
INTRODUCTION
Breast cancer (BC, OMIM#114480) is the most prevalent cancer in the world and accounts for 14.7 million of mortality cases [1]. Approximately, 10% of BC cases have a genetic, inherited etiology referred as Hereditary Breast and Ovarian Cancer (HBOC) with an important impact in genetic counseling and cancer prevention interventions [2].
Pathogenic variants in BRCA1 and BRCA2 genes are the most prevalent in HBOC, collectively contributing to 15-25% of the cases [3]. Pathogenic alleles in these genes frequently have high penetrance and have been found in different populations, including countries from Latin America, such as Mexico, Colombia, Argentina, Chile, Brazil, among others [4, 5]. However, locus heterogeneity has been found in patients without mutations in BRCA1 and BRCA2 [6] together with additional pathogenic variants at lower frequency and in genes that confer moderate risk including CHEK2, PALB2, ATM, FANCM, ATR, STK11, RAD51C, BRIP1, CDH1, NF1, NBN and ERCC3 [7, 8]. The prevalence of these novel, moderate-risk genes in HBOC patients has recently started to be defined by massive parallel sequencing (MPS). Studies indicate that causal variants have very low frequency in most of the populations studied and are spread in a larger array of genes that remain unexplored (Table 1). Until now, the contribution of pathogenic variants in genes other than BRCA1 and BRCA2 has not been entirely defined and studies in Latin American populations are still scarce.
Table 1: Summary of gene panel studies in hereditary breast cancer
Year |
Genes |
Sample |
Country |
Methods |
BRCA Frequency1 |
Non-BRCA Frequency3 |
Non –BRCA Genes |
Ref. |
|---|---|---|---|---|---|---|---|---|
Proportion2 |
Proportion4 |
|||||||
2018 |
143 |
327 |
Mexico |
GeneRead (Qiagen) |
7.3% (24/327) |
8.5% (28/327) |
MSR1, ATM, ERCC3, FANCI, LIG4, PDE11A, ATR, FANCB, FANCC, FANCL, FANCM, RECQL4, SDHB, WRN, MLH1, NBN, RAD51C, CHEK2, FANCF, POLH and PTEN |
This study |
46.1 % (24/52) |
53.8% (28/52) |
|||||||
2018 |
35 |
120 |
Korea |
OncoRisk (Celemics) |
Negative |
7.5% (9/120) |
TP53, PALB2, BARD1 and MRE11A |
[41] |
2017 |
21 |
65,057 |
USA Multicentric |
Multiple |
2.8% (1874/65057) |
5.3% (3422/65057) |
CDH1, PTEN, TP53, ATM, BARD1, CHEK2, PALB2, and RAD51D. |
[37] |
35% (1874/5296) |
64% (3422/5296) |
|||||||
2017 |
10 |
581 |
Germany |
TruSight Cancer |
12.4% (72/581) |
5.5% (32/581) |
CHEK2, PALB2, NBN, RAD51C, ATM, TP53, RAD51D and MSH6 |
[42] |
69% (72/104) |
30%(32/104) |
|||||||
2017 |
16 |
453 |
Palestine |
SureSelect (Agilent) |
6.8% (31/453) |
6.6 (30/453) |
TP53 (founder mutation), ATM, CHEK2, BARD1, BRIP1, PALB2, MRE11A, PTEN, and XRCC2 |
[43] |
50.8% (31/61) |
49.1% (30/61) |
|||||||
2017 |
94 |
255 |
Italy |
Trusight Cancer (Illumina) |
22.3% (57/255) |
6.6% (17/255) |
PALB2, ATM, BRIP1, RAD51D, MSH6, PPM1D, RECQL4, ERCC3, TSC2, SLX4 and other Fanconi anemia genes |
[44] |
77% (57/74) |
22.9% (17/74) |
|||||||
2017 |
27 |
240 |
China |
BGI chip (Blackbird platform) |
5.8%(14/240) |
9.6% (23/240) |
MUTYH, CHEK2, PALB2, ATM, BARD1, NBN, RAD51C, TP53 and BRIP1 |
[45] |
38%(14/37) |
62% (23/37) |
|||||||
2017 |
25 |
85 |
Colombia |
MyRisk (Myriad) |
17.6% (15/85) |
4.7% (4/85) |
PALB2, ATM, MSH2 and PMS2 |
[46] |
79% (15/19) |
21% (4/19) |
|||||||
2016 |
29 |
10,030 |
USA |
SureSelect targeted capture |
2.54% (255/10,030) |
6.7% (682/10,030) |
MLH1, MSH2, MSH6, PMS2, EPCAM, APC, MUTYH, CDH1, PTEN, STK11, and TP53 |
[47] |
27% (255/937) |
73%(682/937) |
|||||||
2016 |
4 |
1427 479=Sanger 948=NGS |
China |
PCR design |
8.8% (126/1427) |
0.49%(7/1427) |
TP53 and PTEN |
[48] |
95%(126/133) |
5% (7/133) |
|||||||
2016 |
19 |
684 BRCA negative patients |
Australia |
Agilent Target Enrichment |
Negative |
11.1% (76/684) |
TP53, PALB2, ATM, CHEK2, CDH1, PTEN and STK11 Segregation study: CDH1, CHEK2, PALB2 and TP53 |
[49] |
2016 |
13 |
141 |
India |
Trusight Cancer |
4.9% (7/141) |
9.9% (14/141) |
ATM, BRIP1, CHEK2, PALB2, RAD51C and TP53 |
[50] |
33% (7/21) |
66% (14/21) |
|||||||
2016 |
68 |
133 |
Taiwan |
NimblGen capture (Roche) |
15% (20/133) |
7.5% (10/133) |
RAD50, TP53, ATM, BRIP1, FANCI, MSH2, MUTYH, and RAD51C |
[51] |
66% (20/30) |
33% (10/30) |
|||||||
2015 |
25 |
2,158 |
USA |
RainDance Thunderstorm emulsion polymerase chain reaction (PCR) system |
Cohort 1 9.3% (165/1781) Cohort 2 NA |
Cohort 1 |
CHEK2, ATM and PALB2 |
[52] |
2015 |
29 |
Total: 1,062 |
USA |
SureSelect and Integrated DNA Technologies |
9% (66/735) |
3.9% (26/735) |
ATM, PALB2, CHEK2, MLH1, MSH2, MSH6, and PMS2 |
[53] |
72% (66/92) |
28% (26/92) |
|||||||
2015 |
29 (Invitae) 25 (Myriad) |
1,046 BRCA negative patients |
USA |
Hereditary Cancer Syndromes test (Invitae) MyRisk test (Myriad Genetics) |
Negative |
3.8% (40/1046) |
CHEK2, ATM, PALB2 |
[12] |
2015 |
94 genes and 284 SNPs |
620 |
Germany |
TruSight (Illumina) and Haloplex |
9.2% (57/620) |
2.9% (18/620) |
CHEK2, ATM, CDH1, NBN, PALB2 and TP53 |
[54] |
76% (57/75) |
24% (18/75) |
|||||||
2015 |
25 |
155 |
Japan |
AmpliSeq Library Kit 2.0 |
7% (11/155) |
1.9% (3/155) |
ATM, MRE11A and MSH6 |
[55] |
78.5% (11/14) |
21.5% (3/14) |
1Absolute frequency of patients with a pathogenic variant in BRCA1 and BRCA2.
