
INTRODUCTION
Tumor progression depends on molecular interactions of malignant cells with their surrounding stroma. After initiation of tumor development, dividing cancer cells need to establish communication with normal cells in order to receive nutrients and adhesion. In vivo modeling of these basic aspects of malignant tumor growth is important to identify novel therapeutic targets or biomarkers. There are only few reports about in vivo models of hepatoblastoma (HBL), [1, 2] even though the Huh6 cell line has been established more than 40 years ago. There is therefore a need for novel cell lines and models for HBL [3]. HBL, the most common pediatric liver cancer, is classified into histological subtypes reflecting different stages of liver development [4]. Etiology of the disease is unclear with few validated information available, but low birth weight and tobacco use of parents are emerging as factors favoring HBL [5]. Treatment options for HBL include chemotherapy, surgery or liver transplantation resulting in a five-year survival up to 85% [6]. Pretreatment staging and treatment outcome are defined by international collaborative research consortiums such as PRETEXT (PRETreatment EXTent of disease) and CHIC (Children’s Hepatic Tumors International Collaboration) [7]. In general, HBL respond well to chemotherapy. However, some tumors are resistant and chemotherapy can have dramatic effects on developing children, such as ototoxicity and severe hematological complications [8]. Therefore, novel therapeutic approaches and targets are needed to overcome limitations of current treatments.
On a molecular level, deregulations of several important signaling pathways have been evidenced in HBL. Signaling networks active during normal development become deregulated in HBL, including sonic hedgehog, MYC and Wnt, [9, 10] the latter being mainly triggered by gain-of-function mutations in the CTNNB1 gene [11, 12]. These molecular alterations, which are thought to drive tumor initiation and progression, might become druggable targets in the future, once potent inhibitors are identified and validated. We have recently shown that microRNAs targeting beta-catenin might be used to inhibit HBL growth in vitro and in vivo, [13] opening the way to target oncogenes which are difficult to block pharmacologically. Previously, microarray analyses of HBL tumors have led to the identification of a 16-gene signature, which distinguishes between high risk and low risk patients [14]. Expression of a few genes can thus be informative for clinical outcome in HBL.
The chick embryo has been used successfully to gain insight into the interactions of malignant human cells of various origins with stromal cells of the host with a particular emphasis on lymphatic and blood vascular cells, including action of tumor-secreted growth factors such as VEGF [15–18]. The combination of a pertinent in vivo model with modern high-throughput microarrays allows discovery of molecular mechanisms driving tumor progression [16, 19]. Cross-species hybridization of experimental tumors grown on the CAM simultaneously on human and chick gene chips has broadened our understanding of tumor-stroma interaction, because tumor-cell regulated genes can be distinguished from stroma-expressed genes. Using this strategy NTN1 and CXCL4V1 have been identified as key tumor-promoting factors produced in pancreatic tumor cells [19, 20].
Here we used high throughput RNA sequencing (RNA-seq) to decipher molecular changes occurring during adaptation of HBL-derived Huh6 cells from in vitro growth condition to in vivo evolution on the CAM, separating tumor-cell gene regulation from the stroma compartment. Several genes identified in this model have never been described in the context of HBL. Their potential role for hepatoblastoma growth is discussed and their contribution to HBL progression warrants further investigation.
RESULTS
Morphological characterization of Huh6 cell growth on the CAM
At T1/E11 (T = Tumor day, E = embryonic day), a defined area on the CAM was covered by the tumor cell/matrigel mix, which adheres to the CAM surface (Figure 1A). Some bleeding was observed due to CAM laceration prior implantation. Tumors were grown until T7/E17, without clear externally visible changes in morphology except progressive covering of the experimental tumor with CAM cells (Figure 1A, T7/E17). HES staining of paraffin-embedded tumors show unorganized Huh6 cells in matrigel on T1/E11, which subsequently change morphology and organize themselves into acini-like structures visible on T4/E14 and T7/E17 (Figure 1B, arrows). Tumor cell organization was accompanied by continuous cell proliferation as evidenced by strong nuclear expression of Ki67 protein in the majority of tumor cells, clearly visible at T4/E14 and T7/E17 (Figure 1C). Importantly, experimental tumors maintained expression of cytoplasmic and nuclear β-catenin (Figure 1D). EpCAM (CD326), a surface receptor implicated in cell adhesion and associated with poor clinical outcome, has been shown to be expressed in a mouse model of HBL [25]. Strong immunoreactivity for this tumor marker was evidenced in experimental tumors grown on the CAM (Figure 1E). CK19, another protein frequently found overexpressed in HBL cells, was found highly expressed at the membrane of tumor cells on CAM (Figure 1F). For all antibodies used, the CAM itself (asterisks) showed no positive reactivity, except for beta-catenin (Figure 1D), which stained CAM epithelial cells. Taken together, these morphological and immunohistological results suggest that the chicken embryo CAM stroma provides a microenvironment favorable for short-term HBL growth.
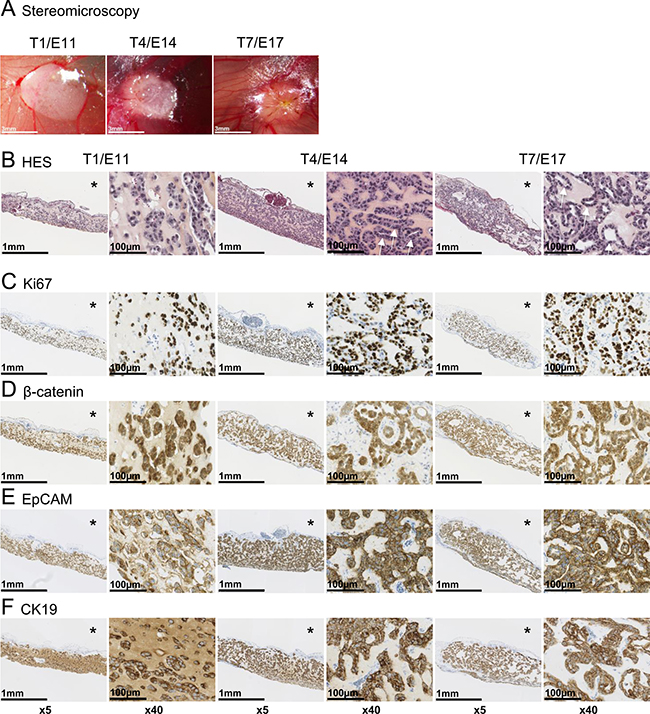
Figure 1: Histological and immunohistochemical analysis of experimental hepatoblastoma on the CAM. (A) Stereomicroscopy monitoring of Huh6 cell growth and tumor formation on the CAM. (B) HES staining of paraffin-embedded tumors from T1 to T7 (arrows: acini-like structures; * = CAM). (C–F) Strong immunoreactivity for the proliferation marker Ki-67 and indicated proteins commonly found in hepatoblastoma tissue. Note that no signal was found in the CAM itself (asterisks), except for beta-catenin at the surface of CAM epithelial cells.
