
INTRODUCTION
The TP53 gene has 11 exons and produces an mRNA of 2.2-2.5 kb. The wild-type (WT) p53 protein has a wide range of functions involved in cell homeostasis, including the cell cycle, DNA maintenance, and apoptosis, while mutant p53 protein is seen in most cancers. Therefore, the TP53 gene has been referred to as the “Guardian of the Genome” [1] or the “Death Star” [2]. In 1981, it was discovered that the p53 protein, which is a 53,000-dalton protein, accumulates along with proto-virus protein [3]. The TP53 gene was initially thought to be an oncogene, since mutant p53 protein was overexpressed in cancer cells. However, it was subsequently confirmed that the TP53 gene is located on the short arm of chromosome 17, which is frequently deleted in human cancer [4, 5]. In addition, mutation of the TP53 gene was confirmed to cause Li-Fraumeni syndrome, which leads to the development of various primary cancers at a young age [6]. Thus, research on the function of p53 protein tended to focus on its role as a tumor suppressor gene.
The canonical functions of the TP53 gene are based on its transcriptional activity [7] and its promotion of the transactivation of the p21WAF1 gene, which is involved in cell cycle arrest [8]. After crystal structure analyses became possible, the DNA binding region of p53 protein was found to be the location of many point mutations associated with tumorigenesis. Therefore, mutation of the DNA binding region of p53 protein was considered to lead to a dysfunction in the transactivation of normal p53 protein [9]. Distinctive functional abnormalities occur depending on the type and locations of TP53 gene mutation. TP53 gene alterations often consist of missense mutations, but infrequent nonsense or frameshift mutations do occur. Missense mutations are usually observed in exons 5-8, while nonsense and frameshift mutations are more frequent in exons 4, 9, and 10 [10]. Single point mutations are located in the TP53 DNA-binding domain for more than 95% of all reported carcinogenic mutations [11]. Mutations at these residues can be categorized as contact (R273H, R248Q and R248W) or structural (R175H, G245S, R249S and R282H) mutants, depending on whether the residues have a role in direct DNA contact or in the maintenance of the p53 structure. Reportedly, contact mutant proteins are destabilizing and often exhibit a nuclear distribution in response to genotoxic stress, while structural mutant proteins are typically distributed around the nucleus of protein aggregates [12].
A detailed sequence analysis revealed the domains that are involved in protein modifications of p53 protein, including a transactivation domain, an apoptosis-related domain within a proline-rich domain, a tetramerization-related domain, and a basic domain [13]. Moreover, the C-terminus contains a nuclear localization signal (NLS) and a nuclear export signal (NES), which are involved in the regulation of the appropriate subcellular localization of p53 function (Figure 1a) [14]. WT p53 protein forms a tetramer and performs transcriptional control by binding to specific sequence sites of genomic DNA (Figure 1b, 1c) [15]. Cross-linking of various post-transcriptional modifications (PTMs) have been reported to affect both the stability and activation of p53, including tetramer formation and localization [16–18], and these PTMs can be functionally divided into two groups: ones that stabilize and activate p53 (phosphorylation, acetylation, and methylation of K372), and ones that, in contrast, lead to the degradation and inactivation of p53 (ubiquitination, neddylation, sumoylation, and methylation of certain lysines). Although the functional analysis of PTMs has been much discussed, the significance of many PTMs, including ubiquitination and acetylation, has largely remained unclear.
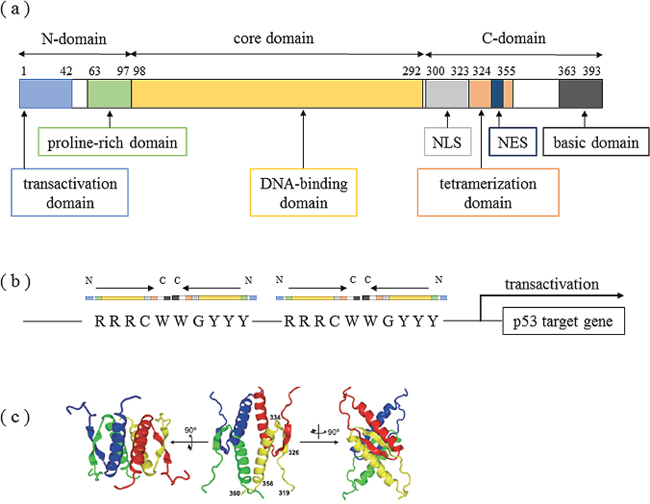
Figure 1: Structure of the tetramer formation domain in p53 protein. (a) Domain structure of p53. Human p53 is composed of 393 amino acid residues and has a transactivation domain, proline-rich domain, DNA-binding domain, nuclear localization signal area, tetramerization domain, nuclear export signal area, and basic domain. Modified from Ref. 10. (b) Transcriptional activation mechanisms of target genes by p53 protein. Various stresses activate p53, which then recognizes the sequence RRRCWWGYYY (R: purine base, Y: pyrimidine base, W: adenine or thymine) in the nucleus and combines with the sequence to form a tetramer. (c) NMR (nuclear magnetic resonance) structure of tetramerization domains. The tetramerization domain contains a [β strand-turn-α helix] structure and forms a tetramer from 2 antiparallel b sheets and 4 helix bundles. Citation from Ref. 15.
In this review article, we would like to provide the rationale for a review on mutant p53 from a clinical point of view, including its clinical relevance and potential therapeutic targets of the TP53 gene in the context of its classically canonical functions and its latest non-canonical functions in human cancer.
Canonical functions of WT p53 protein and its transcriptional target genes
Although normal cells retain only a small amount of p53 protein, levels are increased during times of stress [19]. Under basal conditions, the dysfunctionality of p53 protein occurs mainly by binding to MDM2 and MDM4 [20, 21]. Heterodimers of MDM2 and MDM4 bind to p53 through their N-terminus and activate the E3 ligase activity of MDM2 to induce proteasomal degradation of the p53 protein [22–24]. MDM4 inhibits both the degradation and transactivity of p53, thereby becoming a target of the stress signal to disengage the p53/MDM2 feedback loop for appropriate p53 responses to stresses [23, 25–27]. The balance of p53 activation-related genes can have a very important role in the mechanism of carcinogenesis, since MDM2 gene amplification has been recognized in tumors like sarcoma, in which TP53 alterations are less frequent [28].