2 Proportion of pathogenic variants in BRCA1 and BRCA2 relative to other genes.
3Absolute frequency of patients with a pathogenic variant in non-BRCA cancer-associated genes.
4Proportion of pathogenic variants in non-BRCA cancer-associated genes relative to BRCA1 and BRCA2.
In recent years, important efforts to define common susceptibility loci for breast cancer in large cohorts have identified more than 90 SNPs, which predispose to this disease [9]. However, the risk conferred by these common susceptibility loci can explain up to 14% of hereditary breast cancer aggregation in the European population [10]. Additional SNPs remain to be discovered and association studies need to be conducted in other populations to better define the prevalence and clinical relevance of novel pathogenic alleles [11]. The identification of rare or population specific, high/moderate-risk pathogenic alleles could be translated in better molecular diagnosis, personalized risk assessment and treatment [12].
To determine the prevalence of pathogenic variants in cancer predisposing genes in Mexican patients, an understudied mixed population, and the potential benefit for molecular diagnosis with gene panel testing, we performed a germline genetic analysis in 327 patients with a clinical indication of HBOC. We analyzed all cases using a panel of 143 genes associated with different inherited oncologic diseases, by massive parallel sequencing.
RESULTS
Clinical and epidemiological description of breast cancer cases
Clinical and pathological characteristics of a total of 300 sequenced cases diagnosed with breast cancer are described in Table 2. Mean age at diagnosis was 41 years (range 23-69, SD: 7.3). Seventy one percent of cases had family history of cancer, 85% reported at least one pregnancy and the average parity was 3 children (SD: 1.6), 60% never use of oral contraceptives and 93% reported not being current alcohol drinkers. Importantly, sixty two percent of all cases were overweight, obese or extremely obese. Mutational status was defined as the presence of a pathogenic or likely pathogenic variant (ACMG classification) in any of the 143 genes evaluated. Fifteen percent of this group had a pathogenic or likely pathogenic variant.
Table 2: Clinical and epidemiological characteristics of 300 women with breast cancer and 27 familial breast cancer risk women
Epidemiological Characteristics |
High risk individuals |
Cancer cases |
Clinical Characteristics |
Cancer cases |
|||
|---|---|---|---|---|---|---|---|
n 27 |
(%) (100) |
n 300 |
(%) (100) |
n 300 |
(%) (100) |
||
Age |
Histopathological Subtype |
||||||
20-29 years |
6 |
(22.2) |
13 |
(4.3) |
DCIS |
43 |
(14.4) |
30-39 years |
5 |
(18.5) |
96 |
(32.0) |
LCIS |
18 |
(6.0) |
40-49 years |
10 |
(37.1) |
146 |
(48.7) |
IDC |
189 |
(63.0) |
50-59 years |
2 |
(7.4) |
21 |
(7.0) |
ILC |
16 |
(5.3) |
60-69 years |
4 |
(14.8) |
8 |
(2.7) |
MC |
3 |
(1.0) |
Missing |
0 |
(0.0) |
16 |
(5.3) |
Missing |
31 |
(10.3) |
BMI |
Stage |
||||||
Underweight (<18.5) |
1 |
(3.7) |
1 |
(0.3) |
I |
51 |
(17.0) |
Normal weight (18.5<25) |
11 |
(40.8) |
107 |
(35.7) |
II |
115 |
(38.3) |
Overweight (25.0<30) |
9 |
(33.3) |
118 |
(39.3) |
III |
86 |
(28.7) |
Obese (30.0<40) |
5 |
(18.5) |
66 |
(22.0) |
IV |
10 |
(3.3) |
Extreme obese (>40) |
0 |
(0.0) |
3 |
(1.0) |
Missing |
38 |
(12.7) |
Missing |
1 |
(3.7) |
5 |
(1.7) |
ER status |
||
Current alcohol drinker |
Negative |
38 |
(12.7) |
||||
No |
25 |
(92.6) |
278 |
(92.7) |
Positive |
20 |
(6.6) |
Yes |
2 |
(7.4) |
16 |
(5.3) |
Missing |
242 |
(80.7) |
Missing |
0 |
(0.0) |
6 |
(2.0) |
PR status |
||
Current tobacco smoker |
Negative |
135 |
(45.0) |
||||
No |
0 |
(0.0) |
84 |
(28.0) |
Positive |
22 |
(7.3) |
Yes |
0 |
(0.0) |
74 |
(24.7) |
Missing |
143 |
(47.7) |
Missing |
27 |
(100) |
142 |
(47.3) |
HER2 status |
||
Pregnancy |
Negative |
7 |
(2.3) |
||||
Yes |
18 |
(66.7) |
256 |
(85.4) |
Positive |
45 |
(15.0) |
No |
8 |
(29.6) |
43 |
(14.3) |
Missing |
248 |
(82.7) |
Missing |
1 |
(3.7) |
1 |
(0.3) |
Mutational status |
||
Ever use of oral contraceptives |
Non-mutated |
254 |
(84.7) |
||||
Yes |
7 |
(25.9) |
115 |
(38.3) |
Mutated |
46 |
(15.3) |
No |
20 |
(74.1) |
179 |
(59.7) |
|||
Missing |
0 |
(0.0) |
6 |
(2.0) |
|||
Family History of cancer |
|||||||
Yes |
27 |
(100) |
214 |
(71.3) |
|||
No |
0 |
(0.0) |
80 |
(26.7) |
|||
Missing |
0 |
(0.0) |
6 |
(2.0) |
|||
DCIS= Ductal carcinoma in situ, LCIS=Lobular carcinoma in situ, IDC= Invasive ductal carcinoma, ILC=Invasive lobular carcinoma, MC= Medullary carcinoma. ER= Estrogen receptor, PR= Progesterone receptor. Mutational status is based on the presence of a pathogenic mutation in any of the 143 genes.
Age at diagnosis was the only epidemiological characteristic statistically associated with mutational status (p=0.04). No association was found between stage, histological subtype, hormone receptor status and mutational status in cases. Analysis by gene showed no association between presence of a mutation and a clinical or pathological characteristic (data not shown).
Patients in the older age group (60-69 years) were characterized by presenting early stage tumors (I/II) and absence of mutations.
