
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway is highly conserved throughout evolution. Initial signaling starts upon ligand binding at the cytokine and growth factor receptor, emanating a signal from the cell membrane to the nucleus (Figure 1). The activation of receptor-associated tyrosine kinases – the JAKs – is followed by activation and tyrosine phosphorylation of the STATs [1]. STAT inactivation is largely carried out by tyrosine phosphatase action. Moreover, control by suppressor of cytokine signaling (SOCS), protein inhibitors of activated STAT (PIAS), signal transducing adaptor molecule (STAM), Sprouty-related Ena/VASP homology 1-domain-containing protein (SPRED) and SPROUTY proteins were described as negative regulators of JAK/STAT activation [2, 3]. STAT proteins can regulate proliferation, differentiation, growth and apoptosis [4]. In mammals, seven different STATs are known which share several conserved functional domains, but the transactivation domain (TAD) at the C-terminus is the most diverse part (Figure 2B).
STAT5 biology
Only upon ligand binding to the cytokine receptor, the associated JAK kinase dimer becomes trans-activated and phosphorylates the cytoplasmic part of the receptor on distinct tyrosine residues [5]. Newest findings present a complete model of receptor-linked JAK2 activation after growth hormone (GH) binding [6]. Once the GH receptor dimer is activated, the transmembrane helices rearrange from a parallel to a left-handed cross-over state. This causes the removal of one JAK2 pseudokinase domain from the kinase domain of the respective JAK2 binding partner, trans-activation of the kinases and phosphorylation of the receptor. Another recent study enlightens the interaction between the JAK kinase, tyrosine kinase 2 (Tyk2) and the interferon-α receptor (IFNAR1) [7]. Binding to IFNAR1 resembles a SH2-like phosphopeptide interaction with Tyk2, with a glutamate replacing the usual phosphotyrosine residue when co-crystallized. STAT proteins bind via their N-terminus and SH2 domain to the phosphorylated cytokine receptors and crystal structure analysis revealed their pre-dimerization without the necessity of tyrosine phosphorylation as parallel/anti-parallel dimers [8]. Tyrosine phosphorylated STATs form efficient dimers via their SH2 domains and translocate to the nucleus to bind DNA. The two variants of STAT5 (STAT5A/B) are activated by more than 20 different cytokines, hormones and growth factors. Prominent cytokines include interleukin (IL)-2, 3, 4, 5, 7, 9, 15, 21, erythropoietin (EPO), thrombopoietin (TPO), prolactin (PRL), and granulocyte macrophage colony-stimulating factor (GM-CSF) and GH [5]. Activation is associated with tyrosine 694/699 phosphorylation in human STAT5A/B, which is a prerequisite for stable parallel dimer formation and initiation of transcription of STAT5-regulated genes [5]. Specific isoforms of STAT5A/B were associated with human cancer types, but the exact roles for each isoform in distinct cancer types are not studied yet [4]. Both proteins are widely expressed, but differences became also apparent in single knock-out mice. Loss of Stat5a results in impaired mammary gland development [9], whereas deletion of Stat5b causes stunted body growth and NK cell defects [10]. Stat5a/b double knock-out mice die perinatal on a C57BL/6 and Balb/c genetic background, but Sv129/C57BL/6 Stat5a/b double knock-out mice have a compensatory mechanism via high pYSTAT3 activity and a sub Mendelian fraction of severely sick mice can survive up to 5 weeks [11]. The reversible tyrosine phosphorylation of the STAT proteins is regulated by protein tyrosine phosphatases (PTP) of which 109 different family members are known [12]. SH2-domain-containing protein tyrosine phosphatase-2 (SHP-2), PTP1-B as well as serine protein phosphatase 2A (PP2A) have been reported to be associated with STAT5 regulation but it remains largely unknown which particular phosphatases act on STAT5A/B in specific cell types [13–15]. Interestingly, serine phosphatase PP2A-activating drugs were recently found to kill therapy-resistant chronic myeloid leukemia (CML) stem cells [16].
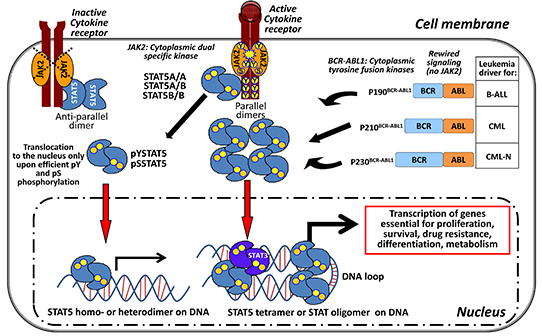
Figure 1: STAT5 – the central signaling node in BCR-ABL1 + leukemia. Canonical activation of STAT5 starts at the cell membrane with cytokine binding to specific receptors. Receptor dimers undergo conformational changes followed by pY phosphorylation and activation of the receptor-associated tyrosine kinase JAK2. JAK2 mediates phosphorylation of the receptor at the cytoplasmic end at specific sites. STAT5 can bind to the receptors as preformed parallel and anti-parallel dimers regardless of its phosphorylation status and is activated by JAK2. The activated STAT5 proteins form parallel homo- or hetero (STAT5A/A; STAT5A/B; STAT5B/B) dimers via their SH2 domains and translocate to the nucleus to bind to the DNA. In BCR-ABL1-induced leukemias a translocation between chromosomes 9 and 22 occurs and results in a constitutive active cytoplasmic tyrosine fusion kinase. Depending on the break-point within the BCR gene three predominant variants of the BCR-ABL1 kinase are known: p190BCR-ABL1, p210BCR-ABL1, p230BCR-ABL1. The associated leukemias are shown next to the different fusion kinases. BCR-ABL1 activity rewires the JAK/STAT signaling in the leukemic cell. STAT5 is directly pY phosphorylated by BCR-ABL1 which makes JAK2 signaling dispensable. STAT5 tetramers and STAT oligomers (with activated STAT3 dimers) bind to the DNA and result in elevated target gene expression for the indicated cellular processes in a leukemic cell.
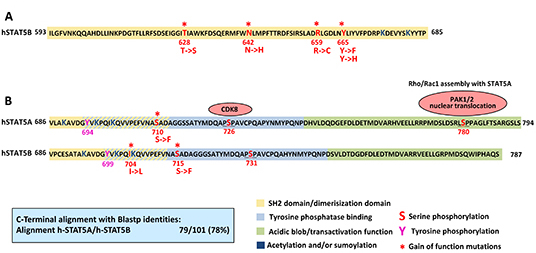
Figure 2: Gain of function mutations in STAT5. (A) The SH2/dimerization domain (yellow) of STAT5B ranges from 593 to 712 amino acids [105]. So far, somatic mutations in the STAT5B SH2 domain have been described in LGL, T-ALL, T-PLL and HSTL. Asterisks indicate the GOF mutation position. (B) The C-terminus of STAT5A and B is the most divergent part and shares 78% sequence identity between the two closely related proteins. Lysines (K- dark blue) nearby and in the tyrosine phosphatase binding domain (light blue) undergo acetylation or sumoylation, which positively or negatively regulates pYSTAT5, respectively [106]. Apart from tyrosines 694/699 (pink), serines sites (red) 726/780 in STAT5A are constitutively phosphorylated and crucial for leukemic transformation. As upstream kinases CDK8 and PAKs have been identified. GOF mutations have been described for S710/S715 in retro virally induced screening methods and I704 in T-ALL. The transactivation domain (green) is rich in aspartic (D) and glutamic acid (E) forming a highly negatively charged region, the acidic blob, which interacts with other factors of the transcriptional machinery.