The major canonical functions of WT p53 involve growth arrest, apoptosis, and DNA repair. Cell cycle arrest by WT p53 is mediated through transcriptional target genes such as p21, 14-3-3σ and reprimo. p21 suppresses both the G1/S and the G2/M phases [8, 29], while 14-3-3σ and reprimo suppress the G2/M phase [30, 31]. During apoptosis, the Bax gene is directly induced by the p53 protein. [32, 33]. Bax promotes the release of cytochrome c in mitochondria and causes apoptosis through the activation of multiple caspases. Moreover, p53 can directly induce the expression of the PUMA gene, the protein of which can bind to Bcl-2 or Bcl-xL through the Bcl-2 homology 3 (BH3) domain to induce Bax [34]. Noxa also encodes a BH3 domain and causes apoptosis through the same mechanism [35]. In addition, p53 can induce the KILLER/DR5 gene, with the resulting DR5 protein activating Bax through FADD [36]. Apoptosis is strictly controlled by the numerous overlapping layers of p53 functions. DNA repair genes are also strictly increased in the p53 canonical pathway through the induction of the expressions of the GADD45, XPC, and p53 R2 genes [37–39]. When the cells suffer minor DNA damage, the aberrations are repaired during cell cycle arrest, enabling the cells to survive while destroying cancer cells that have received major DNA damage.
On the other hand, several recent reports have indicated that WT and mutant p53 protein have many non-canonical functions, the detailed mechanisms of which are not all clear. The targets of the WT p53 protein that are involved in non-canonical functions are the BAI1 gene (brain-specific angiogenesis inhibitor 1) [40, 41], which is associated with the inhibition of angiogenesis; the TIGAR gene (TP53-induced glycolysis and apoptosis regulator), which is associated with the inhibition of glycolysis and protection against oxidative stress [42]; the SCO2 gene (synthesis of cytochrome C oxidase 2), which is associated with mitochondrial respiration [43, 44]; the DRAM gene (damage-regulated autophagy modulator), which is associated with the positive regulation of autophagy [45]; and miR-34s associated with the production of cell-cycle arrest and an increase in apoptosis [46]. A summary of the molecular mechanisms of p53 functions is shown in Figure 2.
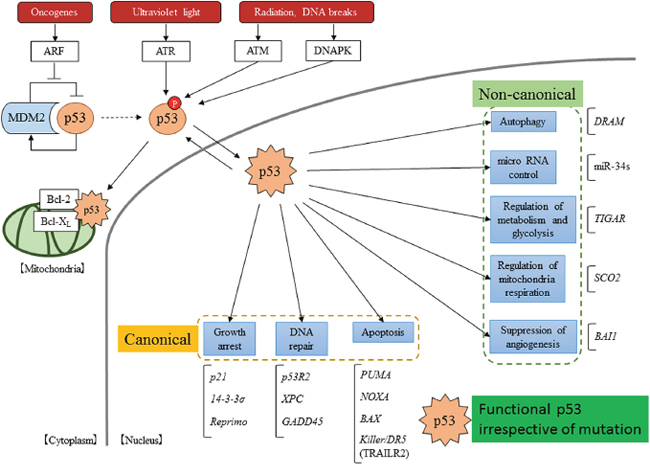
Figure 2: Scheme of the p53 pathway. p53 is activated by a variety of stressors and p53 suppressor genes, such as MDM2. In the nucleus, activated p53 induces the transcriptional activation of many target genes. Moreover, in the cytoplasm, activated p53 has been reported to localize directly in mitochondria and to induce non-transferable dependent apoptosis.
“Gain of function” associated with TP53 mutation explains a novel p53 non-canonical function
Mutant p53 protein is frequently overexpressed in cancer cells [47], and thus the mutation of the TP53 gene results in not only a loss of function of WT p53, but also a gain of function (GOF). Overexpression of the mutant p53 protein has been presumed to be related to the new acquisition of oncogenic functions.
The R175H and R273H mutations, which are mutation hot spots for the TP53 gene, were confirmed to accelerate metastasis via a GOF in various mouse models. Liu et al. reported that mice with the R175H mutation exhibit increased tumor formation and metastasis and a simultaneous loss of WT p53 functions [48]. Lang et al. also reported that embryonic fibroblasts from p53R175H mutant mice showed enhanced cell proliferation, DNA synthesis, and a metastatic status [49]. Similarly, mice with the R273H mutation exhibited tumor growth [50], and Heinlein et al. revealed that genomic instability was not accompanied by such tumorigenic changes [51]. It has become obvious that TP53 mutation elicits GOF effects.
Mutant p53 has been demonstrated to facilitate non-canonical GOF effects, such as the epithelial mesenchymal transition (EMT) [52, 53], the activation of NF-kβ [54], increases in chemo-resistance [55] and radio-resistance [56], assistance in the resolution of the proteasome [57], an increase in the MDM2 isoform [58], the stimulation of aerobic glycolysis [59], the inhibition of anabolic metabolism [60], the promotion of DNA synthesis [61, 62], histone modification [63], and the enhancement of integrin and Rho GTPase signaling [64]. Therefore, some of these non-canonical functions can be explained, at least in part, by a GOF of mutant p53 (Figure 3a).
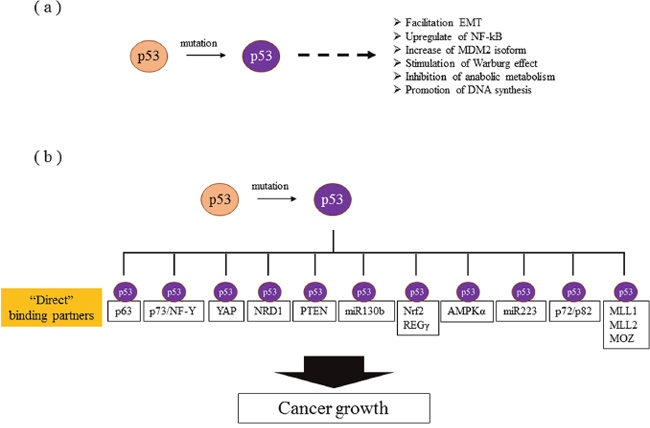
Figure 3: Molecular mechanisms of gain of function. (a) Non-canonical functions whose direct interactions with mutant p53 have not been elucidated. (b) Mutant p53s promote the transcriptional activation of various genes. The molecules reported to bind directly to mutant p53 are listed.
The precise molecular mechanisms of mutant p53 have been lately studied more deeply. Mutant p53, but not WT p53, can directly bind to isoforms of p63, which is a p53 family protein [65, 66]. Muller et al. reported that the direct inhibition of p63 by mutant p53 is involved in cell proliferation and invasion via the enhanced expression of α5β1-integrin and EGFR signals in tumor cells [67]. To identify the mechanism involved in such non-canonical functions accurately, we need to determine the direct binding partner, such as p63, with mutant p53 protein to explain the GOF. p73, a p53 family member other than p63, is also a direct binding partner of mutant p53 [68]. Although p63 and p73 are not directly associated with tumorigenesis in normal cells, in the presence of mutant p53, they can become a transcription target and are related to the progression of tumors. Indeed, there is a report that mutant p53 induces PDGFRb (platelet derived growth factor receptor beta) through a cell-autonomous mechanism involving the inhibition of a p73/NF-Y complex that represses PDGFRb expression in p53-deficient, noninvasive cells [69].
