
INTRODUCTION
Despite significant advancements over the years, cancer remains the second leading cause of death in the US with an estimated 600,920 cancer-related deaths and 1,688,780 new cancer cases in the US in 2017 [1]. As our understanding of the disease improves, it has become clear that the progression of disease is dependent not only on the growth of the malignant cells but also on the behavior of all components of the tumor microenvironment (TME). The interactions between tumor cells and the TME are analogous to ecosystem interactions and crosstalk between tumor cells and non-malignant cells within the TME create supporting networks that are critical for determining disease severity [2–5]. Despite the effectiveness of current therapies which focus on targeting malignant cells, patients continue to recur. Research has suggested that the TME plays a critical role in recurrence [6, 7]. Thus, cancer research has expanded to include efforts to target TME components and supportive networks [8, 9].
The TME includes various immune cells referred to by pathologists as tumor inflammation. While inflammation describes the presence of immune cells, it does not provide information as the how these cells influence the tumor. Multiple types of inflammation exist including Th1 responses which fight bacterial infections and Th2 responses which promote wound healing. The inflammatory response within a tumor depends on the types and activation states of infiltrating immune cells. While the type of tumor inflammation can be critical for prognosis, pathology reports do not differentiate between the inflammatory cell activation states, thus creating a gap in knowledge between the clinic and tumor immunology research.
The macrophage is one such immune cell that has drastically different effects on the tumor depending on its activation or polarization state [10]. Classically-activated macrophages, or M1s, drive Th1 responses and are generally polarized by stimulants such as INFγ, LPS, and other Toll-like receptor ligands. Markers and secreted molecules associated with an M1 polarization include STAT1, STAT3, IRF4, NFκB, CIITA, CD80, CD86, major histocompatibility complex class II receptor (MHC-II), chemokines receptors (CXCs), chemokine ligands (CXCLs and CCLs) including CCL2 (MCP1), COX-2, TNF, IFNγ, nitric oxide (NO), iNOS, reactive oxygen species (ROS), IL1β, IL12B, and IL6. Alternatively-activated macrophages, or M2s, drive Th2 responses and are generally polarized by IL4, IL13, IL10, and glucocorticoids. Markers and secreted molecules associated with an M2 polarization include SOCS1, PPARγ, STAT6, GATA3, c-MYC, HIF-1α, LXR, CD206 (MRC1), CD163, CD36, ARG1, IL10, TGFβ, matrix metalloproteases (MMPs), FIZZ1, and YM1 (Chi3l3). Macrophage phenotypes are plastic and it is recognized that macrophages exist across the polarization spectrum as they respond to new stimuli. Thus, the M1–M2 model of macrophage polarization is an acknowledged oversimplification as altering the expression of M1 markers does not necessitate the alteration of M2 marker expression and vice versa. Tumor-associated macrophages (TAMs) can exhibit either polarization phenotype and studies performed in multiple cancer types reveal a correlation between TAM polarization and prognosis [11–14]. Across all cancer types, higher M1 infiltrate correlates with a better prognosis and higher M2 infiltrate correlates with a poor prognosis.
Epigenetic regulation plays a significant role in controlling macrophage polarization and can be manipulated by pharmacologic modulators or inhibitors, many of which are used clinically against cancer and other diseases. Extensive work has been done to understand epigenetic regulation of macrophages in the context of infectious and chronic inflammatory diseases largely involving M1s. Several exceptional reviews have been published on these topics [15–18]. However, a current perspective of the epigenetic regulation relating to TAMs or M2s, especially in relation to cancer, remains an unmet need. This review aims to communicate the current knowledge of macrophage epigenetics applicable to TAMs and highlight the implications of these regulatory mechanisms for combating cancer.
Epigenetic processes
Epigenetic regulation is the mechanism that allows genetically identical cells to differ phenotypically. This process is crucial for maintaining cell-type and tissue-specific functions and recapitulating them to daughter cells after replication. Conventional epigenetic mechanisms include DNA methylation, histone methylation, and histone acetylation which remodel chromatin to allow differential gene expression.
DNA methylation involves the methylation of the 5′-carbon on cytosine bases located in promoter CpG islands. This mark prevents transcriptional machinery from assembling on the altered promoter and silences gene transcription. Methyl groups are added to CpG islands by DNA methyl transferases (DNMTs) and removed by ten eleven translocation (TET) proteins.
Histone modification is important for activation state of both promoters and enhancers. Histones are modified on lysine and arginine residues on histone tails of primarily the H3 and H4 subunits. While histones can be modified with a number of post-translational modifications (PTMs) including phosphorylation and ubiquitination, the most important PTMs for controlling gene expression are methylation and acetylation. Histone acetylation marks promote gene transcription and are added to histones by histone acetyl transferases (HATs) and removed by histone deacetylases (HDACs). Histone acetylation marks are bound by bromodomain and extra-terminal motif (BET) proteins which initiate cellular processes such as transcription. Conversely, histone methylation marks can either activate or silence gene transcription depending on the residue modified, the number of methyl groups added to the residue, and the co-localization of the modified histone to an enhancer or promoter region of the regulated gene. Histone methylation is facilitated by histone methyl transferases (HMTs) and removed by histone demethylase (HDMs).
As histones remodel to expose enhancers, lineage-determining transcription factors (LDTFs) bind the open enhancers. These LDTFs determine a cell’s lineage and its response to various signals. When stimuli are applied, enhancer regions interact with promoter regions through bridges of scaffold proteins which include LDTFs and signal-dependent transcription factors (SDTFs) to initiate transcription. Well known LDTFs for myeloid cell lineages include PU.1 and C/EBP [15].
Epigenetic regulation of macrophage polarization
While there exists a significant body of work on macrophage epigenetic processes, most of this work has been performed using M1s. Detailed below is the field’s current knowledge of epigenetic regulation in macrophages, emphasizing M2 and TAM biology. The impacts of these epigenetic regulators on macrophage phenotype are also listed in Table 1 and depicted in Figure 1.
