
INTRODUCTION
In adults, T lymphocytes start developing from the pluripotent stem cell, go through the stages of lymphocyte committed stem cell, pre-T cell, which migrate in the thymus and form the thymocyte, later migrating to the periphery and forming the naive T cell. This process follows three main paths to T helper cells, T cytotoxic cells and T memory cells (Figures 1 and 2). MHC complexes are recognized by the T cell receptor (TCR), these stimulated T lymphocytes that have an effect on macrophages and B cells, thus augmenting their activities. Thus, a major structures involved in T cell function is the TCR, a receptor with therapeutic potential in the clinic. TCR is formed in more than 90% of the T cells by an α and a β chain. The domains and sequences that form the TCR are (starting from the N-terminus and form the extracellular domains to the intracellular ones): a leader sequence, a variable region, a constant region, a small connecting peptide, a transmembrane domain and a cytoplasmic region [1]. TCRs functions together with other structures, with which forms the T-cell receptor complex, as seen in Figure 3. One topic of high interest is the management of the ICU patient diagnosed with a hematological malignancy, for which he is under treatment. A special emphasis in hematological patients that are treated together with the ICU team is the clinical management based on cellular therapies or immunotherapy. A state-of-the-art protocol that has seen quick development over the last years and presents a high potential in treating hematological malignancies is the CAR-T cell technology [2-4]. Murine models that assess the effects and toxicity of CAR-T cells represent important areas of research before phase I-III clinical trials, due to the potential of this technology to become tomorrow's therapeutics.
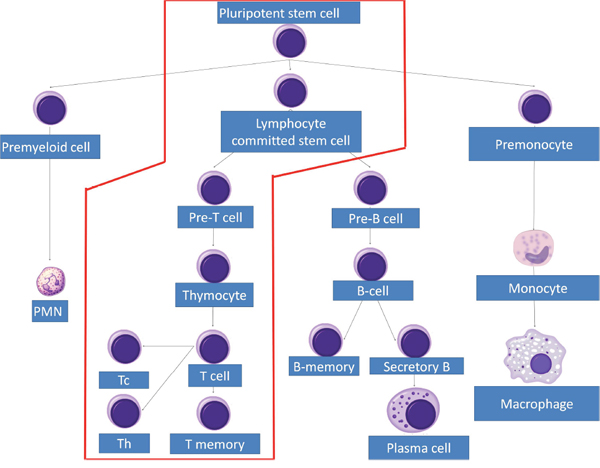
Figure 1: T cell lineage development.
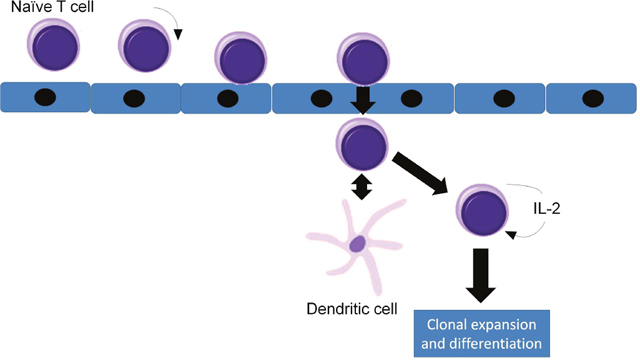
Figure 2: Circulation, rolling and adherence of naïve T cells in the high endothelial venule (HEV), their diapedesis through the HEV wall, interaction with dendritic cells and activation.
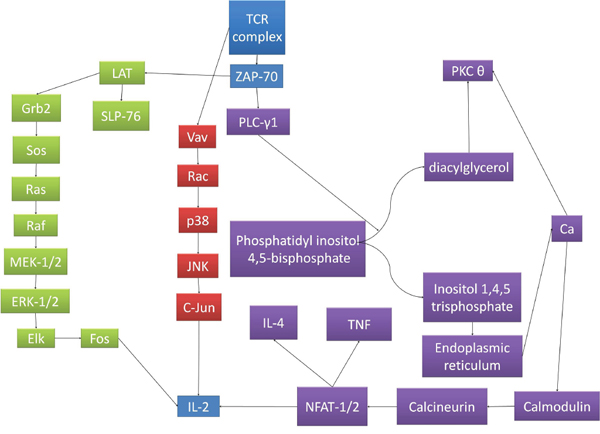
Figure 3: The signaling pathways for TCR complex activation. The pathways converge to IL-2 transcription, which determines T cell clonal expansion and subsequent immune response. TCRs functions together with other structures, with which forms the T-cell receptor complex. After the TCR binds the peptide-MHC complex, the TCR undergoes conformational changes that determine the phosphorylation of the ITAMs (immunoreceptor tyrosine-based activation motifs) located on CD247 and the CD3 polypeptides. ITAM phosphorylation creates binding sites for proteins presenting Src homology 2 (SH2) domains, one of the more important ones being the zeta associated protein of 70 kDa (ZAP-70). After binding and activation, ZAP-70 recruits linker of activation of T cells (LAT). After the binding and activation of LAT, other signaling molecules are recruited. Such is the case of SH2-binding leukocyte phosphoprotein of 76-kDa (SLP-76) and Grb2. Grb2 activates SOS, which catalyzes the exchange of GDP to GTP linked to Ras, activating the MAPK pathway, phosphorylating the extracellular receptor-activated kinase 1 and 2 (ERK1/2), which in turn, phosphorylates Elk. Thus determines the transcription of Fos, important for the transcription of interleukin-2 (IL-2). Other pathway that branches off the TCR and phosphorylated proteins complex determine the activation of Vav, which determines the GDP/GTP exchange on Rac, which in turn activates p38. This action determines, in the end, the activation of c-Jun N-terminal kinase (JNK), which phosphorylates c-Jun, representing the second molecule implicated in the transcription of IL-2.
CAR-T cells therapy basically requires drawing blood from patients and separating out the T cells. Next, by using a disarmed virus, the T cells are genetically engineered to produce receptors on their surface called chimeric antigen receptors, or CARs. The final step is the infusion of the CAR-T cells into the patient (which is preceded by a “lymphodepletion” chemotherapy regimen). The engineered cells further multiply in the patient’s body and, with guidance from their engineered receptor, recognize and kill cancer cells that harbor the antigen on their surfaces [5-9].
As many other therapeutic alternatives, CAR-T cells also have an important toxicity in the host organism. Nevertheless, the clinical response that CAR-T cells can induce is worth the risk [10]. The toxicity induced by CAR-T cells is linked to immune-mediated adverse effects, out of which some last longer than the toxicities induced by conventional pharmaceutical molecules [10, 11]. Such toxicities are mainly represented by cytokine release syndrome (CRS) and B-cell aplasia [12-16]. The expansion of CAR-T cells and the activation of lymphoid and myeloid cells determine the release of high amounts of pro-inflammatory molecules [17], with the cross-reactivity of CAR-T cells with normal tissues leading sometimes to organ damage, caused by similar expression patterns of normal tissues with the target cells [18, 19]. Furthermore, CAR-T cells and CRS can be also linked by the rise in the levels of IL-6, TNF, IL-2 and IL-8 that eventually lead to arterial hypotension and fever [20-23].
Another important protocol in lymphocyte engineering is the use of DLI for cytomegalovirus (CMV) reactivation, as well as other disorders related to immune deficiency. DLI may also improve the graft-versus-tumor (GVT) or graft-versus-leukemia effect of an allogeneic stem cell transplantation [24, 25]. In the current paper we aim to approach the ICU patient that presents following a diagnosis with a hematological disease and treated mainly through the use of CAR-T cells, DLI, as well as similar novel technologies.
Design of CAR-T cells
CAR-T cells are produced by a multistep process that implicates primary T cells harvesting, modification and then use. Primary T cells are harvested from a patient’s peripheral blood and enriched. Their enhancement is possible by the use of lentiviral vectors [26-29]. These vectors integrate in the host genome and determine the expression of the CAR construct. One problem that can occur is that the lentiviral capsid presents a natural tropism against CD4+ cells. The solution comes from pseudotyping the lentiviral capsid with viral glycoproteins, the most common used being the vesicular stomatitis virus glycoprotein (VSV-G). However, VSV-G is not suitable for transfection of B and T lymphocytes. Thus, other glycoproteins have been used for this matter, as is the case of the measles virus, hemagglutinin and fusion glycoproteins [30, 31].
Although CAR-T cells have presented great promise for clinical applications, there are two main problems that can arise: CRS and in the case of anti-CD19 CAR-T cells, B-cell aplasia and subsequent immunodepression [23, 32-34]. One of the solutions used in this regard is the use of switch molecules, like rimiducid, to control CAR-T cells activity [35-37]. Another approach for controlling CAR-T cells activity is represented by using suicide genes in the CAR construct [38].