Direct potential candidate partners with mutant p53 have been recently and rigorously reported, as shown in Figure 3b. PTEN enhances mutant p53 protein levels via the inhibition of mutant p53 degradation by Mdm2, and this is considered to be mediated through direct protein binding between mutant p53 and PTEN [70]. The Rab11 effector protein, a Rab-coupling protein, was also reported to be mediated through a mechanism independent of p63 and to result in the enhancement of α5β1-integrin and EGFR in tumor cells [67]. Coffill et al. proposed a different mechanism for mutant p53-driven invasion: the interaction of p53 R273H with nardilysin (NRD1) promotes an invasive response to heparin binding-epidermal growth factor-like growth factor that is p53R273H-dependant but that does not require Rab coupling protein or p63 [71]. Mutant p53 also reportedly exerts oncogenic functions and promotes EMT in endometrial cancer by binding directly to the promoter of miR-130b, a negative regulator of ZEB1, and inhibiting its transcription [53]. Moreover, mutant p53 protein prevents Smad3/N-CoR complex formation on the REGγ promoter, which enhances the activity of the REGγ-20S proteasome pathway and contributes to mutant p53 GOF [57]. In addition, mutant p53 protein binds the miR-223 promoter and reduces its transcriptional activity, resulting in chemo-resistance via the upregulation of STMN-1 [55]. Furthermore, some reports have indicated that mutant 53 protein, but not WT p53 protein, preferentially binds to the AMPKα subunit and inhibits AMPK activation under conditions of energy stress [60].
p53 mutants also bind to and upregulate chromatin regulatory genes, including the methyltransferases MLL1 (also known as KMT2A), MLL2 (also known as KMT2D), and the acetyltransferase MOZ (also known as KAT6A or MYST3), resulting in genome-wide increases in histone methylation and acetylation [63]. Mutant p53 also binds and sequesters RNA helicases p72/82 from the microprocessor complex, interfering with the Drosha-pri-miRNAs association [72]. Recently, mutant p53 was reported to cooperate with Nrf2 (NFE2L2) to activate proteasome gene transcription, resulting in resistance to the proteasome inhibitor carfilzomib in cancer cells [73]. In addition, the p53 mutant protein physically interacts with Yes-associated protein (YAP), a key transcriptional regulator controlling organ growth, tissue homeostasis, and cancer [74].
Clinical relevance of the TP53 gene mutation during molecular carcinogenesis
Vogelstein et al. proposed that genetic aberrations accumulate in accordance with the precancerous-cancer sequence [75]. Genetic aberration occurs in a multi-step carcinogenesis process that includes genomic mutations as well as other somatic changes such as chromosomal instability (CIN), microsatellite instability (MSI), and a CpG island methylation phenotype (CIMP) [76]. The mutation of the TP53 gene is induced by activation-induced cytidine deaminase (AID) through chronic inflammation in gastric cancer (GC), liver cancer, and colorectal cancer (CRC) [77–82]. Thus, a common molecular mechanism explaining TP53 gene mutagenesis in the context of chronic inflammation is assumed to exist.
Molecular multi-step carcinogenesis has been commonly recognized in CRC. The mutation of the TP53 gene is thought to occur during the latter half of carcinogenesis (highly atypical polyp) and to cause malignant transformation in CRC [75]. Actually, the TP53 mutation was found in 58% of CRC and was accompanied by a high frequency of APC gene mutation (82%) and KRAS gene mutation (46%) [83]. The CIN phenotype includes chromosomal amplification or deletion and has been observed in about 80% of CRCs [84]. The amplification of chromosome 8q, 13q, or 20q and the deletion of chromosome 8p, 15q, 17p, or 18q are correlated with carcinogenesis in high-grade adenoma [85]. Although the TP53 gene mutation coexists with CIN in many cases of CRC and was thought to elicit CIN, experiments with knockout WT TP53 show no change in CIN [86]. Therefore, the loss of p53 function has recently been considered to result from CIN. On the other hand, an inverse correlation between the frequency of the occurrence of CIN and MSI/CIMP has been reported [87]. In other words, the mutation of the TP53 gene is rarely detected in lesions with CIMP, while the mutation of the TP53 gene and CIN are common changes associated with carcinogenesis in CIMP-negative tumors.
This inverse correlation has also been observed in GC, especially intestinal-type cancer [88]. In GC, diffuse type and intestinal type cancers are mixed, and the latter is often triggered by chronic inflammation associated with Helicobacter pylori infection. For this reason, GC has a lower frequency of TP53 gene mutation than CRC [88]. Recently, the comprehensive molecular characteristics of GC have been clarified, a GC can now be classified into four types according to its molecular features (Figure 4a) [89]. These subgroups consist of CIN, genomic stable (GS), MSI, and EBV infection, and TP53 gene mutations are concentrated in the CIN group, which are often intestinal-type cancers. Intestinal-type GC has a similar oncogenic process to CRC and can be triggered by chronic inflammation. In contrast, diffuse-type tumor, the major phenotype of GC, has been reported to occur because of E-cadherin mutation, with only a few tumors carrying TP53 gene mutations.
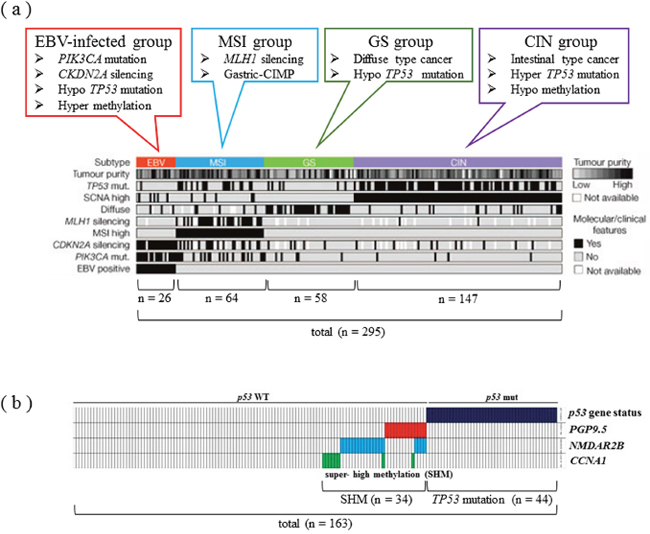
Figure 4: Classification and features of carcinogenic mechanisms through a comprehensive molecular search in GC. (a) Comprehensive molecular search of 295 gastric adenocarcinoma cases. In the EBV-infected group, PI3K mutation and p16 silencing are characteristic genomic features, while the MSI group has low-level gene expressions of MLH1. TP53 gene mutations are concentrated in the CIN group, which are often intestinal-type cancer. Modified from Ref. 88. (b) Epigenetic control of cancer suppressor genes related to the TP53 pathway in GC. The super high methylation of PGP9.5, NMDAR2B, and CCNA1 genes is only seen in GC patients with WT TP53. The TP53 pathway-aberration group, including mutant TP53 and SHM of the 3 genes, is frequently observed in intestinal-type GC. Modified from Ref. 87.