RNA-sequencing, DE genes and bioinformatics results
To understand the development of Huh6 cells on the CAM at a molecular level, we performed RNA-seq transcriptional analysis of tumors on days T1, T4 and T7, and in parallel of CAM on days E11, E14 and E17 (Figure 2). Since each tumoral sample grown on CAM contained fraction of chicken cells we used a conservative experimental strategy allowing us to clearly distinguish the origin of each read. We mapped all RNA-seq reads to both genomes and used for subsequent analysis only those uniquely mapping to either chicken or human genome (Figure 3A). This protocol resulted in 86% of reads coming from Huh6 cell RNA mapping only to the human genome, one percent of reads were common to human and chick and 13% failed to map. Similar proportions were found for reads coming from CAM RNA: 83% mapped to chicken genome, 1% was ambiguously mapped and 15% failed to map. In CAMs bearing Huh6 cells 63% of reads matched to human only, 23% to chicken, 1% to both and 13% did not map.
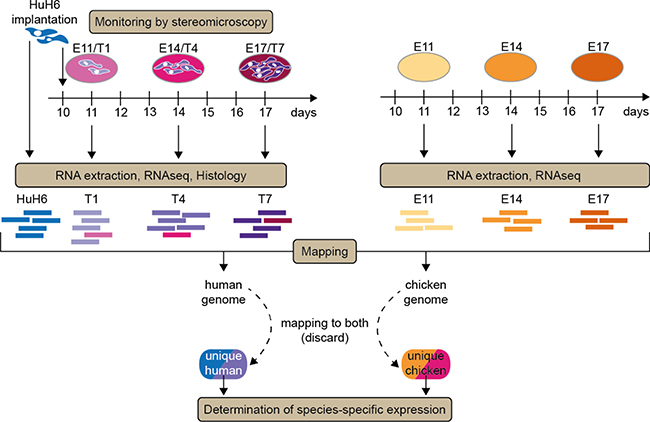
Figure 2: Implantation and sequencing strategy. The schema is presenting the overall model and gene selection set-up. Huh6 cells were implanted on embryonic day 10 (E10) and tumors isolated one (T1, E11), four (T4, E14) and seven (T7, E17) days later. At each day, photos were taken and representative tumors isolated for further processing (histology, RNA extraction). Normal CAMs at the same days were also isolated as reference tissue containing only chick genes as well as Huh6 cell mRNA prior to implantation on the CAM. Species-specific gene regulation strategies are applied at indicated stages/tumor progression days.
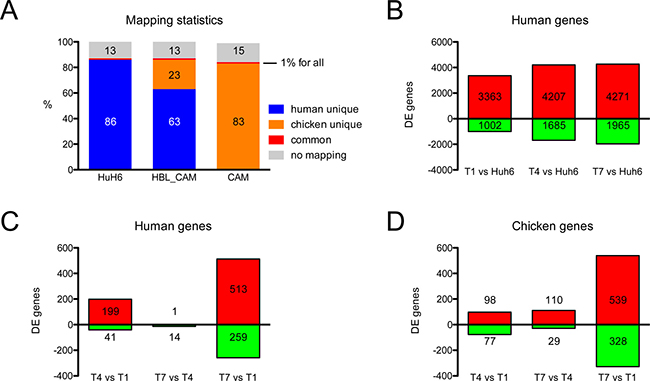
Figure 3: RNA-seq mapping statistics and significantly regulated genes. (A) Due to experimental design there was a need to separate the origin of RNA reads. Therefore, reads were mapped to both human and chicken genomes. Around 1% of them were mapped to both species in any of the samples, whereas around 14% failed to map. See Results for naming of resulting read subsets. (B) A large number of genes were significantly regulated between Huh6 cells in vitro and different days on CAM (T1-7) with many genes upregulated. Regulation of genes expressed by Huh6 cells (C) and chicken host (D) during growth on CAM.
We analyzed differentially expressed (DE) genes for each subset of samples. We found that 3,363 human genes are upregulated in the developing Huh6 tumors as compared to the cell line, whereas 1,002 genes are downregulated (Figure 3B). We compared the numbers of DE genes during the course of experiment separately for human genes originating from Huh6-derived tumors (Figure 3C) and for chicken genes in tumor stroma/adjacent CAM (Figure 3D). We noticed that only 15 human genes were regulated between T7 and T4, whereas the chick stroma transcriptome changes gradually with 139 genes differentially expressed in the same time frame. We then analyzed in details the top DE genes between Huh6 cells before and after implantation (Figure 4A), tumor transcripts (Figure 4B) and chicken transcripts during experiment (Figure 4C). The top genes upregulated in implanted Huh6 belong to a family of MT-RNR2-like genes encoding humanin peptides, whereas some of the most downregulated genes are ribosomal proteins RPL7 and RPS3A (Figure 4A and Supplementary Table 1). In general, Huh6 cells growing on CAM start to express some of the typical liver genes including complement component C3 and many members of the cytochrome p450 family (Supplementary Table 4–6). Ingenuity pathway analysis (IPA) revealed also upregulation of many homeobox proteins including HOXC12, HOXA9 and HOX (Supplementary Figure 1). During the course of tumor development we observed a slight but significant decrease of LIN28A stem cell marker typical for HBL and an increase of 3-hydroxy-3-methylglutaryl-CoA synthase 2 HMGCS2 and MEP1A (Figure 4B, Supplementary Tables 2 and 7–9). On the other hand, the CAM during the course of the experiments expresses typical genes involved in chicken development like TGM4, KRT15 and NEU2 (Figure 4C, Supplementary Tables 3 and 10–12). Because we detected an upregulation of MEP1A, a gene normally expressed in small intestine and colon, in Huh6 cells grown on the CAM, we set out to confirm the relevance of its expression in HBL and confirmed the overexpression of the protein by immunohistochemistry in a patient sample and in Huh6 tumors grown on CAM (Figure 4D).