Other concerns regarding CAR-T cells is the potential of lentiviral vectors to generate insertional mutagenesis [39, 40]. Efforts have been made in the scientific community to generate integration-deficient lentiviral vectors and the addition to those of a scaffold/matrix associated region, so the vector and the CAR construct can persist through subsequent cell divisions as an episome [41]. In addition to lentiviral vectors, there are two more approaches that can be used: transposon and the CRISPR/Cas9 system. Transposons have already been used in clinical trials and their insertion in the human genome has not been associated with any disease [42-45]. Nevertheless, if transposons are to be used, a transposase should also be expressed in the cell, either from the same construct or from a different construct, which affects the experimental setup and add more complexity. Inserting the CAR construct in a T lymphocyte by using the CRISPR/Cas9 system is realized from an endonuclease guided by an RNA, each formed from crRNA (the sequence for recognition of the specific part of the genome) and transcrRNA (the sequence that is recognized and bound by the Cas9 so it can be guided at the specific site). This system generates a double stranded break in the host genome and, through, non-homologous end joining the CAR construct can be inserted [46, 47].
Despite CAR-T cell therapy have shown promise in both the preclinical setting, as well as in clinical trials, various drawbacks have been described due to the rapid expansion. One such side-effects is the severe cytokine release after the antigen recognition [32], as well the inability of anti-CD19 CARs to tell the difference between normal and malignant B lymphocytes. This will lead to the development of a long-term B-cell aplasia [23]. Thus, a control of CAR T cell function may be very important due to a reduction of side-effects that might potentially be life-threatening. To overcome this endeavor, various research groups have used antibody-based switch molecules that are designed to control the immunological synapse between a CAR and a malignant cell. This leads to a highly controlled cytotoxic activity, as well as to increased specificity for cancer cells [48, 49].
Another safety concern is related to insertional mutagenesis potential of integrating vectors. Although an important step was made by switching from onco-retroviral vectors to lentiviral vectors, that are considered as a safer alternative to the former ones due to a relative random insertional pattern. However, the oncogenic potential of lentiviral vectors has been previously reported [40, 50] and this might raise safety issues regarding the use of integrating vectors. Towards this end, efforts have been made to reduce the insertional mutagenesis potential of delivery vectors for CAR into T-cells. Generation of integration-deficient lentiviral vectors and inclusion of a scaffold/matrix associated region (S/MAR) in the vector backbone displayed comparable cytotoxic effect of CAR-T cells engineered with non-integrating vectors to those that have the integration function unaffected [51]. Non-integrating vectors due to the presence of S/MAR element in their design are maintained in subsequent cell generation as an episome.
An alternative to lentiviral vectors could be represented by transposons, as they have been described as efficient gene delivery vectors and has been used for gene therapy applications in clinical trials [42, 44], 45]. DNA transposons have been used as gene delivery vehicles instead of retro-transposons because their genomic insertions have not been associated with any human disease [52] (Figure 4). However delivery of the transgene is mediated by an encoding transposase that must be provided in trans from the same construct or a second construct and this might add an extra level of complexity to the experimental setup.
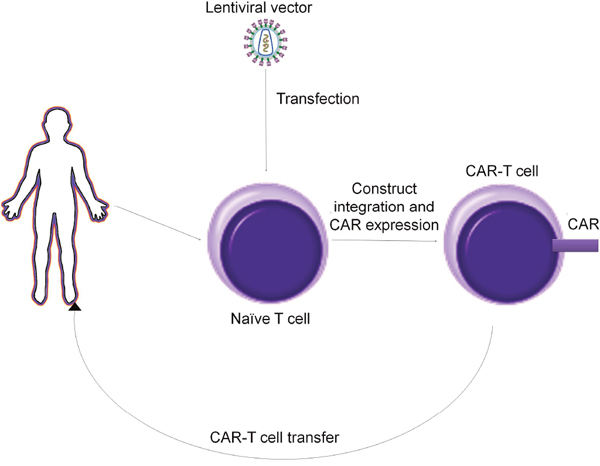
Figure 4: Production alghoritm of CAR T cells.
Yet, another alternative to both viral and non-viral delivery could be represented by the newly descried gene editing tool, named CRISPR/Cas9. This technology offers the possibility to target virtually any genomic site in a RNA-guided manner. The editing complex futures the Cas9 nuclease and a guide RNA, comprised of a CRISPR RNA (crRNA) and a trans-acting crRNA (tracrRNA). Upon hybridization of the crRNA to the target sequence, Cas9 generates a double-strand break (DBS), that can be repaired by non-homologous end joining (NHEJ), an event that can result in a loss-of-function of the genomic locus. In the presence of a donor DNA, by a mechanism of homology-directed recombination, an exogenous sequence can be introduced in the targeted locus [46, 47]. This knock-in capability of CRISPR/Cas9 can be exploited to deliver CAR expression cassette in a desired genomic locus that does not interfere with gene function and therefore minimizing the genotoxic effects experienced with integrating viral vectors. And recent improvements in gRNA and Cas9 have reduced the off-target effects to a minimum, increasing the chances of CRISPR/Cas9 to reach clinical applicability. Up to date, CRISPR/Cas9 already proved its applicability in the field of immunotherapy by enhancing CAR-T cells potency by knock-out diverse genes with importance in target recognition and cytotoxic activity [53]. Therefore, CRISPR/Cas9 will surly make a difference in advancing immunotherapies for malignant disorders, in both hematological and solid cancers. However further improvements in delivery systems are still to be made, and stated above, designing more specific and regulated systems are desirable to achieve a controlled activity of CAR-T cells.
In present-day medicine, four generations of CAR-T cells are described, each presenting a chimeric activated receptor with common regions among them. Starting from the exterior of the cell to the cytoplasm these regions are: single-chain variable fragment (scFv), the hinge region, the transmembrane domain and the CD3ζ intracellular domain of the T cell receptor [54, 55]. The first generation of CAR-T cells presented only these regions and showed good results in vitro, but did not present efficiency in vivo because of the lack of costimulatory signals [56-59]. By trying to overcome these limitations, further generations of CAR-T cells have been developed. The second generation of CAR-T cells present, in addition to the basic construct a costimulatory domain, such as CD28 or 41BB (CD137) close to the CD3ζ domain, which are both associated with clonal expansion and survival of T cells in their activated state [60-62]. The third generation of CAR-T cells can be generated by the addition to the second generation CAR of other costimulatory regions, like CD27, ICOS or OX40 (CD134), which can further improve cell survival [63, 64]. The fourth generation of CAR-T cells (also called TRUCKS) can be built using any of the first three generations and by the addition of a promoter that can be regulated, thus putting CAR-T cell activity under the practitioner’s control [54].
CAR-T cells-based and DLI therapy in the intensive care unit
Indications of using CAR-T cells therapy are acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia and non-Hodgkin lymphoma. CAR-T cell therapies are also being developed for solid tumors but studies are being in the early stages. Still, the first steps in investigating the side-effects of CAR T cells are represented by the use of murine models of the therapy. One of the first documented adverse reactions on CAR T cell therapy in preclinical murine models is the cytokine release syndrome (CRS). It has been shown in a murine model that CAR T-cell infusion associated CRS can be prevented through the administration of the kinase inhibitor ibrutinib [16]. To the present date, graft versus host disease (GVHD) is not a real concern regarding CAR T-Cell therapy side effects [65]. In two clinical reports, patients that underwent allogeneic hematopoietic stem cell transplant (allo HSCT) also received infusions of anti CD19 CAR allogeneic T cells from their initial transplant donors. The first report did not identify any GVHD in any of the eight transplanted patients [66], while the second report showed that one out of twenty patients developed a worsening of a pre-existing chronic GVHD [67].