STAT5, the central signaling node in BCR-ABL1+ leukemias
Persistent activation of STAT5 is found in many solid cancers as well as in the majority of hematologic malignancies [4]. Especially in myeloid malignancies, the role and oncogenic properties of STAT5 are well established [17–20]. The role of STAT5 proteins in solid cancers is less clear due to largely lacking conditional targeting approaches in appropriate genetic cancer models. With regard to most hematopoietic cancers, STAT5 is unmutated and its constitutive activation is a secondary event and triggered by oncogenic upstream tyrosine kinases such as BCR-ABL1 (Figure 1). The requirement of STAT5 in BCR-ABL1+ leukemias for transformation and disease maintenance has been well described [21]. Further, the role of the different STAT5A/B isoforms has recently been addressed, where in a BCR-ABL1-inducible cellular system, knock-down of STAT5B sensitized leukemic cells to imatinib treatment [22] and the attenuation of STAT5A resulted in enhanced basal oxidative stress and DNA damage of normal CD34+ and CML cells as well as growth inhibition of CD34+ cells from patients with acquired imatinib resistance [23].
CML is a prototype of a myeloid neoplasm where a single driver mutant, BCR-ABL1, initiates activation of multiple pro-oncogenic signaling molecules, including STAT5 [24]. In the chronic phase of CML, BCR-ABL1 is essential for the survival and proliferation of leukemic cells. In line with this concept, BCR-ABL1-targeting drugs are able to produce stable complete responses in many patients with Philadelphia chromosome positive (Ph+) CML [25]. Indeed, for these patients, the development of BCR-ABL1 tyrosine kinase inhibitors (TKI) remains a huge success story. The first active compound with large clinical applications in CML was imatinib [26] followed by dasatinib, which binds BCR-ABL1 and other oncogenic kinases, such as SRC family kinases in both configurations of inactive or active kinase state [27]. Further TKI improvement came from the higher binding affinity and selectivity than imatinib through the follow-up substance nilotinib (AMN107) [28]. In addition, bosutinib (SKI-606) was developed, a second line inhibitor which has the broadest target spectrum inhibiting SRC, ABL and TEC, as well as serine kinases CAMK2G and STE20, but bosutinib does not target PDGFR and KIT [29, 30]. Finally, the third line inhibitor ponatinib has been developed and shown to exhibit substantial efficacy in patients with CML in whom the polyresistant BCR-ABL1 T315I mutation is expressed in leukemic cells [31].
STAT5 enhances resistance to therapy
Overall, TKI have changed the fate of patients suffering from BCR-ABL1-driven CML [25] or Ph+ acute lymphoblastic leukemia. Despite this great success,TKI are not curative and in most cases medication needs to be taken life-long to prevent relapse [32]. On the other hand, CML might be cured with TKI in some cases as shown by the Stop imatinib (STIM) trial [33]. Within a cohort of 100 patients who were treated with imatinib for at least two years and had achieved a complete molecular remission (CMR), the probability to persist in CMR was 41%. Nevertheless, CML is a chronic hematopoietic disease derived from transformed long-term hematopoietic stem cells (LT-HSC) that can act as leukemic stem cells (LSC) and maintain the disease by signaling via STAT5 [34]. Majoritarian, TKI treatment results in the decimation of LSC below detection limits but is unable to completely eradicate the LSC [35]. This TKI resistance can be explained by the behavior of LSC like non-transformed stem cells rather than oncogene addicted CML cancer stem cells [36]. Further, CML stem cells are capable of autocrine TNF-α production which supports the expression of IL-3 and GM-CSF receptor and is associated with cell survival and enhanced proliferation [37]. Moreover, CD25/IL-2 receptor high expressing CML stem cells were shown to have a higher capacity to induce leukemia than CD25 negative CML stem cells [38]. Interestingly, a similar important role for STAT5-regulated CD25 expression as a leukemic stem cell marker was demonstrated to be a predictive biomarker in an acute myeloid leukemia (AML) for sensitivity of PIM kinase inhibitors [39]. These new findings further underline the role of STAT5 signaling in CML or AML stem cells.
15–25% of all patients develop resistance towards imatinib mostly mediated by point mutations of the BCR-ABL1 oncogene [40]. In this regard it is of interest that high levels of STAT5 suffice to enhance imatinib resistance in CML cells [41]. High STAT5 expression levels were also shown to correlate with the occurrence of BCR-ABL1 mutations [42]. Formal proof for STAT5 as proto-oncogene in hematopoietic diseases was obtained by retroviral transduction of normal murine bone marrow (BM) cells followed by transplant with gain of function (GOF) Stat5 variants. Thus, hyperactivity of STAT5 as mimicked by expression of a constitutively activated STAT5A (cSTAT5A) variant suffices to induce a multilineage leukemia upon transplantation [43].
Somatic mutations of STAT5 on the rise
Since 2013, somatic STAT5B mutations were discovered in large granula lymphocytic leukemia (LGL) [44], in T-promyelocytic leukemia (T-PLL) [45], in acute T-cell leukemia (T-ALL) [46,47] and hepatosplenic T-cell lymphoma (HSTL) [48], where more mutations can be expected through progress in the cancer genome project [200]. Over-expression of the mutated STAT5B versions resulted in increased transcriptional activity and was associated with poor disease outcome. Surprisingly and interestingly, most mutations occur in the SH2 domain of STAT5B, with the point mutation N642H being the most frequent. How these mutations might affect nuclear shuttling of STAT5 is unclear. A graphical scheme of GOF somatic STAT5B mutations is given in Figure 2A. In addition, in acute promyelocytic leukemia, STAT5B translocates and fuses with the retinoic acid receptor (RAR) alpha with resulting enhanced STAT3-mediated signaling [49]. cSTAT5A triggers leukemogenesis through the N-terminus, which regulates STAT5 tetramer formation and oligomerization among multiple STAT family members [43]. Furthermore, cSTAT5 in hematopoietic stem cells (HSC) was shown to be crucial for development of leukemia and knock-down of STAT5A was associated with inhibiting growth of CML CD34+ cell from patients with acquired resistances towards TKI [23,50]. Interestingly, in leukemic cells, tyrosine phosphorylated STAT5A (pYSTAT5) not only localizes in the nucleus but it is also found predominantly localized in the cytoplasm where it can form a signaling complex with GRB-associated binding protein 2 (Gab2) and phosphoinositide 3-kinase (PI3K) leading to protein kinase B (PKB) activation [51]. It is unclear and hard to dissect whether the cytoplasmic activity of STAT5 contributes to its oncogenic potential, but studying the effects of inhibitors of nuclear STAT5 transport could illuminate the role of cytoplasmic STAT5 in the context of leukemogenesis.
Blocking STAT5 nuclear translocation
Several lines of evidence argue for a critical role of STAT5A/B in leukemia. Based on the essential role of these molecules in leukemia evolution and progression, several targeting approaches have been proposed [52, 53]. We could show that serine phosphorylation of STAT5A is a prerequisite for nuclear translocation [54]. Here, we propose that the inhibition of this transport by direct or indirect interaction with the transcription factor or upstream regulators of nuclear import (summarized in Figure 3) could be a feasible targeting approach. Unbiased cell-based screens with various small-molecule compound libraries are one way to identify new targets of already approved drugs. By employing STAT5 transcriptional activity and luciferase reporter assays, pimozide was identified as a potential STAT5 inhibitor in BCR-ABL1+ leukemic cells and in an AML mouse model [55, 56]. Pimozide was found to inhibit tyrosine phosphorylation of STAT5, but did not target BCR-ABL1. The mechanistic details of STAT5 inhibition by pimozide are unclear but most likely indirect. A direct interaction between STAT5 and pimozide was not demonstrated and the use of high molecular concentrations suggests that the effects of pimozide on STAT5 might be secondary and not potent enough to be considered for clinical application [55]. Still it was the first experimental tool to target STAT5 in clinically relevant cell systems. A current study identifies the synthetic chalone α-Br-TMC as an inhibitor of JAK2/STAT5 signaling [57]. Although, cytotoxic effects are only observed at high concentrations (100 µM) in cells expressing cSTAT5, tyrosine phosphorylation of STAT5 and JAK2 was strongly reduced at 10 µM. STAT5 dependant target gene transcription is altered but is remains to be determined whether STAT5 inhibition is direct or a secondary effect.