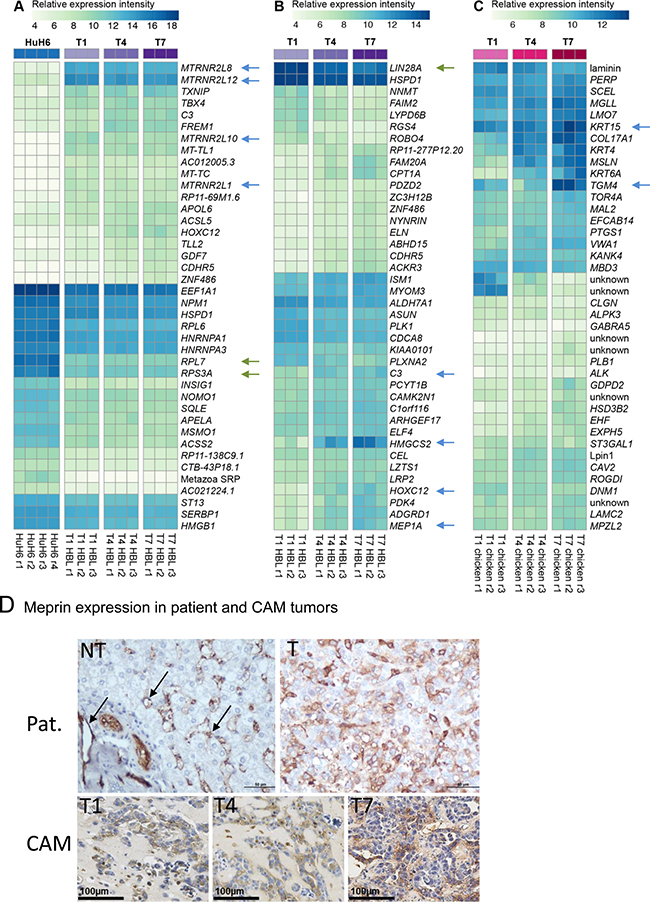
Figure 4: Differentially expressed human and chick genes. (A) Striking differences in gene regulation were observed between in vitro cultured Huh6 cells and CAM-implanted cells. (B) Top 40 differentially expressed human genes during short-term evolution of experimental HBL tumors from T1 to T7. Arrows point to genes strongly induced during tumor growth on CAM. (C) Chicken genes regulated in the tumor stroma and adjacent CAM. (A–C) Upregulated and downregulated human genes are indicated with blue and green arrows, respectively. (D) Comparison of meprin-1α expression in a representative patient (Pat.) hepatoblastoma with experimental HBL grown on the CAM. Arrows point to the wall of blood capillaries in non-tumoral liver (NT), whereas in HBL, cytoplasmic staining is found in tumor cells, in a similar manner as on the CAM.
Lastly we wanted to focus on the transcriptional differences in the CAM tissue that are specifically induced by the growth of Huh6 tumors. We first performed a principal component analysis of the 1,000 most expressed genes in non-implanted CAM (E11, E14 and E17) and in CAM bearing tumors at the corresponding developmental days (Figure 5A). At day 1, CAMs with (T1) or without (E11) Huh6 tumor cells showed a heterogeneous distribution, most likely reflecting differences in development speed, more pronounced in earlier stages of development. At day 4, as the CAM vasculature becomes mature, differences between Huh6-implanted CAMs and non-tumor-bearing (normal) CAMs were minimal (compare T4 and E14). Finally, at day 7 normal CAMs (E17) were different from tumor CAMs of the same day (T7), and T4 and T7 tumors grouped together with the E14 CAMs indicating a more immature phenotype of E17 CAMs with tumors cells (Figure 5A). Then we looked at the most deregulated genes in CAM with tumors and found genes responsible for proliferation like KIF26A, HHIP and FGF10, collagens COL22A1 and COL18A1 and complement components C7 and C3d (Figure 5B and Supplementary Table 13). IPA analysis revealed that many of the deregulated genes in the chick stroma in presence of Huh6 cells are participating in the formation of blood vessels (Supplementary Figure 2). These genes include VEGFA, the major proangiogenic growth factor, together with one of its receptors, FLT1, but also other proangiogenic genes such as ANGPT1, CXCR4, FN1 and HGF.
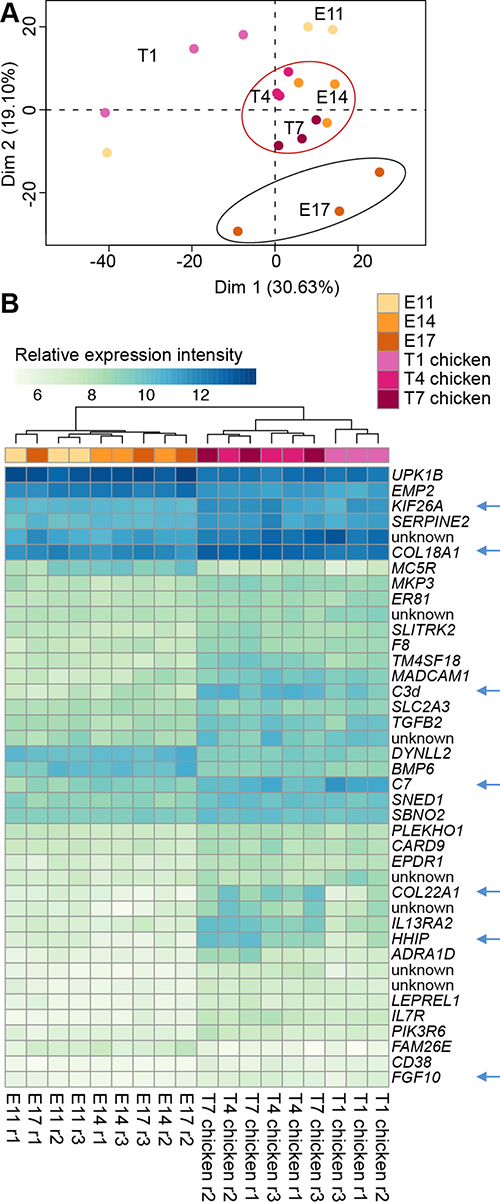
Figure 5: Comparison of CAM transcriptomes during normal development and with Huh6 tumor growth. (A) Principal component analysis (PCA) of 1,000 most expressed genes in CAM samples. Difference between E17 and E14 is more pronounced than between T7 and T4 despite the same time frame. (B) Heatmap of top 40 genes differentially expressed between normal CAMs and corresponding CAMs implanted with tumor cells. Arrows point to interesting candidate genes, whose pro-tumoral functions are discussed in text. Samples on both panels are colored according to the legend. Normal CAM samples are listed as E11, E14 and E17, whereas CAM with Huh6 tumors are listed as T1, T4 and T7 chicken.