Across the large variety and number of preclinical publications focusing on CAR T cells, very few of them document toxicity in animal models as it would seem normal with any new compound that has a potential use in a clinical setting. Paradoxically, there are numerous studies reporting the clinical use of CAR T cells even though their safety has not yet been evaluated extensively in vivo. An explanation for this phenomena could include factors like the large variety of engineered CAR cells, the differences between mouse and human physiology and T-Cell biology and the differences in drug metabolism capacity in each species. An example which would confirm this hypotheses would be the fact that one in vivo study involves CAR T cells targeting the Her2/neu antigen, proving the antineoplastic activity and the biological safety of Her2/neu-specific CAR T cells in transgenic animals with lymphodepletion [68], yet the clinical trial involving the same engineered cells showed that one of the patients died due to a massive cytokine release syndrome [69]. The majority of preclinical studies investigating CAR T cells have focused on verifying their specificity and potency for antineoplastic activity, the key advantage of CARs in vivo being the fact that they possess the ability to redirect T-cell effector function without HLA-restriction. The in vivo testing of CARs expresses several drawbacks. First of all the successful engraftment of T-cells in immunocompromised mice is hard to achieve due to the residual elements of the mouse’s innate immune system; another drawback is the fact that even if the engraftment is successful, most of the mice develop GVHD in long term studies (more than 60 days) [70]. CAR T-Cells target human antigens which are restricted to transplanted tumor cells in mice, rendering the assessment of their effects on healthy tissues in mice models hard to achieve [71]. The humanized NSG mouse has been an indispensable tool for evaluating short-term CAR T cell activity in vivo. CARs that act against ROR1 for mantle cell lymphoma and CD44v6 for acute myeloid leukemia and multiple myeloma have been tested in humanized NSG mice extensively [72, 73]. Humanized mice have been also used to assess the function and efficacy of co-stimulatory domains like CD27, ICOS, CD28 and 4-1BB, due to their potential enhanced efficiency in targeting malignancies and augmenting CARs safety [74, 75]. In a humanized animal model, the Hu-PBL-SCID NSG mouse, engineered T cells showed the ability to destroy a cancer cell line that expressed prostate tumor antigens [76]. Modern genetic engineering methods like messenger RNA transduction have been used to generate CAR NK-Cells and to successful target a non-Hodgkin's lymphoma in a Hu-PBL-SCID NSG model. The study confirmed that activated expanded PBNK became highly cytolytic, eradicating resistant CD20+ B-leukemia/lymphoma after nucleofection with anti-CD20 CAR messenger RNA [77]. Najima et al have successfully transplanted WT-1 specific TCR transduced human HSCs into class I matched transgenic NSG mice. The WT-1 tetramer positive T cells differentiated in the thymus and the splenic cytotoxic lymphocytes of the mice targeted leukemia cells in an antigen-specific HLA restricted manner and destroyed them [78].
Even if current mouse models for CAR T cells have a poor predictive nature, these may relate to the biological differences between species, a barrier which could be overcome by developing new humanized mice models. Studies in the last decade have focused mainly on their clinical applications with toxicity being neglected as a main research aim. Most of the clinical studies report toxic effects on these engineered cells which in turn will cause a stronger will of the researchers to better understand the potential mechanisms of in vivo toxicity by developing better animal models to this purpose.
Cytokine capture system (CCS) is a protocol for isolating different cell populations, stimulating and using them further on. However, the manual method of CCS is time consuming, requiring 10 to 12 hours and has to be undertaken by a skilled operator. These problems have been solved by Prodigy (Miltenyi Biotech), an apparatus that can perform this operation in a closed environment and within 2 hours. The automated protocol consists in four main steps: preparation of materials under sterile conditions, preparation and use of automated cell enrichment system, cell count determination and examination of the separation performance; these steps are described in more detail by Kumaresan et al [79]. The use of this technique, and thus of Prodigy, has been studied in the field of hematopoietic stem cell transplantation (HSCT), specifically offering an infusion of selected T cells which have an effect against opportunistic infections and also improve the GVT effect [80, 81]. A good example of using this assay is the treatment of opportunistic CMV infections in patients that have underwent an allogeneic HSCT. This can be done by incubating overlapping peptides from the CMV pp65 antigen with total nuclear cells provided by CMV positive donors. The pp65 peptide fragments stimulate the T cells to secrete interferon-γ (IFN-γ), after which, antigen-specific T-cells can be selected, using reagents that can recognize CD45 and IFN-γ [82-84].
DLI represents a form of adoptive therapy used after allogeneic stem cell transplant for its anti-tumor and anti-infectious properties and it has been used to restore the patient’s immune function, thus aiding in the prophylaxis or treatment of relapse, in preventing infectious diseases (as is the case of CMV infection) and to establish full donor chimerism [85-87]. The start of the idea of GVT activity was observed after a flare of graft versus host disease (GVHD) or after sudden discontinuation of immunosuppression [88, 89]. GVT can be confirmed by the fact that T cell-depleted graft transplantation are associated with a higher risk of relapse [90-92]. Following these observations, DLI was tested for its antileukemic effects. The first described application was in 1990 by Horowitz et al, who described three patients treated for chronic myelogenous leukemia (CML) with matched-sibling allogeneic HSCT that have relapsed with chronic-phase CML and were treated for the relapse with DLI. The results showed a complete cytogenetic remission in all three patients. Further studies were done for the use of DLI against CML relapse and the majority of the studies have presented long term molecular remission. DLI has been studied as a therapeutic tool in other hematological malignancies, but it’s efficiency was not as good as with CML [93-97].
The complications that occur after DLI include, more notably GVHD and marrow aplasia. GVHD in these patients has been correlated with a GVT effect, although some studies contradict these results [93, 95, 98-101]. Although progress has been made in predicting GVHD, the influence of DLI on these effects remains poorly defined. In this direction, studies working on the hypothesis that lower doses or escalating doses of T cells can minimize GVHD, while still maintaining a significant level of GVT activity, have been made [102-104]. Even if the advantage of unrelated donor grafts depleted of T cells is obvious through the lower incidence of GVHD, there are also marked disadvantages to this approach, one of them being represented by the increased number of opportunistic infections CMV and Epstein-Barr virus infections, but not only [105-111]. Naturally, several studies have been devised to research the application of DLI in preventing or curing these infections [110-114]. A study on 462 recipients of bone marrow transplant from unrelated donors showed that infections were the cause of 30% of deaths, compared to 14% in the case of disease recurrence [115]. Other reports showed that infections accounted for 38 to 75% of the cases of death compared to 8 to 25% in the case of leukemia relapse [108, 109, 116-118]. One study, in particular, has analyzed the immune reconstitution after unrelated HSCT, in this report is has been show that T cell depleted HSCT, is associated with prolonged T cell lymphopenia and CD4 lymphopenia [119-122].
Other refinements or reconfigurations of CAR-T cells are being tested. One approach is the development of CAR-T cells therapies that use immune cells collected not from patients, but from healthy donors. The idea is to create so-called off-the-shelf CAR-T cells therapies that are immediately available for use and don’t have to be manufactured for each patient.
Although adoptive transfer of CAR-T cells is a unique and promising cancer therapeutic, there are significant safety concerns. Clinical trials of this therapy have revealed potential toxic effects of these CARs when healthy tissues express the same target antigens as the tumor cells, leading to outcomes similar to GVHD [65, 123-125]. A potential solution to this problem is engineering a suicide gene into the modified T cells. In this way, administration of a prodrug designed to activate the suicide gene during GVHD triggers apoptosis in the suicide gene-activated CAR-T cells. This method has been used safely and effectively in HSCT. Adoption of suicide gene therapy to the clinical application of CAR-T cells adoptive cell transfer has potential to alleviate GVHD while improving overall anti-tumor efficacy [66, 67, 126].
Early case reports of unexpected organ damage and deaths following CAR-T cells therapy first highlighted the possible dangers of this new treatment. CAR-T cells can potentially damage normal tissues by specifically targeting a tumor-associated antigen that is also expressed on those tissues.
Complications that require ICU admission for the CAR-T cells-treated patient
The main complications for patients that receive a CAR-T cells-based therapy are the CRS and B-cell aplasia, out of which CRS is one of the most severe, requiring ICU admission and treatment. It ranges from mild to life threatening and it is an oncologic emergency. CRS can be observed following the previous administration of immune-based therapy drugs, as is the case of rituximab or other monoclonal antibodies [127, 128]. This condition appears due to a massive release of cytokines (high levels of IL-6 and IL-12) into the bloodstream, followed clinically by high fever and a sudden fall in blood pressure, tachycardia, as well as hemophagocytic lymphohistiocytosis or macrophage-activation syndrome [129], even multi-organ failure with potential fatal outcome [130, 131]. IL-6 is important in neutrophil trafficking, acute phase response, B-cell differentiation, angiogenesis and the production of autoantibodies and is produced by dendritic cells, monocytes, T cells, keratinocytes and fibroblasts. IL-10 also plays an important role, being produced by monocytes/macrophages and it regulates both cell-mediated and innate immunity after the inhibition of the activated macrophages. This interleukin is synthethised by B cells, mastocytes and T helper cells, but not by cytotoxic T cells, thus making it not the ideal target cytokine for treatment options in CRS. Other molecules involved in CRS are the TNF, IL-8 and IL-2, reported in patients treated with CAR-T cells and blinatumomab [132-135].
In a patient with newly developed fever, which is often the first sign, CRS is diagnosed as the day with the first fever over 38 °C related to the infusion of CAR-T cells and the recovery from CRS is diagnosed as 24 hours without fever or vasoactive medication [131, 136, 137]. The clinical management of CRS is oriented according to the grade, as presented in Table 1.