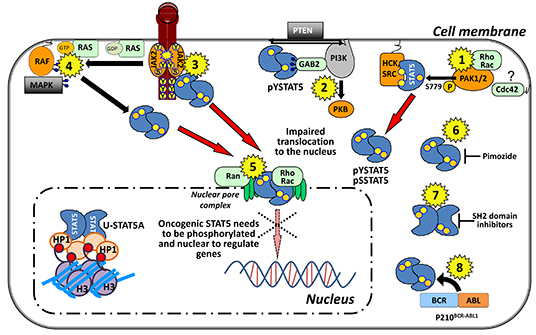
Figure 3: Targeting STAT5A nuclear translocation at a glance in leukemia. (1) PAK1 and 2 phosphorylate STAT5A on S779 in the presence of Rho GTPases and might travel through the nuclear pore complex assembled to those. Here, targeting Rho GTPases and the down-stream effectors, PAKs, seem reasonable. Cdc42 is a close homologue of Rho, but it interacts with membranes via post-translational C-terminal geranylgeranyl lipid modification. It is unclear if Cdc42 is involved in shuttling processes of STAT5 (marked by a ?) and it is thus not found in the nucleus. SRC-family kinases (SFK) retain pYSTAT5A in the cytoplasm. Thus, targeting SFK could enhance STAT5 nuclear localization, which might not be therapeutically beneficial. (2) Targeting the complex formation of pYSTAT5A in the cytoplasm with Gab2 and PI3K which lead to PKB activation. (3) JAK2 inhibition targets the canonical signaling and nuclear translocation which was shown to be relevant in the CML stem cell. (4) Targeting oncogenic Ras which promotes nuclear localization of STAT5 in HSC. (5) Ran GTPases are thought to be essential mediators of the STAT-transporter complex involved in nuclear translocation. (6) Pimozide inhibits pYSTAT5. (7) SH2 domain inhibitors block the effective dimer formation of STAT molecules that bind to DNA. (8) BCR-ABL1 phosphorylates STAT5 directly thus TKI target tyrosine phosphorylation and nuclear translocation of STAT5 indirectly.
SH2 domain inhibitors
The transcriptional activity of STAT5 requires phosphorylation on tyrosine and parallel dimer formation via SH2 domains. Inhibitors targeting the SH2 domain of STAT5B have been identified but very high concentrations of these compounds were required to induce apoptosis in lymphoma cells [58]. In a report by the Gunning group salicylic acid-based small molecules targeting STAT5 SH2 domain also required high IC50 values to counteract growth of BCR-ABL1+ cells (20 µM) [59]. The same group improved STAT5 SH2 domain inhibitors further that now work in vitro in a nanomolar range (Cumaraswamy et al. 2014 accepted in ACS Medicinal Chemistry Letters). Of interest is also a STAT3 SH2 domain-targeting compound that showed low micromolar effects (1 µM) in combination with imatinib on survival of CD34+ cells obtained from CML patients with BCR-ABL1-independent TKI resistance [60]. This is of clinical importance as STAT3 was shown to mediate resistance in these leukemias.
Up-stream regulator JAK2
The receptor-associated tyrosine kinase JAK2 is the main activator of STAT5 in hematopoietic cells and is thus considered an obvious target in JAK-STAT-driven malignancies. Further, the identification of activating mutations in JAK kinases in Ph- myeloproliferative neoplasms , with JAK2 (V617F) being the most frequent [61–63], has changed therapeutic strategies dramatically. Moreover, many JAK mutations were shown to be associated with acute leukemias [64,65]. Indeed, JAK-targeting TKI have reached the clinics recently, several of which also target JAK2 with low micromolar concentration to exert pro-apoptotic effects in CML cells in vitro (reviewed in [66]). However, BCR-ABL1-induced myeloid transformation and disease maintenance is independent of JAK2 in mouse models and STAT5 is highly activated despite deletion of JAK2 in these leukemia models [67]. Off-target effects of JAK2 TKI inhibitors explain this apparent contradiction. Most JAK2-targeting TKI are capable of inducing apoptosis not only in JAK2 wild-type cells but also in JAK2-deficient cells by inhibiting BCR-ABL1 kinase itself. Transformation by BCR-ABL1 rewires signaling and BCR-ABL1 itself phosphorylates and activates STAT5. Another layer of complexity was introduced by the fact that knock-down of BCR-ABL1 in CD34+ human CML stems cells fails to efficiently eradicate these leukemic cells [68]. Reduced colony formation of CD34+ CML cells in the presence of dasatinib was complemented upon administration of cytokines that provide survival signals via JAK2 [69]. On the other hand, the absence of JAK2 dramatically accelerated disease progression in a BCR-ABL1+ mouse model [70]. Competitive transplantation studies showed that loss of JAK2 enabled leukemia initiating cells to outcompete the normal HSC. This again relativizes the possible benefit of JAK2 inhibition in BCR-ABL1+ leukemia, as the kinase is a critical factor in normal hematopoiesis. Thus, JAK2 inhibition might affect HSC and LSC in comparable manner and even abolish the therapeutic efficacy [71]. Nevertheless, early clinical trials aim to assess the possibility to combine the JAK2 inhibitor ruxolitinib in CML patients receiving already treatment with TKI [201, 202]. A recent study demonstrated reduction and apoptosis of CD34+ CML cells upon combined treatment with nilotinib and ruxolitinib, rather than single application [72]. In the mouse model the combination of TKI showed toxicity toward healthy HSC, but to a lesser extent than toward CD34+ CML cells, which suggested that combinatorial TKI therapy is advantageous over TKI monotherapy.
Controversial: targeting SFK
Other kinases that have been reported to activate STAT5 in BCR-ABL1+ disease belong to the SRC-family kinases (SFK). The SFK members HCK and LYN were found to be highly activated in p210BCR-ABL1 cells [73]. SRC interacts with STAT5 and can phosphorylate STAT5 on tyrosine 694 upon EPO receptor stimulation [74]. Similarly, HCK is capable of phosphorylating STAT5B in BCR-ABL1-transformed cells [75]. The effects of the BCR-ABL1 TKI dasatinib, which also inhibits SRC kinase activity and the dual SRC/ABL kinase inhibitor bosutinib on CML stem cells appeared mild and no strong pro-apoptotic response could be achieved [76,77]. Recently, the SRC kinases HCK and SRC, but not LYN, were shown to retain STAT5A in the cytoplasm of BCR-ABL1+ cells [78]. The significance of this observation is indicative of a potential negative control of SFK to retain pYSTAT5 in the cytoplasm blocking or causing diminished nuclear pYSTAT5-induced transcription. Thus, SFKs might balance the high levels of STAT5 which was shown to associate pretty much in a 1:1 stoichiometry in K562 BCR-ABL1+ cells to relieve oncogenic stress induced by STAT5 [67,79]. Thus, inhibition of this control mechanism by targeting SFKs might explain partly why SFK inhibitors failed for clinical applications since their inhibition could accelerate pYSTAT5 nuclear translocation.