Table 1: Effects of epigenetic enzymes on macrophage polarization to M1 or M2
Epigenetic enzyme |
Effect in M1s |
Effect in M2s |
|
|---|---|---|---|
DNMT |
DNMT1 |
↓ SOCS1 [21] |
|
DNMT3B |
↑ TNF [20] |
↑ Pparɣ promoter methylation [20] |
|
↓ Arg1, CD206, Mgl-1, Pparg [20] |
|||
TET |
TET2 |
↓ Il6 [24] |
|
HMT |
ASH1 |
↓ Il6, Tnf [116] |
|
EZH1 |
↓ IL6, TNF, IFNb, TLR signaling [117] |
||
EZH2 |
↓ Ccl2, Ccl8 [118] |
||
G9A (EHMT2) |
|||
MLL1 |
↑ CXCL10 [27] |
||
MLL4 (WBP7) |
↑ Pigp, LPS signaling [120] |
||
PRMT1 |
↓ CIITA [26] |
↑ PPARɣ [25] |
|
SET7 |
↑ TNF, CCL2, IL8 [121] |
||
SETDB1 |
↓ TNF [122] |
||
SETDB2 |
↓ Cxcl1, Il12b, Cxcl2, Ym1 [123] |
||
SMYD2 |
↓ Tnf, Il6, MHC-II, CD40/80 [124] |
||
SMYD5 |
↓ Tnf, Il1a, Il1b, Ccl4, Cxcl10 [125] |
||
SUV39H2 |
↓ IL6, TNF [126] |
||
SUV40H1 |
↑ Tnf, Cxcl10 [125] |
||
SUV40H2 |
↑ Tnf, Cxcl10, Phf2 [125] |
||
HDM |
AOF1 |
↑ NFκB signaling [127] |
|
JMJD2D |
↑ Ccl22, Il12b [128] |
||
JMJD3 |
↑ TNF [30] |
||
LSD1 |
↓ Il6 [33] |
||
UTX (KDM6A) |
↑ IL6, IFNβ [129] |
||
HDAC |
HDAC1 |
||
HDAC2 |
↑ IFN signaling, IRF3 activation [130] ↓ IL6 [24], MHC-II [132] |
||
HDAC3 |
↓ IL4 signaling [38] |
||
HDAC4 |
↑ TNF, IL6 [45] ↓ NFκB signaling [44] |
↑ STAT6 signaling, Arg1 [44] |
|
HDAC5 |
↑ TNF, CCL2, IL10 [133] |
||
HDAC6 |
|||
HDAC7 |
↑ TLR signaling [138] |
||
HDAC11 |
↑ antigen presentation, CD4+ T cell stimulation [42] ↓ IL10 [42]; IL1 [43] |
||
SIRT1 |
↓ NFκB signaling [51]; Ccl2, Il1β, Il6, Nos2 [47]; TNF [47, 48] |
||
SIRT2 |
↓ NFκB signaling, TNF, IL6, CCL2, IL1β [46] |
↑ Gata3, Arg1, Cd11c [46] |
|
SIRT6 |
↓ IL1β [16] |
||
BET |
↑ IL6, IL1b, IFNg, IL12B, Il1a, Ccl5, Cxcl10, Cxcl2/3 [50] |
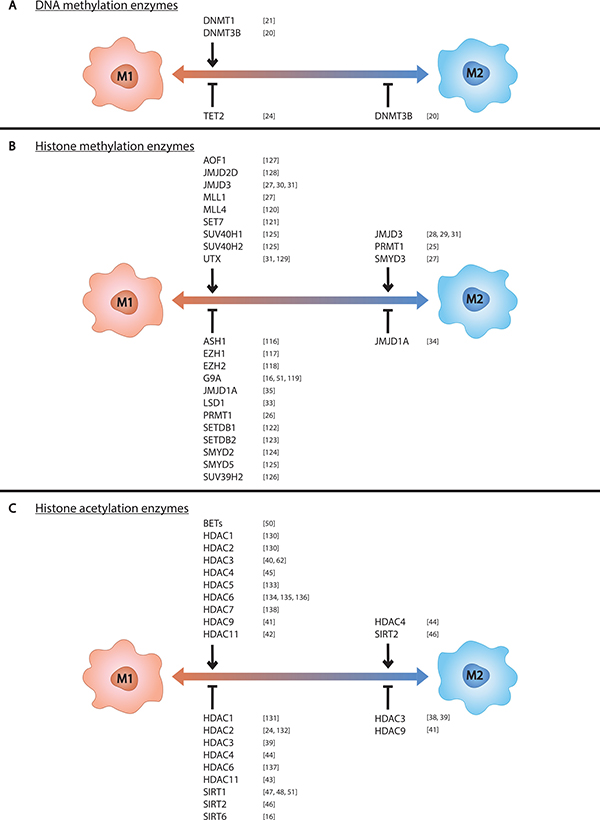
Figure 1: Epigenetic enzymes control macrophage phenotype. Epigenetic enzymes known to control (A) DNA methylation, (B) histone methylation, and (C) histone acetylation in macrophages are listed above the left side of the spectrum if they promote M1 polarization and below if they inhibit M1 polarization. Those known to control M2 polarization are listed likewise on the right side of the spectrum.
DNA methylation
As monocytes differentiate into macrophages, there is a net demethylation of promoter CpG islands [19]. Removing this silencing mechanism presumably primes these genes for transcription.
DNMT3B is the only DNMT with a known role in M2 differentiation and phenotypic control. Knockdown of DNMT3B in RAW264.7 macrophages and mouse bone marrow-derived macrophages (BMDMs) induces M2 polarization and prevents M1 marker expression, LPS-induced TNF secretion, and migratory response to CCL2 gradient [20]. Additionally, overexpression of DNMT3B prevents IL4-induced expression of Arg1 [20]. DNMT3B also methylates the promoter of Pparγ which is reduced in response to IL4 stimulation [20].
In M1s, DNMT1 positively regulates the M1 phenotype by silencing SOCS1 in LPS-stimulated RAW264.7 cells which induces TNF and Il6 expression [21]. If DNMT1 has similar suppressive functions of SOCS1 in M2s, then it likely suppresses the expression of M2 genes. However, functional studies of DNMT1 in M2s are required to implicate this enzyme as a negative regulator of M2 phenotype. Furthermore, DNMT1 is known to be the primary methyltransferase responsible for propagating DNA methylation marks during DNA replication [22]. This function suggests that DNMT1 has an essential role in regulating gene expression beyond SOCS1 in both M1s and M2s.
As for demethylation, TET2 loss of function mutations are implicated in myeloid malignancies, though little is known about the impact of TET proteins on macrophage polarization. Studies involving TET2 knockdown in mouse bone marrow-derived hematopoietic precursor cells revealed an increase in differentiation towards macrophage lineages implicating TET2 as a regulator of myelopoiesis [23]. Additionally, M1 polarization by LPS-stimulation of human and mouse macrophages upregulates expression of TET2 which acts as a co-repressor of Il6 transcription alongside HDAC2 [24]. While this information is important for understanding myeloid- and M1-specific epigenetic regulation, it does not offer insight as to the role of TET2 or TET proteins in M2 polarization.
Histone methylation
PRMT1 methylates the arginine located at residue 3 on the tail of histone H4 (H4R3) and is implicated as a positive regulator of M2 phenotype through its induction of PPARg in IL4-stimulated mouse peritoneal macrophages [25]. Alternatively, a study using IFNg-stimulated RAW264.7 cells revealed PRMT1 negatively regulates the M1 phenotype by repressing CIITA. Additionally, it was observed that PRMT1 is downregulated by IFNγ signaling [26]. Thus, PRMT1 adopts opposing roles in epigenetic regulation of M1s and M2s with its expression driving an M2 polarization.
Another HMT, SMYD3, a H3K4 methyltransferase, is speculated to positively regulate M2 polarization. Its expression levels increase in human monocyte-derived macrophages (HMDMs) with exposure to the combination of M-CSF, IL4, and IL13 (M-CSF + IL4 + IL13) and decrease with exposure to LPS + IFNγ [27]. Its upregulation coincides with methylation and transcriptional activation of ALOX15, a lipoxygenase M2 marker, which SMYD3 is known to regulate in other contexts [27]. However, functional studies are needed in order to make conclusions on these speculations about M2 phenotypic regulation. Additionally, no functional studies have been performed to test its role in M1 polarization.
JMJD3 (KDM6B), an H3K27 demethylase, has been recognized as an essential regulator of M2 polarization through its induction of Irf4, Arg1, CD206, and other M2 markers in IL4- stimulated [28] and IL4 + IL13-stimulated [29] mouse BMDMs. While this study revealed JMJD3 to be unnecessary for M1 polarization of TLR ligand-stimulated mouse peritoneal macrophages, other studies involving IFNg-stimulated HMDMs [27] or LPS + IFNγ-stimulated HMDMs from rheumatoid arthritis patients [30] detail its role in inducing pro-inflammatory cytokine expression. In addition to its upregulation in M2s, JMJD3 is also upregulated in IFNγ-stimulated HMDMs [27, 31]. Due to its positive regulatory role in both M1s and M2s, JMJD3 is attributed as a regulator of general stimulus response in macrophages rather than specific polarization roles [31]. Notably, loss of JMJD3 does not lead to significant changes in H3K27 methylation indicating that JMJD3 may regulate macrophage phenotype through modification of proteins other than histones [27, 31].
