
INTRODUCTION
Effective antitumor immune responses rely on the presence and functionality of T cells that are able to recognize and destroy tumor cells. Tumor-specific cytotoxic T lymphocytes (CTLs) are often present in the periphery and within the tumor nest of cancer patients. However, tumor-infiltrating CTLs frequently lack functional antitumor reactivity. The latter is explained by the hostile tumor environment, which is enriched with immunosuppressive cell types, such as immature myeloid cells as well as immunosuppressive factors, including transforming growth factor-β (TGF-β). These inhibitory mechanisms actively quench the antitumor immune response [1, 2].
Over the years, the injection of various antitumor agents such as cells, proteins, nucleic acids or viral vectors, into the tumor has been thoroughly evaluated in preclinical and clinical immunotherapy studies [3]. These agents differ in their mechanism of action: some lead to (re-)activation of effector immune cells, while others counteract the immunosuppressive environment. Strategies developed to induce immune activation include the direct injection of dendritic cells (DCs), and use of DC-recruiting and/or DC-potentiating factors. Our group recently demonstrated that DCs within the tumor have the capacity to engulf mRNA [4–6]. More importantly, we showed that intratumoral delivery of mRNA encoding DC-modulating stimuli, collectively referred to as TriMix, had a therapeutic effect in tumor bearing mice [4]. In the context of the tumor environment, several therapies targeting immunosuppressive mediators such as myeloid-derived suppressor cells (MDSCs), regulatory T (Treg) cells, and secreted factors like TGF-β have been used. Moreover, fusokines such as the FIST protein, consisting of interleukin-2 (IL-2) and the ectodomain of the TGF-β receptor II, have been developed to combine immune activation and tumor modulation [7, 8]. In addition, it was shown that besides activation of CTLs, modulation of the tumor environment is a prerequisite to support the effector phase of the antitumor immune response.
Given above described factors, we evaluated whether we can exploit tumor-residing DCs as ‘factories’ for the production of a mRNA-encoded fusokine. This novel fusokine consists of interferon-β (IFN-β) and the ectodomain of the TGF-β receptor II, and is referred to as Fβ2. The rationale for fusing IFN-β with a TGF-β antagonist is based on the observation that blockade of TGF-β within the tumor is most effective when combined with an immune activator [9]. In addition, type I IFNs, such as IFN-β are potent activators of adaptive immune responses within the tumor environment [10]. We demonstrate that the fusokine Fβ 2 modulates various cell populations, including tumor cells, DCs, MDSCs and CD8+ T cells, leading to the favorable therapeutic outcome after its intratumoral delivery in the form of mRNA.
RESULTS
Fβ2 mRNA is translated into a functional protein
To address whether mRNA encoding Fβ2, a fusion protein consisting of IFN-β and the ectodomain of the TGF-β receptor II (Fig. 1A), can be translated into a functional protein, we electroporated HEK293T cells with 20 μg of Fβ2 or eGFP mRNA. Supernatants were collected 24 hours later. First, we determined the functionality and estimated the amount of the in vitro generated fusokine. Therefore, we quantified the expression of the type I IFN-inducible gene Mx1 in splenocytes cultured for 6 hours with the supernatants or with varying amounts of recombinant IFN-β. qPCR analysis demonstrated that the CT-values obtained upon treatment of splenocytes with Fβ2 supernatants was comparable to those obtained upon treatment of splenocytes with 17.1 ± 1.7 ng/ml (n = 3) recombinant IFN-β (Fig. 1B). To investigate the capacity of the fusokine to neutralize TGF-β we used the TGF-β reporter HEK293T cell line, which expresses eGFP under the control of a TGF-β responding promoter [11]. Indeed, higher eGFP expression was observed when the cell line was cultured with increased amounts of recombinant TGF-β (Fig. 1C). This was greatly reduced by the presence of Fβ2 in the supernatants (Fig. 1D). Additionally, the neutralization capacity of the fusokine was compared to a commercially available neutralizing anti-TGF-β antibody. The capacity of Fβ2 to neutralize TGF-β was comparable with 20 ng/ml of the commercially available anti-TGF-β antibody (Fig. 1D). These results are consistent with the quantity of Fβ2 estimated based on the expression of the IFN-inducible Mx1 gene (Fig. 1B). Together, these data demonstrate that the mRNA encoding the Fβ2 fusokine is translated into a functional protein.
The Fβ2 fusokine modulates myeloid cells to improve CD8+ T cell responses
To analyze the effect of Fβ2 on DCs, we cultured them for 48 hours in supernatants of HEK293T cells that were electroporated with 20 μg of Fβ2 or eGFP mRNA. DCs cultured with 20 ng/ml of recombinant IFN-β or activated for 4 hours with 100 ng/ml LPS were used as a control. Flow cytometry analysis revealed that DCs cultured with Fβ2 displayed an enhanced expression of co-stimulatory and antigen-presenting molecules (Fig. 2A), and secreted pro-inflammatory cytokines (Fig. 2B). To further evaluate the functionality of these DCs, we performed an in vitro stimulation of OT-I cells. We demonstrated that DCs pulsed with SIINFEKL peptide and cultured in the presence of Fβ2 lead to enhanced production of IFN-γ by antigen-specific CD8+ OT-I cells (Fig. 2C-D). We next analyzed the effect of Fβ2 on MDSCs. To that end, MDSCs that closely resemble those found within tumors were generated in vitro [12, 13]. Of note, these MDSCs produce high levels of TGF-β (Fig. 2E). The MDSCs were cultured for 3 days in supernatants of HEK293T cells that were electroporated with 20 μg of Fβ2 or eGFP mRNA. We found that MDSCs cultured in the presence of Fβ2 were no longer able to fully suppress the functionality of CD8+ T cells as shown by the ability of these T cells to produce IFN-γ (Fig. 2F). This might be explained by the reduced cell viability and the increased expression of the surface marker sca-1 on the MDSCs cultured in Fβ2 supernatants (Fig. 2G-I). Overall, these data suggest that Fβ2 potentiates the antigen-presenting function of DCs, whilst decreasing the suppressive capacity of MDSCs, therefore supporting CD8+ T cell-mediated responses.