STAT5 serine phosphorylation
STAT5 harbours serine phosphorylation sites within the C-terminal TAD, with only partially understood biological functions (reviewed in [80]). Both, STAT5A and STAT5B harbour a serine residue at position 725 or 730 within a conserved PSP motif that becomes phosphorylated upon stimulation with GH, PRL and IL-2 (see graphical scheme Figure 2B). Mutation of the conserved serine site was shown to negatively influence transcriptional activity of STAT1 and STAT3. Interestingly, human STAT5A has a unique serine site at position 780 within an LSSP motif. The role of those serine sites in BCR-ABL1+ leukemia has recently been clarified. STAT5A is highly serine-phosphorylated in human CML cells. Friedbichler et al. showed that loss of serine 725 and 779 phosphorylation impaired leukemogenesis induced by cSTAT5A [81]. The fact that regular hematopoiesis was largely unaffected placed STAT5 serine phosphorylation into the lime light of potential target sites.
In fibroblasts, the serine/threonine kinase inhibitor flavopiridol reduced STAT5 serine 725 phosphorylation triggered by the growth-factor GM-CSF and knock-down experiments identified CDK8 as a responsible up-stream kinase phosphorylating STAT1 on serine 727 [82]. In leukemias this STAT1 serine site is also critical as natural killer (NK) cells lacking phosphorylation on STAT1 serine 727 displayed enhanced cytotoxicity and were capable to eradicate leukemic cells significantly better when compared to wild-type STAT1-expressing cells [83]. Treatment with CDK8 inhibitors could thus improve tumor surveillance by enhancing NK cell-mediated cytotoxicity. Flavopiridol was the first serine/threonine kinase inhibitor in clinical trials and is currently tested in clinical chronic lymphocytic leukemia (CLL), AML, and CML trials. A Phase I clinical trial with combination therapy consisting of imatinib and flavopiridol in CML and BCR-ABL1+ ALL patients has already been conducted with considerable success [84].
Targeting p21-activated kinases
In BCR-ABL1+ leukemias, STAT5A mutants lacking serine phosphorylation on S725 and S779 strongly impaired the viability of the leukemic cells and did not support transformation by Abelson oncogenes [54]. The mutation of a single serine (STAT5S779A) sufficed to prolonged disease onset in BCR-ABL1-driven leukemic mouse model. We identified p21-activated kinases (PAK) as major up-stream kinases and thus triggers of pSTAT5S779. Biochemical studies confirmed the direct interaction and phosphorylation of STAT5S779 by PAK which required the concomitant presence of the Rho GTPase family [54]. Further evidence for a key role of PAK in hematopoietic malignancies stems from a report on c-kit-induced myeloid neoplasms where PAK acts in conjunction with Rac kinase to support leukemogenesis [85]. Interestingly, Rac-dependant signaling has also been implicated in the signaling cascades downstream of p210BCR-ABL1 in CML stem and progenitor cells [86–89]. Rac1 and its up-stream activator MgcRacGAP, a nuclear localizing signal-containing nuclear chaperone, are essential for the nuclear translocation of STAT transcription factors in cytokine-induced and oncogenic (Flt3-ITD) signaling [90,91]. Our recent work bridges the gap and allows us to understand the mechanistic details underlying these observations. In particular, STAT5S779 phosphorylation has been identified as a prerequisite for STAT5A to translocate into the nucleus and nuclear accumulation of STAT5A is prevented by PAK I group kinase inhibitors in BCR-ABL1+ leukemias [54]. The importance of STAT5S779 for nuclear translocation has long not been recognized perhaps because it has been masked by endogenous wild-type STAT5 [92–94]. We introduced a YFP-tagged STAT5S779A mutant into BCR-ABL1-expressing HEK 293T cells and observed no accumulation in the nucleus but an even distribution throughout the cell [54]. Upon co-transfection with untagged STAT5 wild-type, nuclear accumulation of YFP-tagged STAT5 was observed. Thus, with a STAT5 molecule with intact S779 within the dimer, transport to the nucleus is possible. This might also explain why dominant-negative STAT5 lacking the TAD with the serine site 779 is still able to go nuclear [95]. Further, the massive over-expression might also promote STAT5 into the nucleus. Human STAT5B also harbours a serine site at position 779 in the TAD but it shows essential differences to STAT5A as the motif is not flanked by prolines (DSQ) [102]. The role of this phosphorylation site has not been addressed so far. The mechanistic details of STAT5B translocation to the nucleus remain unknown, but it is generally accepted that STAT5B can be transported to the nucleus in a heterodimer with STAT5A and upon GH stimulation STAT5B homodimers efficiently go nuclear [96].
GTPases and STAT cellular shuttling
STAT proteins were also shown to be able to undergo nucleocytoplasmic transport through the cell without being tyrosine phosphorylated (U-STAT), whereas the re-translocation of pYSTAT from the nucleus back into the cytoplasm requires its dephosphorylation. In order to translocate into the nucleus, molecules need to pass the nuclear pore complex (NPC) which consists of about 30 different proteins, the nucleoporins (Nups) (reviewed in [97] and [98]). Large proteins such as the STAT proteins (95-100 kDA) cannot freely pass the NPC. Two modes of transfer through the NPC have been postulated: (i) carrier-independent transport mediated by direct interaction with the Nups or (ii) carrier-dependent transport, where the cargo protein is associated with the transport factors importins for nuclear import and chromosome region maintenance 1 (CRM1) protein for export from the nucleus. The Ras-related nuclear (Ran) proteins are present in the cell in two nucleotide bound forms: GDP- and GTP-bound. The asymmetrical distribution of Ran-GTP, which is highly concentrated in the nucleus and Ran-GDP, with high levels in the cytoplasm, drives nuclear shuttling. Non-tyrosine phosphorylated STAT proteins were shown to travel by carrier-free transportation mediated by direct interactions with the Nups as well as through carrier-dependant transport out of the nucleus mediated by interaction with CRM1 [99]. For nuclear export of STAT proteins, a ternary complex of CRM1-RanGTP with a Leucine-rich nuclear export signal needs to be formed in the nucleus and this complex is then disassembled by GTP hydrolysis by Ran GTPase activating protein in the cytoplasm. These shuttling processes are dependent on small GTPase activity which seems to be in line with the finding that phosphorylation of STAT5A on serine 779 by PAKs requires the presence of members of the Rho GTPase family which might travel in complex with the STAT5 proteins through the nuclear pore. This implies that translocation of STAT5A is regulated by small GTPases, that can be therapeutically targeted with small molecule compounds. However, we want to mention that small GTPases such as oncogenic Ras are difficult to be targeted due to the high affinity of GTP to small GTPases [100]. Interestingly, oncogenic Ras was recently linked to promote enhanced nuclear accumulation of STAT5 in HSC, partly through diminished Socs2 expression promoting renewal and engraftment under competitive transplant situation and accelerated growth of HSC and the myeloid progenitor pool in particular [101]. Inhibitors targeting Rho GTPase family members and their downstream effectors have been developed [102,103] and only a few were adopted for clinical use and therefore data on toxicity in mammals are limited (reviewed in [104]).
CONCLUSIONS
Treatment of TKI-resistant CML is an emerging challenge in experimental and applied hematology. In fact, CML LSC exhibit multiple forms of resistance and may escape treatment with novel TKI. This has accelerated the search for alternative therapies and drug targets in CML. As described in this article, STAT5 fulfils all criteria of a major drug target in BCR-ABL1+ leukemia. However, there are other potential indications for STAT5 inhibitors. Notably, STAT5 is important for efficient generation of many blood lineages and it is essential for proper immune function. Moreover, it is an important regulator in function and differentiation of epithelial cell types. It can be a driver or preventer of solid cancers and STAT5 protein ablation might not turn out favourable, but it was surprisingly well tolerated in genetic mouse models of Stat5a/b ablation. The path to therapeutic success might involve the targeting of modulators such as serine kinases or small GTPases and interacting proteins of STAT5. The specific inhibition of components of the aberrant signaling to which STAT5 is a central mediator can result in differentiation, growth arrest or apoptosis of leukemic cells. Thus, modulation of the irregular signaling in the cancer cell towards the normal levels is a promising avenue to target STAT5-dependent leukemia such as CML.