In vivo growth inhibition of experimental tumors by cisplatin
To validate the model for experimental treatments, we applied standard chemotherapy to HBL CAM tumors. Cisplatin treatment leads to significant growth inhibition in the HBL CAM model as evidenced by HES staining and several biological parameters (Figure 6). First, cisplatin-treated tumors showed signs of necrosis on the surface more frequently compared to control tumors (Figure 6A, 6E). No effect of cisplatin treatment on tumor weight is observed at T1, but at T4 and T7, a significant reduction of tumor weight is evidenced (P < 0.001, Figure 6F). This is further confirmed by HES histology (Figure 6B), as well as the strong presence of apoptotic cells (cleaved caspase 3 staining; Figure 6C) although treated tumors still contained Ki-67 positive cells (Figure 6D). These results suggest that the model is suitable for testing novel anti-tumor drugs in a controlled in vivo environment within one week only.
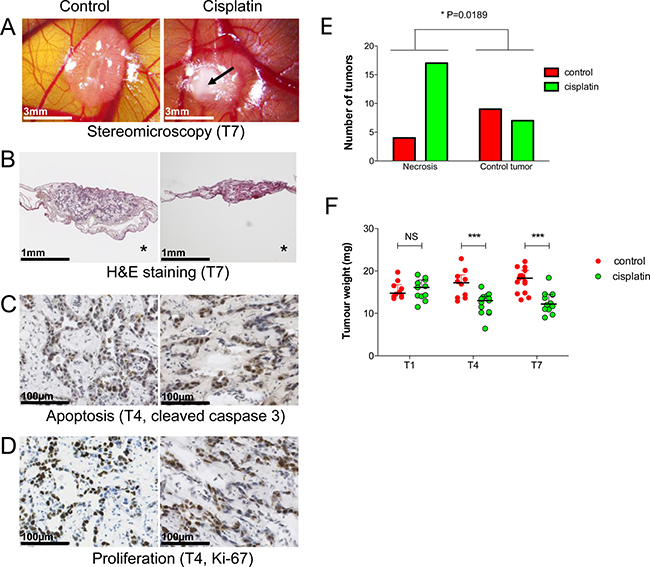
Figure 6: Cisplatin effects on experimental hepatoblastoma on the CAM. (A) Stereomicroscopy of control and cisplatin-treated tumors at T7 on the CAM. Signs of necrosis (arrow), a phenotype used to evaluate treatment efficacy (see E). (B) HES staining confirms tumor growth inhibition after cisplatin treatment. (C, D) Immunohistological demonstration of increased apoptosis and decreased cell proliferation in the cisplatin group. (E) Quantification of cisplatin effects based on phenotypic examination of tumors (signs of necrosis). (F) HBL growth inhibition by cisplatin at T4 and T7 evidenced by tumor weight.
DISCUSSION
To model tumor-host interactions in vivo, we have established several models of highly aggressive tumors using the chick chorioallantoic membrane as host tissue [16, 19, 26, 27]. Cellular and molecular characterization of these models using microarrays revealed expression of genes playing important roles in human tumors as well. To uncover novel gene deregulations using our model, we focused here on HBL, a rare but aggressive childhood tumor. Although high-throughput gene expression studies have been performed to compare HBL to normal liver, [28] such studies cannot distinguish between tumor genes and stroma-expressed genes. However, it has been shown recently that the stroma compartment reacts specifically to tumor growth, and genes expressed by cancer-associated fibroblasts (CAFs) or other stromal cells can predict clinical outcome to anti-cancer therapy [29]. It is therefore of interest to develop strategies to assign gene deregulation either to the cancer cells or their supporting stroma. Few studies have addressed this question so far. We have shown for the first time that such a species-specific high throughput strategy is feasible in vivo using the CAM model and can lead to the discovery of tumor progression genes in cancer cells which are not expressed in vitro, such as NTN1 encoding netrin-1 or the chemokine CXCL4V1 [19, 20]. Other groups have since demonstrated the usefulness of this approach for the study of brain metastasis [30] and colon carcinoma [29] and Bradfort et al. have improved the technique by applying RNA-seq to xenografts in mice to identify tumor and host expression signatures triggered by anti-angiogenic treatment [31]. Valdes et al. have undertaken a systematic RNA-seq and microarray study to investigate the degree of cross-hybridization and cross-alignment between mouse and human cell mixes [32]. Only a few percent of the genes cross-reacted. However, some important cancer progression genes were in these lists. In our system, less than 1% of the genes cross-aligned, most likely due to the greater phylogenetic distance of chick and human (320 MYA) compared to human and mouse (90 MYA). It is surprising that from a morphological point of view, experimental tumors on the CAM shared several important features with orthotopically implanted tumors in mice, such as cytokeratin 19 and beta-catenin expression [2, 33]. Only human tumor cells able to communicate with host cells can form tumors on CAM, through secretion of growth-promoting molecules such as growth factors which activate their receptor on chicken cells. This has been evidenced for vascular endothelial growth factor (VEGF) and its receptor KDR [16].
Chick genes
Increased expression of COL17A1, KRT4, KRT6A and KRT15 in CAM with tumors (T7 CAMs compared to T1 CAMs) most likely reflect normal differentiation processes. However, higher levels of CAV2 in T7 CAMs could be interpreted as a sign of increased activation of the endothelium due to the presence of cancer cells [34]. Angiogenic activity in the CAM tissue decreases in the course of development, [35] and at E13, the CAM vasculature is quiescent. Compared to normal CAMs, Huh6 tumor CAMs show altered expression of a network of genes regulating basic endothelial cell function such as adhesion and differentiation, as revealed by IPA analysis (Supplementary Figure 2). These include genes such as HGF, FN1, CXCR4, ANPT1 and FLT1, amongst others. This program is most likely triggered by VEGFA secreted by Huh6 cells, which display 3.6-fold increase of this gene at T4 and 2.8-fold at T7 compared to in vitro culture conditions (Supplementary Tables 5, 6). The more immature nature of tumor-bearing CAMs is also evidenced by PCA analysis, where T7 CAMs group together with E14 CAMs, whereas E17 CAMs are placed clear away from this group (Figure 5). Among the top 40 regulated genes, we also found increased expression of hedgehog-interacting protein (HHIP), which plays important roles in promoting cancer cell growth in the stromal compartment [36].
Human genes
Interestingly, we found several ribosomal genes such as RPL6, RPL7 and RPS3A downregulated in Huh6 cells on the CAM compared to in vitro culture. Extra-ribosomal functions of ribosomal proteins have now retained attention since they play important roles in cancer progression [37]. In zebrafish, loss of function of ribosomal proteins leads to growth impairment, but also predisposes to cancer development [38]. RPL7 and RPS3A are highly expressed in Huh6 cells in vitro, but become strongly downregulated in tumors on the CAM (Figure 4A). This most likely reflects a profound change in cell behavior in the novel in vivo environment, which is also underpinned by the very high number of genes significantly regulated when Huh6 cells interact with the CAM (>6,000). Low levels of some ribosomal proteins might favor tumor growth, since RPL7A is strongly downregulated in osteosarcoma tissue compared to normal bone and low levels of RPL7A are associated with poor survival [39]. In ovarian cancer, shRNA-mediated knock down of RPS7 accelerated tumor cell proliferation [40] and silencing of RPL41, which is 13.3-fold down-regulated in Huh6 cells on CAM, leads to anchorage-independent growth of fibroblasts and accelerated tumor growth in mice [41].