Though JMJD3 is the only HDM with a known effect on M2 polarization, LSD1 (KDM1A) and JMJD1A (KDM3A) play critical roles in myeloid cells. LSD1, a H3K4 and H3K9 demethylase, is essential for myeloid cell differentiation through silencing of stem and progenitor cell genes [32] and is also involved in LPS tolerance-induced Il6 silencing in mouse BMDMs [33]. Additionally, JMJD1A, a H3K9 demethylase, represses Ccl2, Ccr1, and Ccr5 in mouse peritoneal macrophages and RAW264.7 cells under hypoxic conditions [34]. Furthermore, JMJD1A inhibition decreases macrophage infiltrate in subcutaneous A673 sarcoma tumors in mice [35]. These studies raise the possibility that JMJD1A plays a role in TAM biology and macrophage phenotype control especially in the context of a hypoxic tumor microenvironment.
Histone acetylation
Certain acetylation marks have been discovered to contribute to macrophage phenotypic control. H3 acetylation is important for inducing IFNα, TNF, and IL6 expression in THP-1 cells indicating the importance for H3 acetylation in expression of M1 phenotype [36]. More specifically, the H3K9 and H3K14 acetylation of Tnf, Il6, Nos2, and MHC-II promoters in LPS-stimulated mouse microglia is integral to expression of these genes [37]. However, the importance of these marks on M2 phenotype in IL4- or IL13-stimulated macrophages has yet to be elucidated.
While currently investigations into the importance of HATs in macrophage polarization are lacking, extensive observations have been made concerning the role of HDACs with some of these studies examining their effects in M2s.
HDAC3 negatively regulates M2 polarization by repressing IL4 signaling in mouse BMDMs [38] and TGFβ production in mouse peritoneal macrophages [39]. HDAC3 performs an opposing function in M1s by promoting M1 polarization in LPS-stimulated mouse BMDMs thereby proving critical for LPS signaling [40].
HDAC9 is another negative regulator of M2 polarization as peritoneal macrophages from HDAC9-deficient mice expressed higher levels of M2 genes and lower levels of M1 genes compared to wild type mice [41]. Additionally, the promoter acetylation levels and expression levels of Pparγ were significantly increased in the HDAC9-deficient mice. The observations implicate HDAC9 as a repressor of the M2 phenotype and inducer of the M1 phenotype with Pparγ promoter deacetylation acting as a key component of its regulatory mechanism.
While HDAC11 has not been studied in M2s, its effects in M1s suggest it may act as a negative regulator of M2 phenotype. In RAW264.7 cells, HDAC11 represses IL10 expression [42]. Thus, if HDAC11 acts similarly in M2s as it does in M1s, then it likely opposes M2 polarization. Apart from its effect on IL10, HDAC11 also represses IL1β in LPS-stimulated mouse BMDMs [43] and promotes antigen presentation and CD4+ T cell stimulation in RAW264.7 cells [42] thereby remaining ambiguous as to whether it promotes or inhibits M1 polarization.
HDAC4 induces STAT6 signaling and Arg1 expression in IL4-stimulated mouse bone-marrow derived dendritic cells implicating it as a positive regulator of M2 polarization [44]. In M1s, HDAC4 functions as both a negative and positive regulator. One study using LPS + IFNg-stimulated of mouse BMDMs revealed that HDAC4 inhibited NFκB signaling [44] while another study using the LPS-stimulated mouse microglial BV2 cell line revealed that HDAC4 induces TNF and IL6 secretion [45].
The HDAC SIRT2 acts as another positive M2 phenotype regulator by inducing Gata3, Arg1, and Cd11c expression in IL4-stimulated mouse BMDMs [46]. The same study discovered SIRT2 also represses the M1 phenotype by downregulating NFkB signaling and IL1b and TNF secretion in LPS-stimulated mouse BMDMs [46].
SIRT1 has been tested against M2s but was found to have no impact on M2 polarization in IL4-stimulated mouse BMDMs [47]. It does, however, decrease expression of various M1 markers in LPS + IFNγ-stimulated mouse BMDMs [47] and LPS-stimulated RAW264.7 cells [48].
General BET protein activity promotes inflammatory cytokine production in LPS-stimulated mouse BMDMs [49, 50], although further studies investigating BET proteins in macrophage polarization are lacking.
Epigenetic enzymes as targets for disrupting M2 polarization
Of the enzymes described here, the most relevant for targeting TAMs are those that promote the M2 phenotype, namely PRMT1, JMJD3, HDAC4, SIRT2, and potentially SMYD3. Inhibiting these enzymes in TAMs would prevent these macrophages from polarizing to M2s and supporting the tumor. It is important to note that histone modifying enzymes have secondary functions and act on proteins other than histones as well. Much of their macrophage-polarizing function is speculated to arise from modification of non-histone proteins especially since many histone modifying enzymes are known to act in the cytoplasm [15, 51]. Additionally, not all regulators have opposing function in M1s vs M2s and not all regulators have exclusively positive or negative effects on one polarization state. These discrepancies highlight the complexity of macrophage biology and the challenges of finding effective epigenetic targets in TAMs. There are several enzymes that have known functional roles in M1s but are not discussed here because they have not been studied in M2s. These enzymes and their effects on M1 phenotype are included alongside the enzymes discussed above in Table 1 and Figure 1. This extensive list exemplifies the discrepancy between macrophage research in M1- and M2-related diseases. In order to progress our understanding of the TME’s influences on tumor growth, further investigations into mechanisms of M2 phenotypic control are needed.
Pharmacologic modulators and their effects in macrophages
There are many pharmacologic modulators of epigenetic enzymes, some of which target specific enzymes and others which broadly target multiple enzyme classes. Modulators of inhibitors that target DNMTs are commonly referred to as DNMTis, those that target TET proteins are referred to as TETis, and so on. Not all epigenetic modulators have been tested against macrophages and even less have been tested against M2s specifically. Discussed below are the pharmacologic modulators tested against macrophages and their effect on macrophage polarization. This information is also listed in Tables 2 and 3 and depicted in Figure 2.