Chronic inflammation induced by H. pylori infection has been demonstrated to be related to the DNA methylation of tumor suppressor genes in GC [90]. Moreover, it has been suggested that epigenetic control is associated with the malignant conversion of tumors, and the “super high methylation (SHM)” of 3 tumor suppressor genes (PGP9.5, NMDAR2B, and CCNA1), which are related to the TP53 pathways (Figure 4b), was identified [88]. The SHM of these genes was only recognized in GC patients with WT TP53, which is reminiscent of the inverse correlation between TP53 mutation/CIN and MSI/CIMP in GC, similar to CRC. In this study, the p53 pathway-aberration group, including TP53 mutation and the SHM of the 3 genes, was frequently observed in intestinal-type GC.
Breast cancer is known to have different TP53 mutation rates according to the basic therapeutic subtypes, such as the luminal A, luminal B, HER2-enriched (HER2E) and triple-negative (TN) subtypes [91]. TP53 mutations in TN tumors were the most frequent among the subtypes (80%), while a few mutations of other cancer-related genes including PIK3CA were also present. The luminal A subtype had fewer TP53 mutations (12%), compared with the TN subtype, but had numerous PIK3CA mutations (45%). The luminal B subtype had mutations of TP53 and PIK3CA (29% each), whereas the HER2E subtype had a hybrid pattern with a high frequency of TP53 (72%) and PIK3CA (39%) mutations.
The genome profiles of TN breast cancer and high-grade serous ovarian cancer (HGS-OvCa) were recently reported [91]. HGS-OvCa has a high TP53 mutation rate (96%), but a few somatic mutations of BRCA1/2 are present in an additional 3% of cases [92]. On the other hand, clear-cell OvCa has a low rate of TP53 mutations but exhibits recurrent PIK3CA and ARID1A mutations [93–95]. Endometrioid OvCa has a lower rate of TP53 and more frequent CTTNB1, ARID1A, and PIK3CA mutations, while KRAS mutations are prevalent in mucinous OvCa [94–96].
Similar to TN breast cancer and HGS-OvCa, almost all lung squamous cell carcinoma (lung SqCC) exhibited frequent somatic mutations of TP53 (81%), while both EGFR and KRAS mutations were almost absent [97]. Lung SqCC also had frequent alterations in the CDKN2A/RB1, NFE2L2/KEAP1/CUL3, PI3K/AKT and SOX2/TP63/NOTCH1 pathways and shared many gene mutations in common with head and neck squamous cell carcinomas without evidence of human papilloma virus infection, including PIK3CA, PTEN, TP53, CDKN2A, NOTCH1, and HRAS [98, 99].
In esophageal adenocarcinoma (EAC), significant mutations of TP53 (71%) and CDKN2A (14%) have been reported [100], and these results were consistent with the prominence of TP53 and CDKN2A mutations in Barrett’s esophagus, a precancerous condition of EAC. Likewise, the prominence of TP53 mutation was observed (91%) in esophageal squamous cell carcinoma (ESCC). In the latest detailed molecular classification, ESCC was classified into a high TP53 mutation group, which possess similar somatic alterations to lung SqCC and head-and-neck SCC, and a low TP53 mutation group, which has a higher mutation rate of PIK3CA and SMARCA4 [100].
Nearly half (49%) of urothelial bladder cancers had TP53 mutations, and 76% of the cancers had inactive TP53 functions [101] because their relationship with the amplification (9%) and overexpression (29%) of MDM2 was mutually exclusive.
Treatment strategy for TP53 mutant cancer
Mutant TP53 could therefore be a therapeutic target. Molecular target drugs often exert their effects by suppressing oncogenes that are overexpressed in cancer cells, and oncogenes tend to be the therapeutic targets. In terms of TP53-targeted therapy, however, both WT and mutant TP53 gene-targeted treatments have been proposed [102]. When grouping these treatments according to therapeutic strategy, they can be classified according to 4 objectives: 1) restoration of the normal p53 function lost by genomic mutation [103]; 2) direct attack on p53-deficient cells [104]; 3) enhancement of normal p53 function [105]; and 4) mimicking DNA damage with a virus [106] (Table 1). Many studies examining these possibilities and problems are ongoing, and some interesting studies have successfully achieved their objectives. The major mutant p53 target strategies are shown in Figure 5.
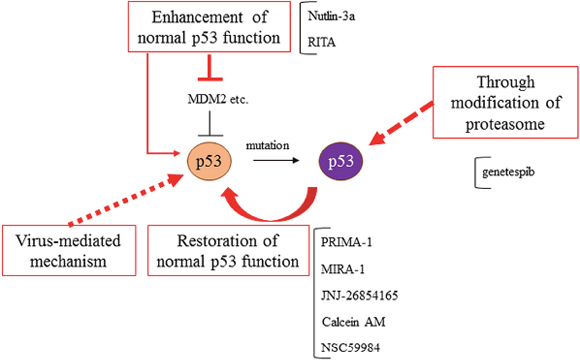
Figure 5: Mutant p53s targeted therapies. Treatment strategies for mutant p53s have been developed, such as the restoration of normal p53 function, through the modification of proteasome and autophagy, the enhancement of normal p53 function, and virus-mediated mechanisms.
Table 1: Treatment strategies targeting mutant p53 cells
Therapeutic concept |
Target |
Reference No. |
|---|---|---|
Restration of normal p53 function |
mutant p53 cell |
103, 108, 122, 125 |
Direct attack to mutant p53 cell |
mutant p53 cell |
104 |
Enhancement of normal p53 function |
normal p53, MDM2, E6, TopBP1 |
105, 118, 119, 123, 124 |
Mimicking of DNA damage using a virus |
mutant p53 cell (DNA) |
106 |
PRIMA-1 has been identified as a small molecule that restores transcriptional activity, DNA binding ability, and the structure of WT p53 and that induces mutant p53-dependent apoptosis [107]. PRIMA-1MET, or methylated PRIMA-1, contributes to the restoration of WT p53 function through a variety of mechanisms [108]. Currently, there is an ongoing phase II trial examining PRIMA-1, and anti-tumor effects against various cancers have been reported [73, 109–117]. Surprisingly, PRIMA-1MET exerts tumor suppressive effects in various cancers and showed a synergistic effect when combined with existing chemotherapy.