Table 1: Clinical grading of cytokine release syndrome
Grade |
Clinical symptoms / Therapy |
|---|---|
Grade 1 |
Not life-threatening symptoms. Only symptomatic therapy is advised – intravenous fluids, antipyretics |
Grade 2 |
Symptoms that require moderate intervention. Oxygen requirement <40% or arterial hypotension treated with fluids or low dose vasopressor or grade 2 organ toxicity |
Grade 3 |
Symptoms that require aggressive intervention. Oxygen requirement >40% or arterial hypotension treated high dose vasopressors or grade 3 organ toxicity or grade 4 transaminitis |
Grade 4 |
Life-threatening symptoms, that require ventilator support or grade 4 organ toxicity, with the exception of transaminitis |
Grade 5 |
Death of the patient |
Cytokine elevations are measurable in most patients and a CRS approach may be by targeting elevated cytokine directly with anti-cytokine directed therapies, as is the case of tocilizumab. This drug is a humanized monoclonal antibody directed against the IL-6 receptor and its use has lead to a dramatic improvement of severe CRS for patients treated with CAR-T cells or blinatumomab (a drug used for Philadelphia chromosome-negative relapsed or refractory acute lymphoblastic leukemia). Still, tocilizumab comes with side-effects such as elevated liver enzymes and cytopenias. IL-6 is a potential target for other new inhibitors, as is the case of siltuximab and the IL-6 trans-signalling blocker sgp130Fc. These small molecules are interesting at this point, but further phase I-III clinical trials are required [138-140].
The use of corticosteroids represents an obvious solution for CRS because they have proven their efficacy in inhibiting activated T cells, as is the case of GVHD. The major drawback that limits their use could be the potential to negatively affect the antitumor effects of the CAR-T cells. Corticosteroids have shown a partial response in a patient who received corticosteroids early after the infusion of CAR-T cells [132, 141, 142]. Brentjens et al have used short-term steroids for the therapy of severe CRS without compromising CAR-T cell proliferation and efficiency. At least in vitro, the steroids take down the cytokine levels without the reduction of T-cell activation, but in vivo the effects may be totally different. Thus, the use of corticosteroids should be reserved for neurological symptoms and CRS unresponsive to tocilizumab [71, 143-145].
Steroids still represents the basic therapy for blinatumomab-induced CRS, as the Food and Drug Administration (FDA) recommends pretreatment with 20 mg of intravenous dexamethasone before the first dose of blinatumomab, as well as before each intracycle dose escalation, as well as when restarting an infusion after the previous interruption of therapy.
CRS management should involve the entire healthcare professional team, not only the hematologist or the ICU doctor, but also the nursing team. Nurses must thus be familiar with the toxicity profile, the type of infusion and with the protocols used in monitoring the vital signs for any indication of a severe reaction [146-148].
Taking all this into consideration, patients receiving CAR-T cell therapies should have limited comorbidities so that they are able to tolerate potentially severe CRS.
CONCLUSIONS
T lymphocytes play an important role in the treatment of cancer. T cells behavior are influenced by the T-cell receptors complex and calcium signaling. CAR-T cells and DLI are novel diagnostic and treatment technologies for hematological malignancies in intensive care units. CAR-T cells are classified as first, second, third and fourth generation cells, depending on the intracellular signaling domain number of T cell receptors. Clinical trials utilizing the first generation CAR-T cells have failed to exhibit significant clinical benefits because lack of costimulatory signals so further generations of CAR-T cells have been developed by trying to overcome this limitation. Although CAR-T cells have presented great promise for clinical applications, there are significant safety concerns. The main complications are the cytokine release syndrome and B-cells aplasia. Another important protocol in lymphocyte engineering is the use of DLI which represents a form of adoptive therapy used after stem cell transplant for its anti-tumor and anti-infectious, thus aiding in the prophylaxis or treatment of relapse, in preventing infectious diseases and to establish full donor chimerism. The complications that occur after DLI include GVHD and marrow aplasia. Preventive strategies which could include using predictive biomarkers for predicting which patients will become critically ill should be researched.
Further studies are necessary to establish clear guidelines for treating hematological malignancies with these therapies and a better collaboration between hematologists and intensive care unit doctors.
Abbreviations
CAR-T cells: chimeric antigen receptor-T cells; DLI: donor lymphocyte infusion; ICU: intensive care unit; CRS: cytokine release syndrome; GVT: graft-versus-tumor; GVHD: graft versus host disease.
ACKNOWLEDGMENTS
Tiberiu Tat, Huming Li and Catalin Constantinescu contributed equally to the current paper. The research on CAR-T cells was funded by an internal grant of the Iuliu Hatieganu University, awarded to Tiberiu Tat, as well as by a National Grant of the Romanian Government, for postdoctoral research, awarded to Delia Dima.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Garcia KC, Teyton L, Wilson IA. Structural basis of T cell recognition. Annu Rev Immunol. 1999; 17:369–97. https://doi.org/10.1146/annurev.immunol.17.1.369.
2. Yeku O, Li X, Brentjens RJ. Adoptive T-Cell Therapy for Solid Tumors. Am Soc Clin Oncol Educ Book. 2017;37:193-204. https://doi.org/10.14694/EDBK_180328.
3. Pan J, Yang JF, Deng BP, Zhao XJ, Zhang X, Lin YH, Wu YN, Deng ZL, Zhang YL, Liu SH, Wu T, Lu PH, Lu DP, et al. High efficacy and safety of low-dose CD19-directed CAR-T cell therapy in 51 refractory or relapsed B acute lymphoblastic leukemia patients. Leukemia. 2017; 31:2587–93. https://doi.org/10.1038/leu.2017.145.
4. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, Bleakley M, Brown C, Mgebroff S, Kelly-Spratt KS, Hoglund V, Lindgren C, Oron AP, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017; 129:3322-3331. https://doi.org/10.1182/blood-2017-02-769208.
5. Frey N. The what, when and how of CAR T cell therapy for ALL. Best Pract Res Clin Haematol. 2017; 30:275–81. https://doi.org/10.1016/j.beha.2017.07.009.
6. Frey NV, Porter DL. CAR T-cells merge into the fast lane of cancer care. Am J Hematol. 2016; 91:146–50. https://doi.org/10.1002/ajh.24238.
7. Liu B, Song Y, Liu D. Clinical trials of CAR-T cells in China. J Hematol Oncol. 2017; 10:166. https://doi.org/10.1186/s13045-017-0535-7.
8. Golubovskaya V. CAR-T cell therapy: from the bench to the bedside. Cancers (Basel). 2017; 9:9. https://doi.org/10.3390/cancers9110150.
9. Avanzi MP, Brentjens RJ. Emerging role of CAR T cells in non-Hodgkin’s Lymphoma. J Natl Compr Canc Netw. 2017; 15:1429–37. https://doi.org/10.6004/jnccn.2017.7045.
10. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016; 3:16011. https://doi.org/10.1038/mto.2016.11.
11. Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, Vogel AN, Kalos M, Riley JL, Deeks SG, Mitsuyasu RT, Bernstein WB, Aronson NE, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012; 4:132ra53. https://doi.org/10.1126/scitranslmed.3003761.
12. Luskin MR, DeAngelo DJ. Chimeric antigen receptor therapy in acute lymphoblastic leukemia clinical practice. Curr Hematol Malig Rep. 2017; 12:370–79. https://doi.org/10.1007/s11899-017-0394-x.
13. Rossig C, Pule M, Altvater B, Saiagh S, Wright G, Ghorashian S, Clifton-Hadley L, Champion K, Sattar Z, Popova B, Hackshaw A, Smith P, Roberts T, et al. Vaccination to improve the persistence of CD19CAR gene-modified T cells in relapsed pediatric acute lymphoblastic leukemia. Leukemia. 2017; 31:1087–95. https://doi.org/10.1038/leu.2017.39.
14. Frey NV, Porter DL. The Promise of Chimeric Antigen Receptor T-Cell Therapy. Oncology (Williston Park). 2016; 30:880-8, 890.
15. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, Grupp SA, Mackall CL. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014; 124:188–95. https://doi.org/10.1182/blood-2014-05-552729.
16. Ruella M, Kenderian SS, Shestova O, Klichinsky M, Melenhorst JJ, Wasik MA, Lacey SF, June CH, Gill S. Kinase inhibitor ibrutinib to prevent cytokine-release syndrome after anti-CD19 chimeric antigen receptor T cells for B-cell neoplasms. Leukemia. 2017; 31:246–48. https://doi.org/10.1038/leu.2016.262.
17. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016; 127:3321–30. https://doi.org/10.1182/blood-2016-04-703751.
18. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, Binder-Scholl GK, Smethurst DP, Gerry AB, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013; 122:863–71. https://doi.org/10.1182/blood-2013-03-490565.
19. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, Phan GQ, Hughes MS, Kammula US, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013; 36:133–51. https://doi.org/10.1097/CJI.0b013e3182829903.
20. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V, Ambrose D, Grupp SA, Chew A, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015; 7:303ra139. https://doi.org/10.1126/scitranslmed.aac5415.
21. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, Robinson E, Steevens NN, Chaney C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016; 126:2123–38. https://doi.org/10.1172/JCI85309.
22. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014; 371:1507–17. https://doi.org/10.1056/NEJMoa1407222.
23. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, Steinberg SM, Stroncek D, Tschernia N, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015; 385:517–28. https://doi.org/10.1016/S0140-6736(14)61403-3.