Table 2: Effects of pharmacologic modulators of DNA methylation on macrophage polarization to M1 or M2
Pharmacologic modulator |
Effect in M1s |
Effect in M2s |
|
|---|---|---|---|
DNMTi |
azacytidine (Vidaza®, AZA) |
↑ Arg1, Fizz1 [55] |
|
decitabine (Dacogen®, DEC) |
↑ ARG1, CD206, STAT3 activation [58]; SOCS1 [21]; PPARɣ, LXRα [57] ↓ TNF, IL6, IL1β, iNOS, CCL2, CCL5 [57, 58]; CCL9 [57]; IL1α, CCL3, CCL4, CCL7, CCL10, CCL12, IL1RN [58] |
||
TETi |
dimethyloxallyl glycine (DMOG) |
↑ IL10 [59] ↓ NFκB activity, iNOS [59] |
↑ Arg1, Fizz1, Ym1 [59] |
Table 3: Effects of pharmacologic modulators of histone modification on macrophage polarization to M1 or M2
Pharmacologic modulator |
Effect in M1s |
Effect in M2s |
|
|---|---|---|---|
HMTi |
3-deazaneplanocin (DZNep) |
↓ TNF [60] |
|
AMI-1 |
↓ PPARɣ, CD36, CD206, CD209, SOCS1 [25] |
||
methylthioadenosine (MTA) |
↑ Il1β [62] ↓ TNF [62, 64]; IL6 [62]; iNOS [64]; CD69, CD86, MHC-II, NFκB signaling [63] |
↑ Arg1 [62] |
|
MI-2-2 |
↓ CXCL10 [27] |
||
HDMi |
GSK-J4 |
↓ CD206 [65] |
|
pargyline (Eutonyl, Eutron) |
|||
HATi |
C646 |
↓ NFκB activation, TNF, IL8, IL12, iNOS, IL1β [76] |
↑ Ym1, Cd36 [75] ↓ Fizz1, Mgl2 [75]; Arg1 [74, 75]; Ym1 [74] |
curcumin |
↑ PPARɣ, CD36 [82]; SOCS-1, SOCS-3 [84] ↓ TNF, IL6 [81, 82, 84]; IL12B [81, 82]; CCL2 [83, 85]; NFκB activation [81]; ROS [85], COX-2 [84] |
↑ IL12 [78] ↓ IL10, TGF-β, MMP2, MMP9, VEGF, STAT3 activation [78] |
|
epigallocatechin-3-gallate (EGCG) |
|||
garcinol |
|||
Histone Acetyltransferase Inhibitor II (HATi II) |
↓ IL1β [43] |
||
roscovitine |
↓ COX-2, iNOS, NO, NFκB activation [66] |
||
HDACi |
butyrate |
||
CAY10603 (BML-281) |
|||
dacinostat (LAQ824) |
↓ TNF, NFκB activation [109]; MCP-2, MCP-3, CCL2, CCL15, CCL23, CCR1, CCR5, CD38 [108]; IL10 [106] |
||
entinostat (MS-275) |
↑ TNF, IL1β, IL12B, NFκB activity [92]; Cox-2 [89]; IL10 [92, 107] ↓ IL1β, iNOS, IFNɣ, IL17, MMP-9 [107] |
||
givinostat (ITF2357) |
|||
trichostatin A (TSA) |
↑ Cox-2 [89]; Stat1 [62]; IL1β [43]; IFNβ [88]; ROS [137] ↓ Tnf [62, 88, 109]; Il6 [62, 88]; Cox-2 [62]; NFκB activation [109]; Ccl7 [89]; IL12B [88, 89]; Ccl8, Ccl12, Cxcl10, Nos2, Irf7 [88] |
||
tubastatin A |
↑ ROS [137] ↓ phagocytosis [137]; TNF, IL6, NO [139]; apoptosis [105] |
||
valproic acid (Depakene®, VPA) |
↑ IL10, CD86 [102] ↓ CD40, CD80 [102]; IL12B [88, 102]; iNOS, TNF, IL6 [88] |
||
vorinostat (Zolinza®, SAHA) |
↑ Cox-2 [89]; Il1β [92]; ROS [137] ↓ NO [90]; TNF, IL6, IL12B [88]; Ccl7 [89] |
||
BETi |
GSK 525768A (I-BET-762) |
↓ Il6, Ifnβ, Il12α, Cxc19, Ccl12 [50] |
|
I-BET151 |
↑ PPARɣ, LXR [112] ↓ IL6 [111]; NFκB signaling [112]; CXCL10, CXCL11, IFNβ [110] |
↓ PPARɣ, ENPP2, MS4A4A, IL7R, ABIN3 [110] |
|
JQ1 |
↓ TNF, IL6 [49, 113]; CCL2 [49]; Il1β [113]; Nos2 [114]; PD-L1 [115] |
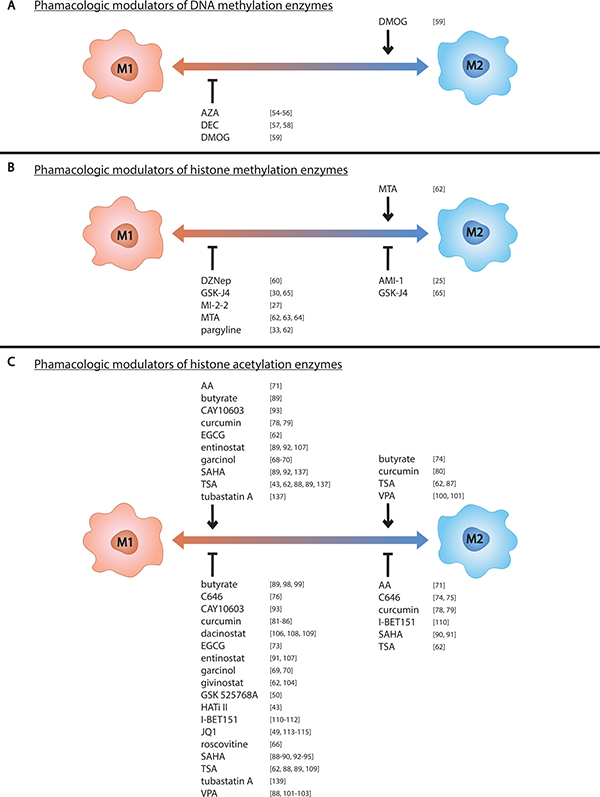
Figure 2: Pharmacologic modulators of epigenetic enzymes influence macrophage phenotype. Pharmacologic modulators known to influence (A) DNA methylation, (B) histone methylation, and (C) histone acetylation in macrophages are listed above the left side of the spectrum if they promote M1 polarization and below if they inhibit M1 polarization. Those known to control M2 polarization are listed likewise on the right side of the spectrum.
DNMTis
Azacytidine (AZA) and decitabine (DEC), otherwise known as Vidaza® and Dacogen®, respectively, are two well-established DNMTis used clinically to treat myelodysplastic syndrome (MDS) [52, 53]. Both are involved in clinical trials against various leukemias and solid tumors (Table 4) [52, 53]. The effects of AZA and DEC have not been investigated in M2s although many studies have examined the effects of these modulators in M1s and unpolarized macrophages.
Table 4: Clinical applications of pharmacologic modulators tested against macrophages
Pharmacologic modulator |
Approved clinical indications a |
Clinical trial cancer indications (total # of trials for cancer) b |
|
|---|---|---|---|
DNMTi |
azacytidine (Vidaza®, AZA) |
acute myeloid leukemia, chronic myelomonocytic leukemia |
various hematologic and solid tumor cancers (520) |
decitabine (Dacogen®, DEC) |
acute myeloid leukemia, chronic myelomonocytic leukemia, myelodysplastic syndrome |
various hematologic and solid tumor cancers (241) |
|
TETi |
dimethyloxallyl glycine (DMOG) |
||
HMTi |
3-deazaneplanocin (DZNep) |
||
AMI-1 |
|||
methylthioadenosine (MTA) |
lung cancer and sarcoma (2) |
||
MI-2-2 |
|||
HDMi |
GSK-J4 |
||
pargyline (Eutonyl, Eutron) |
hypertension c |
||
HATi |
anacardic acid (AA) |
||
C646 |
|||
curcumin |
various hematologic and solid tumor cancers (55) |
||
epigallocatechin-3-gallate (EGCG) |
various solid tumor cancers (28) |
||
garcinol |
|||
Histone Acetyltransferase Inhibitor II (HATi II) |
|||
roscovitine |
breast cancer, non-small cell lung cancer, advanced solid tumors (3) |
||
HDACi |
butyrate |
various hematologic and solid tumor cancers (114) |
|
CAY10603 (BML-281) |
|||
dacinostat (LAQ824) |
|||
entinostat (MS-275) |
various hematologic and solid tumor cancers (47) |
||
givinostat (ITF2357) |
various hematologic cancers (6) |
||
trichostatin A (TSA) |
various hematologic and solid tumor cancers (8) |
||
tubastatin A |
|||
valproic acid (Depakene®, VPA) |
epilepsy, migraine, schizophrenia, seizures, acute manic episodes |
various hematologic and solid tumor cancers (82) |
|
vorinostat (Zolinza®, SAHA) |
cutaneous T-cell lymphoma |
various hematologic and solid tumor cancers (242) |
|
BETi |
GSK 525768A (I-BET-762) |
breast cancer, prostate cancer, hematologic cancers, NUT midline carcinoma (4) |
|
I-BET151 |
|||
JQ1 |
aInformation acquired from www.drugbank.ca.
bInformation acquired from clinicaltrials.gov.
cAll FDA-approved drugs have been discontinued [140].