Formerly, transcription factors like the STAT family members were considered to be undruggable as they lack a catalytic activity. Today, there are many possibilities to target STAT proteins indirectly via interfering with its up-stream regulators thereby negatively influencing the nuclear translocation of STAT proteins and inhibiting their transcriptional activity. The direct inhibition via SH2 domains has been a long discussed issue considering that formerly developed SH2 domain inhibitors lacked potency and specificity. However, continuous improvements were achieved in this field and the development of the first inhibitor displaying low micromolar antileukemic potency might soon have consequences for the clinics.
FINANCIAL & COMPETING INTERESTS DISCLOSURE
Authors were partly supported by the Austrian Science Fund (FWF), grants SFB F47 and SFB F28 and the Austrian Federal Ministry of Science and Research GENAU grant ‘PLACEBO’. P.V. has received honoraria from Novartis, BMS, Pfizer and a research grant from Novartis.
REFERENCES
1. Stark GR, Darnell JE. The JAK-STAT pathway at twenty. Immunity. 2012 Apr 20; 36:503–14.
2. Hou SX, Zheng Z, Chen X, Perrimon N. The Jak/STAT pathway in model organisms: emerging roles in cell movement. Dev Cell. 2002 Dec; 3:765–78.
3. Yoshimura A. Regulation of cytokine signaling by the SOCS and Spred family proteins. Keio J Med. 2009 Jun; 58:73–83.
4. Ferbeyre G, Moriggl R. The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochim Biophys Acta. 2011 Jan; 1815:104–14.
5. Paukku K, Silvennoinen O. STATs as critical mediators of signal transduction and transcription: lessons learned from STAT5. Cytokine Growth Factor Rev. 2004 Dec; 15:435–55.
6. Brooks AJ, Dai W, O’Mara ML, Abankwa D, Chhabra Y, Pelekanos RA, Gardon O, Tunny KA, Blucher KM, Morton CJ, Parker MW, Sierecki E, Gambin Y, Gomez GA, Alexandrov K, Wilson IA, Doxastakis M, Mark AE, Waters MJ. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science. 2014 May 16; 344:1249783.
7. Wallweber HJA, Tam C, Franke Y, Starovasnik MA, Lupardus PJ. Structural basis of recognition of interferon-α receptor by tyrosine kinase 2. Nat Struct Mol Biol. Nature Publishing Group; 2014 May 6; 21:443–8.
8. Zhong M, Henriksen MA, Takeuchi K, Schaefer O, Liu B, ten Hoeve J, Ren Z, Mao X, Chen X, Shuai K, Darnell JE. Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation-inactivation cycle. Proc Natl Acad Sci U S A. 2005 Mar 15; 102:3966–71.
9. Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997 Jan 15; 11:179–86.
10. Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci U S A. 1997 Jul 8; 94:7239–44.
11. Hoelbl A, Kovacic B, Kerenyi MA, Simma O, Warsch W, Cui Y, Beug H, Hennighausen L, Moriggl R, Sexl V. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood. 2006 Jun 15; 107:4898–906.
12. Hendriks WJAJ, Pulido R. Protein tyrosine phosphatase variants in human hereditary disorders and disease susceptibilities. Biochim Biophys Acta. 2013 Oct; 1832:1673–96.
13. Yokoyama N, Reich NC, Miller WT. Involvement of protein phosphatase 2A in the interleukin-3-stimulated Jak2-Stat5 signaling pathway. J Interferon Cytokine Res. 2001 Jun; 21:369–78.
14. Chen Y, Wen R, Yang S, Schuman J, Zhang EE, Yi T, Feng G-S, Wang D. Identification of Shp-2 as a Stat5A phosphatase. J Biol Chem. 2003 May 9; 278:16520–7.
15. Aoki N, Matsuda T. A cytosolic protein-tyrosine phosphatase PTP1B specifically dephosphorylates and deactivates prolactin-activated STAT5a and STAT5b. J Biol Chem. 2000 Dec 15; 275:39718–26.
16. Neviani P, Harb JG, Oaks JJ, Santhanam R, Walker CJ, Ellis JJ, Ferenchak G, Dorrance AM, Paisie CA, Eiring AM, Ma Y, Mao HC, Zhang B, Wunderlich M, May PC, Sun C, Saddoughi SA, Bielawski J, Blum W, Klisovic RB, Solt JA, Byrd JC, Volinia S, Cortes J, Huettner CS, Koschmieder S, Holyoake TL, Devine S, Caligiuri MA, Croce CM, Garzon R, Ogretmen B, Arlinghaus RB, Chen C-S, Bittman R, Hokland P, Roy D-C, Milojkovic D, Apperley J, Goldman JM, Reid A, Mulloy JC, Bhatia R, Marcucci G, Perrotti D. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest. 2013 Oct 1; 123:4144–57.
17. Cain JA, Xiang Z, O’Neal J, Kreisel F, Colson A, Luo H, Hennighausen L, Tomasson MH. Myeloproliferative disease induced by TEL-PDGFRB displays dynamic range sensitivity to Stat5 gene dosage. Blood. 2007 May 1; 109:3906–14.
18. Choudhary C, Brandts C, Schwable J, Tickenbrock L, Sargin B, Ueker A, Böhmer F-D, Berdel WE, Müller-Tidow C, Serve H. Activation mechanisms of STAT5 by oncogenic Flt3-ITD. Blood. 2007 Jul 1; 110:370–4.
19. Lacronique V, Boureux A, Monni R, Dumon S, Mauchauffé M, Mayeux P, Gouilleux F, Berger R, Gisselbrecht S, Ghysdael J, Bernard OA. Transforming properties of chimeric TEL-JAK proteins in Ba/F3 cells. Blood. 2000 Mar 15; 95:2076–83.
20. Walz C, Ahmed W, Lazarides K, Betancur M, Patel N, Hennighausen L, Zaleskas VM, Van Etten RA. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2(V617F) in mice. Blood. 2012 Apr 12; 119:3550–60.
21. Hoelbl A, Schuster C, Kovacic B, Zhu B, Wickre M, Hoelzl MA, Fajmann S, Grebien F, Warsch W, Stengl G, Hennighausen L, Poli V, Beug H, Moriggl R, Sexl V. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol Med. 2010 Mar; 2:98–110.
22. Schaller-Schönitz M, Barzan D, Williamson AJK, Griffiths JR, Dallmann I, Battmer K, Ganser A, Whetton AD, Scherr M, Eder M. BCR-ABL affects STAT5A and STAT5B differentially. PLoS One. 2014 Jan; 9:e97243.
23. Casetti L, Martin-Lannerée S, Najjar I, Plo I, Augé S, Roy L, Chomel J-C, Lauret E, Turhan AG, Dusanter-Fourt I. Differential contributions of STAT5A and STAT5B to stress protection and tyrosine kinase inhibitor resistance of chronic myeloid leukemia stem/progenitor cells. Cancer Res. 2013 Apr 1; 73:2052–8.
24. Ilaria RL, Van Etten RA. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J Biol Chem. 1996 Dec 6; 271:31704–10.
25. Hochhaus A, O’Brien SG, Guilhot F, Druker BJ, Branford S, Foroni L, Goldman JM, Müller MC, Radich JP, Rudoltz M, Mone M, Gathmann I, Hughes TP, Larson RA. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia. 2009 Jun; 23:1054–61.
26. Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001 Apr 5; 344:1031–7.