Pathogenic variants in the breast cancer cases
In the group diagnosed with breast cancer (300 cases), we detected 46 pathogenic or likely pathogenic variants in 46 patients (Figure 1; Supplementary Table 1), including 22 frameshift changes, 13 stop gain/loss mutations and 4 splicing variants. Fifty-six percent (26/46) of mutations detected were already reported in ClinVar as pathogenic. Twenty six percent (12/46) of the pathogenic variants were recurrent, and no genetic alteration was found in 73.6% (221/300) of the patients (Figure 1).
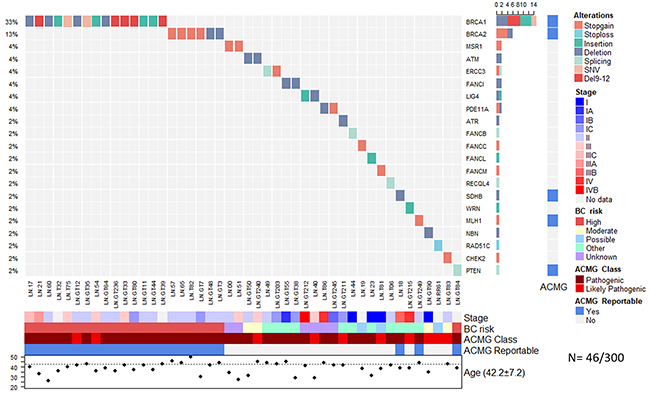
Figure 1: Allelic distribution of the pathogenic variants in patients with cancer. The grid panel depicts the pathogenic mutations found in each patient color-coded for each type. Right panel: gene reportable by the suggestion of the ACMG (light blue=yes, gray=no). Bottom axis: patient ID. Left axis: relative frequency of mutations per gene. Right axis: gene mutated. Right bar plot: absolute frequency and type of pathogenic mutation per gene. Bottom panel: stage (I-IVB); risk associated with a pathogenic variant; ACMG variant class (pathogenic, likely pathogenic); gene reportable by the suggestion of the ACMG (light blue=yes, gray=no); gender (pink=female, blue=male); age distribution.
Notably, BRCA1 (5%, 15/300), BRCA2 (2%, 6/300) and PTEN (0.3%, 1/300) were the only high-risk mutated genes associated with HBOC (Figure 1). No Ashkenazi founder mutations were detected and no pathogenic variants were found in other high-risk HBOC genes such as TP53, CDH1, PALB2 and STK11. Pathogenic alterations in moderate-risk genes were found in ATM (0.6%, 2/300), CHEK2 (0.3% 1/300) and NBN (0.3%) (Supplementary Table 1) (Figure 1).
Pathogenic variants in familial breast cancer risk individuals without cancer diagnosis
In the group of 27 individuals without cancer and with suspicion of familial breast cancer risk, we found pathogenic variants in 6 individuals (22%) (Figure 2). The affected genes were BRCA1 (2/27), BRCA2 (1/27), FANCF (1/27), PDE11A (1/27) and POLH (1/27).
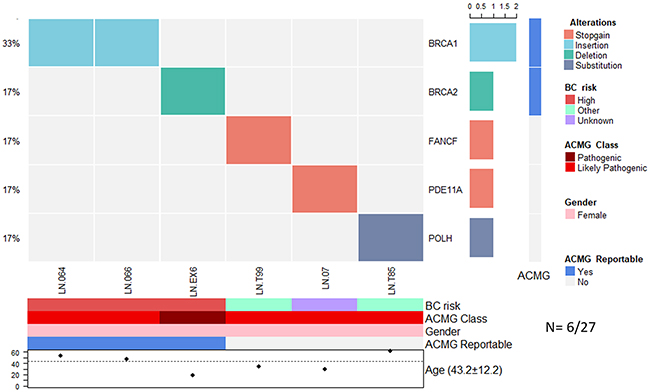
Figure 2: Allelic distribution of the pathogenic variants in high-risk patients with a severe family history of cancer. The grid panel depicts the pathogenic mutations found in each patient color-coded for each type. Bottom axis: patient ID. Left axis: relative frequency of mutations per gene. Right axis: gene mutated. Right bar plot: absolute frequency and type of pathogenic mutation per gene. Right panel: gene reportable by the suggestion of the ACMG (light blue=yes, gray=no). Bottom panel: risk associated with a pathogenic variant; ACMG variant class (pathogenic, likely pathogenic); gender (pink=female, blue=male); gene reportable by the suggestion of the ACMG (light blue=yes, gray=no); age distribution.
Recurrent mutations in BRCA1 and BRCA2 in both groups
There were 2 recurrent pathogenic alleles in BRCA1, including p.G228fs in five individuals (29%, 5/17), and the Mexican founder mutation in BRCA1 (deletion of exons 9-12), also present in 29% (5/17) (Figure 1, Figure 2, Figure 3). One recurrent mutation was found in BRCA2: p.R2494X, which was detected in 2 patients (Supplementry Table 1).
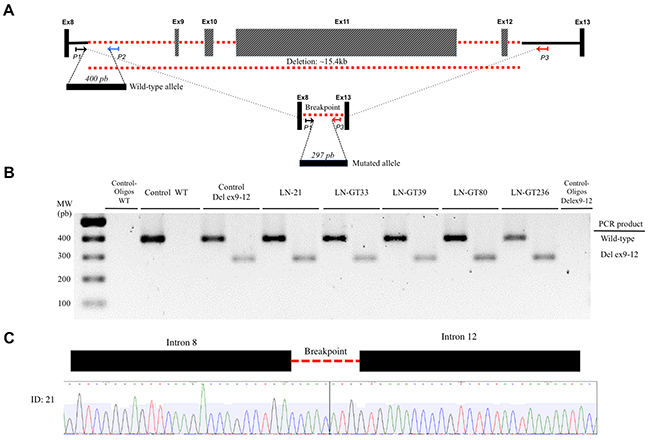
Figure 3: Detection of BRCA1 deletion of exons 9-12. (A) The locus of exons BRCA1 8 to 13 is indicated. Exons (not to scale) are depicted as boxes and introns as lines, where the discontinuous red line indicates the deleted exons 9-12. The location and orientation of the primers used for amplification of the wild-type (P1, P2) and the mutant alleles (P1, P3) are shown. The PCR products for both amplicons are depicted as horizontal boxes, with their respective number of bp. (B) The resolved PCR products of the patients with the deletion are shown, along with the wild-type and negative controls. (C) The electropherogram of the sequence shows the intron-intron junction in the deletion.