Tumor cells treated with Fβ2 show lower proliferation rates and increased expression levels of MHC I and PD-L1
To investigate the effect of Fβ2 on tumor cells, we cultured tumor cells of various histological origin for 1 or 4 days with Fβ2 supernatants. Subsequently, we evaluated their phenotype and proliferation respectively. Tumor cells exposed to Fβ2 showed decreased proliferation (Fig. 3A) and enhanced expression of the antigen-presenting molecule MHC I as well as the co-inhibitory molecule PD-L1 (Fig. 3B-C). Next we analyzed whether exposure of E.G7-OVA cells to Fβ2 facilitated their recognition by activated CD8+ OT-I cells (Fig. 3D) despite PD-L1 up-regulation. These T cells showed an increased cytolytic activity (Fig. 3E-F), indicating that the expression of PD-L1 on tumor cells did not completely abrogate their recognition by T cells.
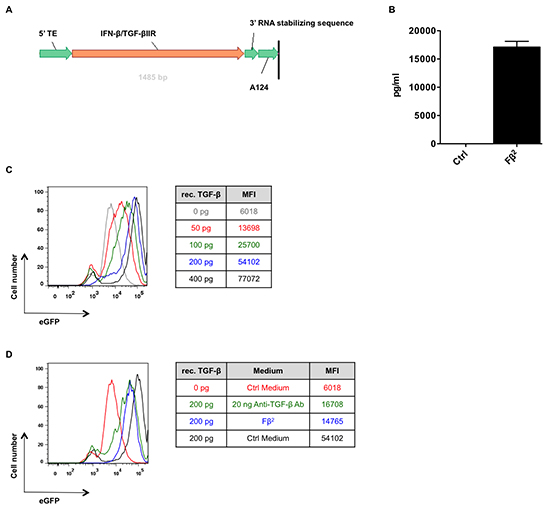
Figure 1: Fβ2 mRNA is translated to a functional protein. (A) Schematic representation of the Fβ2 mRNA construct. (B) Splenocytes were cultured for 5 hours in Fβ2, control (Ctrl) supernatants or exposed to increasing amounts of recombinant IFN-β. qPCR was performed to determine Mx1 expression. The graph depicts the amount of fusokine (pg/ml). The quantity was determined based on the expression of Mx1 upon treatment with recombinant IFN-β and normalized to 2−Δct for Ppia (n=3). (C) The TGF-β reporter HEK293T cell line was cultured for 24 hours in 0 to 400 pg/ml of recombinant TGF-β (rec. TGF-β). The histogram overlay depicts the eGFP expression. Representative plots are shown (n=4) (D) The TGF-β reporter HEK293T cell line was cultured for 24 hours in Fβ2, Ctrl supernatants or exposed to 20 ng of a commercial available anti-TGF-β antibody, and supplemented with 200 pg of recombinant TGF-β. The histogram overlay depicts the eGFP expression. Representative plots are shown (n=3).
Simultaneous exposure of tumor cells and CD8+ T cells to Fβ2 significantly enhances the killing capacities of antigen-specific T cells
To investigate the direct effect of Fβ2 on CD8+ T cells, we cultured CD8+ OT-I spleen cells for 3 days in supernatants of HEK293T cells that were electroporated with 20 μg of Fβ2 or eGFP mRNA. Simultaneously, E.G7-OVA tumor cells were cultured in Fβ2 containing supernatants. Subsequently, co-cultures were set up according to the scheme shown in figure 4A, and the degranulation ability of the CD8+ OT-I cells was evaluated (Fig. 4B-D). We demonstrated that CD8+ OT-I cells pre-treated with Fβ 2 recognized tumor cells more efficiently and that this recognition was enhanced when tumor cells were pretreated with Fβ2 (Fig. 4C-D). In summary, we showed that the Fβ2 fusokine potentiates the killing capacity of CD8+ T cells.
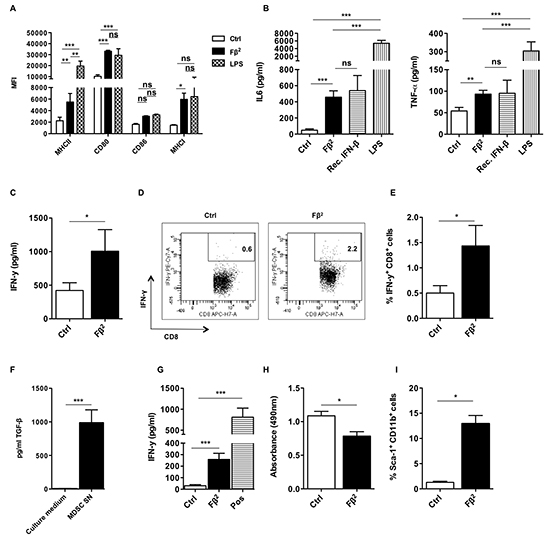
Figure 2: The Fβ2 fusokine modulates myeloid cells to improve CD8+ T cell responses. (A-E) DCs were cultured for 48 hours in Fβ2 or Control (Ctrl) supernatants. (A) Expression of MHC II, CD80, CD86 and MHC I was evaluated by flow cytometry. The column graph depicts the median fluorescence intensity (MFI) (n=6). (B) Supernatants were analyzed for the presence of IL-6 and TNF-α (n=3-12). (C-E) DCs were pulsed with the peptide SIINFEKL and co-cultured for 6 days with CD8+ OT-I cells at a 1:10 ratio. (C) The production of IFN-γ by OT-I cells was determined by ELISA (n=9). (D) The production of IFN-γ by OT-I cells was confirmed via flow cytometry. Representative plots are show. (E) The column graph depicts the results of the experiments (n=6). (F) The TGF-β reporter HEK293T cell line was cultured for 24 hours in the conditioned medium used to generate MDSCs or in supernatants of in vitro generated day 5 MDSCs (n=3). (G-I) MDSCs were cultured for 3 days in Fβ2 or Ctrl supernatants. (G) These MDSCs were co-cultured for 3 days with CD8+ splenocytes that were activated with anti-CD3/anti-CD28 beads. Activated CD8+ splenocytes cultured in the absence of MDSCs served as a positive control (Pos). Supernatants were screened for the presence of IFN-γ (n=4). (H) MDSC viability was assessed using a colorimetric assay. The colorimetric assay reflects the amount of viable cells in the plate, and is measured as absorbance. The graph depicts the absorbance (n=3). (I) Flow cytometry was performed to assess the percentage of CD11b and sca-1+ cells (n=3).
