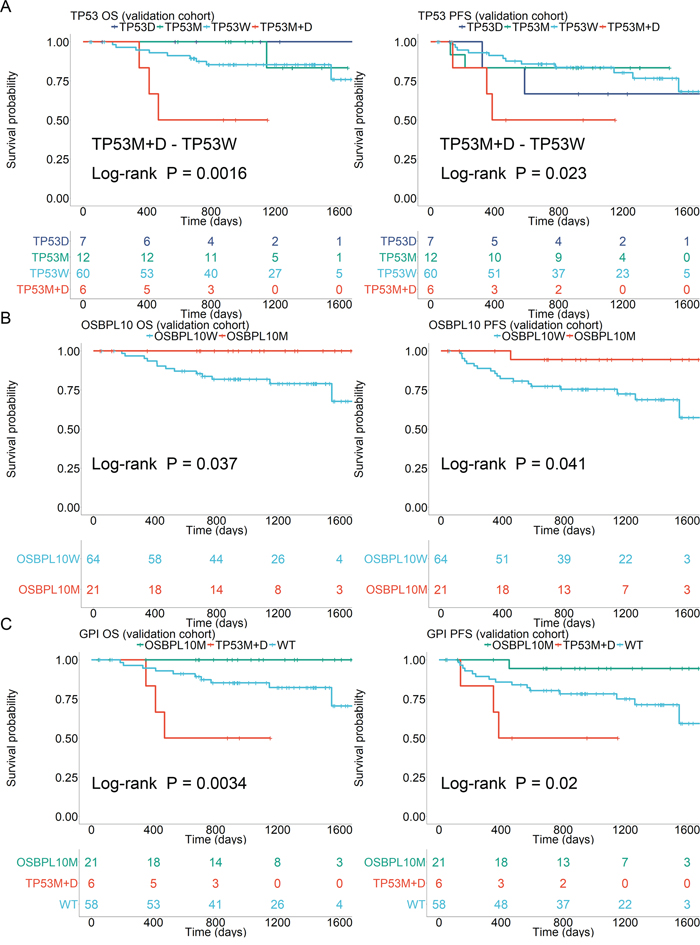
Figure 4: Survival analyses stratified by TP53 and OSBPL10 aberrations. (A) Survival stratified by TP53 status in the validation cohort. (B) Survival stratified by OSBPL10 status in the validation cohort. (C) Survival stratified by Genetic Prognostic Index (GPI) in the validation cohort. TP53W: TP53 wild-type; TP53M: TP53 mutation; 17pD: 17p deletion; OSBPL10W: OSBPL10 wild-type; and OSBPL10M: OSBPL10 mutation.
In other clinicopathological factors listed in Table 1 and Supplementary Table 1, patients harboring both TP53 mutations and deletions showed significantly lower ECOG performance status (ECOG-PS) (P = 0.004). Therefore, we applied the IPW method [21] to reduce the effects of IPI factors and conflicting gene mutations. Patients harboring both TP53 mutation and deletion still had significantly poorer OS (P < 0.01) and PFS (P < 0.01) (Supplementary Figure 4A). The presence of both TP53 mutation and deletion was found to be an independent poor prognostic factor from IPI in OS and PFS. Patients harboring OSBPL10 mutations showed extremely good prognoses and tended to have better OS (P = 0.05) and PFS (P = 0.05) after applying the IPW method (Supplementary Figure 4B).
DISCUSSION
In the present study, we showed that TP53 mutation and 17p/TP53 deletion were poor prognostic factors for OS and PFS in DLBCL patients treated with R-CHOP or similar regimens only when both aberrations were present. It is interesting to clarify whether the poor prognosis of patients harboring both aberrations is caused by loss of TP53 function alone or is augmented by deletions in other genes in the 17p region. Liu et al. suggested that the selective advantage of tumors is produced by the combined effects of TP53 loss and the reduced levels of tumor suppressor genes linked to 17p deletion [22]. They reported that acute myeloid leukemia (AML) patients harboring both TP53 mutation and 17p deletion showed a significantly poorer prognosis than patients with only one of these two genetic aberrations [22]. Liu et al. also demonstrated that heterozygous deletion of mouse chromosome 11B3, which corresponds to human 17p13.1, resulted in more aggressive lymphoma and leukemia than that produced by Trp53 deletion because of the combined effect of Trp53 loss and co-deletion of tumor suppressor genes in 11B3 [22]. The poor prognoses observed in our patients and the three refractory patients in a previous Korean study [13] who harbored both TP53 mutation and 17p/TP53 deletion were consistent with the observations reported by Liu et al. Meanwhile, no patients with biallelic 17p/TP53 deletions were detected in the present study. Biallelic deletion (complete loss) of some genes in 17p may be lethal to lymphoma cells.