Some genes strongly induced by tumors on the CAM encode for members of the mitochondrial humanin family (Figure 4A, arrows). MTRNR2L8 is induced more than 80-fold on the CAM compared to Huh6 cells in vitro and is also one of the most abundant genes. Humanins (HNs) are short peptides with strong anti-apoptotic properties. HN specifically interacts with the apoptosis activating protein BAX by preventing its translocation from the cytosol to mitochondria [42] and also inhibits the extra-long form of the proapoptotic protein BimEL [43]. It is tempting to speculate that exposure of HBL cells to the in vivo CAM environment triggers an adaptation program which involves reduction of specific ribosomal proteins to favor tumor cell proliferation and increased expression of anti-apoptotic peptides for survival.
Other proteins strongly affected by exposing Huh6 cells to the in vivo microenvironment of the CAM belong to the HOX transcription factor family. Members of this family regulate key morphological events during organogenesis and are often deregulated in human cancer progression [44]. The fact that Hox genes such as HOXD10, HOXA9, HOXD9, HOXD13 are strongly upregulated in Huh6 cells on the CAM reflect a profound reprogramming of cancer cells in a permissive, growth promoting in vivo microenvironment (Supplementary Figure 1).
Huh6 cells organize themselves into acini-like structures after several days on the CAM could be regarded as a differentiation process. Interestingly, mitochondrial HMGCS2, which encodes for an enzyme of the ketogenic pathway, increases 27-fold from T1 to T7 and its expression controls differentiation in colon and breast cancer cells [45, 46]. HMGCS2 plays a critical role in tumor progression, since overexpression increases ketone body production, thereby favoring growth and motility of cancer cells [47].
We validated expression of meprin-1α encoded by the MEP1A gene, which strongly increases in tumors on the CAM, in a HBL patient tumor by immunohistochemistry. Meprin-1α has not yet been described in this pathology, but strong overexpression in poorly differentiated hepatocellular carcinoma has been reported [48]. In normal liver, meprin-1α is expressed in blood vascular and sinusoidal endothelial cells (Figure 4D), whereas in HBL, additional expression occurs in the cytoplasm of cancer cells. Meprin-1α expression is associated with increased invasion and angiogenesis in colon cancer [49]. Further studies should address the protumoral role of MEP1A in HBL given its strong induction in the model and expression in patient tumors.
Taken together, our morphological and molecular characterization of HBL growth using the CAM model sheds new light on the dynamics of the adaptation potential of tumor cells to the microenvironment. Our model should be useful to validate novel therapeutic strategies for HBL with bad prognosis. It should be emphasized that the CAM model system could now be used to implant freshly isolated patient tumor material including HBL tissue [27, 50]. Probing the growing patient-derived tumor with RNA-seq could identify the molecular landscape of tumor regrowth individually and reveal novel, personnel therapeutic targets. Subsequent treatment of patient-derived tumors on the CAM with adequate therapeutics could personalize pediatric anti-cancer treatment.
MATERIALS AND METHODS
Cell culture and cell line authentication
Huh6 cells were received from the Japan Health Sciences Foundation, Osaka, Japan. They were grown at 37° C in a humidified atmosphere of 5% CO2 in D-MEM Glutamax, 1 g/L D-glucose (Gibco) supplemented with 10% fetal calf serum (FCS, Sigma), 100 U/ml of penicillin and 100 μg/ml streptomycin (Gibco). The identity of the cells was verified once a year by Short Tandem Repeat (STR) profiling (LGC Standards) and found to be identical to original Huh6 markers.
Chick embryos and implantation on CAM
Fertilized chick embryos were handled as described [16]. For implantation 1x106 cells were included in 40μL Matrigel® (50% culture medium/50% Matrigel®, growth factor reduced, Corning), incubated 45 minutes at 37° C for polymerization and then deposited on the CAM after mild laceration of the membrane. Three to four independent transplantation experiments with at least 10–30 embryos per group were carried out. Experimental tumors were photographed in vivo using a Nikon SMZ800N Stereomicroscope, connected to a digital image camera (DS Fi2) piloted by Nikon’s Digital Sight DS-U3 microscope camera controller and processed using Nikon NIS Elements software (version D4 20.00).
Tumor isolation and RNA extraction
At indicated days tumors or non-implanted CAMs were isolated, washed in PBS and snap frozen in liquid nitrogen. RNA extraction was carried out using the mirVanaTM Isolation Kit (Ambion) according to the manufacturer’s protocol. RNAs of Huh6 cells in culture were also extracted with the same kit according to the manufacturer’s protocol. RNA quality was verified using Agilent RNA 6000 Nano kit on an Agilent 2100 Bioanalyzer (Agilent Technologies).
RNA sequencing and analysis
Four μg of RNA were used to generate sequencing library using the TruSeq Stranded mRNA Sample Preparation kit (Illumina) according to the standard protocol. Library preparation and sequencing were performed by the MGX-Montpellier GenomiX platform. We sequenced three biological replicates of CAMs and Huh6 deposited on CAM on three time points: day 11, 14 and 17, named E11, E14, E17 (embryonic development day - CAM) and T1, T4, T7 (tumor day - Huh6), see Figure 1. Thus altogether 18 samples were sequenced on HiSeq 2500 (Illumina) using three lanes of a flow cell resulting in on average 40 million 50-nt single-end raw reads per sample. Additionally, four Huh6 RNA samples were sequenced on HiSeq 2500 (Illumina) using half of a flow cell lane, resulting in on average 34 million paired-end raw reads per sample (named Huh6 r1-4), For each sample all resulting reads were mapped to both human (GR38) and chicken (GalGal4) genomes plus transcripts by hisat2 (2.0.3-beta) with default settings [21]. We excluded reads or read pairs that simultaneously mapped to both genomes and kept for subsequent analysis only those that mapped uniquely to either human or chicken genome. Reads were summed-up at the gene level (Ensembl 82) by feature Counts [22]. Differential gene expression analysis was performed by DESeq2 [23]. We included all genes with adjusted P-value < 0.05 and absolute value of log2-fold change >1 (Figure 3, Supplementary Tables 4–12). All heat maps were generated by plotting the number of reads with variance stabilizing transformation (DESeq2) using heatmap.2 function of the R package and gplots or pheatmap using Euclidian distance and complete clustering method. Differential gene expression results from DESeq2 were also analyzed using QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity), focusing on upstream transcriptional regulators and affected signaling networks.