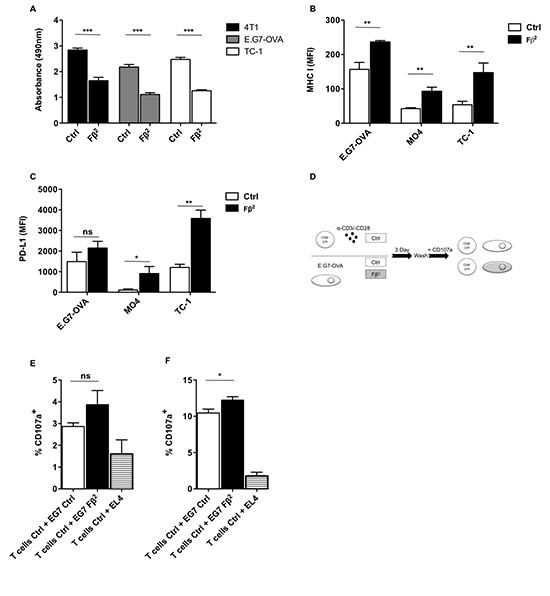
Figure 3: Tumors treated with Fβ2 show lower proliferative rates and express increased levels of MHC I and PD-L1. (A) 4T1, E.G7-OVA and TC-1 tumor cells were cultured for 4 days in Fβ2 or Ctrl supernatants. Cell proliferation was assessed using a colorimetric assay (n=3). (B-C) E.G7-OVA, MO4 and TC-1 cells were cultured for 24 hours in Fβ2 or Ctrl supernatants, after which expression of (B) MHC I and (C) PD-L1 was evaluated by flow cytometry. The column graphs depict the MFI (n=3). (D-F) E.G7-OVA tumor cells were cultured for 3 days in Fβ2 or Ctrl supernatants, after which they were co-cultured with CD8+ OT-I spleen cells in the presence of anti-CD107 antibodies for 4 hours at a tumor:T cell ratio of 1:10 (E) and 1:1 (F). Four hours later cells were additionally stained for CD3 and CD8. The graphs depict the amount of CD107a and CD8+ T cells (n=3).
Intratumoral delivery of Fβ2 mRNA delays tumor growth
To analyze the therapeutic potential of mRNA encoding the Fβ2 fusokine, we first treated mice bearing E.G7-OVA tumors with a single intratumoral injection of 10 μg of Fβ2 mRNA. Mice treated with a single injection of 10 μg tNGFR mRNA served as a control. Tumor growth was delayed over a period of three days (Fig. 5A), after which the effect was no longer detected (data not shown). Therefore, we subsequently treated tumor-bearing mice with three intratumoral injections of 10 μg Fβ2 or tNGFR mRNA at a three-day interval, which resulted in a prolonged survival of the mice (Fig. 5B). To further improve this therapeutic outcome, we further increased the dose of mRNA to 50 μg and delivered the mRNA continuously at a three-day interval (Fig. 5C). This regimen was compared to the delivery of 10 μg of mRNA. Mice treated with repeated injections of the vehicle (0.8 Lactated Ringer’s solution) were used as a control. Notably, the treatment with 50 μg of tNGFR mRNA prolonged mice survival when compared to the treatment with 10 μg of tNGFR mRNA. In addition, we observed that in the groups treated with 10 μg of mRNA, the non-stop treatment regimen did not improve the survival as compared to the three-day regimen (Fig. 5B-C). Importantly, four out of eight mice treated with 50 μg of Fβ2 mRNA demonstrated a long-term survival when compared to mice treated with 10 μg Fβ2 mRNA or 50 μg of tNGFR mRNA (Fig. 5C). Based on these data, an optimal treatment regimen consisting of three immunizations at a three-day interval with 50 μg of mRNA was defined and then applied to the treatment of mice bearing TC-1 tumors (Fig. 6A). Consistent with the E.G7-OVA model, we observed an immediate and a prolonged delay in TC-1 tumor growth (Fig. 6B-C). Consequently, these mice showed an increased survival (Fig. 6D). To investigate the role of CD8+ T cells in this therapeutic outcome, we depleted this cell population prior and during the treatment (Fig. 6E). The results showed that CD8+ T cells are the predominant cell type involved in the observed antitumor activity, since their depletion abrogated the therapeutic effect of the Fβ2 fusokine (Fig. 6F-G).
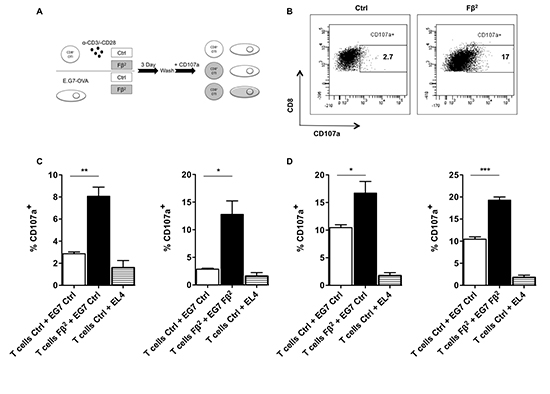
Figure 4: Simultaneous exposure of tumor cells and CD8 T cells to Fβ2 significantly enhances the killing capacities of antigen-specific T cells. (A) CD8+ OT-I spleen cells were cultured for 3 days in Fβ2 or Ctrl supernatants. Simultaneously, E.G7-OVA tumor cells were cultured for 3 days in these supernatants. Subsequently, both cell populations were washed and mixed together as depicted. These were co-cultured in the presence of anti-CD107 antibodies for 4 hours at a tumor:T cell ratio of (C) 10:1 or (D) 1:1, after which the T cells were labeled with anti-CD3 and anti-CD8 antibodies. (B) The flow cytometry graphs are representative for the experiments. (C-D) The graphs depict the percentage of CD107a within CD8+ T cells in the co-cultures (n=3 and n=4, respectively).