Research on p53 activation in cancer cells with normal p53 has led to the generation of Nutlin-3a and RITA (reactivation of p53 and induction of tumor cell apoptosis) [118, 119]. Nutlin-3a inhibits the binding site of MDM2-p53, promotes p53 function, and increases chemotherapy-induced apoptosis in cancer cells lacking functional p53 by activating E2F2, while RITA avoids MDM2-induced p53 repression by binding to p53 [119]. A phase I trial of Nutlin has been started [120].
Other studies have identified factors that restore normal p53 function [121], such as MIRA-1 [122], JNJ-26854165 [123], Calcein AM [124], and NSC59984 [125]; thus, various clinical trials are expected in the future. MIRA-1 induces apoptosis via the restoration of p53-dependent transcriptional transactivation, such as p21, MDM2, and PUMA, by shifting the equilibrium between the native and unfolded conformation of p53 toward the native conformation [122]. JNJ-26854165 induces p53-mediated apoptosis in leukemia cells and has the potential to be an attractive chemotherapeutic as either a single agent or in combination with AraC or anthracyclines [123]. Calcein AM blocks the oligomerization of TopBP, a key mediator for the oncogenic GOF activity of mutant p53, and blocks p53 binding, resulting in the reactivation of E2F1-dependent apoptosis and a GOF of mutant p53 [124]. Especially, NSC59984 has been reported to exert interesting functions that not only restore normal p53 function through p73 activation, but also deplete the non-canonical functions of mutant p53 via MDM2 and ubiquitin-proteasome activation. On the other hand, the Hsp90 inhibitor ganetespib showed a therapeutic effect only for mutant p53 in the context of an HSP90/HDAC6 chaperone mechanism related to the stability of mutant p53 [126]. Genomic therapy using a recombinant oncolytic adenovirus incorporating WT p53 has been attempted in clinical studies in China [127]. Hence, various studies targeting mutant p53 and its correlation with a poor prognosis have been progressing.
Future prospects
The functions and mechanisms of the TP53 gene have gradually become clear over a period of more than 30 years. However, there are still many areas that lack clarity. The next strategy for cancer treatment is expected to become the “Precision Medicine Initiative” announced by the President of the United States, Barack Obama, in 2015. This strategy aims to establish optimal cancer prevention and treatment by grouping patients according to not only genomic phenotypes, but also environments and lifestyles based on information obtained from a cohort study of more than 100 million people. Even using this new approach, the TP53 gene and its mutations will never be excluded from the interest of cancer researchers, since they have a deep impact on molecular carcinogenesis and the potential for new cancer treatments. We have high expectations that research on the TP53 gene will continue to advance steadily.
Abbreviations
GOF, gain of function; WT, wild type; PTMs, post-transcriptional modifications; CIN, chromosomal instability; MSI, microsatellite instability; CIMP, CpG island methylation phenotype; AID, activation-induced cytidine deaminase; CRC, colorectal cancer; GC, gastric cancer; GS, genomic stable; SHM, super high methylation; SCCs, squamous cell carcinomas; ACs, adenocarcinomas; EAC, esophageal adenocarcinoma; ESCC, esophageal squamous cell carcinoma
Author contributions
Conception and design: T.T, K.Y, M.W
Acquisition of data: T.T, K.Y
Analysis and interpretation of data: T.T, K.Y
Drafting the article: T.T, K.Y
Critical revision for important intellectual content: T.T, K.Y, M.W
All the authors have reviewed the manuscript.
ACKNOWLEDGMENTS
We thank FORTE Science Communications, Tokyo, Japan, and International Medical Information Center, Tokyo, Japan, for their professional editorial assistance.
CONFLICTS OF INTEREST
The authors have no conflicts of interest.
FUNDING
There is no financial support.
REFERENCES
1. Lane DP. Cancer. p53, guardian of the genome. Nature. 1992; 358:15-16.
2. Vousden KH. p53: death star. Cell. 2000; 103:691-694.
3. Crawford LV, Pim DC, Gurney EG, Goodfellow P, Taylor-Papadimitriou J. Detection of a common feature in several human tumor cell lines--a 53,000-dalton protein. Proc Natl Acad Sci U S A. 1981; 78:41-45.
4. Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF, Nakamura Y, White R, Vogelstein B. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989; 244:217-221.
5. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991; 253:49-53.
6. Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature. 1990; 348:747-749.
7. Fields S, Jang SK. Presence of a potent transcription activating sequence in the p53 protein. Science. 1990; 249:1046-1049.
8. el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993; 75:817-825.
9. Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994; 265:346-355.
10. Soussi T. p53 alterations in human cancer: more questions than answers. Oncogene. 2007; 26:2145-2156.
11. Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002; 19:607-614.
12. Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, Cornelis A, Rozenski J, Zwolinska A, Marine JC, Lambrechts D, Suh YA, Rousseau F, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol. 2011; 7:285-295.
13. Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004; 4:793-805.
14. Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999; 18:1660-1672.
15. Kamada R, Sakaguchi K. [Tetramerization of tumor suppressor protein p53]. [Article in Japanese]. Seikagaku. 2010; 82:484-493.
16. Marouco D, Garabadgiu AV, Melino G, Barlev NA. Lysine-specific modifications of p53: a matter of life and death? Oncotarget. 2013; 4:1556–71. https://doi.org/10.18632/oncotarget.1436.
17. Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010; 16:528-536.
18. Olsson A, Manzl C, Strasser A, Villunger A. How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression? Cell Death Differ. 2007; 14:1561-1575.
19. Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991; 51:6304-6311.
20. Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009; 137:609-622.
21. Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014; 25:304-317.
22. Perry ME. The regulation of the p53-mediated stress response by MDM2 and MDM4. Cold Spring Harb Perspect Biol. 2010; 2: a000968.
23. Wang X, Jiang X. Mdm2 and MdmX partner to regulate p53. FEBS Lett. 2012; 586:1390-1396.
24. Pei D, Zhang Y, Zheng J. Regulation of p53: a collaboration between Mdm2 and Mdmx. Oncotarget. 2012; 3:228-35. https://doi.org/10.18632/oncotarget.443.
25. Sabbatini P, McCormick F. MDMX inhibits the p300/CBP-mediated acetylation of p53. DNA Cell Biol. 2002; 21:519-525.
26. Marine JC, Dyer MA, Jochemsen AG. MDMX: from bench to bedside. J Cell Sci. 2007; 120:371-378.
27. Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993; 362:857-860.