In M1s, AZA decreases M1 marker expression and increases M2 marker expression. AZA treatment lowers iNOS expression and NO production in LPS- or PGN-stimulated RAW264.7 cells [54], PGN-stimulated mouse BMDMs [55], and mouse peritoneal macrophages stimulated with LPS and IFNγ [56]. The mouse peritoneal macrophages also exhibited decreased TNF expression [56]. Additionally, in the PGN-stimulated RAW264.7 cells, AZA increased expression of M2 markers Arg1 and Fizz1 [55]. A similar effect was observed in mouse macrophages from a myocardial infarction mouse model with cardiac macrophages from untreated mice highly expressing iNOS while cardiac macrophages from AZA-treated mice highly expressing ARG1 [55].
DEC has similar effects to AZA in M1s and unstimulated macrophages. DEC lowers expression of M1 markers TNF, IL6, IL1a, IL1b, iNOS, and a number of chemokines including CCL2, CCL5, and CCL9 in various LPS-stimulated macrophage models and macrophages from atherosclerotic plaques in Ldl-deficient mice [57, 58]. DEC also increases expression of the M2 markers ARG1 in lung macrophages from LPS-treated mice and CD206 in LPS-stimulated mouse BMDMs [58]. This study also found the effects of DEC on Nos2, Arg1, and CD206 expression to be accentuated when in combination with trichostatin A (TSA), an HDAC inhibitor [58]. Additionally, when used in combination with TSA in LPS-stimulated mouse BMDMs, DEC increases STAT3 phosphorylation which downregulates the M1 inflammatory response [58]. Furthermore, DEC increases promoter demethylation and expression of SOCS1 in LPS-stim RAW264.7 cells which promotes the STAT3 pathway [21] and PPARγ and LXRα in unstimulated RAW264.7 cells which causes a decrease in M1 markers TNF, IL6, CCL2, and CCL5, an effect that is reversed by PPARγ and LXRα knockdown [57].
Taken together, these studies suggest that these DNMT modulators would likely promote the M2-TAM phenotype if used in the context of cancer therapy, though no definitive conclusions can be made due to the lack of studies of these modulators in M2s or TAMs.
TETis
The TET protein inhibitor dimethyloxallyl glycine (DMOG) promotes M2 polarization by upregulating Arg1, Fizz1, and Ym1 in mouse peritoneal macrophages in vivo when exposed to chitin, an M2 stimulatory molecule, or in vitro when exposed to IL10 or IL4 + IL13 [59]. In M1s, DMOG downregulates the M1 phenotype by inhibiting LPS-induced NFkβ activity and iNOS expression but upregulates M2 marker IL10 expression [59]. Notably, in unstimulated mouse peritoneal macrophages, DMOG increases expression of the M1 marker iNOS as well as NFkβ activity [59]. These findings are significant for understanding the role of TET proteins in macrophages but suggest that using DMOG in a tumor would promote M2 polarization of TAMs.
HMTis
Few HMTis have been tested against macrophages. 3-Deazaneplanocin (DZNep), an EZH2 inhibitor, inhibits TNF expression in LPS-stimulated RAW264.7 cells [60]. MI-2-2, an inhibitor of MLL-Menin interactions, decreases CXCL10 expression in IFNg-stimulated HMDM [27]. While these inhibitors downregulate M1 phenotype, they have not been tested against M2s.
On the other hand, methylthioadenosine (MTA), an HMT inhibitor, currently involved in clinical trials for oral health [61], has been tested against M2s. In IL4-stimulated mouse BMDMs, pretreatment with MTA upregulated the M2 phenotype by increasing Arg1 expression although it did not significantly affect M2 markers Fizz1, Ym1, Cdh1, and CD206 [62]. The same study found pretreatment with MTA partially downregulated the M1 phenotype by decreasing Tnf mRNA levels as well as TNF and IL6 secretion in LPS + INFγ-stimulated mouse BMDMs while increasing Il1b mRNA levels but exerting no effect on Il6, Nos2, and Stat1 mRNA levels [62]. Other studies which test MTA against M1s have found the same compound downregulates the M1 phenotype in IFNg-stimulated RAW264.7 cells [63] and LPS-stimulated mouse BMDMs [63, 64]. Though MTA has not been tested with in vivo tumor models, its promotion of the M2 phenotype makes it an unlikely candidate for targeting M2 TAMs.
AMI-1, a PRMT1 inhibitor [25], downregulates the M2 phenotype in M2s by decreasing expression of PPARγ and PPARγ-dependent genes CD36, CD206, CD209, and SOCS1 during IL4-induced differentiation of mouse peritoneal macrophages [25]. Though no studies have been performed with M1s, this is a promising compound for altering TAM phenotype since it inhibits normal M2 processes and thus carries the potential to attenuate immunosuppressive, tumor-promoting function of M2-TAMs.
HDMis
Two HDMis, pargyline and GSK-J4, have been tested against macrophages, including M2s. Pargyline, a monamine oxidase and pan-HDM inhibitor previously sold as Eutonyl or Eutron, did not significantly affect the expression of M2 markers Arg1, Fizz1, Ym1, Cdh1, and CD206 in IL4-stimulated mouse BMDMs. Alternatively, the same study found that although pargyline decreases TNF secretion in LPS-challenged mouse BMDMs, it does not significantly affect expression of M1 markers Tnf, Il6, Nos2, Stat1, and Il1b in IFNγ + LPS-stimulated mouse BMDMs [62]. Thus, it appears that pargyline has weak control over macrophage polarization. A separate study found that pargyline prevents LPS tolerance-induced Il6 silencing in mouse BMDMs [33].
GSK-J4, which targets JMJD3 and other KDM6 enzymes, reduces CD206 expression in IL4-stimulated HMDMs [65] and inhibits expression of TNF and other M1 inflammatory cytokines in LPS-stimulated [30] and IFNγ-stimulated [65] HMDMs. These effects are consistent with expectations for a JMJD3 inhibitor due to JMJD3’s vital role in both M1 and M2 polarization. Regardless of its effects on M1s this inhibitor may be a promising compound for decreasing the immunosuppressive and tumor-promoting functions of TAMs.