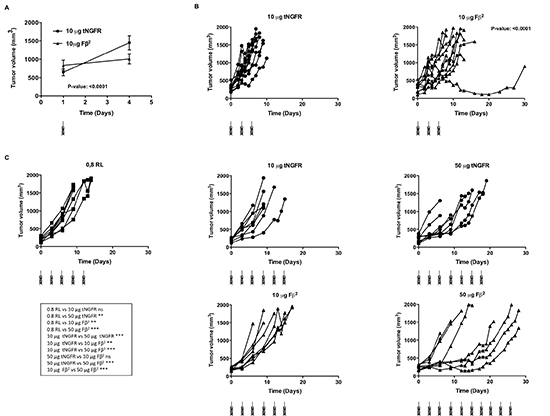
Figure 5: Intratumoral delivery of Fβ2 mRNA to E.G7-OVA bearing mice delays tumor growth. (A-C) E.G7-OVA cells (5 × 106) were transplanted subcutaneously in C57BL/6 mice. (A) Mice were treated with a single intratumoral injection of 10 μg mRNA encoding tNGFR (circle) or Fβ2 (triangle). The graph shows the average tumor size at the day of treatment and 3 days later for a total of 11 mice and summarizes the results of 4 independent experiments. (B) Mice (10 per group) were treated with 3 intratumoral injections of 10 μg of tNGFR (circle) or Fβ2 (triangle) mRNA at three-day intervals. The graph depicts the size of the tumor of individual mice. (C) Mice (8 per group) were treated with consecutive intratumoral injections of 0.8 RL (square), 10 or 50 μg of tNGFR (circle) or Fβ2 (triangle) mRNA at three-day interval. The graph depicts the size of the tumor of individual mice.
Blockade of the PD-1/PD-L1 interaction combined with Fβ2 mRNA treatment improves therapeutic responses
Although intratumoral injection of Fβ2 mRNA induces therapeutic responses, we demonstrated both in vitro (Fig. 3C) and in vivo (data not shown) that Fβ2 mRNA induces PD-L1 up-regulation. We therefore wondered whether additional targeting of the PD-1/PD-L1 pathway could improve therapeutic responses. To test this, we combined intratumoral delivery of Fβ2 mRNA with three intraperitoneal injections of anti-PD1 monoclonal antibodies (Fig. 7A-B). This resulted in an inhibition of TC-1 tumor growth (Fig. 7B). We next increased the amount of tumor cells injected, hereby mimicking a faster growing tumor setting. In this case, no effect was observed for the single treatment with Fβ2 mRNA or anti-PD1 alone (Fig. 7C). However, the combination of Fβ2 mRNA and anti-PD1 resulted in a delayed tumor growth and thus in an increased survival.
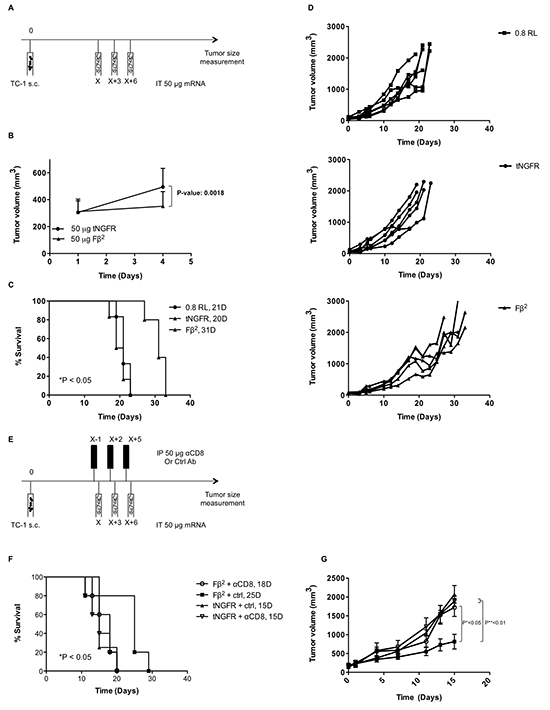
Figure 6: Intratumoral delivery of Fβ2 mRNA to TC-1 bearing mice delays tumor growth. (A-G) TC-1 cells were transplanted subcutaneously in C57BL/6 mice. Once the tumors reached an average size of 100 mm3 the mice were treated according to the scheme shown in A. Tumor growth was monitored. (B) The graph shows the average tumor size at the day of treatment and 3 days later (n=11). (C-D) Mice (5-6 per group) were treated with 3 intratumoral injections of 0.8 RL (square), 50 μg of tNGFR (circle) or Fβ2 (triangle) mRNA at three-day intervals. (C) The graph depicts the median survival. (D) The graph depicts the growth of tumors in individual mice. (E) Treatment regimen with CD8+ T cell depletion. (F) The graph depicts the survival of the mice treated as in E. (G) The graph depicts the mean of the tumor sizes of the respective groups (5 mice per group).
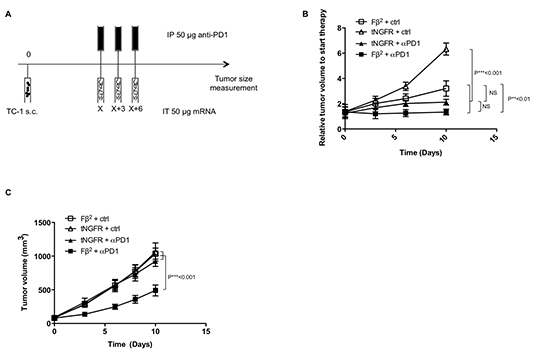
Figure 7: Blockade of PD-1/PD-L1 interactions combined with Fβ2 mRNA treatment improves therapeutic responses. TC-1 cells were transplanted subcutaneously in C57BL/6 mice (5 per group). (A-C) Once the tumors reached a size of 100-500 mm3 mice were treated according to the scheme shown in A. Subsequently, tumor growth was monitored. (B) The graph depicts the mean of the tumor sizes of the respective groups when applying a less aggressive model using 2 × 104 TC-1 cells. (C) The graph depicts the mean of the tumor sizes of the respective groups when applying a more aggressive model using 2 × 105 TC-1 cells.
DISCUSSION
In this study, we demonstrated the feasibility of intratumoral delivery of mRNA encoding immunomodulating proteins to fine-tune the tumor environment and confer antitumor immunity. More specifically, we evaluated the ability of a fusokine called Fβ2, composed of IFN-β and the ectodomain of the TGF-β receptor II.
Over the years, much effort has been put into priming and generating highly potent tumor-specific CTLs by means of cancer vaccination [14]. Although this strategy leads to the priming of tumor-specific CTLs [15], these T cells display reduced functionality due to exposure to the inhibitory mechanisms at the tumor site, which might explain poor results in the control of established tumors upon cancer vaccination. Several approaches are currently under development to tackle this problem. The success of anti-CTLA-4, anti-PD-L1 and anti-PD-1 antibodies in the treatment of various cancer types undoubtedly supports the idea of targeting inhibitory mechanisms that drive CTL dysfunctionality [16–18].