28. Leach FS, Tokino T, Meltzer P, Burrell M, Oliner JD, Smith S, Hill DE, Sidransky D, Kinzler KW, Vogelstein B. p53 Mutation and MDM2 amplification in human soft tissue sarcomas. Cancer Res. 1993; 53:2231-2234.
29. Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998; 282:1497-1501.
30. Ohki R, Nemoto J, Murasawa H, Oda E, Inazawa J, Tanaka N, Taniguchi T. Reprimo, a new candidate mediator of the p53-mediated cell cycle arrest at the G2 phase. J Biol Chem. 2000; 275:22627-22630.
31. Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B. 14-3-3sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell. 1997; 1:3-11.
32. Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem. 2000; 275:16202-16212.
33. Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995; 80:293-299.
34. Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001; 7:673-682.
35. Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000; 288:1053-1058.
36. Sheikh MS, Burns TF, Huang Y, Wu GS, Amundson S, Brooks KS, Fornace AJ Jr, el-Deiry WS. p53-dependent and -independent regulation of the death receptor KILLER/DR5 gene expression in response to genotoxic stress and tumor necrosis factor alpha. Cancer Res. 1998; 58:1593-1598.
37. Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ Jr. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992; 71:587-597.
38. Adimoolam S, Ford JM. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc Natl Acad Sci U S A. 2002; 99:12985-12990.
39. Nakano K, Balint E, Ashcroft M, Vousden KH. A ribonucleotide reductase gene is a transcriptional target of p53 and p73. Oncogene. 2000; 19:4283-4289.
40. Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994; 265:1582-1584.
41. Nishimori H, Shiratsuchi T, Urano T, Kimura Y, Kiyono K, Tatsumi K, Yoshida S, Ono M, Kuwano M, Nakamura Y, Tokino T. A novel brain-specific p53-target gene, BAI1, containing thrombospondin type 1 repeats inhibits experimental angiogenesis. Oncogene. 1997; 15:2145-2150.
42. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006; 126:107-120.
43. Olovnikov IA, Kravchenko JE, Chumakov PM. Homeostatic functions of the p53 tumor suppressor: regulation of energy metabolism and antioxidant defense. Semin Cancer Biol. 2009; 19:32-41.
44. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006; 312:1650-1653.
45. Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006; 126:121-134.
46. He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network--another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007; 7:819-822.
47. Finlay CA, Hinds PW, Tan TH, Eliyahu D, Oren M, Levine AJ. Activating mutations for transformation by p53 produce a gene product that forms an hsc70-p53 complex with an altered half-life. Mol Cell Biol. 1988; 8:531-539.
48. Liu G, McDonnell TJ, Montes de Oca Luna R, Kapoor M, Mims B, El-Naggar AK, Lozano G. High metastatic potential in mice inheriting a targeted p53 missense mutation. Proc Natl Acad Sci U S A. 2000; 97:4174-4179.
49. Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, El-Naggar AK, Lozano G. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004; 119:861-872.
50. Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004; 119:847-860.
51. Heinlein C, Krepulat F, Lohler J, Speidel D, Deppert W, Tolstonog GV. Mutant p53(R270H) gain of function phenotype in a mouse model for oncogene-induced mammary carcinogenesis. Int J Cancer. 2008; 122:1701-1709.
52. Kogan-Sakin I, Tabach Y, Buganim Y, Molchadsky A, Solomon H, Madar S, Kamer I, Stambolsky P, Shelly A, Goldfinger N, Valsesia-Wittmann S, Puisieux A, Zundelevich A, et al. Mutant p53(R175H) upregulates Twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell Death Differ. 2011; 18:271-281.
53. Dong P, Karaayvaz M, Jia N, Kaneuchi M, Hamada J, Watari H, Sudo S, Ju J, Sakuragi N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene. 2013; 32:3286-3295.
54. Cooks T, Pateras IS, Tarcic O, Solomon H, Schetter AJ, Wilder S, Lozano G, Pikarsky E, Forshew T, Rosenfeld N, Harpaz N, Itzkowitz S, Harris CC, et al. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell. 2013; 23:634-646.
55. Masciarelli S, Fontemaggi G, Di Agostino S, Donzelli S, Carcarino E, Strano S, Blandino G. Gain-of-function mutant p53 downregulates miR-223 contributing to chemoresistance of cultured tumor cells. Oncogene. 2014; 33:1601-1608.
56. Bristow RG, Hu Q, Jang A, Chung S, Peacock J, Benchimol S, Hill R. Radioresistant MTp53-expressing rat embryo cell transformants exhibit increased DNA-dsb rejoining during exposure to ionizing radiation. Oncogene. 1998; 16:1789-1802.
57. Ali A, Wang Z, Fu J, Ji L, Liu J, Li L, Wang H, Chen J, Caulin C, Myers JN, Zhang P, Xiao J, Zhang B, et al. Differential regulation of the REGgamma-proteasome pathway by p53/TGF-beta signalling and mutant p53 in cancer cells. Nat Commun. 2013; 4:2667.
58. Zheng T, Wang J, Zhao Y, Zhang C, Lin M, Wang X, Yu H, Liu L, Feng Z, Hu W. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nat Commun. 2013; 4:2996.
59. Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, Lin M, Yu H, Liu L, Levine AJ, Hu W, Feng Z. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013; 4:2935.
60. Zhou G, Wang J, Zhao M, Xie TX, Tanaka N, Sano D, Patel AA, Ward AM, Sandulache VC, Jasser SA, Skinner HD, Fitzgerald AL, Osman AA, et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol Cell. 2014; 54:960-974.
61. Polotskaia A, Xiao G, Reynoso K, Martin C, Qiu WG, Hendrickson RC, Bargonetti J. Proteome-wide analysis of mutant p53 targets in breast cancer identifies new levels of gain-of-function that influence PARP, PCNA, and MCM4. Proc Natl Acad Sci U S A. 2015; 112: E1220-1229.
62. Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, Carrero ZI, Ramakrishnan G, Watabe K, Haupt Y, Haupt S, Pochampally R, Boss GR, et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat Commun. 2015; 6:7389.
63. Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, Barsyte-Lovejoy D, Al-awar R, Katona BW, Shilatifard A, Huang J, Hua X, Arrowsmith CH, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 2015; 525:206-211.
64. Mizuarai S, Yamanaka K, Kotani H. Mutant p53 induces the GEF-H1 oncogene, a guanine nucleotide exchange factor-H1 for RhoA, resulting in accelerated cell proliferation in tumor cells. Cancer Res. 2006; 66:6319-6326.
65. Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol. 2001; 21:1874-1887.