24. Stamouli M, Gkirkas K, Tsirigotis P. Strategies for improving the efficacy of donor lymphocyte infusion following stem cell transplantation. Immunotherapy. 2016; 8:57–68. https://doi.org/10.2217/imt.15.100.
25. Yanagisawa R, Nakazawa Y, Sakashita K, Saito S, Tanaka M, Shiohara M, Shimodaira S, Koike K. Intrathecal donor lymphocyte infusion for isolated leukemia relapse in the central nervous system following allogeneic stem cell transplantation: a case report and literature review. Int J Hematol. 2016; 103:107–11. https://doi.org/10.1007/s12185-015-1902-1.
26. Qian L, Li D, Ma L, He T, Qi F, Shen J, Lu XA. The novel anti-CD19 chimeric antigen receptors with humanized scFv (single-chain variable fragment) trigger leukemia cell killing. Cell Immunol. 2016; 304-305:49–54. https://doi.org/10.1016/j.cellimm.2016.03.003.
27. Morgan RA, Boyerinas B. Genetic Modification of T Cells. Biomedicines; 2016; 4. https://doi.org/10.3390/biomedicines4020009.
28. Maude S, Barrett DM. Current status of chimeric antigen receptor therapy for haematological malignancies. Br J Haematol. 2016; 172:11–22. https://doi.org/10.1111/bjh.13792.
29. Li S, Yang Z, Shen J, Shan J, Qian C. Adoptive therapy with CAR redirected T cells for hematological malignancies. Sci China Life Sci. 2016; 59:370–78. https://doi.org/10.1007/s11427-016-5036-3.
30. Frecha C, Levy C, Cosset FL, Verhoeyen E. Advances in the field of lentivector-based transduction of T and B lymphocytes for gene therapy. Mol Ther. 2010; 18:1748-57. https://doi.org/10.1038/mt.2010.178.
31. Lévy C, Frecha C, Costa C, Rachinel N, Salles G, Cosset FL, Verhoeyen E. Lentiviral vectors and transduction of human cancer B cells. Blood. 2010; 116:498–500. author reply https://doi.org/10.1182/blood-2010-03-276014.
32. Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther. 2010; 18:666-8. https://doi.org/10.1038/mt.2010.31.
33. Junghans RP. Is it safer CARs that we need, or safer rules of the road? Mol Ther. 2010; 18:1742-3. https://doi.org/10.1038/mt.2010.162.
34. Holohan DR, Lee JC, Bluestone JA. Shifting the evolving CAR T cell platform into higher gear. Cancer Cell. 2015; 28:401–02. https://doi.org/10.1016/j.ccell.2015.09.014.
35. Nellan A, Lee DW. Paving the road ahead for CD19 CAR T-cell therapy. Curr Opin Hematol. 2015; 22:516–20. https://doi.org/10.1097/MOH.0000000000000182.
36. Kim MS, Ma JS, Yun H, Cao Y, Kim JY, Chi V, Wang D, Woods A, Sherwood L, Caballero D, Gonzalez J, Schultz PG, Young TS, Kim CH. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J Am Chem Soc. 2015; 137:2832–35. https://doi.org/10.1021/jacs.5b00106.
37. Caruana I, Weber G, Ballard BC, Wood MS, Savoldo B, Dotti G. K562-Derived Whole-Cell Vaccine Enhances Antitumor Responses of CAR-Redirected Virus-Specific Cytotoxic T Lymphocytes
38. Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. 2014; 5:235. https://doi.org/10.3389/fphar.2014.00235.
39. Ranzani M, Annunziato S, Adams DJ, Montini E. Cancer gene discovery: exploiting insertional mutagenesis. Mol Cancer Res. 2013; 11:1141–58. https://doi.org/10.1158/1541-7786.MCR-13-0244.
40. Wei Y, Tsang CK, Zheng XF. Mechanisms of regulation of RNA polymerase III-dependent transcription by TORC1. EMBO J. 2009; 28:2220–30. https://doi.org/10.1038/emboj.2009.179.
41. Jin C, Yu D, Essand M. Prospects to improve chimeric antigen receptor T-cell therapy for solid tumors. Immunotherapy. 2016; 8:1355–61. https://doi.org/10.2217/imt-2016-0125.
42. Aronovich EL, Bell JB, Belur LR, Gunther R, Koniar B, Erickson DC, Schachern PA, Matise I, McIvor RS, Whitley CB, Hackett PB. Prolonged expression of a lysosomal enzyme in mouse liver after Sleeping Beauty transposon-mediated gene delivery: implications for non-viral gene therapy of mucopolysaccharidoses. J Gene Med. 2007; 9:403–15. https://doi.org/10.1002/jgm.1028.
43. Bell JB, Podetz-Pedersen KM, Aronovich EL, Belur LR, McIvor RS, Hackett PB. Preferential delivery of the Sleeping Beauty transposon system to livers of mice by hydrodynamic injection. Nat Protoc. 2007; 2:3153–65. https://doi.org/10.1038/nprot.2007.471.
44. Belur LR, Frandsen JL, Dupuy AJ, Ingbar DH, Largaespada DA, Hackett PB, McIvor RS. Gene insertion and long-term expression in lung mediated by the Sleeping Beauty transposon system. Mol Ther. 2003; 8:501-7. https://doi.org/10.1016/S1525-0016(03)00211-9.
45. Hudecek M, Izsvák Z, Johnen S, Renner M, Thumann G, Ivics Z. Going non-viral: the Sleeping Beauty transposon system breaks on through to the clinical side. Crit Rev Biochem Mol Biol. 2017; 52:355–80. https://doi.org/10.1080/10409238.2017.1304354.
46. Chira S, Gulei D, Hajitou A, Zimta AA, Cordelier P, Berindan-Neagoe I. CRISPR/Cas9: transcending the reality of genome editing. Mol Ther Nucleic Acids. 2017; 7:211–22. https://doi.org/10.1016/j.omtn.2017.04.001.
47. Sánchez-Rivera FJ, Jacks T. Applications of the CRISPR-Cas9 system in cancer biology. Nat Rev Cancer. 2015; 15:387–95. https://doi.org/10.1038/nrc3950.
48. Ma JS, Kim JY, Kazane SA, Choi SH, Yun HY, Kim MS, Rodgers DT, Pugh HM, Singer O, Sun SB, Fonslow BR, Kochenderfer JN, Wright TM, et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci USA. 2016; 113:E450–58. https://doi.org/10.1073/pnas.1524193113.
49. Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, Schulman A, Du J, Wang F, Singer O, Ma J, Nunez V, Shen J, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci USA. 2016; 113:E459–68. https://doi.org/10.1073/pnas.1524155113.
50. Ranzani M, Cesana D, Bartholomae CC, Sanvito F, Pala M, Benedicenti F, Gallina P, Sergi LS, Merella S, Bulfone A, Doglioni C, von Kalle C, Kim YJ, et al. Lentiviral vector-based insertional mutagenesis identifies genes associated with liver cancer. Nat Methods. 2013; 10:155–61. https://doi.org/10.1038/nmeth.2331.
51. Jin C, Fotaki G, Ramachandran M, Nilsson B, Essand M, Yu D. Safe engineering of CAR T cells for adoptive cell therapy of cancer using long-term episomal gene transfer. EMBO Mol Med. 2016; 8:702–11. https://doi.org/10.15252/emmm.201505869.
52. Belancio VP, Hedges DJ, Deininger P. Mammalian non-LTR retrotransposons: for better or worse, in sickness and in health. Genome Res. 2008; 18:343–58. https://doi.org/10.1101/gr.5558208.
53. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, Marson A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017; 7:737. https://doi.org/10.1038/s41598-017-00462-8.
54. Kalaitsidou M, Kueberuwa G, Schütt A, Gilham DE. CAR T-cell therapy: toxicity and the relevance of preclinical models. Immunotherapy. 2015; 7:487–97. https://doi.org/10.2217/imt.14.123.
55. Abate-Daga D, Davila ML. CAR models: next-generation CAR modifications for enhanced T-cell function. Mol Ther Oncolytics. 2016; 3:16014. https://doi.org/10.1038/mto.2016.14.
56. Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J Gene Med. 2012; 14:405–15. https://doi.org/10.1002/jgm.2604.
57. Jena B, Moyes JS, Huls H, Cooper LJ. Driving CAR-based T-cell therapy to success. Curr Hematol Malig Rep. 2014; 9:50–56. https://doi.org/10.1007/s11899-013-0197-7.
58. Urba WJ, Longo DL. Redirecting T cells. N Engl J Med. 2011; 365:754–57. https://doi.org/10.1056/NEJMe1106965.
59. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011; 365:725–33. https://doi.org/10.1056/NEJMoa1103849.
60. Gill S, Maus MV, Porter DL. Chimeric antigen receptor T cell therapy: 25years in the making. Blood Rev. 2016; 30:157–67. https://doi.org/10.1016/j.blre.2015.10.003.