HATis
Several HATis have been tested in M1s or unstimulated macrophages with various effects observed to the M1 phenotype. The M1 phenotype was downregulated in M1s by Histone Acetyltransferase Inhibitor II (HATi II) which decreased IL1b secretion [43] and by roscovitine which decreased COX-2 and iNOS expression, NO production, and NFκB activation [66]. However, roscovitine, a p300 inhibitor, is better known for its CDK-inhibiting functions [67]. Thus, these effects on macrophage polarization may be due to its influence on cell cycle regulation rather than epigenetic regulation. Garcinol, another p300 inhibitor, has mixed results on M1s; it increases TNF and IL6 expression while decreasing iNOS expression, NO production, COX-2 expression, and NFκB activation in LPS-stimulated RAW264.7 cells [68–70]. Another HATi, anacardic acid (AA), has a negative effect on the M2 phenotype as it decreases IL4 and IL10 secretion when tested against unstimulated macrophages [71]. In these same cells, it has mixed effects on the M1 phenotype as it increases NFκB phosphorylation, migration, phagocytosis, and secretion of NO, IL6, and TNF [71]. Though these results are informative for a number of inflammatory diseases, none of these HATis have been tested against M2s. Therefore, inferences as to these modulators’ efficacy against TAMs cannot be drawn with the potential exception of AA which appears to inhibit the M2 phenotype.
Currently, only three HATis have been tested against M2s: epigallocatechin-3-gallate (EGCG), C646, and curcumin. EGCG, which is involved in a number of clinical trials for indications such as type 2 diabetes, obesity, and various cancers [72], does not significantly affect expression of M2 markers Arg1, Fizz1, Ym1, Cdh1, and CD206 in IL4-stimulated mouse BMDMs [62]. Additionally, its effects on M1s are ambiguous. The same study found that EGCG increases Tnf expression without affecting expression of Il6, Nos2, Stat1, and Il1b in LPS + IFNγ-stimulated BMDMs [62] whereas another study found EGCG inhibits TNF expression in LPS-stimulated HMDMs [73]. Due to this ambiguity and lack of effect on M2 polarization, EGCG exhibits little promise as a targeting agent against M2 TAMs.
Studies implicating C646, a second p300 inhibitor, in macrophage polarization have produced slightly more conclusive results. In M-CSF + IL4-stimulated mouse BMDMs, C646 downregulates Arg1 and Ym1 expression [74]. Another study also found C646 downregulates Arg1, Fizz1, and Mgl2 expression but conversely upregulates Ym1 and CD36 in IL4-stimulated mouse BMDMs [75]. In M1s, C646 decreases NFkB activation and expression of TNF, IL8, IL12B, iNOS, and IL1b [76]. C646’s effects on M2s is somewhat ambiguous but suggests that it might decrease the tumor-promoting functions of M2 TAMs within the context of a tumor.
Curcumin is another p300 inhibitor that is often used therapeutically for various cancers, parasitic infections, inflammatory diseases, & other indications [77]. In a study involving nanoparticles containing the synthetic curcumin derivative hydrazinocurcumin (HC), HC exposure altered the phenotypes of RAW264.7 cells previously co-cultured with 4T1 breast cancer cell line cells. After co-culture but before HC exposure, the RAW264.7 cells exhibited an IL10hi, IL12lo, TGF-βhi M2-like phenotype with high STAT3 activity and high expression of STAT3 downstream genes MMP2, MMP9, and VEGF. After HC exposure, these cells exhibited an IL10lo, IL12hi, TGF-βlo M1-like phenotype with low STAT3 activity and expression of STAT3 downstream genes MMP9, MMP2, and VEGF [78]. These HC-exposed RAW264.7 cells also decreased 4T1 cell proliferation and migration when co-cultured and prolonged survival and reduced tumor burden when administered in vivo to mice co-injected with RAW264.7 cell and 4T1 cells subcutaneously [78]. Others have observed results similar to this with increased levels of Stat4 and Il12 as well as decreased levels of Stat3, Il10, and Arg1 in tumor and spleen tissue of mice with subcutaneous 4T1 breast cancer cell line tumors suggesting a shift from M2- to M1-prominent TAM populations [79]. This study also observed reduced tumor volume and weight with curcumin treatment. Notably, this study analyzed cytokine secretion of whole tissue rather than isolated macrophages, therefore it cannot be concluded that the change in cytokine levels is due to a shift in macrophage phenotype. However, studies using unstimulated macrophages also found curcumin promoted an M2 phenotype with curcumin-treated RAW264.7 cells expressing higher levels of IL4, IL13, CD206, ARG1, PPARg, and phosphorylated STAT6 [80]. Alternatively, studies testing curcumin against M1s show it decreases M1 phenotype by downregulating NFκB activity, ROS production, and expression of TNF, IL6, IL12B, CCL2, and COX-2 while increasing expression of the M2 markers PPARγ and CD36 [81–85]. Similar observations have been made in renal tissue of mice with daunorubicin-induced nephrotoxicity [86]. In vivo curcumin treatment in this model increased M2 markers Il10, CD163, and CD36 while decreasing M1 markers CD80, CD86, IFNγ, IL6, TNF, TNF-R1, COX-2, and ICAM-1 expression and NFκB activation [86]. While various macrophage studies and models produce deviating effects, the in vivo tumor model experiments suggest that curcumin may be an effective pharmacologic modulator for targeting M2 TAMs.
HDACis
HDACis are the most extensively studied epigenetic modulator in macrophages, however, nearly all of this work has been performed in M1s in the context of infectious or inflammatory disease. Trichostatin A (TSA) is one of a few HDACis tested against M2s. In IL4-stimulated mouse BMDMs, TSA inhibited Arg1, Fizz1, and Ym1 expression while increasing Cdh1 expression without affecting Cd206 expression [62]. Additionally, it was found to upregulate c-Myc, an important regulator of the M2 phenotype, in GM-CSF-stimulated mouse BMDMs [87]. Because of these opposing effects on M2 phenotype, it is difficult to predict whether TSA would successfully target M2s in the tumor. TSA has mixed effects in M1s across multiple studies reporting decreased expression of some M1 markers such as Tnf and Il6 with increased expression of other markers such as Stat1, IFNb, and a number of chemokine ligands [62, 88]. Additionally, a number of these studies report conflicting results regarding Cox-2, IL1b, and iNOS expression [43, 62, 88, 89]. These discrepancies in TSA’s influence on LPS-induced effect may be attributed to variations in concentration as has been reported [89]. As mentioned previously, the immunomodulatory effects of TSA in M1s is enhanced by combination treatment with the DNMTi AZA [58].
Another prominent pan-HDACi, vorinostat, otherwise known as Zolinza® or suberoylalanide hydroxamic acid (SAHA), is clinically used to treat cutaneous T cell lymphoma (Table 4). SAHA has yet to be tested using in vitro M2 models but has been used in mouse tumor models. In PyMT mice, SAHA delays tumor growth and reduces tumor burden and inhibits TAM infiltration of estrogen receptor-negative (ER-) mammary tumors while decreasing M-CSF and MMP-9 levels in these tumors [90]. Additionally, SAHA inhibits the increase of F4/80+ and ARG1+ macrophages in mouse pancreatic cancer tumors [91] making it a promising agent for targeting M2 TAMs. SAHA has been studied much more extensively in M1 models and has been found to generally inhibit LPS- and IFNγ-induced signaling and polarization [90, 92–95].
Butyrate, a pan-HDACi involved in clinical trials for schizophrenia [96], increases expression of M2 markers Arg1, Fizz1, Ym1, and CD206 & STAT6 phosphorylation in unstimulated mouse BMDMs [74]. Additionally, oral administration reduces progression of atherosclerosis by reducing migration and adhesion of macrophages [97]. Experiments using in vitro M1 models found decreased LPS-induced Ccl7 and pro-inflammatory mediator expressions in butyrate-treated M1s [89, 98]. Butyrate also decreases expression of M1 markers NO, IL12B, and IL6 without affecting TNF or CCL2 in RAW264.7 cells co-cultured with 3T3-L1 adipocytes [99]. Because it generally inhibits the M1 phenotype and promotes the M2 phenotype, it would likely fail as a M2 TAM-targeting agent.