27. Lindauer M, Hochhaus A. Dasatinib. Recent Results Cancer Res. 2010 Jan; 184:83–102.
28. Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W, Bochinski K, Hochhaus A, Griffin JD, Hoelzer D, Albitar M, Dugan M, Cortes J, Alland L, Ottmann OG. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006 Jun 15; 354:2542–51.
29. Puttini M, Coluccia AML, Boschelli F, Cleris L, Marchesi E, Donella-Deana A, Ahmed S, Redaelli S, Piazza R, Magistroni V, Andreoni F, Scapozza L, Formelli F, Gambacorti-Passerini C. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006 Dec 1; 66:11314–22.
30. Remsing Rix LL, Rix U, Colinge J, Hantschel O, Bennett KL, Stranzl T, Müller A, Baumgartner C, Valent P, Augustin M, Till JH, Superti-Furga G. Global target profile of the kinase inhibitor bosutinib in primary chronic myeloid leukemia cells. Leukemia. 2009 Mar; 23:477–85.
31. Frankfurt O, Licht JD. Ponatinib–a step forward in overcoming resistance in chronic myeloid leukemia. Clin Cancer Res. 2013 Nov 1; 19:5828–34.
32. Cortes J, O’Brien S, Kantarjian H. Discontinuation of imatinib therapy after achieving a molecular response. Blood. 2004 Oct 1; 104:2204–5.
33. Mahon F-X, Réa D, Guilhot J, Guilhot F, Huguet F, Nicolini F, Legros L, Charbonnier A, Guerci A, Varet B, Etienne G, Reiffers J, Rousselot P. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010 Nov; 11:1029–35.
34. Kovacic B, Hoelbl A, Litos G, Alacakaptan M, Schuster C, Fischhuber KM, Kerenyi MA, Stengl G, Moriggl R, Sexl V, Beug H. Diverging fates of cells of origin in acute and chronic leukaemia. EMBO Mol Med. 2012 Apr; 4:283–97.
35. Graham SM. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002 Jan 1; 99:319–25.
36. Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. American Society for Clinical Investigation 2011 Jan 4; 121:396–409.
37. Gallipoli P, Pellicano F, Morrison H, Laidlaw K, Allan EK, Bhatia R, Copland M, Jørgensen HG, Holyoake TL. Autocrine TNF-α production supports CML stem and progenitor cell survival and enhances their proliferation. Blood. 2013 Nov 7; 122:3335–9.
38. Kobayashi CI, Takubo K, Kobayashi H, Nakamura-Ishizu A, Honda H, Kataoka K, Kumano K, Akiyama H, Sudo T, Kurokawa M, Suda T. The IL-2/CD25 axis maintains distinct subsets of chronic myeloid leukemia-initiating cells. Blood. 2014 Apr 17; 123:2540–9.
39. Guo Z, Wang A, Zhang W, Levit M, Gao Q, Barberis C, Tabart M, Zhang J, Hoffmann D, Wiederschain D, Rocnik J, Sun F, Murtie J, Lengauer C, Gross S, Zhang B, Cheng H, Patel V, Schio L, Adrian F, Dorsch M, Garcia-Echeverria C, Huang S-MA. PIM inhibitors target CD25-positive AML cells through concomitant suppression of STAT5 activation and degradation of MYC oncogene. Blood. 2014 Jul 8.
40. Gambacorti-Passerini CB, Gunby RH, Piazza R, Galietta A, Rostagno R, Scapozza L. Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet Oncol. 2003 Feb; 4:75–85.
41. Warsch W, Kollmann K, Eckelhart E, Fajmann S, Cerny-Reiterer S, Hölbl A, Gleixner K V, Dworzak M, Mayerhofer M, Hoermann G, Herrmann H, Sillaber C, Egger G, Valent P, Moriggl R, Sexl V. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood. 2011 Mar 24; 117:3409–20.
42. Warsch W, Grundschober E, Berger A, Gille L, Cerny-Reiterer S, Tigan A-S, Hoelbl-Kovacic A, Valent P, Moriggl R, Sexl V. STAT5 triggers BCR-ABL1 mutation by mediating ROS production in chronic myeloid leukaemia. Oncotarget. 2012 Dec; 3:1669–87.
43. Moriggl R, Sexl V, Kenner L, Duntsch C, Stangl K, Gingras S, Hoffmeyer A, Bauer A, Piekorz R, Wang D, Bunting KD, Wagner EF, Sonneck K, Valent P, Ihle JN, Beug H. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell. 2005 Jan; 7:87–99.
44. Rajala HLM, Eldfors S, Kuusanmäki H, van Adrichem AJ, Olson T, Lagström S, Andersson EI, Jerez A, Clemente MJ, Yan Y, Zhang D, Awwad A, Ellonen P, Kallioniemi O, Wennerberg K, Porkka K, Maciejewski JP, Loughran TP, Heckman C, Mustjoki S. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013 May 30; 121:4541–50.
45. Kiel MJ, Velusamy T, Rolland D, Sahasrabuddhe AA, Chung F, Bailey NG, Schrader A, Li B, Li JZ, Ozel AB, Betz BL, Miranda RN, Medeiros LJ, Zhao L, Herling M, Lim MS, Elenitoba-Johnson KSJ. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. 2014 May 13;
46. Kontro M, Kuusanmäki H, Eldfors S, Burmeister T, Andersson EI, Bruserud O, Brümmendorf TH, Edgren H, Gjertsen BT, Itälä-Remes M, Lagsrtöm S, Lohi O, Lundán T, Martí JML, Majumder MM, Parsons A, Pemovska T, Rajala H, Vettenranta K, Kallioniemi O, Mustjoki S, Porkka K, Heckman CA. Novel activating STAT5B mutations as putative drivers of T-cell acute lymphoblastic leukemia. Leukemia. 2014 Feb 27;
47. Bandapalli OR, Schuessele S, Kunz JB, Rausch T, Stütz AM, Tal N, Geron I, Gershman N, Izraeli S, Eilers J, Vaezipour N, Kirschner-Schwabe R, Hof J, von Stackelberg A, Schrappe M, Stanulla M, Zimmermann M, Koehler R, Avigad S, Handgretinger R, Frismantas V, Bourquin JP, Bornhauser B, Korbel JO, Muckenthaler MU, Kulozik AE. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica. 2014 Jun 27.
48. Nicolae A, Xi L, Pittaluga S, Abdullaev Z, Pack SD, Chen J, Waldmann TA, Jaffe ES, Raffeld M. Frequent STAT5B mutations in γδ hepatosplenic T-cell lymphomas. Leukemia. 2014 Jun 20;
49. Dong S, Tweardy DJ. Interactions of STAT5b-RARalpha, a novel acute promyelocytic leukemia fusion protein, with retinoic acid receptor and STAT3 signaling pathways. Blood. 2002 Apr 15; 99:2637–46.
50. Kato Y, Iwama A, Tadokoro Y, Shimoda K, Minoguchi M, Akira S, Tanaka M, Miyajima A, Kitamura T, Nakauchi H. Selective activation of STAT5 unveils its role in stem cell self-renewal in normal and leukemic hematopoiesis. J Exp Med. 2005 Jul 4; 202:169–79.
51. Harir N, Pecquet C, Kerenyi M, Sonneck K, Kovacic B, Nyga R, Brevet M, Dhennin I, Gouilleux-Gruart V, Beug H, Valent P, Lassoued K, Moriggl R, Gouilleux F. Constitutive activation of Stat5 promotes its cytoplasmic localization and association with PI3-kinase in myeloid leukemias. Blood. 2007 Feb 15; 109:1678–86.