The relationship between TP53 status and prognosis in DLBCL patients has been previously reported in 11 studies written in English (Table 3) [14, 23–32]. TP53 mutation and 17p/TP53 deletion showed variable prognostic impacts on DLBCL, although both tended to be poor prognostic factors. Notably, TP53 mutations and deletions tend to coexist, and TP53 deletion is frequently associated with 17p deletion, which frequently involves all or most of the chromosomal arm [22]. Accordingly, in the present study, 75% (6/8) of TP53 mutations and 55% (6/11) of 17p deletions coexisted in the discovery cohort (Figure 1C), and 33% (6/18) of TP53 mutations and 46% (6/13) of TP53 deletions coexisted in the validation cohort. This pattern could be the cause of the variable prognostic impacts of TP53 status as reported in literature. Among the 11 studies (Table 3), four examined both TP53 mutations and 17p/TP53 deletions and reported the number of patients having both genetic aberrations. However, only one study conducted during the CHOP era analyzed the impact of the coexistence of TP53 mutation and deletion; the presence of both aberrations, but not only one of them, was determined to be a poor prognostic factor for DLBCL [27]. Our study is the first to provide data demonstrating the prognostic impacts of the coexistence of TP53 mutation and deletion during the R-CHOP era.
Table 3: Literature review of TP53 variant and prognostic analysis in DLBCL
TP53 mutation |
17p deletion/TP53 loss |
Mutation with17p deletion/TP53 loss |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Treatment |
Study |
Year |
Total cases |
OS |
PFS/DFS |
OS |
PFS/DFS |
OS |
PFS/DFS |
|
CHOP era |
Ichikawa A et al. |
1997 [23] |
102 |
poor |
||||||
Stokke T et al. |
2001 [24] |
94 |
poor |
|||||||
Leroy K et al. |
2002 [25] |
69 |
poor |
|||||||
Young KH et al. |
2007 [26] |
113 |
poor |
NS |
NS |
|||||
Stöcklein H et al. |
2008 [27] |
40 |
NS |
NS |
poor |
|||||
Young KH et al. |
2008 [28] |
477 |
poor |
|||||||
R-CHOP era |
Xu-Monette ZY et al. |
2012 [29] |
506 |
poor |
poor |
NS |
NS |
|||
Asmar F et al. |
2014 [30] |
62 |
poor |
|||||||
Fiskvik I et al. |
2015 [31] |
43 |
poor |
poor |
||||||
Cao Y et al. |
2016 [14] |
165 |
poor |
poor |
poor |
poor |
||||
Zenz T et al. |
2017 [32] |
265 |
poor |
poor |
||||||
Present study |
2017 |
120 |
35 (Discovery cohort) |
NS* |
NS* |
NS# |
NS# |
poor |
poor |
|
85 (Validation cohort) |
NS* |
NS* |
NS# |
NS# |
poor |
poor |
||||
NS: not significant, *: TP53 mutation only, #: 17p deletion/TP53 loss only.
In a previous study, OSBPL10 mutation was reported in three out of nine primary central nervous system lymphomas (PCNSLs) and was indicated as a novel target gene of SHM in PCNSL [33]. The authors suggested that aberrant SHM had a major impact on PCNSL pathogenesis, but the clinical impacts of OSBPL10 mutation were not discussed [33]. In the present study, we confirmed that OSBPL10 is also a target gene of SHM in non-central nervous system DLBCL and identified OSBPL10 mutation as a biomarker for DLBCL with excellent prognosis. SHM, through which multiple somatic mutations may be generated in a single gene, is an important mechanism underlying the pathogenesis of B-cell neoplasms. Some mutation analysis pipelines employ filtering steps that discard candidate mutations when they are detected with several other mutations in a single read. It should be noted that applying such filtering steps to genetic alteration studies in B-cell neoplasms can potentially disregard relevant mutations generated by aberrant SHM.