The datasets generated and/or analyzed during the current study are available at the Gene Expression Omnibus repository, (https://www.ncbi.nlm.nih.gov/geo/) under the number GSE101413.
Histology and immunohistology
One day (T1), four days (T4) and seven days (T7) after the implantation of the cells (Figure 1), the CAM containing the tumors were fixed in situ for 1 hour with formalin (Diapath). In parallel, non-implanted CAMs of corresponding developmental days were fixed using the same protocol. After fixation CAMs were cut out, washed in PBS and stored in 70% ethanol. Huh6 cells were washed with PBS, fixed with formalin (Diapath), suspended in 1% agarose and stored in 70% ethanol. CAMs and cells were then embedded in paraffin and cut into 5 μm sections using a microtome. Hematoxylin/eosin/safran (HES) staining and processing of slides prior to immunostaining was performed using standard procedures. The following primary antibodies were used: anti-β-catenin (1:5000, BD Biosciences, Clone 14, 610154), anti-EpCAM (1:250, Dako, Clone Ber-EP-4, M0804), anti-CK19 (1:50, Dako, Clone RCK108, M0888), anti-Ki67 (1:100, Dako, Clone MIB-1, M7240), anti-Caspase 3 activated (1:200, R&D Systems, AF835) and anti-Meprin-1α (1:100, R&D Systems, AF3220). HRP-coupled secondary antibodies were from the EnVisionTM FLEX kit (Dako) and for anti-Meprin1A, an anti-goat antibody (HRP ImPRESSTM anti-goat IgG, Vector Laboratories) was used. Images were taken using a Nanozoomer PLC (Hamamatsu) at the Bordeaux Imaging Center (BIC http://www.bic.u-bordeaux.fr).
Experimental tumor treatment and phenotypic quantification
Cisplatin (Sigma) was lyophilized and suspended in 0.9% saline solution. The drug was added to the cells together with the Matrigel® to a final concentration of 15 μM, prior to implantation on the CAM. Tumor growth was monitored daily and experiments terminated at E17/T7. Tumors were isolated and weighted at T1, T4 and T7 and tumor bearing CAMs were isolated for histology at T1, T4 and T7. A total of 35 control and 36 cisplatin-treated tumors have been analyzed, embryos that died during incubation were excluded. To evaluate biological effects of cisplatin treatment, a semi-quantitative approach was used, adapted from Auf et al. [24]. Stereomicroscopy photos of all treated and control tumors were randomized using a randomizer script generated by the Script Editor of Apple MacOS El Capitain system. Randomized photos were presented to three observers familiar with the model but not implicated in the design of this study. Observers noted yes (=1) if presence of white areas on the tumor or not (=0) and if tumors resembled to a normal tumor (=1) or not (=0). Data were only processed for tumors where two out of three observers concluded the same result. A contingency table was generated and analyzed using the Chi-square test (GraphPad Prism 5, GraphPad Software).
Author contributions
EI and AD performed CAM experiments, cell culture, histological and molecular biology experiments. KBH did RNA-seq and bioinformatic analysis. CC performed biochemical assays and cell culture. MF, SL and AM analyzed histological slides. NDS and EI did immunohistological stainings and EI scanned the slides. CFG, KBH and MH obtained funding grants. CFG supervised experiments. MH designed and supervised experiments and wrote the paper.
ACKNOWLEDGMENTS
The authors like to thank the Bordeaux Imaging Center (BIC, a service unit of the CNRS-INSERM and Bordeaux University) for providing access to imaging platform and the MGX-Montpellier GenomiX platform for help with RNA sequencing.
CONFLICTS OF INTEREST
The authors declare no conflicts of Interest.
FUNDING
This study was supported by grants from the French National Cancer Institute (project INCa_TRANSLA_2013_209), the French Ministry of Health (Reference n°DGOS/PF4/0013/105), the Fondation pour la Recherche Médicale (FRM; project n°DBI20131228566), the Ligue Nationale contre le Cancer (LNCC), the Agence Nationale de la Recherche in the frame of the ”Investments for the future” Programme IdEx Bordeaux (Reference ANR-10-IDEX-03-02) and the following charity associations: Eva Pour La Vie, Aidons Marina, Fondation Flavien, E.S.C.A.P.E., Les Amis de Marius, Kaëna et les Lapinours foundation and the Cassandra Contre la Leucémie association.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Ellerkamp V, Armeanu-Ebinger S, Wenz J, Warmann SW, Schafer J, Ruck P, Fuchs J. Successful establishment of an orthotopic hepatoblastoma in vivo model in NOD/LtSz-scid IL2Rgammanull mice. PLoS One. 2011; 6:e23419. https://doi.org/10.1371/journal.pone.0023419.
2. Schnater JM, Bruder E, Bertschin S, Woodtli T, de Theije C, Pietsch T, Aronson DC, von Schweinitz D, Lamers WH, Kohler ES. Subcutaneous and intrahepatic growth of human hepatoblastoma in immunodeficient mice. J Hepatol. 2006; 45:377–86. https://doi.org/10.1016/j.jhep.2006.03.018.
3. Rikhi RR, Spady KK, Hoffman RI, Bateman MS, Bateman M, Howard LE. Hepatoblastoma: A Need for Cell Lines and Tissue Banks to Develop Targeted Drug Therapies. Front Pediatr. 2016; 4:22. https://doi.org/10.3389/fped.2016.00022.
4. López-Terrada D, Alaggio R, de Dávila MT, Czauderna P, Hiyama E, Katzenstein H, Leuschner I, Malogolowkin M, Meyers R, Ranganathan S, Tanaka Y, Tomlinson G, Fabrè M, et al, and Children’s Oncology Group Liver Tumor Committee. Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod Pathol. 2014; 27:472–91. https://doi.org/10.1038/modpathol.2013.80.
5. Spector LG, Birch J. The epidemiology of hepatoblastoma. Pediatr Blood Cancer. 2012; 59:776–9. https://doi.org/10.1002/pbc.24215.
6. Haeberle B, Schweinitz D. Treatment of hepatoblastoma in the German cooperative pediatric liver tumor studies. Front Biosci (Elite Ed). 2012; 4:493–8.
7. Hiyama E. Pediatric hepatoblastoma: diagnosis and treatment. Transl Pediatr. 2014; 3:293–9. https://doi.org/10.3978/j.issn.2224-4336.2014.09.01.