61. Mato A, Porter DL. A drive through cellular therapy for CLL in 2015: allogeneic cell transplantation and CARs. Blood. 2015; 126:478–85. https://doi.org/10.1182/blood-2015-03-585091.
62. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, Liu H, Grilley B, Rooney CM, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011; 121:1822–26. https://doi.org/10.1172/JCI46110.
63. Barrett DM, Singh N, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Relation of clinical culture method to T-cell memory status and efficacy in xenograft models of adoptive immunotherapy. Cytotherapy. 2014; 16:619–30. https://doi.org/10.1016/j.jcyt.2013.10.013.
64. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014; 257:107–26. https://doi.org/10.1111/imr.12131.
65. Tomuleasa C, Fuji S, Cucuianu A, Kapp M, Pileczki V, Petrushev B, Selicean S, Tanase A, Dima D, Berindan-Neagoe I, Irimie A, Einsele H. MicroRNAs as biomarkers for graft-versus-host disease following allogeneic stem cell transplantation. Ann Hematol. 2015; 94:1081–92. https://doi.org/10.1007/s00277-015-2369-0.
66. Cruz CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, Diouf O, Liu E, Barrett AJ, Ito S, Shpall EJ, Krance RA, Kamble RT, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013; 122:2965–73. https://doi.org/10.1182/blood-2013-06-506741.
67. Brudno JN, Somerville RP, Shi V, Rose JJ, Halverson DC, Fowler DH, Gea-Banacloche JC, Pavletic SZ, Hickstein DD, Lu TL, Feldman SA, Iwamoto AT, Kurlander R, et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J Clin Oncol. 2016; 34:1112–21. https://doi.org/10.1200/JCO.2015.64.5929.
68. Wang LX, Westwood JA, Moeller M, Duong CP, Wei WZ, Malaterre J, Trapani JA, Neeson P, Smyth MJ, Kershaw MH, Darcy PK. Tumor ablation by gene-modified T cells in the absence of autoimmunity. Cancer Res. 2010; 70:9591–98. https://doi.org/10.1158/0008-5472.CAN-10-2884.
69. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, Morgan RA, Rosenberg SA. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010; 116:4099–102. https://doi.org/10.1182/blood-2010-04-281931.
70. Alcantar-Orozco EM, Gornall H, Baldan V, Hawkins RE, Gilham DE. Potential limitations of the NSG humanized mouse as a model system to optimize engineered human T cell therapy for cancer. Hum Gene Ther Methods. 2013; 24:310–20. https://doi.org/10.1089/hgtb.2013.022.
71. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, Qu J, Wasielewska T, He Q, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014; 6:224ra25. https://doi.org/10.1126/scitranslmed.3008226.
72. Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19:3153-64. https://doi.org/10.1158/1078-0432.CCR-13-0330.
73. Casucci M, Nicolis di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, Gentner B, Gullotta F, Ponzoni M, Bernardi M, Marcatti M, Saudemont A, Bordignon C, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013; 122:3461–72. https://doi.org/10.1182/blood-2013-04-493361.
74. Song DG, Powell DJ. Pro-survival signaling via CD27 costimulation drives effective CAR T-cell therapy. Oncoimmunology. 2012; 1:547–49. https://doi.org/10.4161/onci.19458.
75. Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, Lee J, Posey AD Jr, Scholler J, Scholler N, Bonneau R, June CH. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. 2014; 124:1070–80. https://doi.org/10.1182/blood-2013-10-535245.
76. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013; 31:71–75. https://doi.org/10.1038/nbt.2459.
77. Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, Czuczman M, Cairo MS. Targeting CD20+ aggressive B-cell non-Hodgkin Lymphoma by anti-CD20 CAR mRNA-modified expanded natural killer cells
78. Najima Y, Tomizawa-Murasawa M, Saito Y, Watanabe T, Ono R, Ochi T, Suzuki N, Fujiwara H, Ohara O, Shultz LD, Yasukawa M, Ishikawa F. Induction of WT1-specific human CD8+ T cells from human HSCs in HLA class I Tg NOD/SCID/IL2rgKO mice. Blood. 2016; 127:722–34. https://doi.org/10.1182/blood-2014-10-604777.
79. Kumaresan P, Figliola M, Moyes JS, Huls MH, Tewari P, Shpall EJ, Champlin R, Cooper LJ. Automated cell enrichment of cytomegalovirus-specific T cells for clinical applications using the cytokine-capture system. J Vis Exp. 2015; 104:e52808. https://doi.org/10.3791/52808.
80. Maus MV, Fraietta JA, Levine BL, Kalos M, Zhao Y, June CH. Adoptive immunotherapy for cancer or viruses. Annu Rev Immunol. 2014; 32:189–225. https://doi.org/10.1146/annurev-immunol-032713-120136.
81. Kumaresan PR, Manuri PR, Albert ND, Maiti S, Singh H, Mi T, Roszik J, Rabinovich B, Olivares S, Krishnamurthy J, Zhang L, Najjar AM, Huls MH, et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc Natl Acad Sci USA. 2014; 111:10660–65. https://doi.org/10.1073/pnas.1312789111.
82. Blyth E, Clancy L, Simms R, Ma CK, Burgess J, Deo S, Byth K, Dubosq MC, Shaw PJ, Micklethwaite KP, Gottlieb DJ. Donor-derived CMV-specific T cells reduce the requirement for CMV-directed pharmacotherapy after allogeneic stem cell transplantation. Blood. 2013; 121:3745–58. https://doi.org/10.1182/blood-2012-08-448977.
83. Gerdemann U, Katari UL, Papadopoulou A, Keirnan JM, Craddock JA, Liu H, Martinez CA, Kennedy-Nasser A, Leung KS, Gottschalk SM, Krance RA, Brenner MK, Rooney CM, et al. Safety and clinical efficacy of rapidly-generated trivirus-directed T cells as treatment for adenovirus, EBV, and CMV infections after allogeneic hematopoietic stem cell transplant. Mol Ther. 2013; 21:2113-21. https://doi.org/10.1038/mt.2013.151.
84. Mehta RS, Dave H, Bollard CM, Shpall EJ. Engineering cord blood to improve engraftment after cord blood transplant. Stem Cell Investig. 2017; 4:41. https://doi.org/10.21037/sci.2017.05.01.
85. Porter DL. The graft-versus-tumor potential of allogeneic cell therapy: an update on donor leukocyte infusions and nonmyeloablative allogeneic stem cell transplantation. J Hematother Stem Cell Res. 2001; 10:465–80. https://doi.org/10.1089/15258160152509082.
86. Champlin R, Khouri I, Kornblau S, Marini F, Anderlini P, Ueno NT, Molldrem J, Giralt S. Allogeneic hematopoietic transplantation as adoptive immunotherapy. Induction of graft-versus-malignancy as primary therapy. Hematol Oncol Clin North Am. 1999; 13:1041–57. https://doi.org/10.1016/S0889-8588(05)70108-8.
87. Small TN, Papadopoulos EB, Boulad F, Black P, Castro-Malaspina H, Childs BH, Collins N, Gillio A, George D, Jakubowski A, Heller G, Fazzari M, Kernan N, et al. Comparison of immune reconstitution after unrelated and related T-cell-depleted bone marrow transplantation: effect of patient age and donor leukocyte infusions. Blood. 1999; 93:467–80.
88. Collins RH Jr, Rogers ZR, Bennett M, Kumar V, Nikein A, Fay JW. Hematologic relapse of chronic myelogenous leukemia following allogeneic bone marrow transplantation: apparent graft-versus-leukemia effect following abrupt discontinuation of immunosuppression. Bone Marrow Transplant. 1992; 10:391–95.
89. Odom LF, Githens JH, Morse H, Sharma B, August CS, Humbert JR, Peakman D, Rusnak SL, Johnson FB. Remission of relapsed leukaemia during a graft-versus-host reaction. A “graft-versus-leukaemia reaction” in man? Lancet. 1978; 312:537–40. https://doi.org/10.1016/S0140-6736(78)92879-9.
90. Naparstek E, Or R, Nagler A, Cividalli G, Engelhard D, Aker M, Gimon Z, Manny N, Sacks T, Tochner Z, Weiss L, Samuel S, Brautbar C, et al. T-cell-depleted allogeneic bone marrow transplantation for acute leukaemia using Campath-1 antibodies and post-transplant administration of donor’s peripheral blood lymphocytes for prevention of relapse. Br J Haematol. 1995; 89:506–15. https://doi.org/10.1111/j.1365-2141.1995.tb08356.x.
91. Apperley JF, Mauro FR, Goldman JM, Gregory W, Arthur CK, Hows J, Arcese W, Papa G, Mandelli F, Wardle D, Gravett P, Franklin IM, Bandini G, et al. Bone marrow transplantation for chronic myeloid leukaemia in first chronic phase: importance of a graft-versus-leukaemia effect. Br J Haematol. 1988; 69:239–45. https://doi.org/10.1111/j.1365-2141.1988.tb07628.x.