52. Bibi S, Arslanhan MD, Langenfeld F, Jeanningros S, Cerny-Reiterer S, Hadzijusufovic E, Tchertanov L, Moriggl R, Valent P, Arock M. Co-operating STAT5 and AKT signaling pathways in chronic myeloid leukemia and mastocytosis: possible new targets of therapy. Haematologica. 2014 Mar 1; 99:417–29.
53. Dorritie KA, McCubrey JA, Johnson DE. STAT transcription factors in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia. Macmillan Publishers Limited. 2014 Feb; 28:248–57.
54. Berger A, Hoelbl-Kovacic A, Bourgeais J, Hoefling L, Warsch W, Grundschober E, Uras IZ, Menzl I, Putz EM, Hoermann G, Schuster C, Fajmann S, Leitner E, Kubicek S, Moriggl R, Gouilleux F, Sexl V. PAK-dependent STAT5 serine phosphorylation is required for BCR-ABL-induced leukemogenesis. Leukemia. 2013 Nov 22.
55. Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, Gashin LB, Terrell S, Klitgaard JL, Santo L, Addorio MR, Ebert BL, Griffin JD, Frank DA. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2011 Mar 24; 117:3421–9.
56. Nelson EA, Walker SR, Xiang M, Weisberg E, Bar-Natan M, Barrett R, Liu S, Kharbanda S, Christie AL, Nicolais M, Griffin JD, Stone RM, Kung AL, Frank DA. The STAT5 Inhibitor Pimozide Displays Efficacy in Models of Acute Myelogenous Leukemia Driven by FLT3 Mutations. Genes Cancer. 2012 Jul; 3:503–11.
57. Pinz S, Unser S, Brueggemann S, Besl E, Al-Rifai N, Petkes H, Amslinger S, Rascle A. The synthetic α-bromo-2’,3,4,4′-tetramethoxychalcone (α-Br-TMC) inhibits the JAK/STAT signaling pathway. PLoS One. 2014 Jan; 9:e90275.
58. Müller J, Schust J, Berg T. A high-throughput assay for signal transducer and activator of transcription 5b based on fluorescence polarization. Anal Biochem. 2008 Apr 15; 375:249–54.
59. Page BDG, Khoury H, Laister RC, Fletcher S, Vellozo M, Manzoli A, Yue P, Turkson J, Minden MD, Gunning PT. Small molecule STAT5-SH2 domain inhibitors exhibit potent antileukemia activity. J Med Chem. American Chemical Society 2012 Feb 9; 55:1047–55.
60. Eiring AM, Page BDG, Kraft IL, Mason CC, Vellore NA, Resetca D, Zabriskie MS, Zhang TY, Khorashad JS, Engar AJ, Reynolds KR, Anderson DJ, Senina A, Pomicter AD, Arpin CC, Ahmad S, Heaton WL, Tantravahi SK, Todic A, Moriggl R, Wilson DJ, Baron R, O’Hare T, Gunning PT, Deininger MW. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia. 2014 Aug 19;
61. Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005 Apr 28; 352:1779–90.
62. James C, Ugo V, Le Couédic J-P, Staerk J, Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005 Apr 28; 434:1144–8.
63. Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJP, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D’Andrea A, Fröhling S, Döhner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005 Apr; 7:387–97.
64. Jeong EG, Kim MS, Nam HK, Min CK, Lee S, Chung YJ, Yoo NJ, Lee SH. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin Cancer Res. 2008 Jun 15; 14:3716–21.
65. Mullighan CG. The molecular genetic makeup of acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program. 2012 Jan 1; 2012:389–96.
66. Warsch W, Walz C, Sexl V. JAK of all trades: JAK2-STAT5 as novel therapeutic targets in BCR-ABL1+ chronic myeloid leukemia. Blood. 2013 Sep 26; 122:2167–75.
67. Hantschel O, Warsch W, Eckelhart E, Kaupe I, Grebien F, Wagner K-U, Superti-Furga G, Sexl V. BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat Chem Biol. 2012 Mar; 8:285–93.
68. Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, Nicolini FE, Müller-Tidow C, Bhatia R, Brunton VG, Koschmieder S, Holyoake TL. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012 Mar 9; 119:1501–10.
69. Hiwase DK, White DL, Powell JA, Saunders VA, Zrim SA, Frede AK, Guthridge MA, Lopez AF, D’Andrea RJ, To LB, Melo J V, Kumar S, Hughes TP. Blocking cytokine signaling along with intense Bcr-Abl kinase inhibition induces apoptosis in primary CML progenitors. Leukemia. 2010 Apr; 24:771–8.
70. Grundschober E, Hoelbl-Kovacic A, Bhagwat N, Kovacic B, Scheicher R, Eckelhart E, Kollmann K, Keller M, Grebien F, Wagner K-U, Levine RL, Sexl V. Acceleration of Bcr-Abl(+) leukemia induced by deletion of JAK2. Leukemia. 2014 May 5.
71. Akada H, Akada S, Hutchison RE, Sakamoto K, Wagner K-U, Mohi G. Critical role of jak2 in the maintenance and function of adult hematopoietic stem cells. Stem Cells. 2014 Jul; 32:1878–89.
72. Gallipoli P, Cook A, Rhodes S, Hopcroft L, Wheadon H, Whetton AD, Jørgensen HG, Bhatia R, Holyoake TL. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of chronic phase CML CD34+ cells in vitro and in vivo. Blood. 2014 Jun 23.
73. Danhauser-Riedl S, Warmuth M, Druker BJ, Emmerich B, Hallek M. Activation of Src kinases p53/56lyn and p59hck by p210bcr/abl in myeloid cells. Cancer Res. 1996 Aug 1; 56:3589–96.
74. Okutani Y, Kitanaka A, Tanaka T, Kamano H, Ohnishi H, Kubota Y, Ishida T, Takahara J. Src directly tyrosine-phosphorylates STAT5 on its activation site and is involved in erythropoietin-induced signaling pathway. Oncogene. 2001 Oct 4; 20:6643–50.
75. Klejman A, Schreiner SJ, Nieborowska-Skorska M, Slupianek A, Wilson M, Smithgall TE, Skorski T. The Src family kinase Hck couples BCR/ABL to STAT5 activation in myeloid leukemia cells. EMBO J. 2002 Nov 1; 21:5766–74.
76. Konig H, Holyoake TL, Bhatia R. Effective and selective inhibition of chronic myeloid leukemia primitive hematopoietic progenitors by the dual Src/Abl kinase inhibitor SKI-606. Blood. 2008 Mar 15; 111:2329–38.
77. Konig H, Copland M, Chu S, Jove R, Holyoake TL, Bhatia R. Effects of dasatinib on SRC kinase activity and downstream intracellular signaling in primitive chronic myelogenous leukemia hematopoietic cells. Cancer Res. 2008 Dec 1; 68:9624–33.
78. Chatain N, Ziegler P, Fahrenkamp D, Jost E, Moriggl R, Schmitz-Van de Leur H, Müller-Newen G. Src family kinases mediate cytoplasmic retention of activated STAT5 in BCR-ABL-positive cells. Oncogene. 2013 Aug 1; 32:3587–97.
79. Hantschel O, Rix U, Schmidt U, Bürckstümmer T, Kneidinger M, Schütze G, Colinge J, Bennett KL, Ellmeier W, Valent P, Superti-Furga G. The Btk tyrosine kinase is a major target of the Bcr-Abl inhibitor dasatinib. Proc Natl Acad Sci U S A. 2007 Aug 14; 104:13283–8.
80. Friedbichler K, Hoelbl A, Li G, Bunting KD, Sexl V, Gouilleux F, Moriggl R. Serine phosphorylation of the Stat5a C-terminus is a driving force for transformation. Front Biosci. 2011 Jan; 17:3043–56.