Pathogenic variants in genes with unknown risk in breast cancer
Deleterious variants in low risk HBOC genes were found in two cancer patients in the genes FANCI (0.6%, 2/300), ERCC3 (0.6%), and in one patient in the genes ATR, FANCB, FANCC, FANCF, FANCL, FANCM, MLH1, RAD51C, POLH, RECQL4, SDHB and WRN (0.3%, 1/300) (Figure 1, Figure 2). Furthermore, analysis of genes associated with risk to other inherited neoplastic syndromes different to HBOC identified heterozygous pathogenic variants for MSR1 (4%, 2/52), LIG4 (4% 2/52) and PDE11A (6% 3/52) in the affected women in both groups (Figure 1, Figure 2, Table 3).
Table 3: Syndromes associated with the pathogenic variants detected
Gene |
Frequency |
Syndromes (OMIM) |
Breast cancer risk |
Inherited pattern |
Signaling pathways |
Reportable in ACMG* |
|---|---|---|---|---|---|---|
BRCA1 |
17 |
Hereditary Breast and Ovarian Cancer |
High |
AD |
Double strand damage (HR) |
Yes |
BRCA2 |
11 |
Fanconi Anemia / Hereditary Breast and Ovarian Cancer/ Familiar Pancreatic Cancer / Hereditary Prostate Cancer |
High |
AD/AR |
Double strand damage (HR) |
Yes |
PDE11A |
3 |
Pigmented nodular adrenocortical disease |
Novel |
AD |
Catalyze the hydrolysis of cAMP and cGMP, Metabolism of purines |
No |
ATM |
2 |
Susceptibility to breast cancer / Ataxia Telangiectasia |
Moderate |
AD/AR |
Double strand damage (HR) |
No |
ERCC3 |
2 |
Xeroderma Pigmentosum |
Not established |
AR |
Transcription initiation of RNA Pol II |
No |
FANCI |
2 |
Fanconi Anemia |
Not established |
AR |
Anemia Fanconi Pathway and Double strand damage response |
No |
LIG4 |
2 |
LIG4 Syndrome |
Novel |
AR |
Nucleotide excision DNA repair |
No |
MSR1 |
2 |
Hereditary Barret Esophagus/ Esophagus carcinoma / Hereditary prostate cancer |
Novel |
AD |
Vesicle-mediated transport and AGE/RAGE pathway |
No |
ATR |
1 |
Cutaneous telangiectasia and familial cancer syndrome / Seckel syndrome 1 |
Not established |
AD/AR |
Cell cycle checkpoint regulator |
No |
CHEK2 |
1 |
Li-Fraumeni syndrome /Susceptibility to breast, colorectal and prostate cancer |
Moderate |
AD |
Cell cycle checkpoint regulator |
No |
FANCB |
1 |
Fanconi Anemia |
Not established |
XLR |
Anemia Fanconi Pathway and Double strand damage response |
No |
FANCC |
1 |
Fanconi Anemia |
Not established |
AD/AR |
Anemia Fanconi Pathway |
No |
FANCF |
1 |
Fanconi Anemia |
Not established |
AR |
Anemia Fanconi Pathway |
No |
FANCL |
1 |
Fanconi Anemia |
Not established |
AR |
Anemia Fanconi Pathway, DNA damage, Cell cycle checkpoint regulator |
No |
FANCM |
1 |
Fanconi Anemia |
Not established |
AD/AR |
Anemia Fanconi Pathway, Double strand DNA damage |
No |
MLH1 |
1 |
Hereditary nonpolyposis colorectal cancer, type 2 / Mismatch repair cancer syndrome / Muir-Torre syndrome |
Not established |
AD/AR |
Mismatch repair system |
Yes |
NBN |
1 |
Aplastic Anemia / Acute lymphoblastic Leukemia / Nijmegen breakage syndrome |
Moderate |
AD/AR |
Double strand damage respond in DNA |
No |
POLH |
1 |
Xeroderma pigmentosum |
Not established |
AR |
Homologous DNA recombination and strand interchange |
No |
PTEN |
1 |
Bannayan-Riley-Ruvalcaba syndrome/ Cowden syndrome |
High risk |
AD |
Antagonizes the PI3K signaling pathway and negatively regulates the MAPK pathway |
Yes |
RAD51C |
1 |
Fanconi Anemia / Susceptibility to breast and ovarian cancer |
Not established |
AD/AR |
Double strand damage (HR) |
No |
RECQL4 |
1 |
Rothmund-Thompson Syndrome |
Not established |
AR |
DNA Damage response |
No |
SDHB |
1 |
Carney-Stratakis Syndrome |
Not established |
AD |
Metabolism ( Krebs Cycle) |
Yes |
WRN |
1 |
Werner Syndrome |
Not established |
AR |
C strand synthesis in telomere and cell cycle checkpoint |
No |
*From reference [13].
Description of variants with unknown clinical significance by phosphorylation site disruption analysis
We found 38 VUS in 21 genes, 4 of which were found in homozygosity (Supplementry Table 2). These VUS have MAF < 0.001 in ExAC, and 1000 Genomes databases and not all of them are classified as VUS in ClinVar. To better define the potential effect of VUS in gene functionality, we evaluated the impact of the amino acid change in the context of phosphorylation sites. There was no enrichment in these sites for the occurrence of VUS. However, changes that potentially affect the phosphorylation regulation were found in the AIP and APC genes (Supplementry Figure 2). The changes affected the FKBP C domain and APC basic domain of AIP and APC, respectively.
DISCUSSION
In this work, we evaluated genetic alterations in an expanded panel of 143 genes associated with oncologic inherited diseases, including breast, colon, gastric, among others, by MPS in two groups of high-risk HBOC patients. This is the first study in a Latin American population that analyzes a large cancer risk gene panel by MPS. Overall, in all the individuals included in this study, we detected pathogenic variants in 16% (52/327), including 7% (24/327) of variants in BRCA1/2, and 8% (28/327) in genes other than BRCA1/2 (Table 1). These mutations were found in 21 genes previously associated with more than 25 inherited conditions related to cancer (Table 3). Globally, 8% (27/327) of patients had a pathogenic mutation in one of the genes categorized by the American College of Medical Genetics ACMG as a secondary finding with clinical validity and utility to improve medical outcome [13]. Half of the pathogenic variants, 50% (26/52), have not been reported before in any Latin-American population, which highlights the current need to expand the evaluation of the genetic diversity of under-studied, mixed populations such as Mexicans and its association to HBOC. These results also confirm the high level of locus heterogeneity that has been described for HBOC [6, 12, 14] (Table 1).