OSBPL10 is a member of a family of sterol and phosphoinositide binding proteins, which consist of oxysterol-binding proteins (OSBPs) and OSBP-related proteins (ORPs). The mechanisms underlying their function remain to be fully elucidated [34]. In one breast cancer study, OSBPL10 mutations, which have a prevalence of 5.2%, have been suggested as potential drivers of mutations; however, the clinical impacts of OSBPL10 mutations were not described [35]. It is unclear whether OSBPL10 and/or its mutants play functionally important roles in DLBCL. The biological significance of OSBPL10 mutations remains to be clarified by further studies.
Results of our present study showed that the presence of both TP53 mutation and 17p/TP53 deletion is associated with poor prognosis in DLBCL patients treated with an R-CHOP-like regimen. We also identified OSBPL10 mutations as biomarkers for excellent prognosis in DLBCL patients during the R-CHOP era. In the clinical setting, reduced-intensity treatments may be delivered to patients with excellent prognoses. Further validation studies on larger cohorts, particularly both Asian and non-Asian groups, is warranted.
MATERIALS AND METHODS
Case selection
We selected 35 DLBCL cases as part of the discovery cohort according to the following criteria: (1) individuals diagnosed between January 2006 and December 2011 in The Cancer Institute Hospital (Tokyo, Japan), (2) individuals for whom frozen tissues or extracted DNA from frozen or fresh tissues were available, and (3) individuals with extremely poor prognosis [stable disease (SD) or progressive disease (PD) after first treatment] or with excellent prognosis [progression-free survival during the observation period for at least 3 years (until November 2016)]. Extracted DNA from matched fresh bone marrow specimens without lymphoma infiltration were available from 33 out of 35 cases. For the remaining two cases (Dg24 and Dg25), peripheral blood samples were used as matched normal samples.
All cases (85 DLBCL) that met the following criteria were included in the validation cohort: (1) individuals diagnosed between January 2012 to December 2014 in The Cancer Institute Hospital, (2) individuals for whom frozen tissues or extracted DNA from frozen or fresh materials were available, and (3) individuals treated with R-CHOP-like regimen.
All specimens were examined by pathologists (N. Tsuyama and K. Takeuchi), and DLBCL diagnoses were made according to the 4th edition of the WHO classification [36]. This study was approved by the institutional review board.
Sequencing analysis
Screening for gene mutations was performed via WES, using a customized capture probe set based on SureSelect XT Human All Exon V5 (Agilent, Santa Clara, USA). Libraries was prepared with a SureSelect Target Enrichment kit (Agilent) and sequenced on a HiSeq 2000 instrument (Illumina, San Diego, USA). Whole coding regions of TP53 and OSBPL10 exon 1 were amplified using TruSeq Custom Amplicon Low Input Kit (Illumina) and sequenced on a MiSeq platform (Illumina). Primers used for PCR and direct sequencing are listed in Supplementary Table 7. TP53 copy number variations (CNVs) were determined via real-time quantitative genomic PCR by the 2−ΔΔCT method [37] and using GAPDH as a reference gene. The primers used for real-time quantitative PCR are listed in Supplementary Table 8 [38, 39].