Here we evaluated the use of mRNA encoding the fusokine Fβ2 as a tool to deliver proteins with immunomodulating capacity. This work builds on the previous finding that mRNA can be delivered to the tumor and is engulfed by DCs [4, 5]. Based on these results we hypothesized that DCs could be exploited as ‘factories’ for the production of immunomodulating proteins. Moreover we prove the feasibility of using mRNA encoding a novel fusokine Fβ2, consisting of IFN-β and the ectodomain of the TGF-β receptor II. The combination of IFN-β and a TGF-β signaling antagonist was recently shown to have promising antitumor effects [19].
Type I IFNs have been described as stimulants of DC differentiation and maturation [20, 21]. In addition, it was recently demonstrated by Fuertes et al [22] that IFN-β plays an essential role in priming of T cells by attracting CD8α+ DCs to the tumor environment. This subset of DCs is important for the cross-presentation of tumor antigens to CD8+ T cells and lack of this population results in defective CD8+ T cell immune responses. Importantly, Van Lint et al proved that CD8α+ DCs are the main cell population involved in the uptake of mRNA in the tumor [5]. We demonstrated that Fβ2 induces DC activation. Surprisingly, Fβ2 matured DCs did not secrete enhanced IL-12 levels (data not shown). It has been shown that IL-6 secretion by DCs stimulated with type I IFNs plays a major role in the protection of T cell effector functions from the suppressive capacity of Treg cells [23]. In accordance with their maturation status, these DCs were able to stimulate antigen-specific CD8+ T cells.
Tumors have the capacity to take advantage of myeloid cell plasticity in order to skew them towards an immunosuppressive phenotype [24], which is confirmed by the presence of MDSCs within tumors. To study the effects of Fβ2 on MDSCs, we generated these cells in vitro as it was recently shown that large numbers of MDSCs that closely resemble MDSCs found within the tumor, could be induced [12, 13]. This resemblance was phenotypically based on inter alia the high expression of PD-L1, Arg-1 and CD86 and the lower expression of CD62L [13]. This is of a particular importance given difficulties in obtaining high numbers of pure MDSCs from tumors. Moreover it was also established that splenic MDSCs from tumor-bearing mice are phenotypically and functionally distinct from tumor MDSCs [24]. In accordance with previous reports, we demonstrated that in vitro generated MDSCs secrete high levels of TGF-β, a characteristic feature of in vivo tumor-infiltrating MDSCs [12, 13]. This local accumulation of TGF-β was shown to be accompanied with different immunosuppressive cell types, making TGF-β a core soluble factor in skewing the immunosuppressive balance [25–27]. In addition, type I IFNs have direct effects on the phenotype and function of myeloid cells. TGF-β antagonists and type I IFNs hamper MDSC-mediated suppressive functions [28, 29]. This effect could be linked to the induction of apoptosis as shown by Ellermeier et al [29]. MDSCs exposed to both TGF-β blockade and type I IFNs display an increased expression of CD11c, CD80 and a decrease in the granulocytic fraction [28, 29]. Interestingly, sca-1 up-regulation on MDSCs inversely correlates with their suppressive capacity [29], and our fusokine increases the expression of this marker on treated MDSCs.
Consistent with a previous study showing that MHC I on tumor cells is up-regulated upon treatment with IFN-β, we demonstrated that Fβ2 enhanced MHC I expression [30]. Loss of MHC I expression on cancer cells allows them to escape immunosurveillance, by making them invisible to CD8+ T cells [31]. By enhancing MHC I expression, Fβ2 showed to increase tumor cell recognition by CD8+ T cells. This was confirmed in a co-culture of CD8+ OT-I cells with tumor cells, exposed or not to the Fβ2 fusokine. However, the increase in tumor cell recognition was moderate, which might be explained by the enhanced PD-L1 expression on tumor cells treated with Fβ2 [32, 33]. Up-regulation of PD-L1 is a major obstacle in IFN-β based cancer immunotherapy as it can lead to T cell anergy and apoptosis through interaction with PD-1 [34, 35], but can also down-modulate T cell effector activities [36]. Targeting of this PD-1/PD-L1 interaction in type I IFN based therapy was shown to improve the therapeutic outcome [37, 38]. Importantly, we demonstrated that recognition of tumor cells pretreated with Fβ2 was augmented when T cells were exposed to Fβ2. Taken together, our in vitro findings point out that delivery of Fβ2 to the tumor environment might lead to a ‘CTL-supporting’ rather than a ‘CTL-suppressive’ environment. This was further confirmed by our in vivo data. We showed that intratumoral delivery of Fβ2 mRNA results in a delayed tumor outgrowth. The benefit of the Fβ2 mRNA therapy was dependent on CD8+ T cells, as the antitumor effect of the therapy was abrogated when mice were depleted of this cell population. We showed that the prolonged delay in growth was dependent on the repeated delivery of Fβ2 mRNA. The latter is consistent with the findings of Narumi et al [23], who indicated that low levels of type I IFNs were needed for at least 10 days to obtain successful antitumor immunity. Finally, we demonstrated in vivo that additional targeting of the PD-1/PD-L1 interaction improved the Fβ2 therapy, confirming the detrimental impact of PD-1 engagement on T cells in the tumor environment.
In conclusion, we established the feasibility of using mRNA encoding immunomodulating proteins to modulate the tumor environment. We provide evidence that Fβ2 works via a multistep process acting on DCs, MDSCs, CD8+ T cells as well as tumor cells. Its effect can be further enhanced through additional blockade of PD-1/PD-L1 interactions. This study supports therefore the paradigm that combining immunotherapeutic antitumor strategies might tackle the immunosuppressive tumor environment and lead to an improved outcome [39].
MATERIALS AND METHODS
Mice
6–12 week old female C57BL/6 mice (Charles River, Wilmington, USA) were housed and handled according to the regulations of the Animal Care Committee of the Vrije Universiteit Brussel (VUB). OT-I mice that carry a transgenic CD8+ T cell receptor (TCR) specific for the MHC class I-restricted ovalbumin (OVA257–264) peptide SIINFEKL were provided by B. Lambrecht (University of Ghent, Ghent, Belgium).