Age at diagnosis was the only epidemiological or clinical variable associated to the presence of a pathogenic mutation in breast cancer cases, supporting the NCCN criteria for HBOC (https://www.nccn.org/). Additional risk factors modifying penetrance in BRCA1/2 mutation carriers have been identified in several studies [15]. However, to our knowledge there is no information on modifiable risk factors for HBOC mutation carriers in Latin America available to compare to findings from our study. A large meta-analysis showed that the loss of at least 10 pounds of body weight before the age of 30 was associated to a reduced risk of breast cancer between 30 to 49 years in BRCA1 mutation carriers [15]. Interestingly, in our analysis around 60% of BC cases were overweight or obese at time of diagnosis but only 15% of those had a pathogenic mutation in any of the 143 genes evaluated. Larger prospective studies on HBOC mutation carriers that incorporate information on a variety of environmental exposures, ancestry and lifestyle factors are required in order to identify modifying risk factors in Latin America.
Pathogenic alterations in the HBOC moderate-risk genes ATM, CHEK2 and NBN, were found in a frequency of 0.6%, 0.3%, and 0.3%, respectively. Additionally, we found 7 monoallelic pathogenic variants (2%, 7/327) in 6 genes (besides BRCA1/2) of the interstrand crosslink DNA repair Fanconi anemia pathway (FANCB, FANCC, FANCF, FANCI, FANCL, FANCM) and 1 in RAD51C, a Fanconi-like phenotype gene [16]. The allelic frequency of these variants in the Latin American population spans the 0-0.0015 interval (ExAC). These results confirm findings from other multi-gene panel studies in HBOC patients from other populations, which found mutations in Fanconi genes (Table 1). Although, strong evidence regarding the contribution of mutations in some Fanconi anemia genes to HBOC is still limited [17, 18], our results provide additional support for this potential association.
Interestingly, we detected pathogenic variants in MSR1, LIG4 and PDE11A, genes not previously associated with HBOC, both in BC patients and high-risk cases. Moreover, the mutation MSR1 p.R293X was found in two unrelated patients. This mutation has been associated with Barrett’s esophagus and esophageal adenocarcinoma in European families [19], and with hereditary prostate cancer [20]; although contradictory results also exist [21, 22]. The mutation p.R505fs in the Non-homologous end joining (NHEJ) ligase LIG4, found in one patient, has an allele frequency of 0.0000247 in ExAC. This mutation is located in the ATP dependent DNA ligase C terminal region, and produces a protein lacking both of BRCT-I and BRCT-II domains that are required for chromatin binding [23], abrogating LIG4 function. Recently, germline mutations in LIG4 have been suggested to predispose to diffuse large B-cell lymphomas [24]. Moreover, alterations in LIG4 sensitize cell lines to ionizing radiation, causing immunodeficiency and delay in growth and development in homozygous or compound heterozygous carriers (OMIM#606593) [25]. Given the biochemical function of LIG4 in NHEJ and the low prevalence of its mutations, germline monoallelic mutations could influence HBOC risk, although additional studies are needed to establish this association. In one patient we found a frameshift pathogenic variant p.G57fs in PDE11A, a gene previously associated with different neoplasms including Carney multiple neoplasia complex, prostate cancer and testicular germ cell tumors [26, 27].
Overall, 10.8% of patients that were negative for a pathogenic mutation in any of the 143 genes tested had VUS defined following the ACMG criteria. VUS constitute a universal concern in cancer genetics diagnostic settings. The risk conferred by VUS must be addressed by generating more evidence of their allelic frequency in different populations, and by conducting co-segregation analyses, as well as efforts to define their function at protein level using experimental models. Remarkably, we found 2 VUS -AIP p.V49M in homozygosis and APC p.S2535G in heterozygosis- that potentially affect the phosphorylation regulation of the protein. In fact, mutations in the chaperone aryl hydrocarbon receptor-interacting protein (AIP) have been found in familial cases of pituitary adenomas [28]. Experimental in vitro evidence showed that AIP V49M interferes with AIP activity and stability [29]. APC p.S2535G was predicted to disrupt a phosphorylation site in the protein basic domain, which interacts with the microtubules [30]. Neither this amino acid change nor any other change in this position has been reported in COSMIC, OMIM or ClinVar. Consequently, further functional studies are needed to determine the impact of the APC p.S2535G variant.
On the other hand, seventy-four percent of all patients did not harbor alterations in any of the 143 genes studied. Even though we tested the deletion of exons 9-12 in BRCA1, a mutation with a founder effect and the highest frequency reported in Mexican population [31], additional larger rearrangements, undetectable by MPS could account for the lack of mutation detection in this group of patients. The frequency of large genomic rearrangements in BRCA1/2 varies considerably among populations but higher frequencies are related to founder effect variants [32]. In our study, we found that almost one out of three patients (5/17) with BRCA1 pathogenic variants had the Mexican founder mutation (deletion of exons 9-12), which highlights the additional value of evaluating this alteration through a rapid test, such as the one we used, for screening Mexican cases with suspicion of HBOC. It is also possible that patients who tested negative for any of the genes evaluated may harbor variants in noncoding regions that we did not analyzed. Additional mechanisms of pathogenesis that may play a role in susceptibility to BC might include pathogenic variants affecting splicing mechanisms that disrupt RNA-binding protein (RBBSs) and splicing regulatory (SRBSs) binding sites as well as transcription factor binding site disruption or promoter mutations [14, 33].
An additional limitation of this study is the lack of population paired-controls, which could lead to the wrong attribution of common, non-deleterious variants as pathogenic, and which also could eliminate an important amount of common VUS present in this population. It has been described that each person carries up to 100 loss-of-function variants, thirty of which could be in homozygosis, and these are not necessarily disease-causing variants [34, 35]. To exclude for non-pathogenic natural variation, we used the largest international databases (ExAC, 1000G, ESP) available in our filtering algorithm. These repositories contain whole genome and exome information from a large number (N= 60,706) of sequenced individuals, with broad ancestral diversity, including 5,789 Latinos (2,254 males and 3,535 females). It has been reported that ExAC is not overrepresented for pathogenic variants, which supports its use to estimate normal variation [36]. We estimate that this strategy may have helped to palliate the effect of lack of paired-controls on our study.
Future germline analyses of cohort studies and population based case-control studies specially focused on underrepresented populations such as the Latin American region and including women with BC with and without HBOC susceptibility are necessary to validate our results.
Overall, we found 54% of pathogenic variants in genes other than BRCA1 and BRCA2. Consistently, other studies using a panel sequencing approach have found a proportion of 5-64% of pathogenic variants in non-BRCA genes (Table 1). The rate of pathogenic variant detection in these studies tend to be dependent on the total number of genes analyzed, rather than the number of individuals studied. For example, the largest study evaluated a panel of 21 genes in 65,057 patients with breast cancer and found pathogenic variants in 8 non-BRCA genes [37], and our study, using a panel of 143 genes in 327 individuals with high risk and breast cancer, detected pathogenic variants in 21 non-BRCA cancer-associated genes. Therefore, to further elucidate the wider variation in genes with pathogenic variants that influence HBOC, more studies that investigate larger panels, or ultimately the whole exome or genome, are needed. Likewise, penetrance and polygenic analyses of rare and common variation will aid to provide a more accurate assessment of genetic cancer risk in the clinical setting.