8. Zsiros J, Brugieres L, Brock P, Roebuck D, Maibach R, Zimmermann A, Childs M, Pariente D, Laithier V, Otte JB, Branchereau S, Aronson D, Rangaswami A, et al, and International Childhood Liver Tumours Strategy Group (SIOPEL). Dose-dense cisplatin-based chemotherapy and surgery for children with high-risk hepatoblastoma (SIOPEL-4): a prospective, single-arm, feasibility study. Lancet Oncol. 2013; 14:834–42. https://doi.org/10.1016/S1470-2045(13)70272-9.
9. Cairo S, Armengol C, Buendia MA. Activation of Wnt and Myc signaling in hepatoblastoma. Front Biosci (Elite Ed). 2012; 4:480–6.
10. Eichenmuller M, Gruner I, Hagl B, Haberle B, Muller-Hocker J, von Schweinitz D, Kappler R. Blocking the hedgehog pathway inhibits hepatoblastoma growth. Hepatology. 2009; 49:482–90. https://doi.org/10.1002/hep.22649.
11. Blaker H, Hofmann WJ, Rieker RJ, Penzel R, Graf M, Otto HF. Beta-catenin accumulation and mutation of the CTNNB1 gene in hepatoblastoma. Genes Chromosomes Cancer. 1999; 25:399–402.
12. Jia D, Dong R, Jing Y, Xu D, Wang Q, Chen L, Li Q, Huang Y, Zhang Y, Zhang Z, Liu L, Zheng S, Xia Q, et al. Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology. 2014; 60:1686–96. https://doi.org/10.1002/hep.27243.
13. Indersie E, Lesjean S, Hooks KB, Sagliocco F, Ernault T, Cairo S, Merched-Sauvage M, Rullier A, Le Bail B, Taqu e S, Grotzer M, Branchereau S, Guettier C, et al. MicroRNA therapy inhibits hepatoblastoma growth in vivo by targeting β-catenin and Wnt signaling. Hepatol Commun. 2017; 1:168–83. https://doi.org/10.1002/hep4.1029.
14. Cairo S, Armengol C, De Reynies A, Wei Y, Thomas E, Renard CA, Goga A, Balakrishnan A, Semeraro M, Gresh L, Pontoglio M, Strick-Marchand H, Levillayer F, et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell. 2008; 14:471–84. https://doi.org/10.1016/j.ccr.2008.11.002.
15. Hagedorn M, Balke M, Schmidt A, Bloch W, Kurz H, Javerzat S, Rousseau B, Wilting J, Bikfalvi A. VEGF coordinates interaction of pericytes and endothelial cells during vasculogenesis and experimental angiogenesis. Dev Dyn. 2004; 230:23–33. https://doi.org/10.1002/dvdy.20020.
16. Hagedorn M, Javerzat S, Gilges D, Meyre A, de Lafarge B, Eichmann A, Bikfalvi A. Accessing key steps of human tumor progression in vivo by using an avian embryo model. Proc Natl Acad Sci USA. 2005; 102:1643–48. https://doi.org/10.1073/pnas.0408622102.
17. Exertier P, Javerzat S, Wang B, Franco M, Herbert J, Platonova N, Winandy M, Pujol N, Nivelles O, Ormenese S, Godard V, Becker J, Bicknell R, et al. Impaired angiogenesis and tumor development by inhibition of the mitotic kinesin Eg5. Oncotarget. 2013; 4:2302–16. https://doi.org/10.18632/oncotarget.1490.
18. Hecht M, Papoutsi M, Tran HD, Wilting J, Schweigerer L. Hepatocyte growth factor/c-Met signaling promotes the progression of experimental human neuroblastomas. Cancer Res. 2004; 64:6109–18. https://doi.org/10.1158/0008-5472.CAN-04-1014.
19. Dumartin L, Quemener C, Laklai H, Herbert J, Bicknell R, Bousquet C, Pyronnet S, Castronovo V, Schilling MK, Bikfalvi A, Hagedorn M. Netrin-1 mediates early events in pancreatic adenocarcinoma progression, acting on tumor and endothelial cells. Gastroenterology. 2010; 138:1595–606, 1606 e1–8. https://doi.org/10.1053/j.gastro.2009.12.061.
20. Quemener C, Baud J, Boye K, Dubrac A, Billottet C, Soulet F, Darlot F, Dumartin L, Sire M, Grepin R, Daubon T, Rayne F, Wodrich H, et al. Dual roles for CXCL4 chemokines and CXCR3 in angiogenesis and invasion of pancreatic cancer. Cancer Res. 2016; 76:6507–19. https://doi.org/10.1158/0008-5472.CAN-15-2864.
21. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015; 12:357–60. https://doi.org/10.1038/nmeth.3317.
22. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923–30. https://doi.org/10.1093/bioinformatics/btt656.
23. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. https://doi.org/10.1186/s13059-014-0550-8.
24. Auf G, Jabouille A, Guerit S, Pineau R, Delugin M, Bouchecareilh M, Magnin N, Favereaux A, Maitre M, Gaiser T, von Deimling A, Czabanka M, Vajkoczy P, et al. Inositol-requiring enzyme 1alpha is a key regulator of angiogene sis and invasion in malignant glioma. Proc Natl Acad Sci USA. 2010; 107:15553–8. https://doi.org/10.1073/pnas.0914072107.
25. Armeanu-Ebinger S, Hoh A, Wenz J, Fuchs J. Targeting EpCAM (CD326) for immunotherapy in hepatoblastoma. OncoImmunology. 2013; 2:e22620. https://doi.org/10.4161/onci.22620.
26. Balke M, Neumann A, Kersting C, Agelopoulos K, Gebert C, Gosheger G, Buerger H, Hagedorn M. Morphologic characterization of osteosarcoma growth on the chick chorioallantoic membrane. BMC Res Notes. 2010; 3:58. https://doi.org/10.1186/1756-0500-3-58.
27. Balke M, Neumann A, Szuhai K, Agelopoulos K, August C, Gosheger G, Hogendoorn PC, Athanasou N, Buerger H, Hagedorn M. A short-term in vivo model for giant cell tumor of bone. BMC Cancer. 2011; 11:241. https://doi.org/10.1186/1471-2407-11-241.
28. Yamada S, Ohira M, Horie H, Ando K, Takayasu H, Suzuki Y, Sugano S, Hirata T, Goto T, Matsunaga T, Hiyama E, Hayashi Y, Ando H, et al. Expression profiling and differential screening between hepatoblastomas and the corresponding normal livers: identification of high expression of the PLK1 oncogene as a poor-prognostic indicator of hepatoblastomas. Oncogene. 2004; 23:5901–11. https://doi.org/10.1038/sj.onc.1207782.
29. Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, Mellano A, Senetta R, Cassenti A, Sonetto C, Inghirami G, Trusolino L, Fekete Z, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. 2015; 47:312–9. https://doi.org/10.1038/ng.3224.