Valproic acid (VPA), also known as Depakene®, is another pan-HDACi used to clinically treat seizure disorders, mania, and migraine headaches and is involved in multiple clinical trials for neurological indications (Table 4). Its effects on M2s have not been elucidated although it has been shown to inhibit tumor growth enhancement induced by decoy receptor 3 which promotes M2 TAM infiltration [100]. Additionally, in a nitrogen mustard-induced lung injury model, VPA decreases iNOS+CCR2+ macrophages and increases CD68+, CD163+, and ATR-1a+ macrophages in lung tissue [101]. In M1 in vitro models, VPA has been shown to generally decrease M1 markers and phenotype [88, 102, 103]. However, VPA’s promotion of M2 polarization in vivo makes it a poor M2 TAM-targeting agent.
Several other selective HDACis and pan-HDACis including CAY10603 (BML-281), dacinostat (LAQ824), entinostat (MS-275), givinostat (ITF2357), and tubastatin A have been tested against macrophages, however, these have only been tested against M1s. Givinostat, tubastatin A, and CAY10603 decrease the pro-inflammatory phenotype [62, 93, 104, 105] while entinostat and dacinostat both reduce and enhance the M1 phenotype [89, 91, 92, 106–109]. Additionally, of note, dacinostat decreases IL10 expression in LPS-stimulated mouse peritoneal macrophages [106].
BETis
Of the three BETis tested against macrophages, only I-BET151 has been tested against M2s. I-BET151 inhibited expression of M2 marker genes PPARγ, ENPP2, MS4A4A, IL-7R, and ABIN3 in HMDMs stimulated with IL4 or IL10 [110] implicating it as a promising M2 TAM-targeting agent. In M1s, I-BET151 decreases M1 marker expression [110–112] similar to the other BETis tested against M1s, GSK525768A (I-BET-762) [50] and JQ1 [49, 113–115].
Epigenetic modulators as TAM-targeting agents
Of the pharmacologic modulators described, the most promising as M2 TAM-targeting agents are those that inhibit M2 polarization or decrease TAM infiltrate, namely, HMTi AMI-1; HDMi GSK-J4; HATis C646, curcumin, and potentially AA; HDACis SAHA and potentially TSA; and BETi I-BET151. Many epigenetic modulators have been and are being tested in clinical trials to treat various cancers. These modulators are listed in Table 3 alongside their approved clinical indications and clinical trial indications. While these pharmacologic modulators undoubtedly target the malignant cells, they also likely impact the rest of the tumor microenvironment including TAMs. The effect of these epigenetic modulators on the TME as a whole and TAMs in particular remains largely undetermined and it is possible that a significant portion of their anti-tumor effect is a result of modulating TAM tumor-promoting support networks. Understanding pharmacologic modulators in the context of their effect on all cells within the tumor microenvironment may expand their scope as anti-cancer agents. Their indications, for example, could be expanded to include cancers that are supported by TAM infiltration and not just cancers with significant epigenetic dysregulation.
CONCLUSIONS
The epigenetic mechanisms controlling macrophage polarization are complex. There has been much progress in elucidating these mechanisms in inflammatory macrophages but TAMs have largely been overlooked. It is clear that pharmacologic modulators of epigenetic enzymes have effects that are not cell-specific affecting all cells in the TME and throughout the body and conferring therapeutic effects as well as different levels of toxicity. However, many of these modulators have already been tested clinically and deemed safe for therapeutic use. Therefore, epigenetic modulators provide a promising method for targeting TAMs and, due to their current clinical availability, can easily be repurposed for cancers with high M2 TAM infiltrate. Exploiting the differences in M1 and M2 biology using these modulators would provide a means for targeting M2 TAMs thereby eliminating these key tumor-supporting cells from the TME.
Abbreviations
AZA = azacytidine; BET = bromodomain and extra-terminal motif; BETi = BET protein modulator/inhibitor; BMDM = bone marrow-derived macrophage; DEC = decitabine; DNMT = DNA methyltransferase; DNMTi = DNMT modulator/inhibitor; EGCG = epigallocatechin-3-gallate; HATi = HAT modulator/inhibitor; HATi II = Histone Acetyltransferase Inhibitor II; HC = hydrazinocurcumin; HDAC = histone deacetylase; HDACi = HDAC modulator/inhibitor; HDM = histone demethylase; HDMi = HDM modulator/inhibitor; HMDM = human monocyte-derived macrophage; HMT = histone methyltransferase; HMTi = HMT modulator/inhibitor; LDTF = lineage-determining transcription factor; MHC-II = major histocompatibility complex class II receptor; MMP = matrix metalloprotease; NO = nitric oxide; PTM = post-translational modification; ROS = reactive oxygen species; SAHA = suberoylalanide hydroxamic acid (otherwise known as vorinostat or Zolinza®); SDTF = signal-dependent transcription factor; TAM = tumor-associated macrophage; TET = ten eleven translocation; TETi = TET protein modulator/inhibitor; TME = tumor microenvironment; TSA = trichostatin A; VPA = valproic acid.
ACKNOWLEDGMENTS
The authors thank the members of the Brady Urological Institute, especially Dr. Donald S. Coffey and the members of the Pienta Laboratory, for thoughtful discussion.
CONFLICTS OF INTEREST
The authors have no conflicts to declare.
FUNDING
This work was supported by National Institutes of Health Grants U54CA143803, CA163124, CA093900, and CA143055 as well as the Prostate Cancer Foundation.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67:7–30. https://doi.org/10.3322/caac.21387.
2. Amend SR, Pienta KJ. Ecology meets cancer biology: the cancer swamp promotes the lethal cancer phenotype. Oncotarget. 2015; 6:9669–78. https://doi.org/10.18632/oncotarget.3430.
3. Pienta KJ, Robertson BA, Coffey DS, Taichman RS. The cancer diaspora: Metastasis beyond the seed and soil hypothesis. Clin Cancer Res. 2013; 19:5849–55. https://doi.org/10.1158/1078-0432.CCR-13-2158.
4. Yang KR, Mooney SM, Zarif JC, Coffey DS, Taichman RS, Pienta KJ. Niche inheritance: a cooperative pathway to enhance cancer cell fitness through ecosystem engineering. J Cell Biochem. 2014; 115:1478–85. https://doi.org/10.1002/jcb.24813.
5. de Groot AE, Roy S, Brown JS, Pienta KJ, Amend SR. Revisiting Seed and Soil: Examining the Primary Tumor and Cancer Cell Foraging in Metastasis. Mol Cancer Res. 2017; 15:361–70. https://doi.org/10.1158/1541-7786.MCR-16-0436.
6. Predina J, Eruslanov E, Judy B, Kapoor V, Cheng G, Wang LC, Sun J, Moon EK, Fridlender ZG, Albelda S, Singhal S. Changes in the local tumor microenvironment in recurrent cancers may explain the failure of vaccines after surgery. Proc Natl Acad Sci U S A. 2013; 110:E415–24. https://doi.org/10.1073/pnas.1211850110.
7. Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. 2015; 15:409–25. https://doi.org/10.1038/nrc3958.
8. Camacho DF, Pienta KJ. Disrupting the networks of cancer. Clin Cancer Res. 2012; 18:2801–8. https://doi.org/10.1158/1078-0432.CCR-12-0366.