CONCLUSIONS
Our results show that 16% of the patients with suspicion of HBOC carried a pathogenic mutation in at least one of the 143 genes tested. Fifty-four percent of all pathogenic alterations were not present in BRCA1 and BRCA2, highlighting the locus heterogeneity of this disease. We found 10% of patients with VUS, which require further studies to establish their significance. The genetic information derived from this study could guide the treatment, appropriate follow-up and prophylactic measures in these families and our findings emphasize the benefit of gene panel sequencing service for candidate patients. Although currently clinical guidelines for patients with the pathogenic mutations detected in several of these genes are lacking, the detection of these variants together with a suggestive family history may warrant for a post-test management change, including close follow-up and monitoring. Future efforts will collectively provide enough evidence of the clinical impact of these variants and will foster the development of consensus population-specific guidelines for clinical management.
MATERIALS AND METHODS
Study population and data collection
A total of 327 patients were enrolled based on criteria established in the Genetic/Familial High-Risk Assessment: Breast and Ovarian of the National Comprehensive Cancer Network (NCCN) guidelines, version 2.2015 (https://www.nccn.org/). A transversal series of 300 Mexican female patients diagnosed with primary breast cancer (stages I-IV) were recruited at 4 centers from different states in Mexico: Instituto de Salud del Estado de México, Instituto Estatal de Cancerología de Guerrero, Centro de Investigación Biomédica, Torreón, Coahuila and Hospital de Oncología del CMN Siglo XXI de la Ciudad de México. A second group of 27 individuals without cancer diagnosis who meet NCCN criteria for HBOC susceptibility were additionally included. The protocol was approved by the Ethics Committee of each center. Patients provided written informed consent for participation in this study, and their samples were anonymized and sent to the Laboratorio Nacional en Salud: Diagnóstico Molecular y Efecto Ambiental en Enfermedades Crónico-Degenerativas, Facultad de Estudios Superiores Iztacala, UNAM. Epidemiological and clinical information was obtained from hospital records when available. After revision of their clinical records and age of onset (<45 years), all patients with suspicion of HBOC were invited to participate in this study. After a complete and detailed explanation of the study and written informed consent, a questionnaire of enrollment was used to evaluate the fulfillment of inclusion criteria.
Sample preparation and DNA extraction
For all patients enrolled, 4 mL samples of blood were collected and stored locally at -80°C. The period between sample collection and freezing never exceeded 36 hours. Peripheral blood DNA was extracted with the DNeasy Blood & Tissue Kit (Qiagen) following manufacturer’s instructions. DNA concentration was quantified with the Qubit dsDNA HS Assay Kit (Invitrogen) and the integrity and purity of the material was verified by agarose gel electrophoresis and spectrophotometry, respectively.
Library preparation and Massive parallel sequencing
Peripheral Blood DNA was used for library preparation with the GeneRead Cancer Predisposition V2 Kit (Qiagen), which targets 143 genes, which loss of function is a well-known mechanism associated with 88 inherited oncologic diseases based on data from the College of American Pathologists (CAP) guidelines, NCCN guidelines, late-stage clinical trials, The Cancer Genome Atlas (TCGA), and Ingenuity® Knowledge Base. The amplification is divided in 4-pool PCR reactions with a total of 6582 amplicons. Pair-end sequencing was performed with the MiSeq System platform (Illumina). Briefly, 40±2.5 ng of DNA were amplified with the GeneRead DNAseq Gene Panel Kit (Qiagen) and purified with Agencourt AMPure XP magnetic Beads (Beckman Coulter). The amplified fragments were end-repaired, dA-tailed and the adapter GeneRead Adapter 1 Set plex (Qiagen) was ligated using the GeneRead DNA Library I Core Kit. Amplified segments were then size-selected (200-300 bp) using Agencourt AMPure XP magnetic beads (Beckman Coulter). New England Biolabs barcodes were incorporated by PCR amplification in 10 PCR cycles. The libraries were diluted to 4.0 nM and were pooled in batches of 60-80 samples. Library quality was evaluated by DNA quantification with Qubit after size-selection, and by Bioanalyzer (Agilent) profiling with the High Sensitivity DNA Kit after adaptor-ligated molecules amplification and final library pooling. Pooled barcoded libraries were diluted to 12-14 pM and sequenced with a MiSeq Reagent Kit V2 2X150 cycles (Illumina) to reach a theoretical average coverage of 100X for each sample.
Pathogenic variant detection
Variant detection, alignment and variant calling were performed with BWA and GATK (Broad Institute). FastQC files were aligned to the human genome reference hg19 with BWA-MEM; indels were realigned and bases recalibrated. Adaptors were soft-clipped and reads with <20 bp were eliminated. The overall (327) mean sequencing depth of all samples was 70.3X (SD: 21.35) with a range 30-156X, excluding one sample with depth 20X (Supplementry Figure 1). Variant calling was done with HaplotypeCaller (Broad Institute). Variants were annotated with ANNOVAR and InterVar [38, 39]. Mutation description follows Human Genome Variation Society (HGVS) nomenclature (http://www.hgvs.org/). Variant classification followed the five-tier criteria of the American College of Medical Genetics and Genomics (ACMG) [40] and was manually curated. We excluded variants that were synonymous, with depth <5.0X or with mutant allele fraction <20% and those present in homopolymeric tracts >8 bp. All splicing and null variants (stop-gain/loss, frameshift indels) and missense variants defined as pathogenic in ClinVar were considered unequivocally pathogenic (https://www.ncbi.nlm.nih.gov/clinvar). Null variants present at the 3’ extreme end of the gene that were reported as conflicting in ClinVar were classified as unknown clinical significance (VUS). Minor allelic frequency <0.001 in either the ExAC database, 1000 Genomes (1000G) project or the Exome Sequencing Project (ESP6500) was used to capture rare, potentially pathogenic null and missense variants. Low frequency (<0.001) missense variants predicted as deleterious alleles by SIFT or PolyPhen-2 but with no further evidence of pathogenicity in vitro/vivo or clinically were classified as VUS. All filtered variants were manually curated by inspection of the BAM files with the IGV software (Broad Institute). All pathogenic variants were confirmed by two independent assays of Sanger sequencing. Variants in BRCA1 and BRCA2 were further assessed in the Huntsman Cancer Institute Breast Cancer Genes Prior Probabilities site (http://priors.hci.utah.edu/PRIORS/index.php) to evaluate their potential impact. Variants in MLH1, MSH2, MSH6, PMS2 were also investigated in the Leiden Open Variation Database (http://hci-lovd.hci.utah.edu/home.php).