Whole-exome sequencing data analysis
Analysis was performed as previously described, with several modifications [40]. NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/) and Integrative Japanese Genome Variation Database (iJGVD) (https://ijgvd.megabank.tohoku.ac.jp/) were additionally included in the mutation reference data. For analysis of mutation overview and somatic hypermutation (SHM) targets, only annotated variants that met all the following conditions were selected: variants located in coding regions; variants detected from 10 or more reads; and variants called as somatic variants by more than one analysis tool. For analysis of somatic mutations, annotated variants that met at least one of the following conditions were discarded: exonic synonymous single nucleotide variants (SNVs); variants registered in dbSNP version 131; frequently observed variants (≥ 5%) in 1000 Genomes Project; frequently observed variants (≥ 5%) in NHLBI Exome Sequencing Project esp6500siv2; frequently observed variants (≥ five samples) in HGVD; frequently observed variants (≥ 5%) in iJGVD; and somatic variants called by more than one analysis tool. Copy number variation (CNV) and tumor content analyses were performed using ExomeCNV [41]. CNV was plotted with CIRCOS version 0.69-2, [42] Gviz, [43] and R version 3.3.2.
Amplicon sequencing data analysis
After quality control of sequence reads, read mapping on hg19 was performed following the same method used in WES. SNVs and indel calling was performed using GATK Haplotype Caller and MiSeq Reporter v2. Mutations called by either one of the tools were manually selected using IGV [16].
Propensity score analysis
To reduce bias during patient selection, inverse probability weighting (IPW) using propensity score was performed to investigate the causality of genetic variation and clinical outcomes in the validation cohort. In TP53 mutation analysis, the variables entered in the propensity score model were IPI items [age, clinical stage, lactate dehydrogenase (LDH), ECOG-PS, and extranodal lesion] and OSBPL10 mutation; similarly, in OSBPL10 mutation analysis, the IPI items in addition to the TP53 mutation were included as variables in the propensity score model. Next, analysis of adjusted survival curves and log-rank test were performed based on the IPW method, using R version 3.3.2 and the IPWsurvival package (http://www.divat.fr/en/softwares/ipwsurvival).
Statistical analyses of clinical data
The Mann-Whitney test, Student’s t-test, Welch two-sample t-test, Fisher's exact test, and log-rank test were performed using R version 3.3.2, coin, survminer (version 0.3.1), and survival (version 2.38).
Availability of data and materials
Data has been deposited at the DDBJ Japanese Genotype-phenotype Archive (https://www.ddbj.nig.ac.jp/jga) under the accession JGAS00000000087.
Abbreviations
ABC: activated B cell, AML: acute myeloid leukemia, CNV: copy number variation, CR: complete response, CRu: complete response unconfirmed, Dg: good prognosis in the discovery cohort, DLBCL: diffuse large B-cell lymphoma, Dp: poor prognosis in the discovery cohort, ECOG-PS: ECOG performance status, EFS: event-free survival, GCB: germinal center B cell, GPI: genomic prognostic index, IGV: integrative genomics viewer, iJGVD: integrative Japanese genome variation database, IPI: international prognostic index, IPW: inverse probability weighting, LDH: lactate dehydrogenase, ORPs: OSBP-related proteins, OS: overall survival, OSBPs: oxysterol-binding proteins, PCNSLs: primary central nervous system lymphomas, PD: progressive disease, PFS: progression-free survival, R-CHOP: (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), SD: stable disease, SHM: somatic hypermutation, SNVs: single nucleotide variants, V: the validation cohort, WES: whole-exome sequencing.
Author contributions
A. Dobashi, T. Yamaguchi, T. Noda, and K. Takeuchi conceived the study. A. Dobashi, and K. Takeuchi designed the study and wrote the manuscript. A. Dobashi and Y. Togashi performed molecular experiments. S. Baba performed histopathological experiments. M. Yokoyama and K. Hatake administered the treatments and collected clinical data. N. Tsuyama, and K. Takeuchi reviewed the patient materials. A. Dobashi, N. Tanaka, and S. Mori analyzed whole-exome sequencing data. A. Dobashi performed sequence and statistical analyses.
ACKNOWLEDGMENTS
We thank Ms. Sayuri Sengoku for her managerial assistance in this research project.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
FUNDING
This work was supported by the Japan Agency for Medical Research and Development (AMED) Grant Number 9011327.
REFERENCES
1. Pfreundschuh M, Kuhnt E, Trumper L, Osterborg A, Trneny M, Shepherd L, Gill DS, Walewski J, Pettengell R, Jaeger U, Zinzani PL, Shpilberg O, Kvaloy S, et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol. 2011; 12: 1013-22. https://doi.org/10.1016/S1470-2045(11)70235-2.