Cell lines and Reagents
The human embryonal kidney (HEK) 293T, the breast cancer 4T1, the melanoma B16-F0 and the T-cell lymphoma E.G7-OVA cell lines were obtained from the American Type Culture Collection (Rockville, Maryland, USA). The TGF-β reporter HEK293T cell line was previously described [40]. The mouse melanoma cell line MO4 and the lung carcinoma cell line TC-1 were kindly provided by K. Rock (University of Massachusetts Medical Center, Massachussetts, USA) and T.C. Wu (Johns Hopkins Medical Institution, Baltimore, Maryland, USA). The recombinant IFN-β was purchased from Biolegend. The neutralizing anti-TGF-β antibody was purchased from eBioscience (clone 1D11.16.8). The anti-CD8 (2.43), anti-PD1 (J43) and isotype control (Hamster IgG) monoclonal antibodies were purchased form BioXCell (New Hampshire, USA).
mRNA production
mRNA encoding Fβ2 (1485 base pairs) is comprised of a 5’ terminal cap, the genetic sequence encoding a fusion protein consisting of mouse IFN-β and the ectodomain of TGF-β receptor II, a 3’ RNA stabilizing sequence and a poly(A) tail. The pEtheRNA-Fβ2 vector used to produce the mRNA encoding the Fβ2 fusokine (produced by eTheRNA, Kortenberg, Belgium) was linearized at the 3’ end of the poly(A) tail using the restriction enzyme Bsp M1. The mRNA was produced by in vitro transcription as previously described [41]. The production of mRNA encoding the reporter eGFP or truncated nerve growth factor receptor (tNGFR) was previously described [41].
Electroporation of HEK293T cells with mRNA
Electroporation of HEK293T cells with mRNA was performed according to the protocol described for mRNA electroporation of DCs [42].
Intratumoral administration of mRNA
C57BL/6 mice were inoculated subcutaneously with 5 × 105 E.G7-OVA cells, 2 × 104 or 2 × 105 TC-1 cells. Tumors with a volume of 100-500 mm3 were injected intratumorally with 50 μl of the indicated amount of mRNA dissolved in 0.8 Lactated Ringer’s solution (0.8RL).
RNA isolation, cDNA synthesis and real-time PCR
Total RNA was isolated using the SV Total RNA Isolation System (Promega, Madison, USA). The extracted RNA was reverse transcribed into cDNA with the RevertAidTM H Minus First Strand cDNA Synthesis Kit (Fermentas Inc., Maryland, USA) using the random hexamer primers. Samples were subjected to real-time PCR analysis on an ABI PRISM 7700 Sequence Detection System (Applied Biosystems, Life Technologies, Ghent, Belgium). Primers for amplification of Mx1 were as follows: 5’-CGAGAAGTCCGGAAGCTTGT-3’ and 5’-CATGTACTGAGAAGTTTCATGCA-3’; Probe FAM-CAATTGCCGTCACCGTTCGTTTTCA -TAMRA (Eurogentec, Seraing, Belgium). Primers for the amplification of peptidylprolyl isomerase A (Ppia) were as follows: 5’-TTCACCTTCCCAAAGACCAC-3’ and 5’CAAACACAAACGGTTCCCAG-3’; Probe FAM-TGCTTGCCA/ZEN/TCCAGCCATTCAG-3IABkFQ (Integrated DNA Technologies, Leuven, Belgium).
In vitro generation of dendritic cells and myeloid-derived suppressor cells
Bone marrow-derived DCs were generated and activated with 20 ng/ml recombinant IFN-β or 100 ng/ml lipopolysaccharide (LPS) as described [42].
Bone marrow-derived MDSCs that resemble tumor MDSCs were generated as described [12]. Briefly, bone marrow cells were collected from femur and tibia, treated with red blood cell lyses buffer and washed with Dulbecco's Phosphate Buffered Saline (DPBS, Sigma-Aldrich, Zwijndrecht, Belgium). Bone marrow cells were cultured in 75% conditioned medium obtained from mouse GM-CSF secreting B16-F0 cells and 25% Iscove’s Modified Dulbecco’s Medium (IMDM, Lonza, Verviers, Belgium). The culture was supplemented with 10% fetal clone I (Harlan, Horst, the Netherlands), 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-Glutamine (Sigma-Aldrich). The MDSC cultures were refreshed at day 3 and 5 and ready for use at day 5–7.
In vitro OT-I stimulation assay
In vitro generated DCs were pulsed with 5 μg/ml SIINFEKL (Eurogentec) for 90 minutes at 37°C in a humidified incubator. Subsequently, DCs were co-cultured at a 1:10 ratio with CD8+ T lymphocytes that were enriched from the spleen of OT-I mice using the CD8a+ T cell Isolation Kit II (Miltenyi Biotec, Gladbach, Germany). Supernatants of HEK293T cells that were electroporated with mRNA encoding eGFP or Fβ2 were added to the co-cultures. On day 6, supernatants were collected and analyzed via ELISA for IFN-γ production (eBioScience, San Diego, California, USA). GolgiPlug-containing medium was added and intracytoplasmatic staining for IFN-γ was performed 24 hours later.
CD8+ T lymphocyte suppression assay
CD8+ T lymphocytes were isolated from the spleen of C57BL/6 mice using the CD8α+ T cell Isolation Kit II and were co-cultured with in vitro generated MDSCs at the indicated ratios in the presence of anti-CD3/anti-CD28 antibody coated microbeads (Invitrogen, Oslo, Norway) and 100 U/ml IL-2 (Peprotech, Rocky Hill, New Jersey, USA). Non-stimulated CD8+ T lymphocytes and CD8+ T lymphocytes stimulated in the absence of MDSCs were used as a control. Three days after the start of the co-culture the supernatants were collected and the production of IFN-γ was analyzed by ELISA (eBioScience).
CD107a assay
Isolated CD8+ OT-I cells were stimulated with anti-CD3/anti-CD28 coated microbeads and cultured in Fβ2 or control supernatants for three days. Simultaneously E.G7-OVA tumor cells were cultured in Fβ2 or control supernatants and EL4 cells were used as a negative control. After three days, the cells were thoroughly washed, mixed and co-cultured in the presence of fluorescein isothiocyanate (FITC)-conjugated CD107a antibodies (Becton Dickinson [BD], Erembodegem, Belgium). Four hours later, the cells were washed and additionally stained for CD3 and CD8, after which all samples were acquired on the LSRFortessa (BD).