66. Strano S, Fontemaggi G, Costanzo A, Rizzo MG, Monti O, Baccarini A, Del Sal G, Levrero M, Sacchi A, Oren M, Blandino G. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J Biol Chem. 2002; 277:18817-18826.
67. Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, Cromer A, Brugge JS, Sansom OJ, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009; 139:1327-1341.
68. Marin MC, Jost CA, Brooks LA, Irwin MS, O’Nions J, Tidy JA, James N, McGregor JM, Harwood CA, Yulug IG, Vousden KH, Allday MJ, Gusterson B, et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat Genet. 2000; 25:47-54.
69. Weissmueller S, Manchado E, Saborowski M, Morris JP 4th, Wagenblast E, Davis CA, Moon SH, Pfister NT, Tschaharganeh DF, Kitzing T, Aust D, Markert EK, Wu J, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell. 2014; 157:382-394.
70. Li Y, Guessous F, Kwon S, Kumar M, Ibidapo O, Fuller L, Johnson E, Lal B, Hussaini I, Bao Y, Laterra J, Schiff D, Abounader R. PTEN has tumor-promoting properties in the setting of gain-of-function p53 mutations. Cancer Res. 2008; 68:1723-1731.
71. Coffill CR, Muller PA, Oh HK, Neo SP, Hogue KA, Cheok CF, Vousden KH, Lane DP, Blackstock WP, Gunaratne J. Mutant p53 interactome identifies nardilysin as a p53R273H-specific binding partner that promotes invasion. EMBO Rep. 2012; 13:638-644.
72. Garibaldi F, Falcone E, Trisciuoglio D, Colombo T, Lisek K, Walerych D, Del Sal G, Paci P, Bossi G, Piaggio G, Gurtner A. Mutant p53 inhibits miRNA biogenesis by interfering with the microprocessor complex. Oncogene. 2016; 35:3760-3770.
73. Walerych D, Lisek K, Sommaggio R, Piazza S, Ciani Y, Dalla E, Rajkowska K, Gaweda-Walerych K, Ingallina E, Tonelli C, Morelli MJ, Amato A, Eterno V, et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat Cell Biol. 2016; 18:897-909.
74. Di Agostino S, Sorrentino G, Ingallina E, Valenti F, Ferraiuolo M, Bicciato S, Piazza S, Strano S, Del Sal G, Blandino G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2016; 17:188-201.
75. Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988; 319:525-532.
76. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997; 386:623-627.
77. Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T, Morisawa T, Azuma T, Okazaki IM, Honjo T, Chiba T. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007; 13:470-476.
78. Komori J, Marusawa H, Machimoto T, Endo Y, Kinoshita K, Kou T, Haga H, Ikai I, Uemoto S, Chiba T. Activation-induced cytidine deaminase links bile duct inflammation to human cholangiocarcinoma. Hepatology. 2008; 47:888-896.
79. Endo Y, Marusawa H, Kou T, Nakase H, Fujii S, Fujimori T, Kinoshita K, Honjo T, Chiba T. Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology. 2008; 135:889-898, 898.e1-3.
80. Takai A, Toyoshima T, Uemura M, Kitawaki Y, Marusawa H, Hiai H, Yamada S, Okazaki IM, Honjo T, Chiba T, Kinoshita K. A novel mouse model of hepatocarcinogenesis triggered by AID causing deleterious p53 mutations. Oncogene. 2009; 28:469-478.
81. Nose H, Imazeki F, Ohto M, Omata M. p53 gene mutations and 17p allelic deletions in hepatocellular carcinoma from Japan. Cancer. 1993; 72:355-360.
82. Holzmann K, Klump B, Borchard F, Hsieh CJ, Kuhn A, Gaco V, Gregor M, Porschen R. Comparative analysis of histology, DNA content, p53 and Ki-ras mutations in colectomy specimens with long-standing ulcerative colitis. Int J Cancer. 1998; 76:1-6.
83. Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MD, Niu B, McLellan MD, Uzunangelov V, Zhang J, Kandoth C, Akbani R, et al, and Cancer Genome Atlas Research Network. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014; 158:929-944.
84. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330-337.
85. Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer. 2003; 3:695-701.
86. Bunz F, Fauth C, Speicher MR, Dutriaux A, Sedivy JM, Kinzler KW, Vogelstein B, Lengauer C. Targeted inactivation of p53 in human cells does not result in aneuploidy. Cancer Res. 2002; 62:1129-1133.
87. Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, Hernandez NS, Chen X, Ahmed S, Konishi K, Hamilton SR, Issa JP. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci U S A. 2007; 104:18654-18659.
88. Waraya M, Yamashita K, Ema A, Katada N, Kikuchi S, Watanabe M. Exclusive Association of p53 Mutation with Super-High Methylation of Tumor Suppressor Genes in the p53 Pathway in a Unique Gastric Cancer Phenotype. PLoS One. 2015; 10: e0139902.
89. Bass AJ, Thorsson V, Shmulevich I, Reynolds SM, Miller M, Bernard B, Hinoue T, Laird PW, Curtis C, Shen H, Weisenberger DJ, Schultz N, Shen R, et al, and Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014; 513:202-209.
90. Maekita T, Nakazawa K, Mihara M, Nakajima T, Yanaoka K, Iguchi M, Arii K, Kaneda A, Tsukamoto T, Tatematsu M, Tamura G, Saito D, Sugimura T, et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res. 2006; 12:989-995.
91. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61-70.
92. Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, Godwin AK, Gross J, Hartmann L, et al, and Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609-615.
93. Ho ES, Lai CR, Hsieh YT, Chen JT, Lin AJ, Hung MH, Liu FS. p53 mutation is infrequent in clear cell carcinoma of the ovary. Gynecol Oncol. 2001; 80:189-193.
94. Kuo KT, Mao TL, Jones S, Veras E, Ayhan A, Wang TL, Glas R, Slamon D, Velculescu VE, Kuman RJ, Shih Ie M. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol. 2009; 174:1597-1601.
95. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, Yang W, Heravi-Moussavi A, Giuliany R, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010; 363:1532-1543.
96. Cuatrecasas M, Villanueva A, Matias-Guiu X, Prat J. K-ras mutations in mucinous ovarian tumors: a clinicopathologic and molecular study of 95 cases. Cancer. 1997; 79:1581-1586.
97. Hammerman PS, Lawrence MS, Voet D, Jing R, Cibulskis K, Sivachenko A, Stojanov P, McKenna A, Lander ES, Gabriel S, Getz G, Sougnez C, Imielinski M, et al, and Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012; 489:519-525.
98. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011; 333:1157-1160.
99. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, Zhang N, El-Naggar AK, Jasser SA, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011; 333:1154-1157.