Detection of exon 9-12 deletion in BRCA1
The deletion in exons 9-12 founder mutation was detected by PCR amplification of the mutant and wildtype allele, using specific primers based on Weitzel et al method [31]. The PCR products were resolved in 1.5% agarose gels to identify the amplification of the truncated allele and sequenced.
Phosphorylation site disruption analysis
To evaluate the impact of missense changes in phosphorylation sites, the protein sequences and amino acid changes of all VUS variants (Supplementry Table 2) were submitted to the ReKINect portal (http://rekinect.science/home). Only mutations predicted to disrupt the phosphorylation site and those which have previous evidence of functional impact in experimental studies were considered.
Statistical analyses
Characteristics of cases with confirmed diagnosis of breast cancer were summarized with descriptive statistics. The association between demographic and clinical characteristics on the presence of pathological mutations was assessed using unadjusted logistic regression. The logistic regression model utilized all available data (complete and missing). All the analyses were conducted using STATA 13.0.
Abbreviations
Gene names per standard nomenclature.
Author contributions
FVP and RG conceived the study. FVP and SP wrote the manuscript. FVP, RQU performed the bioinformatic analyses. LERC processed bioinformatic data. SP analyzed the clinical data. RQU, JO, CF, CLAH, LIT, YIC, RMAG, GTM, MD and IR contributed to critical review of the manuscript. RG, GTM, IR, MPRC, MST, AFM, OMG, IDE, HODL, FRL, VJ, VHGB, PRF, JHSC, PKES, LGE applied the questionnaires, revised the clinical records of patients and participated in recruitment. RQU and CEDV extracted the DNA, and prepared and sequenced the libraries. RQU, CEDV, HMG, ERJ, VFO and RMAG performed the validation of mutations and the BRCA1 del ex9-12 detection. LAH, RG, LIT, GTM, IR and FVP contributed with reagents, materials and analysis tools.
ACKNOWLEDGMENTS
We are thankful with M. C. Laura Margarita Márquez Valdemar for her support with the Sanger sequencing assays used for validation.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
FUNDING
This work was supported by the National Autonomous University of Mexico (UNAM: PAPIIT IA204215) and by the National Council for Science and Technology (CONACyT: 232630, 252854, 264410, 271685). Rosalía Quezada-Urban received a scholarship from CONACyT and in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute.
REFERENCES
1. Ervik M LF, Ferlay J, Mery L, Soerjomataram I, Bray F. Cancer Today.
2. Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet. 1998; 62:676–89.
3. Kast K, Rhiem K, Wappenschmidt B, Hahnen E, Hauke J, Bluemcke B, et al. Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J Med Genet. 2016; 53:465–71.
4. Torres-Mejia G, Royer R, Llacuachaqui M, Akbari MR, Giuliano AR, Martinez-Matsushita L, et al. Recurrent BRCA1 and BRCA2 mutations in mexican women with breast cancer. Cancer Epidemiol Biomarkers Prev. 2015; 24:498–505.
5. Dutil J, Golubeva VA, Pacheco-Torres AL, Diaz-Zabala HJ, Matta JL, Monteiro AN. The spectrum of BRCA1 and BRCA2 alleles in latin america and the caribbean: a clinical perspective. Breast Cancer Res Treat. 2015; 154:441–53.
6. Nielsen FC, van Overeem Hansen T, Sorensen CS. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat Rev Cancer. 2016; 16:599–612.
7. Lu C, Xie M, Wendl MC, Wang J, McLellan MD, Leiserson MD, et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat Commun. 2015; 6:10086.
8. Maxwell KN, Hart SN, Vijai J, Schrader KA, Slavin TP, Thomas T, et al. Evaluation of ACMG-guideline-based variant classification of cancer susceptibility and non-cancer-associated genes in families affected by breast cancer. Am J Hum Genet. 2016; 98:801–17.
9. Maxwell KN, Nathanson KL. Common breast cancer risk variants in the post-COGS era: a comprehensive review. Breast Cancer Res. 2013; 15:212.
10. Michailidou K, Beesley J, Lindstrom S, Canisius S, Dennis J, Lush MJ, et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet. 2015; 47:373–80.
11. Kar SP, Beesley J, Amin Al Olama A, Michailidou K, Tyrer J, Kote-Jarai Z, et al. Genome-wide meta-analyses of breast, ovarian, and prostate cancer association studies identify multiple new susceptibility loci shared by at least two cancer types. Cancer Discov. 2016; 6:1052–67.
12. Desmond A, Kurian AW, Gabree M, Mills MA, Anderson MJ, Kobayashi Y, et al. Clinical actionability of multigene panel testing for hereditary breast and ovarian cancer risk assessment. JAMA Oncol. 2015; 1:943–51.
13. Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the american college of medical genetics and genomics. Genet Med. 2017; 19:249–55.
14. Caminsky NG, Mucaki EJ, Perri AM, Lu R, Knoll JH, Rogan PK. Prioritizing variants in complete hereditary breast and ovarian cancer genes in patients lacking known BRCA mutations. Hum Mutat. 2016; 37:640–52.
15. Friebel TM, Domchek SM, Rebbeck TR. Modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: systematic review and meta-analysis. J Natl Cancer Inst. 2014; 106:dju091.
16. Ceccaldi R, Sarangi P, D’Andrea AD. The fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016; 17:337–49.
17. Berwick M, Satagopan JM, Ben-Porat L, Carlson A, Mah K, Henry R, et al. Genetic heterogeneity among fanconi anemia heterozygotes and risk of cancer. Cancer Res. 2007; 67:9591–6.
18. Garcia MJ, Fernandez V, Osorio A, Barroso A, Fernandez F, Urioste M, Benitez J. Mutational analysis of FANCL, FANCM and the recently identified FANCI suggests that among the 13 known fanconi anemia genes, only FANCD1/BRCA2 plays a major role in high-risk breast cancer predisposition. Carcinogenesis. 2009; 30:1898–902.
19. Orloff M, Peterson C, He X, Ganapathi S, Heald B, Yang YR, et al. Germline mutations in MSR1, ASCC1, and CTHRC1 in patients with barrett esophagus and esophageal adenocarcinoma. JAMA. 2011; 306:410–9.
20. Xu J, Zheng SL, Komiya A, Mychaleckyj JC, Isaacs SD, Hu JJ, et al. Germline mutations and sequence variants of the macrophage scavenger receptor 1 gene are associated with prostate cancer risk. Nat Genet. 2002; 32:321–5.
21. Hope Q, Bullock S, Evans C, Meitz J, Hamel N, Edwards SM, et al. Macrophage scavenger receptor 1 999c>t (R293X) mutation and risk of prostate cancer. Cancer Epidemiol Biomarkers Prev. 2005; 14:397–402.