Flow cytometry
Staining of cell surface markers was previously described [43]. The following antibodies were used: FITC-conjugated antibodies against CD11b (BD), AlexaFluor647 (AF647)-conjugated antibodies against CD11c and Ly6G (BioLegend, San Diego, California, USA), PE-conjugated antibodies against CD4 (BD), CD11b, MHC II (BioLegend), CD25, PD-L1 and PD-1 (eBioScience), PE-Cy7-conjugated antibodies against Ly6C (BioLegend), PercP-Cy5.5-conjugated antibodies against CD3 (BioLegend) and CD4 (BD), Pacific Blue-conjugated antibodies against CD3 (BioLegend), Brilliant Violet 605-conjugated antibodies against CD45 (BD), APC-conjugated antibodies against Foxp3 (eBioScience), APC-H7-conjugated antibodies against CD8 (BD), Brilliant Violet-conjugated antibodies against CD3 (BioLegend) and FITC-conjugated MHC class I (H2-Kb, prepared in-house), PE-Cy7-conjugated antibodies against IFN-γ (eBioScience). Data were collected using the FACSCanto or LSRFortessa Flow Cytometer (BD) and analysis was performed with FACSDiva (BD) or Flow Jo 7 software (Tree Star Inc., Ashland, Oregon, USA).
Enzyme-linked Immunosorbent Assay
Supernatants were screened in a sandwich ELISA for the presence of IL-6, IL-12, TNF-α or IFN-γ (eBioscience).
Colorimetric assay (MTS assay)
To assess the viability of MDSCs and tumor cell proliferation, an MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay was performed according to the manufacturer’s protocol (Promega, Wisconsin, USA).
Statistical analysis
Comparison of two data sets was performed using the unpaired student’s t-test. For the comparison of more than three groups, we performed a one-way ANOVA followed by a Bonferroni’s multiple comparison test. Number of asterisks in the figures indicates the level of statistical significance as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. The results are shown in a column graph or table as the mean ± standard error of the mean (SEM). Survival was visualized in a Kaplan-Meier plot. Differences in survival were analyzed by the log-rank test.
ACKNOWLEDGEMENTS
We thank Dr. Federico Sandoval for reviewing the manuscript and Petra Roman, Elsy Vaeremans, Xavier Debaere and Angelo Willems for their technical assistance.
GRANT SUPPORT
This work was supported by grants from the Interuniversity Attraction Poles Program-Belgian State-Belgian Science Policy, the National Cancer Plan of the Federal Ministry of Health, the Stichting tegen Kanker, the Vlaamse Liga tegen Kanker, an Integrated Project and an EU FP7-funded Network of Excellence, an IWT-TBM program, the FWO-Vlaanderen and the Scientific Fund Willy Gepts of the University Hospital Brussels. KVdJ and LB are supported by the IWT. KB is a postdoctoral fellow of the FWO-Vlaanderen. DE is funded by a Miguel Servet Fellowship from the Instituto de Salud Carlos III, Spain. TL received an overseas research scholarship, UCL, United Kingdom.
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
1. Frey AB. Signaling defects in anti-tumor T cells. 2008: 192–205.
2. Emeagi PU, Goyvaerts C, Maenhout S, Pen J, Thielemans K, Breckpot K. Lentiviral vectors: a versatile tool to fight cancer. Curr. Mol. Med. 2013; 13:602–25.
3. Marabelle a, Kohrt H, Caux C, Levy R. Intratumoral Immunization: A New Paradigm for Cancer Therapy. Clin. Cancer Res. 2014; 20:1747–1756.
4. Van Lint S, Renmans D, Benteyn D, Goyvaerts C, Maenhout S, Broos K, Du four S, Goethals L, Van der Jeught K, Bialkowski L, Heirman C, Thielemans K, Breckpot K. Intratumoral immunotherapy using mRNA encoding CD40 Ligand, active TLR4 and CD70. JNCI 2014. Under revision.
5. Van Lint S, Renmans D, Broos K, Dewitte H, Lentacker I, Heirman C, Breckpot K, Thielemans K. The ReNAissanCe of mRNA-based cancer therapy. Exp. Rev. Vaccines. Accepted 2014.
6. Van Lint S, Maenhout S, Benteyn D, Heirman C, Breckpot K, Thielemans K. Intratumoral and Intranodal Administration of Trimix and Antigen MRNA Results in Effective Anti-tumor Immunity. J. Immunother. 2012; 35:745–745.
7. Penafuerte C, Bautista-Lopez N, Bouchentouf M, Birman E, Forner K, Galipeau J. Novel TGF-beta antagonist inhibits tumor growth and angiogenesis by inducing IL-2 receptor-driven STAT1 activation. J. Immunol. 2011; 186:6933–6944.
8. Penafuerte C, Galipeau J. FIST, a sword and shield fusokine for cancer immunotherapy. Oncoimmunology. 2012; 1:224–226.
9. Terabe M, Ambrosino E, Takaku S, O'Konek JJ, Venzon D, Lonning S, McPherson JM, Berzofsky JA. Synergistic enhancement of CD8+ T cell-mediated tumor vaccine efficacy by an anti-transforming growth factor-beta monoclonal antibody. Clin. Cancer Res. 2009; 15:6560–9.
10. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011; 208:1989–2003.
11. Arce F, Breckpot K, Stephenson H, et al. Selective ERK activation differentiates mouse and human tolerogenic dendritic cells, expands antigen-specific regulatory T cells, and suppresses experimental inflammatory arthritis. Arthritis Rheum. 2011; 63:84–95.
12. Liechtenstein T, Perez-Janices N, Blanco-Luquin I, Goyvaerts C, Schwarze J, Dufait I, Lanna A, De Ridder M, Guerrero-Setas D, Breckpot K, Escors D. Anti-melanoma vaccines engineered to simultaneously modulate cytokine priming and silence PD-L1 characterized using ex vivo myeloid-derived suppressor cells as a readout of therapeutic efficacy. Oncoimmunology. 2014; 3:e29178.
13. Liechtenstein T, Perez-Janices N, Gato M, Caliendo F, Kochan G, Blanco-Luquin I, Van der Jeught K, Arce F, Guerrero-Setas, Fernandez-Irigoyen J, Santamaria E, Breckpot K, Escors D. A highly efficient tumorinfiltrating MDSC differentiation system for discovery of anti-neoplastic targets, which circumvents the need for tumor establishment in mice. Oncotarget; Adv. Online Publ. 2014.
14. Lizée G, Overwijk WW, Radvanyi L, Gao J, Sharma P, Hwu P. Harnessing the power of the immune system to target cancer. Annu. Rev. Med. 2013; 64:71–90.