92. Tomuleasa C, Dima D, Frinc I, Patiu M, Petrushev B, Cucuianu A, Berindan-Neagoe I. BCR-ABL1 T315I mutation, a negative prognostic factor for the terminal phase of chronic myelogenous leukemia treated with first- and second-line tyrosine kinase inhibitors, might be an indicator of allogeneic stem cell transplant as the treatment of choice. Leuk Lymphoma. 2015; 56:546–47. https://doi.org/10.3109/10428194.2014.940582.
93. Kolb HJ, Mittermüller J, Clemm C, Holler E, Ledderose G, Brehm G, Heim M, Wilmanns W. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood. 1990; 76:2462–65.
94. Porter DL, Roth MS, McGarigle C, Ferrara JL, Antin JH. Induction of graft-versus-host disease as immunotherapy for relapsed chronic myeloid leukemia. N Engl J Med. 1994; 330:100–06. https://doi.org/10.1056/NEJM199401133300204.
95. Kolb HJ, Schattenberg A, Goldman JM, Hertenstein B, Jacobsen N, Arcese W, Ljungman P, Ferrant A, Verdonck L, Niederwieser D, van Rhee F, Mittermueller J, de Witte T, et al, and European Group for Blood and Marrow Transplantation Working Party Chronic Leukemia. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood. 1995; 86:2041–50.
96. Ho VT, Kim HT, Kao G, Cutler C, Levine J, Rosenblatt J, Joyce R, Antin JH, Soiffer RJ, Ritz J, Avigan D, Alyea EP 3rd. Sequential infusion of donor-derived dendritic cells with donor lymphocyte infusion for relapsed hematologic cancers after allogeneic hematopoietic stem cell transplantation. Am J Hematol. 2014; 89:1092–96. https://doi.org/10.1002/ajh.23825.
97. Eefting M, von dem Borne PA, de Wreede LC, Halkes CJ, Kersting S, Marijt EW, Veelken H, Falkenburg JF. Intentional donor lymphocyte-induced limited acute graft-versus-host disease is essential for long-term survival of relapsed acute myeloid leukemia after allogeneic stem cell transplantation. Haematologica. 2014; 99:751–58. https://doi.org/10.3324/haematol.2013.089565.
98. Salama M, Nevill T, Marcellus D, Parker P, Johnson M, Kirk A, Porter D, Giralt S, Levine JE, Drobyski W, Barrett AJ, Horowitz M, Collins RH. Donor leukocyte infusions for multiple myeloma. Bone Marrow Transplant. 2000; 26:1179–84. https://doi.org/10.1038/sj.bmt.1702685.
99. Baron F, Maris MB, Sandmaier BM, Storer BE, Sorror M, Diaconescu R, Woolfrey AE, Chauncey TR, Flowers ME, Mielcarek M, Maloney DG, Storb R. Graft-versus-tumor effects after allogeneic hematopoietic cell transplantation with nonmyeloablative conditioning. J Clin Oncol. 2005; 23:1993–2003. https://doi.org/10.1200/JCO.2005.08.136.
100. Levine JE, Braun T, Penza SL, Beatty P, Cornetta K, Martino R, Drobyski WR, Barrett AJ, Porter DL, Giralt S, Leis J, Holmes HE, Johnson M, et al. Prospective trial of chemotherapy and donor leukocyte infusions for relapse of advanced myeloid malignancies after allogeneic stem-cell transplantation. J Clin Oncol. 2002; 20:405–12. https://doi.org/10.1200/JCO.2002.20.2.405.
101. Porter DL, Collins RH Jr, Shpilberg O, Drobyski WR, Connors JM, Sproles A, Antin JH. Long-term follow-up of patients who achieved complete remission after donor leukocyte infusions. Biol Blood Marrow Transplant. 1999; 5:253-61. https://doi.org/10.1053/bbmt.1999.v5.pm10465105.
102. Mackinnon S, Papadopoulos EB, Carabasi MH, Reich L, Collins NH, Boulad F, Castro-Malaspina H, Childs BH, Gillio AP, Kernan NA. Adoptive immunotherapy evaluating escalating doses of donor leukocytes for relapse of chronic myeloid leukemia after bone marrow transplantation: separation of graft-versus-leukemia responses from graft-versus-host disease. Blood. 1995; 86:1261–68.
103. Dazzi F, Szydlo RM, Craddock C, Cross NC, Kaeda J, Chase A, Olavarria E, van Rhee F, Kanfer E, Apperley JF, Goldman JM. Comparison of single-dose and escalating-dose regimens of donor lymphocyte infusion for relapse after allografting for chronic myeloid leukemia. Blood. 2000; 95:67–71.
104. Guglielmi C, Arcese W, Dazzi F, Brand R, Bunjes D, Verdonck LF, Schattenberg A, Kolb HJ, Ljungman P, Devergie A, Bacigalupo A, Gomez M, Michallet M, et al. Donor lymphocyte infusion for relapsed chronic myelogenous leukemia: prognostic relevance of the initial cell dose. Blood. 2002; 100:397–405. https://doi.org/10.1182/blood.V100.2.397.
105. Chown SR, Marks DI, Cornish JM, Pamphilon DH, Potter MN, Steward CG, Oakhill A. Unrelated donor bone marrow transplantation in children and young adults with acute myeloid leukaemia in remission. Br J Haematol. 1997; 99:36–40. https://doi.org/10.1046/j.1365-2141.1997.3593173.x.
106. Hongeng S, Krance RA, Bowman LC, Srivastava DK, Cunningham JM, Horwitz EM, Brenner MK, Heslop HE. Outcomes of transplantation with matched-sibling and unrelated-donor bone marrow in children with leukaemia. Lancet. 1997; 350:767–71. https://doi.org/10.1016/S0140-6736(97)03098-5.
107. Oakhill A, Pamphilon DH, Potter MN, Steward CG, Goodman S, Green A, Goulden P, Goulden NJ, Hale G, Waldmann H, Cornish JM. Unrelated donor bone marrow transplantation for children with relapsed acute lymphoblastic leukaemia in second complete remission. Br J Haematol. 1996; 94:574–78. https://doi.org/10.1046/j.1365-2141.1996.d01-1834.x.
108. Casper J, Camitta B, Truitt R, Baxter-Lowe LA, Bunin N, Lawton C, Murray K, Hunter J, Pietryga D, Garbrecht F. Unrelated bone marrow donor transplants for children with leukemia or myelodysplasia. Blood. 1995; 85:2354–63.
109. Marks DI, Cullis JO, Ward KN, Lacey S, Syzdlo R, Hughes TP, Schwarer AP, Lutz E, Barrett AJ, Hows JM, Batchelor JR, Goldman JM. Allogeneic bone marrow transplantation for chronic myeloid leukemia using sibling and volunteer unrelated donors. A comparison of complications in the first 2 years. Ann Intern Med. 1993; 119:207–14. https://doi.org/10.7326/0003-4819-119-3-199308010-00005.
110. O’Reilly RJ, Small TN, Papadopoulos E, Lucas K, Lacerda J, Koulova L. Biology and adoptive cell therapy of Epstein-Barr virus-associated lymphoproliferative disorders in recipients of marrow allografts. Immunol Rev. 1997; 157:195–216. https://doi.org/10.1111/j.1600-065X.1997.tb00983.x.
111. Heslop HE, Rooney CM. Adoptive cellular immunotherapy for EBV lymphoproliferative disease. Immunol Rev. 1997; 157:217–22. https://doi.org/10.1111/j.1600-065X.1997.tb00984.x.
112. Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995; 333:1038–44. https://doi.org/10.1056/NEJM199510193331603.
113. Papadopoulos EB, Ladanyi M, Emanuel D, Mackinnon S, Boulad F, Carabasi MH, Castro-Malaspina H, Childs BH, Gillio AP, Small TN, Young JW, Kernan NA, O’Reilly RJ. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med. 1994; 330:1185–91. https://doi.org/10.1056/NEJM199404283301703.
114. Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, Ponzoni M, Rossini S, Mavilio F, Traversari C, Bordignon C. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997; 276:1719–24. https://doi.org/10.1126/science.276.5319.1719.
115. Kernan NA, Bartsch G, Ash RC, Beatty PG, Champlin R, Filipovich A, Gajewski J, Hansen JA, Henslee-Downey J, McCullough J, McGlave P, Perkins HA, Phillips GL, et al. Analysis of 462 transplantations from unrelated donors facilitated by the National Marrow Donor Program. N Engl J Med. 1993; 328:593–602. https://doi.org/10.1056/NEJM199303043280901.
116. Nademanee A, Schmidt GM, Parker P, Dagis AC, Stein A, Snyder DS, O’Donnell M, Smith EP, Stepan DE, Molina A. The outcome of matched unrelated donor bone marrow transplantation in patients with hematologic malignancies using molecular typing for donor selection and graft-versus-host disease prophylaxis regimen of cyclosporine, methotrexate, and prednisone. Blood. 1995; 86:1228–34.