2. Gisselbrecht C, Glass B, Mounier N, Singh Gill D, Linch DC, Trneny M, Bosly A, Ketterer N, Shpilberg O, Hagberg H, Ma D, Briere J, Moskowitz CH, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010; 28: 4184-90. https://doi.org/10.1200/JCO.2010.28.1618.
3. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000; 403: 503-11. https://doi.org/10.1038/35000501.
4. Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, Jackman S, Krzywinski M, Scott DW, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011; 476: 298-303. https://doi.org/10.1038/nature10351.
5. Morin RD, Mungall K, Pleasance E, Mungall AJ, Goya R, Huff RD, Scott DW, Ding J, Roth A, Chiu R, Corbett RD, Chan FC, Mendez-Lago M, et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood. 2013; 122: 1256-65. https://doi.org/10.1182/blood-2013-02-483727.
6. Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, Dunphy C, Choi W, Au WY, Srivastava G, Lugar PL, Rizzieri DA, Lagoo AS, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013; 110: 1398-403. https://doi.org/10.1073/pnas.1205299110.
7. Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, Novak AJ, Dogan A, Ansell SM, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012; 109: 3879-84. https://doi.org/10.1073/pnas.1121343109.
8. Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, Chan J, Bhagat G, Chadburn A, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011; 43: 830-7. https://doi.org/10.1038/ng.892.
9. Dobashi A. Molecular Pathogenesis of Diffuse Large B-Cell Lymphoma. J Clin Exp Hematop. 2016; 56: 71-8. https://doi.org/10.3960/jslrt.56.71.
10. Dubois S, Viailly PJ, Mareschal S, Bohers E, Bertrand P, Ruminy P, Maingonnat C, Jais JP, Peyrouze P, Figeac M, Molina TJ, Desmots F, Fest T, et al. Next-Generation Sequencing in Diffuse Large B-Cell Lymphoma Highlights Molecular Divergence and Therapeutic Opportunities: a LYSA Study. Clin Cancer Res. 2016; 22: 2919-28. https://doi.org/10.1158/1078-0432.CCR-15-2305.
11. Mareschal S, Dubois S, Viailly PJ, Bertrand P, Bohers E, Maingonnat C, Jais JP, Tesson B, Ruminy P, Peyrouze P, Copie-Bergman C, Fest T, Jo Molina T, et al. Whole exome sequencing of relapsed/refractory patients expands the repertoire of somatic mutations in diffuse large B-cell lymphoma. Genes Chromosomes Cancer. 2016; 55: 251-67. https://doi.org/10.1002/gcc.22328.
12. Morin RD, Assouline S, Alcaide M, Mohajeri A, Johnston RL, Chong L, Grewal J, Yu S, Fornika D, Bushell K, Nielsen TH, Petrogiannis-Haliotis T, Crump M, et al. Genetic Landscapes of Relapsed and Refractory Diffuse Large B-Cell Lymphomas. Clin Cancer Res. 2016; 22: 2290-300. https://doi.org/10.1158/1078-0432.CCR-15-2123.
13. Park HY, Lee SB, Yoo HY, Kim SJ, Kim WS, Kim JI, Ko YH. Whole-exome and transcriptome sequencing of refractory diffuse large B-cell lymphoma. Oncotarget. 2016; 7:86433–86445. https://doi.org/10.18632/oncotarget.13239.
14. Cao Y, Zhu T, Zhang P, Xiao M, Yi S, Yang Y, Li Q, Ling S, Wang Y, Gao L, Zhu L, Wang J, Wang N, et al. Mutations or copy number losses of CD58 and TP53 genes in diffuse large B cell lymphoma are independent unfavorable prognostic factors. Oncotarget. 2016; 7:83294–83307. https://doi.org/10.18632/oncotarget.13065.
15. Tsuyama N, Sakata S, Baba S, Mishima Y, Nishimura N, Ueda K, Yokoyama M, Terui Y, Hatake K, Kitagawa M, Ishizuka N, Tomita N, Takeuchi K. BCL2 expression in DLBCL: reappraisal of immunohistochemistry with new criteria for therapeutic biomarker evaluation. Blood. 2017; 130: 489-500. https://doi.org/10.1182/blood-2016-12-759621.
16. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011; 29: 24-6. https://doi.org/10.1038/nbt.1754.
17. Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003; 31: 3812-4.
18. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010; 7: 248-9. https://doi.org/10.1038/nmeth0410-248.
19. Spencer J, Dunn-Walters DK. Hypermutation at A-T Base Pairs: The A Nucleotide Replacement Spectrum Is Affected by Adjacent Nucleotides and There Is No Reverse Complementarity of Sequences Flanking Mutated A and T Nucleotides. J Immunol. 2005; 175: 5170-7. https://doi.org/10.4049/jimmunol.175.8.5170.
20. Khodabakhshi AH, Morin RD, Fejes AP, Mungall AJ, Mungall KL, Bolger-Munro M, Johnson NA, Connors JM, Gascoyne RD, Marra MA, Birol I, Jones SJ. Recurrent targets of aberrant somatic hypermutation in lymphoma. Oncotarget. 2012; 3: 1308-19. https://doi.org/10.18632/oncotarget.653.
21. Xie J, Liu C. Adjusted Kaplan-Meier estimator and log-rank test with inverse probability of treatment weighting for survival data. Stat Med. 2005; 24: 3089-110. https://doi.org/10.1002/sim.2174.
22. Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J, Spitzer B, Bosbach B, Kastenhuber ER, Baslan T, Ackermann S, Cheng L, Wang Q, et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature. 2016; 531: 471-5. https://doi.org/10.1038/nature17157.
23. Ichikawa A, Kinoshita T, Watanabe T, Kato H, Nagai H, Tsushita K, Saito H, Hotta T. Mutations of the p53 gene as a prognostic factor in aggressive B-cell lymphoma. N Engl J Med. 1997; 337: 529-34. https://doi.org/10.1056/NEJM199708213370804.
24. Stokke T, DeAngelis P, Smedshammer L, Galteland E, Steen HB, Smeland EB, Delabie J, Holte H. Loss of chromosome 11q21-23.1 and 17p and gain of chromosome 6p are independent prognostic indicators in B-cell non-Hodgkin's lymphoma. Br J Cancer. 2001; 85: 1900-13. https://doi.org/10.1054/bjoc.2001.2164.
25. Leroy K, Haioun C, Lepage E, Le Metayer N, Berger F, Labouyrie E, Meignin V, Petit B, Bastard C, Salles G, Gisselbrecht C, Reyes F, Gaulard P, et al. p53 gene mutations are associated with poor survival in low and low-intermediate risk diffuse large B-cell lymphomas. Ann Oncol. 2002; 13: 1108-15.
26. Young KH, Weisenburger DD, Dave BJ, Smith L, Sanger W, Iqbal J, Campo E, Delabie J, Gascoyne RD, Ott G, Rimsza L, Muller-Hermelink HK, Jaffe ES, et al. Mutations in the DNA-binding codons of TP53, which are associated with decreased expression of TRAILreceptor-2, predict for poor survival in diffuse large B-cell lymphoma. Blood. 2007; 110: 4396-405. https://doi.org/10.1182/blood-2007-02-072082.
27. Stocklein H, Smardova J, Macak J, Katzenberger T, Holler S, Wessendorf S, Hutter G, Dreyling M, Haralambieva E, Mader U, Muller-Hermelink HK, Rosenwald A, Ott G, et al. Detailed mapping of chromosome 17p deletions reveals HIC1 as a novel tumor suppressor gene candidate telomeric to TP53 in diffuse large B-cell lymphoma. Oncogene. 2008; 27: 2613-25. https://doi.org/10.1038/sj.onc.1210901.
28. Young KH, Leroy K, Moller MB, Colleoni GW, Sanchez-Beato M, Kerbauy FR, Haioun C, Eickhoff JC, Young AH, Gaulard P, Piris MA, Oberley TD, Rehrauer WM, et al. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: an international collaborative study. Blood. 2008; 112: 3088-98. https://doi.org/10.1182/blood-2008-01-129783.
29. Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu WM, Montes-Moreno S, Dybkaer K, Chiu A, Orazi A, Zu Y, Bhagat G, Richards KL, et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood. 2012; 120: 3986-96. https://doi.org/10.1182/blood-2012-05-433334.
30. Asmar F, Hother C, Kulosman G, Treppendahl MB, Nielsen HM, Ralfkiaer U, Pedersen A, Moller MB, Ralfkiaer E, de Nully Brown P, Gronbaek K. Diffuse large B-cell lymphoma with combined TP53 mutation and MIR34A methylation: Another “double hit” lymphoma with very poor outcome? Oncotarget. 2014; 5: 1912-25. https://doi.org/10.18632/oncotarget.1877.
31. Fiskvik I, Beiske K, Delabie J, Yri O, Spetalen S, Karjalainen-Lindsberg ML, Leppa S, Liestol K, Smeland EB, Holte H. Combining MYC, BCL2 and TP53 gene and protein expression alterations improves risk stratification in diffuse large B-cell lymphoma. Leuk Lymphoma. 2015; 56: 1742-9. https://doi.org/10.3109/10428194.2014.970550.
32. Zenz T, Kreuz M, Fuge M, Klapper W, Horn H, Staiger AM, Winter D, Helfrich H, Huellein J, Hansmann ML, Stein H, Feller A, Moller P, et al. TP53 mutation and survival in aggressive B cell lymphoma. Int J Cancer. 2017; 141:1381–1388. https://doi.org/10.1002/ijc.30838.
33. Vater I, Montesinos-Rongen M, Schlesner M, Haake A, Purschke F, Sprute R, Mettenmeyer N, Nazzal I, Nagel I, Gutwein J, Richter J, Buchhalter I, Russell RB, et al. The mutational pattern of primary lymphoma of the central nervous system determined by whole-exome sequencing. Leukemia. 2015; 29: 677-85. https://doi.org/10.1038/leu.2014.264.
34. Olkkonen VM, Li S. Oxysterol-binding proteins: sterol and phosphoinositide sensors coordinating transport, signaling and metabolism. Prog Lipid Res. 2013; 52: 529-38. https://doi.org/10.1016/j.plipres.2013.06.004.
35. Pongor L, Kormos M, Hatzis C, Pusztai L, Szabo A, Gyorffy B. A genome-wide approach to link genotype to clinical outcome by utilizing next generation sequencing and gene chip data of 6,697 breast cancer patients. Genome Med. 2015; 7: 104. https://doi.org/10.1186/s13073-015-0228-1.
36. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. (2008). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Fourth Edition.
37. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25: 402-8. https://doi.org/10.1006/meth.2001.1262.
38. Yamashita Y, Minoura K, Taya T, Fujiwara SI, Kurashina K, Watanabe H, Choi YL, Soda M, Hatanaka H, Enomoto M, Takada S, Mano H. Analysis of chromosome copy number in leukemic cells by different microarray platforms. Leukemia. 2007; 21: 1333-7. https://doi.org/10.1038/sj.leu.2404636.
39. Wang X, Spandidos A, Wang H, Seed B. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 2012; 40: D1144-9. https://doi.org/10.1093/nar/gkr1013.
40. Dobashi A, Tsuyama N, Asaka R, Togashi Y, Ueda K, Sakata S, Baba S, Sakamoto K, Hatake K, Takeuchi K. Frequent BCOR aberrations in extranodal NK/T-Cell lymphoma, nasal type. Genes Chromosomes Cancer. 2016; 55: 460-71. https://doi.org/10.1002/gcc.22348.
41. Sathirapongsasuti JF, Lee H, Horst BA, Brunner G, Cochran AJ, Binder S, Quackenbush J, Nelson SF. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics. 2011; 27: 2648-54. https://doi.org/10.1093/bioinformatics/btr462.
42. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009; 19: 1639-45. https://doi.org/10.1101/gr.092759.109.
43. Hahne F, Ivanek R. Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol Biol. 2016; 1418: 335-51. https://doi.org/10.1007/978-1-4939-3578-9_16.
44. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014; 505: 495-501. https://doi.org/10.1038/nature12912.