15. Benteyn D, Van Nuffel AMT, Wilgenhof S, Corthals J, Heirman C, Neyns B, Thielemans K, Bonehill A. Characterization of CD8+ T-cell responses in the peripheral blood and skin injection sites of melanoma patients treated with mRNA electroporated autologous dendritic cells (TriMixDC-MEL). Biomed Res. Int. 2013; 2013:976383.
16. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012; 366:2443–2454.
17. Brahmer JR, Tykodi SS, Chow LQM, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012; 366:2455–2465.
18. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den EertWegh AJM, Lutzky J, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010; 363:711–723.
19. Kim S, Buchlis G, Fridlender ZG, Sun J, Kapoor V, Cheng G, Haas A, Cheung HK, Zhang X, Corbley M, Kaiser LR, Ling L, Albelda SM. Systemic blockade of transforming growth factor-beta signaling augments the efficacy of immunogene therapy. Cancer Res. 2008; 68:10247–10256.
20. Breckpot K, Corthals J, Bonehill A, Michiels A, Tuyaerts S, Aerts C, Heirman C, Thielemans K. Dendritic cells differentiated in the presence of IFN-beta and IL-3 are potent inducers of an antigen-specific CD8+ T cell response. J. Leukoc. Biol. 2005; 78:898–908.
21. Santini SM, Lapenta C, Logozzi M, Parlato S, Spada M, Di Pucchio T, Belardelli F. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J. Exp. Med. 2000; 191:1777–88.
22. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8alpha+ dendritic cells. J. Exp. Med. 2011; 208:2005–16.
23. Narumi K, Udagawa T, Kondoh A, Kobayashi A, Hara H, Ikarashi Y, Ohnami S, Takeshita F, Ochiya T, Okada T, Yamagishi M, Yoshida T, Aoki K. In vivo delivery of interferon-α gene enhances tumor immunity and suppresses immunotolerance in reconstituted lymphopenic hosts. Gene Ther. 2012; 19:34–48.
24. Maenhout SK, Van Lint S, Emeagi PU, Thielemans K, Aerts JL. Enhanced suppressive capacity of tumor-infiltrating myeloid-derived suppressor cells compared with their peripheral counterparts. Int. J. Cancer. 2014; 134:1077–90.
25. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006; 66:1123–31.
26. Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, Ouyang GF, Okada M, Balazs L, Adany R, Shibata T, Takami T. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J. Leukoc. Biol. 2008; 83:1136–44.
27. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J. Immunol. 2009; 182:4499–506.
28. Zoglmeier C, Bauer H, Nörenberg D, Wedekind G, Bittner P, Sandholzer N, Rapp M, Anz D, Endres S, Bourquin C. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin. Cancer Res. 2011; 17:1765–75.
29. Ellermeier J, Wei J, Duewell P, Hoves S, Stieg MR, Adunka T, Noerenberg D, Anders HJ, Mayr D, Poeck H, Hartmann G, Endres S, Schnurr M. Therapeutic efficacy of bifunctional siRNA combining TGF-β1 silencing with RIG-I activation in pancreatic cancer. Cancer Res. 2013; 73:1709–20.
30. Hofbauer GF, Geertsen R, Laine E, Burg G, Dummer R. Impact of interferons on the expression of melanoma-associated antigens in melanoma short-term cell cultures. Melanoma Res. 2001; 11:213–8.
31. Mapara MY, Sykes M. Tolerance and cancer: mechanisms of tumor evasion and strategies for breaking tolerance. J. Clin. Oncol. 2004; 22:1136–51.
32. Pen JJ, Keersmaecker BD, Heirman C, Corthals J, Liechtenstein T, Escors D, Thielemans K, Breckpot K. Interference with PD-L1/PD-1 co-stimulation during antigen presentation enhances the multifunctionality of antigen-specific T cells. Gene Ther. 2014; 21:262–271.
33. Liechtenstein T, Dufait I, Bricogne C, Lanna A, Pen J, Breckpot K, Escors D. PD-L1 / PD-1 Co-Stimulation, a Brake for T cell Activation and a T cell Differentiation Signal. J. Clin. Cell. Immunol. 2013:1–14.
34. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon V, Celis E, Chen L. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 2002; 8:793–800.
35. Shi F, Shi M, Zeng Z, Qi RZ, Liu ZW, Zhang JY, Yang YP, Tien P, Wang FS. PD-1 and PD-L1 upregulation promotes CD8(+) T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer. 2011; 128:887–96.
36. Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, Escors D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol. Med. 2011; 3:581–92.
37. Yang X, Zhang X, Fu ML, Weichselbaum RR, Gajewski TF, Guo Y, Fu YX. Targeting the Tumor Microenvironment with Interferon-β Bridges Innate and Adaptive Immune Responses. Cancer Cell. 2014; 25:37–48.
38. Bald T, Landsberg J, Lopez-Ramos D, Renn M, Glodde N, Jansen P, Gaffal E, Steitz J, Tolba R, Kalinke U, Limmer A, Jönsson G, Hölzel M, et al. Immune-cell poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014. doi:10.1158/2159-8290. CD-13-0458.
39. Van den Boorn JG, Hartmann G. Turning tumors into vaccines: co-opting the innate immune system. Immunity. 2013; 39:27–37.
40. Arce F, Breckpot K, Stephenson H, Karwacz K, Ehrenstein MR, Collins M, Escors D. Selective ERK Activation Differentiates Mouse and Human Tolerogenic Dendritic Cells, Expands Antigen-Specific Regulatory T Cells, and Suppresses Experimental Inflammatory Arthritis. Arthritis Rheum. 2011; 63:84–95.
41. Van Lint S, Goyvaerts C, Maenhout S, Goethals L, Disy A, Benteyn D, Pen J, Bonehill A, Heirman C, Breckpot K, Thielemans K. Preclinical Evaluation of TriMix and Antigen mRNA-Based Antitumor Therapy. Cancer Res. 2012; 72:1661–1671.
42. Van Meirvenne S, Straetman L, Heirman C, Dullaers M, De Greef C, Van Tendeloo V, Thielemans K. Efficient genetic modification of murine dendritic cells by electroporation with mRNA. Cancer Gene Ther. 2002; 9:787–97.
43. Breckpot K, Dullaers M, Bonehill A, van Meirvenne S, Heirman C, De Greef C, van der Bruggen P, Thielemans K. Lentivirally transduced dendritic cells as a tool for cancer immunotherapy. J. Gene Med. 2003; 5:654–667.