81. Friedbichler K, Kerenyi MA, Kovacic B, Li G, Hoelbl A, Yahiaoui S, Sexl V, Müllner EW, Fajmann S, Cerny-Reiterer S, Valent P, Beug H, Gouilleux F, Bunting KD, Moriggl R. Stat5a serine 725 and 779 phosphorylation is a prerequisite for hematopoietic transformation. Blood. 2010 Sep 2; 116:1548–58.
82. Bancerek J, Poss ZC, Steinparzer I, Sedlyarov V, Pfaffenwimmer T, Mikulic I, Dölken L, Strobl B, Müller M, Taatjes DJ, Kovarik P. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity. 2013 Feb 21; 38:250–62.
83. Putz EM, Gotthardt D, Hoermann G, Csiszar A, Wirth S, Berger A, Straka E, Rigler D, Wallner B, Jamieson AM, Pickl WF, Zebedin-Brandl EM, Müller M, Decker T, Sexl V. CDK8-mediated STAT1-S727 phosphorylation restrains NK cell cytotoxicity and tumor surveillance. Cell Rep. 2013 Aug 15; 4:437–44.
84. Bose P, Perkins EB, Honeycut C, Wellons MD, Stefan T, Jacobberger JW, Kontopodis E, Beumer JH, Egorin MJ, Imamura CK, Douglas Figg W, Karp JE, Koc ON, Cooper BW, Luger SM, Colevas AD, Roberts JD, Grant S. Phase I trial of the combination of flavopiridol and imatinib mesylate in patients with Bcr-Abl+ hematological malignancies. Cancer Chemother Pharmacol. 2012 Jun; 69:1657–67.
85. Martin H, Mali RS, Ma P, Chatterjee A, Ramdas B, Sims E, Munugalavadla V, Ghosh J, Mattingly RR, Visconte V, Tiu R V, Vlaar CP, Dharmawardhane S, Kapur R. Pak and Rac GTPases promote oncogenic KIT-induced neoplasms. J Clin Invest. 2013 Oct 1; 123:4449–63.
86. Thomas EK, Cancelas JA, Chae H-D, Cox AD, Keller PJ, Perrotti D, Neviani P, Druker BJ, Setchell KDR, Zheng Y, Harris CE, Williams DA. Rac guanosine triphosphatases represent integrating molecular therapeutic targets for BCR-ABL-induced myeloproliferative disease. Cancer Cell. 2007 Nov; 12:467–78.
87. Thomas EK, Cancelas JA, Zheng Y, Williams DA. Rac GTPases as key regulators of p210-BCR-ABL-dependent leukemogenesis. Leukemia. 2008 May; 22:898–904.
88. Sengupta A, Arnett J, Dunn S, Williams DA, Cancelas JA. Rac2 GTPase deficiency depletes BCR-ABL+ leukemic stem cells and progenitors in vivo. Blood. 2010 Jul 8; 116:81–4.
89. Tala I, Chen R, Hu T, Fitzpatrick ER, Williams DA, Whitehead IP. Contributions of the RhoGEF activity of p210 BCR/ABL to disease progression. Leukemia. 2013 Apr; 27:1080–9.
90. Kawashima T, Bao YC, Minoshima Y, Nomura Y, Hatori T, Hori T, Fukagawa T, Fukada T, Takahashi N, Nosaka T, Inoue M, Sato T, Kukimoto-Niino M, Shirouzu M, Yokoyama S, Kitamura T. A Rac GTPase-activating protein, MgcRacGAP, is a nuclear localizing signal-containing nuclear chaperone in the activation of STAT transcription factors. Mol Cell Biol. 2009 Apr; 29:1796–813.
91. Kawashima T, Bao YC, Nomura Y, Moon Y, Tonozuka Y, Minoshima Y, Hatori T, Tsuchiya A, Kiyono M, Nosaka T, Nakajima H, Williams DA, Kitamura T. Rac1 and a GTPase-activating protein, MgcRacGAP, are required for nuclear translocation of STAT transcription factors. J Cell Biol. 2006 Dec 18; 175:937–46.
92. Clark DE, Williams CC, Duplessis TT, Moring KL, Notwick AR, Long W, Lane WS, Beuvink I, Hynes NE, Jones FE. ERBB4/HER4 potentiates STAT5A transcriptional activity by regulating novel STAT5A serine phosphorylation events. J Biol Chem. 2005 Jun 24; 280:24175–80.
93. Yamashita H, Nevalainen MT, Xu J, LeBaron MJ, Wagner KU, Erwin RA, Harmon JM, Hennighausen L, Kirken RA, Rui H. Role of serine phosphorylation of Stat5a in prolactin-stimulatedbeta-casein gene expression. Mol Cell Endocrinol. 2001Oct 25; 183:151–63.
94. Beuvink I, Hess D, Flotow H, Hofsteenge J, Groner B, Hynes NE. Stat5a serine phosphorylation. Serine 779 is constitutively phosphorylated in the mammary gland, and serine 725 phosphorylation influences prolactin-stimulated in vitro DNA binding activity. J Biol Chem. 2000 Apr 7; 275:10247–55.
95. Moriggl R, Gouilleux-Gruart V, Jähne R, Berchtold S, Gartmann C, Liu X, Hennighausen L, Sotiropoulos A, Groner B, Gouilleux F. Deletion of the carboxyl-terminal transactivation domain of MGF-Stat5 results in sustained DNA binding and a dominant negative phenotype. Mol Cell Biol. 1996 Oct; 16:5691–700.
96. Cella N, Groner B, Hynes NE. Characterization of Stat5a and Stat5b homodimers and heterodimers and their association with the glucocortiocoid receptor in mammary cells. Mol Cell Biol. 1998 Apr; 18:1783–92.
97. Vinkemeier U. Getting the message across, STAT! Design principles of a molecular signaling circuit. J Cell Biol. 2004 Oct 25; 167:197–201.
98. Güttler T, Görlich D. Ran-dependent nuclear export mediators: a structural perspective. EMBO J. 2011 Aug 31; 30:3457–74.
99. Marg A, Shan Y, Meyer T, Meissner T, Brandenburg M, Vinkemeier U. Nucleocytoplasmic shuttling by nucleoporins Nup153 and Nup214 and CRM1-dependent nuclear export control the subcellular distribution of latent Stat1. J Cell Biol. 2004 Jun 21; 165:823–33.
100. Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011 Oct; 3:1787–808.
101. Li Q, Bohin N, Wen T, Ng V, Magee J, Chen S-C, Shannon K, Morrison SJ. Oncogenic Nras has bimodal effects on stem cells that sustainably increase competitiveness. Nature. Nature Publishing Group 2013 Dec 5; 504:143–7.
102. Montalvo-Ortiz BL, Castillo-Pichardo L, Hernández E, Humphries-Bickley T, De la Mota-Peynado A, Cubano LA, Vlaar CP, Dharmawardhane S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J Biol Chem. 2012 Apr 13; 287:13228–38.
103. Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004 May 18; 101:7618–23.
104. Mardilovich K, Olson MF, Baugh M. Targeting Rho GTPase signaling for cancer therapy. Future Oncol. 2012 Feb; 8:165–77.
105. Neculai D, Neculai AM, Verrier S, Straub K, Klumpp K, Pfitzner E, Becker S. Structure of the unphosphorylated STAT5a dimer. J Biol Chem. 2005 Dec 9; 280:40782–7.
106. Krämer OH, Moriggl R. Acetylation and sumoylation control STAT5 activation antagonistically. JAK-STAT. 2012 Jul 1; 1:203–7.
HOMEPAGES
[200] http://www.sanger.ac.uk/research/projects/cancergenome/
[201] http://clinicaltrials.gov/show/NCT01702064
[202] http://clinicaltrials.gov/show/NCT01751425