117. Davies SM, Shu XO, Blazar BR, Filipovich AH, Kersey JH, Krivit W, McCullough J, Miller WJ, Ramsay NK, Segall M. Unrelated donor bone marrow transplantation: influence of HLA A and B incompatibility on outcome. Blood. 1995; 86:1636–42.
118. Speiser DE, Tiercy JM, Rufer N, Grundschober C, Gratwohl A, Chapuis B, Helg C, Löliger CC, Siren MK, Roosnek E, Jeannet M. High resolution HLA matching associated with decreased mortality after unrelated bone marrow transplantation. Blood. 1996; 87:4455–62.
119. Kook H, Goldman F, Giller R, Goeken N, Peters C, Comito M, Rumelhart S, Holida M, Lee N, Trigg M. Reconstruction of the immune system after unrelated or partially matched T-cell-depleted bone marrow transplantation in children: functional analyses of lymphocytes and correlation with immunophenotypic recovery following transplantation. Clin Diagn Lab Immunol. 1997; 4:96–103.
120. Chang X, Zang X, Xia CQ. New strategies of DLI in the management of relapse of hematological malignancies after allogeneic hematopoietic SCT. Bone Marrow Transplant. 2016; 51:324–32. https://doi.org/10.1038/bmt.2015.288.
121. Loren AW, Porter DL. Donor leukocyte infusions after unrelated donor hematopoietic stem cell transplantation. Curr Opin Oncol. 2006; 18:107–14. https://doi.org/10.1097/01.cco.0000208781.61452.d3.
122. Barge RM, Osanto S, Marijt WA, Starrenburg CW, Fibbe WE, Nortier JW, Falkenburg JH, Willemze R. Minimal GVHD following in-vitro T cell-depleted allogeneic stem cell transplantation with reduced-intensity conditioning allowing subsequent infusions of donor lymphocytes in patients with hematological malignancies and solid tumors. Exp Hematol. 2003; 31:865–72. https://doi.org/10.1016/S0301-472X(03)00200-5.
123. Singh N, Shi J, June CH, Ruella M. Genome-editing technologies in adoptive T cell immunotherapy for cancer. Curr Hematol Malig Rep. 2017; 12:522–29. https://doi.org/10.1007/s11899-017-0417-7.
124. Liu J, Zhang X, Zhong JF, Zhang C. CAR-T cells and allogeneic hematopoietic stem cell transplantation for relapsed/refractory B-cell acute lymphoblastic leukemia. Immunotherapy. 2017; 9:1115–25. https://doi.org/10.2217/imt-2017-0072.
125. Liu J, Zhong JF, Zhang X, Zhang C. Allogeneic CD19-CAR-T cell infusion after allogeneic hematopoietic stem cell transplantation in B cell malignancies. J Hematol Oncol. 2017; 10:35. https://doi.org/10.1186/s13045-017-0405-3.
126. Ghosh A, Smith M, James SE, Davila ML, Velardi E, Argyropoulos KV, Gunset G, Perna F, Kreines FM, Levy ER, Lieberman S, Jay HV, Tuckett AZ, et al. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat Med. 2017; 23:242–49. https://doi.org/10.1038/nm.4258.
127. Rafei H, Nassereddine S, Garcia IF. Disseminated intravascular coagulation-like reaction following rituximab infusion. BMJ Case Rep. 2017; 2017. https://doi.org/10.1136/bcr-2016-218443.
128. Williams M, Khalid T, Hughes S, Bonney D, Wynn R. Rituximab-induced cytokine storm in the absence of overt lymphoproliferative disease. J Pediatr Hematol Oncol. 2016; 38:e29–31. https://doi.org/10.1097/MPH.0000000000000485.
129. Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014; 20:119–22. https://doi.org/10.1097/PPO.0000000000000035.
130. Kulkarni HS, Kasi PM. Rituximab and cytokine release syndrome. Case Rep Oncol. 2012; 5:134–41. https://doi.org/10.1159/000337577.
131. Smith L, Venella K. Cytokine release syndrome: inpatient care for side effects of CAR T-cell therapy. Clin J Oncol Nurs. 2017; 21:29–34. https://doi.org/10.1188/17.CJON.S2.29-34.
132. Frey NV, Porter DL. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program. 2016; 2016:567–72. https://doi.org/10.1182/asheducation-2016.1.567.
133. Wang L, Ma N, Okamoto S, Amaishi Y, Sato E, Seo N, Mineno J, Takesako K, Kato T, Shiku H. Efficient tumor regression by adoptively transferred CEA-specific CAR-T cells associated with symptoms of mild cytokine release syndrome. Oncoimmunology. 2016; 5:e1211218. https://doi.org/10.1080/2162402X.2016.1211218.
134. Klaver Y, van Steenbergen SC, Sleijfer S, Debets R, Lamers CH. Plasma IFN-γ and IL-6 levels correlate with peripheral T-cell numbers but not toxicity in RCC patients treated with CAR T-cells. Clin Immunol. 2016; 169:107–13. https://doi.org/10.1016/j.clim.2016.06.014.
135. Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, Klichinsky M, Aikawa V, Nazimuddin F, Kozlowski M, Scholler J, Lacey SF, Melenhorst JJ, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest. 2016; 126:3814–26. https://doi.org/10.1172/JCI87366.
136. Tanyi JL, Stashwick C, Plesa G, Morgan MA, Porter D, Maus MV, June CH. Possible compartmental cytokine release syndrome in a patient with recurrent ovarian cancer after treatment with mesothelin-targeted CAR-T cells. J Immunother. 2017; 40:104–07. https://doi.org/10.1097/CJI.0000000000000160.
137. Fitzgerald JC, Weiss SL, Maude SL, Barrett DM, Lacey SF, Melenhorst JJ, Shaw P, Berg RA, June CH, Porter DL, Frey NV, Grupp SA, Teachey DT. Cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Crit Care Med. 2017; 45:e124–31. https://doi.org/10.1097/CCM.0000000000002053.
138. Calabrese LH, Rose-John S. IL-6 biology: implications for clinical targeting in rheumatic disease. Nat Rev Rheumatol. 2014; 10:720–27. https://doi.org/10.1038/nrrheum.2014.127.
139. Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, Kakehi T. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood. 2008; 112:3959–64. https://doi.org/10.1182/blood-2008-05-155846.
140. Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci. 2012; 8:1237–47. https://doi.org/10.7150/ijbs.4989.
141. Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, Pequignot E, Gonzalez VE, Chen F, Finklestein J, Barrett DM, Weiss SL, Fitzgerald JC, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor t-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016; 6:664–79. https://doi.org/10.1158/2159-8290.CD-16-0040.
142. Kalos M. Chimeric antigen receptor-engineered T cells in CLL: the next chapter unfolds. J Immunother Cancer. 2016; 4:5. https://doi.org/10.1186/s40425-016-0108-2.
143. Namuduri M, Brentjens RJ. Medical management of side effects related to CAR T cell therapy in hematologic malignancies. Expert Rev Hematol. 2016; 9:511–13. https://doi.org/10.1080/17474086.2016.1183479.
144. Sadelain M, Brentjens R, Riviere I, Park J. CD19 CAR Therapy for Acute Lymphoblastic Leukemia. Am Soc Clin Oncol Educ Book. 2015:e360-3. https://doi.org/10.14694/EdBook_AM.2015.35.e360.
145. Davila ML, Sauter C, Brentjens R. CD19-targeted T Cells for hematologic malignancies: clinical experience to date. Cancer J. 2015; 21:470–74. https://doi.org/10.1097/PPO.0000000000000153.
146. Wilke AC, Gökbuget N. Clinical applications and safety evaluation of the new CD19 specific T-cell engager antibody construct blinatumomab. Expert Opin Drug Saf. 2017; 16:1191–202. https://doi.org/10.1080/14740338.2017.1338270.
147. Marini BL, Sun Y, Burke PW, Perissinotti AJ. Successful reintroduction of blinatumomab in a patient with relapsed/refractory acute lymphoblastic leukemia following grade 4 cytokine release syndrome. J Oncol Pharm Pract. 2018; 24:67-73. J Oncol Pharm Pract. 2018; 24:67-73. https://doi.org/10.1177/1078155216676633.
148. Martinelli G, Boissel N, Chevallier P, Ottmann O, Gökbuget N, Topp MS, Fielding AK, Rambaldi A, Ritchie EK, Papayannidis C, Sterling LR, Benjamin J, Stein A. Complete hematologic and molecular response in adult patients with relapsed/refractory Philadelphia chromosome-positive b-precursor acute lymphoblastic leukemia following treatment with blinatumomab: results from a phase II, single-arm, multicenter study. J Clin Oncol. 2017; 35:1795–802. https://doi.org/10.1200/JCO.2016.69.3531.