30. Park ES, Kim SJ, Kim SW, Yoon SL, Leem SH, Kim SB, Kim SM, Park YY, Cheong JH, Woo HG, Mills GB, Fidler IJ, Lee JS. Cross-species hybridization of microarrays for studying tumor transcriptome of brain metastasis. Proc Natl Acad Sci USA. 2011; 108:17456–61. https://doi.org/10.1073/pnas.1114210108.
31. Bradford JR, Farren M, Powell SJ, Runswick S, Weston SL, Brown H, Delpuech O, Wappett M, Smith NR, Carr TH, Dry JR, Gibson NJ, Barry ST. RNA-Seq Differentiates Tumour and Host mRNA Expression Changes Induced by Treatment of Human Tumour Xenografts with the VEGFR Tyrosine Kinase Inhibitor Cediranib. PLoS One. 2013; 8:e66003. https://doi.org/10.1371/journal.pone.0066003.
32. Valdes C, Seo P, Tsinoremas N, Clarke J. Characteristics of cross-hybridization and cross-alignment of expression in pseudo-xenograft samples by RNA-Seq and microarrays. J Clin Bioinforma. 2013; 3:8. https://doi.org/10.1186/2043-9113-3-8.
33. Fuchs J, Schmidt D, Pietsch T, Miller K, von Schweinitz D. Successful transplantation of human hepatoblastoma into immunodeficient mice. J Pediatr Surg. 1996; 31:1241–6.
34. Liu Y, Sowa G. Role of caveolin-2 in subcutaneous tumor growth and angiogenesis associated with syngeneic mouse Lewis lung carcinoma and B16 melanoma models. Cancer Cell Microenviron. 2014; 1:e439. https://doi.org/10.14800/ccm.439.
35. Javerzat S, Franco M, Herbert J, Platonova N, Peille AL, Pantesco V, De Vos J, Assou S, Bicknell R, Bikfalvi A, Hagedorn M. Correlating global gene regulation to angiogenesis in the developing chick extra-embryonic vascular system. PLoS One. 2009; 4:e7856. https://doi.org/10.1371/journal.pone.0007856.
36. Meerang M, Berard K, Felley-Bosco E, Lauk O, Vrugt B, Boss A, Kenkel D, Broggini-Tenzer A, Stahel RA, Arni S, Weder W, Opitz I. Antagonizing the Hedgehog Pathway with Vismodegib Impairs Malignant Pleural Mesothelioma Growth In Vivo by Affecting Stroma. Mol Cancer Ther. 2016; 15:1095–105. https://doi.org/10.1158/1535-7163.MCT-15-0583.
37. Zhou X, Liao WJ, Liao JM, Liao P, Lu H. Ribosomal proteins: functions beyond the ribosome. J Mol Cell Biol. 2015; 7:92–104. https://doi.org/10.1093/jmcb/mjv014.
38. Lai K, Amsterdam A, Farrington S, Bronson RT, Hopkins N, Lees JA. Many ribosomal protein mutations are associated with growth impairment and tumor predisposition in zebrafish. Dev Dyn. 2009; 238:76–85. https://doi.org/10.1002/dvdy.21815.
39. Zheng SE, Yao Y, Dong Y, Lin F, Zhao H, Shen Z, Sun YJ, Tang LN. Down-regulation of ribosomal protein L7A in human osteosarcoma. J Cancer Res Clin Oncol. 2009; 135:1025–31. https://doi.org/10.1007/s00432-008-0538-4.
40. Wang Z, Hou J, Lu L, Qi Z, Sun J, Gao W, Meng J, Wang Y, Sun H, Gu H, Xin Y, Guo X, Yang G. Small ribosomal protein subunit S7 suppresses ovarian tumorigenesis through regulation of the PI3K/AKT and MAPK pathways. PLoS One. 2013; 8:e79117. https://doi.org/10.1371/journal.pone.0079117.
41. Wang S, Huang J, He J, Wang A, Xu S, Huang SF, Xiao S. RPL41, a small ribosomal peptide deregulated in tumors, is essen tial for mitosis and centrosome integrity. Neoplasia. 2010; 12:284–93. https://doi.org/10.1593/neo.91610.
42. Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, Reed JC. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003; 423:456–61. https://doi.org/10.1038/nature01627.
43. Luciano F, Zhai D, Zhu X, Bailly-Maitre B, Ricci JE, Satterthwait AC, Reed JC. Cytoprotective peptide humanin binds and inhibits proapoptotic Bcl-2/Bax family protein BimEL. J Biol Chem. 2005; 280:15825–35. https://doi.org/10.1074/jbc.M413062200.
44. Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat Rev Cancer. 2010; 10:361–71. https://doi.org/10.1038/nrc2826.
45. Gromov P, Espinoza JA, Talman ML, Honma N, Kroman N, Timmermans Wielenga V, Moreira JM, Gromova I. FABP7 and HMGCS2 are novel protein markers for apocrine differentiation categorizing apocrine carcinoma of the breast. PLoS One. 2014; 9:e112024. https://doi.org/10.1371/journal.pone.0112024.
46. Camarero N, Mascaro C, Mayordomo C, Vilardell F, Haro D, Marrero PF. Ketogenic HMGCS2 Is a c-Myc target gene expressed in differentiated cells of human colonic epithelium and down-regulated in colon cancer. Mol Cancer Res. 2006; 4:645–53. https://doi.org/10.1158/1541-7786.MCR-05-0267.
47. Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Ketone body utilization drives tumor growth and metastasis. Cell Cycle. 2012; 11:3964–71. https://doi.org/10.4161/cc.22137.
48. OuYang HY, Xu J, Luo J, Zou RH, Chen K, Le Y, Zhang YF, Wei W, Guo RP, Shi M. MEP1A contributes to tumor progression and predicts poor clinical outcome in human hepatocellular carcinoma. Hepatology. 2016; 63:1227–39. https://doi.org/10.1002/hep.28397.
49. Lottaz D, Maurer CA, Noel A, Blacher S, Huguenin M, Nievergelt A, Niggli V, Kern A, Muller S, Seibold F, Friess H, Becker-Pauly C, Stocker W, et al. Enhanced activity of meprin-alpha, a pro-migratory and pro-angiogenic protease, in colorectal cancer. PLoS One. 2011; 6:e26450. https://doi.org/10.1371/journal.pone.0026450.
50. Sys G, Van Bockstal M, Forsyth R, Balke M, Poffyn B, Uyttendaele D, Bracke M, De Wever O. Tumor grafts derived from sarcoma patients retain tumor morphology, viability, and invasion potential and indicate disease outcomes in the chick chorioallantoic membrane model. Cancer Lett. 2012; 326:69–78. https://doi.org/10.1016/j.canlet.2012.07.023.