100. Cancer Genome Atlas Research Network, Analysis Working Group: Asan University, BC Cancer Agency, Brigham and Women’s Hospital, Broad Institute, Brown University, Case Western Reserve University, Dana-Farber Cancer Institute, Duke University, Greater Poland Cancer Centre, Harvard Medical School, Institute for Systems Biology, KU Leuven, et al. Integrated genomic characterization of oesophageal carcinoma. Nature. 2017; 541:169-175.
101. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014; 507:315-322.
102. Vogelstein B, Kinzler KW. Achilles’ heel of cancer? Nature. 2001; 412:865-866.
103. Roth JA, Grammer SF, Swisher SG, Nemunaitis J, Merritt J, Meyn RE Jr. Gene replacement strategies for treating non-small cell lung cancer. Semin Radiat Oncol. 2000; 10:333-342.
104. Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, Ng L, Nye JA, Sampson-Johannes A, Fattaey A, McCormick F. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996; 274:373-376.
105. Hietanen S, Lain S, Krausz E, Blattner C, Lane DP. Activation of p53 in cervical carcinoma cells by small molecules. Proc Natl Acad Sci U S A. 2000; 97:8501-8506.
106. Raj K, Ogston P, Beard P. Virus-mediated killing of cells that lack p53 activity. Nature. 2001; 412:914-917.
107. Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG, Selivanova G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002; 8:282-288.
108. Lambert JM, Gorzov P, Veprintsev DB, Soderqvist M, Segerback D, Bergman J, Fersht AR, Hainaut P, Wiman KG, Bykov VJ. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009; 15:376-388.
109. Grellety T, Laroche-Clary A, Chaire V, Lagarde P, Chibon F, Neuville A, Italiano A. PRIMA-1(MET) induces death in soft-tissue sarcomas cell independent of p53. BMC Cancer. 2015; 15:684.
110. Li XL, Zhou J, Chan ZL, Chooi JY, Chen ZR, Chng WJ. PRIMA-1met (APR-246) inhibits growth of colorectal cancer cells with different p53 status through distinct mechanisms. Oncotarget. 2015; 6:36689–99. https://doi.org/10.18632/oncotarget.5385.
111. Sobhani M, Abdi J, Manujendra SN, Chen C, Chang H. PRIMA-1Met induces apoptosis in Waldenstrom’s Macroglobulinemia cells independent of p53. Cancer Biol Ther. 2015; 16:799-806.
112. Aryee DN, Niedan S, Ban J, Schwentner R, Muehlbacher K, Kauer M, Kofler R, Kovar H. Variability in functional p53 reactivation by PRIMA-1(Met)/APR-246 in Ewing sarcoma. Br J Cancer. 2013; 109:2696-2704.
113. Patyka M, Sharifi Z, Petrecca K, Mansure J, Jean-Claude B, Sabri S. Sensitivity to PRIMA-1MET is associated with decreased MGMT in human glioblastoma cells and glioblastoma stem cells irrespective of p53 status. Oncotarget. 2016; 7:60245–69. https://doi.org/10.18632/oncotarget.11197.
114. Deben C, Lardon F, Wouters A, Op de Beeck K, Van den Bossche J, Jacobs J, Van Der Steen N, Peeters M, Rolfo C, Deschoolmeester V, Pauwels P. APR-246 (PRIMA-1(MET)) strongly synergizes with AZD2281 (olaparib) induced PARP inhibition to induce apoptosis in non-small cell lung cancer cell lines. Cancer Lett. 2016; 375:313-322.
115. Krayem M, Journe F, Wiedig M, Morandini R, Najem A, Sales F, van Kempen LC, Sibille C, Awada A, Marine JC, Ghanem G. p53 Reactivation by PRIMA-1(Met) (APR-246) sensitises (V600E/K)BRAF melanoma to vemurafenib. Eur J Cancer. 2016; 55:98-110.
116. Liu DS, Read M, Cullinane C, Azar WJ, Fennell CM, Montgomery KG, Haupt S, Haupt Y, Wiman KG, Duong CP, Clemons NJ, Phillips WA. APR-246 potently inhibits tumour growth and overcomes chemoresistance in preclinical models of oesophageal adenocarcinoma. Gut. 2015; 64:1506-1516.
117. Tessoulin B, Descamps G, Moreau P, Maiga S, Lode L, Godon C, Marionneau-Lambot S, Oullier T, Le Gouill S, Amiot M, Pellat-Deceunynck C. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood. 2014; 124:1626-1636.
118. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. in vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004; 303:844-848.
119. Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, Pramanik A, Selivanova G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004; 10:1321-1328.
120. Zauli G, Celeghini C, Melloni E, Voltan R, Ongari M, Tiribelli M, di Iasio MG, Lanza F, Secchiero P. The sorafenib plus nutlin-3 combination promotes synergistic cytotoxicity in acute myeloid leukemic cells irrespectively of FLT3 and p53 status. Haematologica. 2012; 97:1722-1730.
121. Saha MN, Qiu L, Chang H. Targeting p53 by small molecules in hematological malignancies. J Hematol Oncol. 2013; 6:23.
122. Bykov VJ, Issaeva N, Zache N, Shilov A, Hultcrantz M, Bergman J, Selivanova G, Wiman KG. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J Biol Chem. 2005; 280:30384-30391.
123. Kojima K, Burks JK, Arts J, Andreeff M. The novel tryptamine derivative JNJ-26854165 induces wild-type p53- and E2F1-mediated apoptosis in acute myeloid and lymphoid leukemias. Mol Cancer Ther. 2010; 9:2545-2557.
124. Chowdhury P, Lin GE, Liu K, Song Y, Lin FT, Lin WC. Targeting TopBP1 at a convergent point of multiple oncogenic pathways for cancer therapy. Nat Commun. 2014; 5:5476.
125. Zhang S, Zhou L, Hong B, van den Heuvel AP, Prabhu VV, Warfel NA, Kline CL, Dicker DT, Kopelovich L, El-Deiry WS. Small-Molecule NSC59984 Restores p53 Pathway Signaling and Antitumor Effects against Colorectal Cancer via p73 Activation and Degradation of Mutant p53. Cancer Res. 2015; 75:3842-3852.
126. Alexandrova EM, Yallowitz AR, Li D, Xu S, Schulz R, Proia DA, Lozano G, Dobbelstein M, Moll UM. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature. 2015; 523:352-356.
127. Yang ZX, Wang D, Wang G, Zhang QH, Liu JM, Peng P, Liu XH. Clinical study of recombinant adenovirus-p53 combined with fractionated stereotactic radiotherapy for hepatocellular carcinoma. J Cancer Res Clin Oncol. 2010; 136:625-630.