22. Wang L, McDonnell SK, Cunningham JM, Hebbring S, Jacobsen SJ, Cerhan JR, Slager SL, Blute ML, Schaid DJ, Thibodeau SN. No association of germline alteration of MSR1 with prostate cancer risk. Nat Genet. 2003; 35:128–9.
23. Liu S, Liu X, Kamdar RP, Wanotayan R, Sharma MK, Adachi N, Matsumoto Y. C-Terminal region of dna ligase iv drives XRCC4/DNA ligase IV complex to chromatin. Biochem Biophys Res Commun. 2013; 439:173–8.
24. Leeksma OC, de Miranda NF, Veelken H. Germline mutations predisposing to diffuse large B-cell lymphoma. Blood Cancer J. 2017; 7:e532.
25. O’Driscoll M, Cerosaletti KM, Girard PM, Dai Y, Stumm M, Kysela B, et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell. 2001; 8:1175–85.
26. Faucz FR, Horvath A, Rothenbuhler A, Almeida MQ, Libe R, Raffin-Sanson ML, et al. Phosphodiesterase 11a (PDE11A) genetic variants may increase susceptibility to prostatic cancer. J Clin Endocrinol Metab. 2011; 96:E135–40.
27. Horvath A, Korde L, Greene MH, Libe R, Osorio P, Faucz FR, et al. Functional phosphodiesterase 11a mutations may modify the risk of familial and bilateral testicular germ cell tumors. Cancer Res. 2009; 69:5301–6.
28. Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006; 312:1228–30.
29. Formosa R, Vassallo J. Aryl hydrocarbon receptor-interacting protein (AIP) N-terminus gene mutations identified in pituitary adenoma patients alter protein stability and function. Horm Cancer. 2017; 8:174–84.
30. Deka J, Kuhlmann J, Muller O. A domain within the tumor suppressor protein APC shows very similar biochemical properties as the microtubule-associated protein tau. Eur J Biochem. 1998; 253:591–7.
31. Weitzel JN, Lagos VI, Herzog JS, Judkins T, Hendrickson B, Ho JS, et al. Evidence for common ancestral origin of a recurring BRCA1 genomic rearrangement identified in high-risk hispanic families. Cancer Epidemiol Biomarkers Prev. 2007; 16:1615–20.
32. Ewald IP, Ribeiro PL, Palmero EI, Cossio SL, Giugliani R, Ashton-Prolla P. Genomic rearrangements in BRCA1 and BRCA2: a literature review. Genet Mol Biol. 2009; 32:437–46.
33. Zhou L, Yao F, Luan H, Wang Y, Dong X, Zhou W, Wang Q. Three novel functional polymorphisms in the promoter of FGFR2 gene and breast cancer risk: a HuGE review and meta-analysis. Breast Cancer Res Treat. 2012; 136:885–97.
34. Cassa CA, Tong MY, Jordan DM. Large numbers of genetic variants considered to be pathogenic are common in asymptomatic individuals. Hum Mutat. 2013; 34:1216–20.
35. MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012; 335:823–8.
36. Song W, Gardner SA, Hovhannisyan H, Natalizio A, Weymouth KS, Chen W, et al. Exploring the landscape of pathogenic genetic variation in the exac population database: insights of relevance to variant classification. Genet Med. 2016; 18:850–4.
37. Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol 2017.
38. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164.
39. Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. 2017; 100:267–80.
40. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015; 17:405–24.
41. Park JS, Lee ST, Nam EJ, Han JW, Lee JY, Kim J, Kim TI, Park HS. Variants of cancer susceptibility genes in korean BRCA1/2 mutation-negative patients with high risk for hereditary breast cancer. BMC Cancer. 2018; 18:83.
42. Kraus C, Hoyer J, Vasileiou G, Wunderle M, Lux MP, Fasching PA, et al. Gene panel sequencing in familial breast/ovarian cancer patients identifies multiple novel mutations also in genes others than BRCA1/2. Int J Cancer. 2017; 140:95–102.
43. Lolas Hamameh S, Renbaum P, Kamal L, Dweik D, Salahat M, Jaraysa T, et al. Genomic analysis of inherited breast cancer among palestinian women: genetic heterogeneity and a founder mutation in TP53. Int J Cancer 2017.
44. Tedaldi G, Tebaldi M, Zampiga V, Danesi R, Arcangeli V, Ravegnani M, et al. Multiple-gene panel analysis in a case series of 255 women with hereditary breast and ovarian cancer. Oncotarget 2017.
45. Jian W, Shao K, Qin Q, Wang X, Song S, Wang X. Clinical and genetic characterization of hereditary breast cancer in a chinese population. Hered Cancer Clin Pract. 2017; 15:19.
46. Cock-Rada AM, Ossa CA, Garcia HI, Gomez LR. A multi-gene panel study in hereditary breast and ovarian cancer in colombia. Fam Cancer 2017.
47. Susswein LR, Marshall ML, Nusbaum R, Vogel Postula KJ, Weissman SM, Yackowski L, et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med. 2016; 18:823–32.
48. Kwong A, Shin VY, Au CH, Law FB, Ho DN, Ip BK, et al. Detection of germline mutation in hereditary breast and/or ovarian cancers by next-generation sequencing on a four-gene panel. J Mol Diagn. 2016; 18:580–94.
49. Li J, Meeks H, Feng BJ, Healey S, Thorne H, Makunin I, et al. Targeted massively parallel sequencing of a panel of putative breast cancer susceptibility genes in a large cohort of multiple-case breast and ovarian cancer families. J Med Genet. 2016; 53:34–42.
50. Mannan AU, Singh J, Lakshmikeshava R, Thota N, Singh S, Sowmya TS, et al. Detection of high frequency of mutations in a breast and/or ovarian cancer cohort: implications of embracing a multi-gene panel in molecular diagnosis in india. J Hum Genet. 2016; 61:515–22.
51. Lin PH, Kuo WH, Huang AC, Lu YS, Lin CH, Kuo SH, et al. Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget. 2016; 7:8310–20.
52. Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, Bowles K, et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer. 2015; 121:25–33.
53. Lincoln SE, Kobayashi Y, Anderson MJ, Yang S, Desmond AJ, Mills MA, et al. A systematic comparison of traditional and multigene panel testing for hereditary breast and ovarian cancer genes in more than 1000 patients. J Mol Diagn. 2015; 17:533–44.
54. Schroeder C, Faust U, Sturm M, Hackmann K, Grundmann K, Harmuth F, et al. HBOC multi-gene panel testing: comparison of two sequencing centers. Breast Cancer Res Treat. 2015; 152:129–36.
55. Hirotsu Y, Nakagomi H, Sakamoto I, Amemiya K, Oyama T, Mochizuki H, Omata M. Multigene panel analysis identified germline mutations of DNA repair genes in breast and ovarian cancer. Mol Genet Genomic Med. 2015; 3:459–66.