9. Pienta KJ, McGregor N, Axelrod R, Axelrod DE. Ecological therapy for cancer: defining tumors using an ecosystem paradigm suggests new opportunities for novel cancer treatments. Transl Oncol. 2008; 1:158–64.
10. Zarif JC, Taichman RS, Pienta KJ. TAM macrophages promote growth and metastasis within the cancer ecosystem. Oncoimmunology. 2014; 3:e941734. https://doi.org/10.4161/21624011.2014.941734.
11. Yuan A, Hsiao YJ, Chen HY, Chen HW, Ho CC, Chen YY, Liu YC, Hong TH, Yu SL, Chen JJ, Yang PC. Opposite Effects of M1 and M2 Macrophage Subtypes on Lung Cancer Progression. Sci Rep. 2015; 5:14273. https://doi.org/10.1038/srep14273.
12. Zhang M, He Y, Sun X, Li Q, Wang W, Zhao A, Di W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. Journal of Ovarian Research. 2014; 7:19. https://doi.org/10.1186/1757-2215-7-19.
13. Almatroodi SA, McDonald CF, Darby IA, Pouniotis DS. Characterization of M1/M2 Tumour-Associated Macrophages (TAMs) and Th1/Th2 Cytokine Profiles in Patients with NSCLC. Cancer Microenvironment. 2016; 9:1–11. https://doi.org/10.1007/s12307-015-0174-x.
14. Lanciotti M, Masieri L, Raspollini MR, Minervini A, Mari A, Comito G, Giannoni E, Carini M, Chiarugi P, Serni S. The Role of M1 and M2 Macrophages in Prostate Cancer in relation to Extracapsular Tumor Extension and Biochemical Recurrence after Radical Prostatectomy. BioMed Research International. 2014; 2014:486798. https://doi.org/10.1155/2014/486798.
15. Hoeksema MA, de Winther MP. Epigenetic Regulation of Monocyte and Macrophage Function. Antioxid Redox Signal. 2016; 25:758–74. https://doi.org/10.1089/ars.2016.6695.
16. Kapellos TS, Iqbal AJ. Epigenetic Control of Macrophage Polarisation and Soluble Mediator Gene Expression during Inflammation. Mediators Inflamm. 2016; 2016:6591703. https://doi.org/10.1155/2016/6591703.
17. Gosselin D, Glass CK. Epigenomics of macrophages. Immunol Rev. 2014; 262:96–112. https://doi.org/10.1111/imr.12213.
18. Ivashkiv LB. Epigenetic regulation of macrophage polarization and function. Trends in Immunology. 2013; 34:216–23. https://doi.org/10.1016/j.it.2012.11.001.
19. Wallner S, Schroder C, Leitao E, Berulava T, Haak C, Beisser D, Rahmann S, Richter AS, Manke T, Bonisch U, Arrigoni L, Frohler S, Klironomos F, et al. Epigenetic dynamics of monocyte-to-macrophage differentiation. Epigenetics Chromatin. 2016; 9:33. https://doi.org/10.1186/s13072-016-0079-z.
20. Yang X, Wang X, Liu D, Yu L, Xue B, Shi H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol Endocrinol. 2014; 28:565–74. https://doi.org/10.1210/me.2013-1293.
21. Cheng C, Huang C, Ma TT, Bian EB, He Y, Zhang L, Li J. SOCS1 hypermethylation mediated by DNMT1 is associated with lipopolysaccharide-induced inflammatory cytokines in macrophages. Toxicol Lett. 2014; 225:488–97. https://doi.org/10.1016/j.toxlet.2013.12.023.
22. Vandiver AR, Idrizi A, Rizzardi L, Feinberg AP, Hansen KD. DNA methylation is stable during replication and cell cycle arrest. Sci Rep. 2015; 5:17911. https://doi.org/10.1038/srep17911.
23. Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, Liu XS, Aravind L, Agarwal S, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010; 468:839–43. https://doi.org/10.1038/nature09586.
24. Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, Zhao D, Liu Y, Wang C, Zhang X, Su X, Liu J, Ge W, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015; 525:389–93. https://doi.org/10.1038/nature15252.
25. Tikhanovich I, Zhao J, Olson J, Adams A, Taylor R, Bridges B, Marshall L, Roberts B, Weinman SA. Protein arginine methyltransferase 1 modulates innate immune responses through regulation of peroxisome proliferator-activated receptor gamma-dependent macrophage differentiation. J Biol Chem. 2017; 292:6882–94. https://doi.org/10.1074/jbc.M117.778761.
26. Fan Z, Li J, Li P, Ye Q, Xu H, Wu X, Xu Y. Protein arginine methyltransferase 1 (PRMT1) represses MHC II transcription in macrophages by methylating CIITA. Sci Rep. 2017; 7:40531. https://doi.org/10.1038/srep40531.
27. Kittan NA, Allen RM, Dhaliwal A, Cavassani KA, Schaller M, Gallagher KA, Carson WF 4th, Mukherjee S, Grembecka J, Cierpicki T, Jarai G, Westwick J, Kunkel SL, et al. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PLoS One. 2013; 8:e78045. https://doi.org/10.1371/journal.pone.0078045.
28. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, Honma K, Matsuyama T, Yui K, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010; 11:936–44. https://doi.org/10.1038/ni.1920.
29. Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM, Carson WF 4th, Cavassani KA, Li X, Lukacs NW, Hogaboam CM, Dou Y, Kunkel SL. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood. 2009; 114:3244–54. https://doi.org/10.1182/blood-2009-04-217620.
30. Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, Eberhard D, Hutchinson S, Jones E, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012; 488:404–8. https://doi.org/10.1038/nature11262.
31. Van den Bossche J, Neele AE, Hoeksema MA, de Winther MP. Macrophage polarization: the epigenetic point of view. Curr Opin Lipidol. 2014; 25:367–73. https://doi.org/10.1097/MOL.0000000000000109.
32. Kerenyi MA, Shao Z, Hsu YJ, Guo G, Luc S, O’Brien K, Fujiwara Y, Peng C, Nguyen M, Orkin SH. Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. Elife. 2013; 2:e00633. https://doi.org/10.7554/eLife.00633.
33. Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007; 447:972–8. https://doi.org/10.1038/nature05836.
34. Tausendschon M, Dehne N, Brune B. Hypoxia causes epigenetic gene regulation in macrophages by attenuating Jumonji histone demethylase activity. Cytokine. 2011; 53:256–62. https://doi.org/10.1016/j.cyto.2010.11.002.
35. Osawa T, Tsuchida R, Muramatsu M, Shimamura T, Wang F, Suehiro J, Kanki Y, Wada Y, Yuasa Y, Aburatani H, Miyano S, Minami T, Kodama T, et al. Inhibition of histone demethylase JMJD1A improves anti-angiogenic therapy and reduces tumor-associated macrophages. Cancer Res. 2013; 73:3019–28. https://doi.org/10.1158/0008-5472.CAN-12-3231.
36. Feng D, Sangster-Guity N, Stone R, Korczeniewska J, Mancl ME, Fitzgerald-Bocarsly P, Barnes BJ. Differential requirement of histone acetylase and deacetylase activities for IRF5-mediated proinflammatory cytokine expression. J Immunol. 2010; 185:6003–12. https://doi.org/10.4049/jimmunol.1000482.
37. Chauhan A, Quenum FZ, Abbas A, Bradley DS, Nechaev S, Singh BB, Sharma J, Mishra BB. Epigenetic Modulation of Microglial Inflammatory Gene Loci in Helminth-Induced Immune Suppression: Implications for Immune Regulation in Neurocysticercosis. ASN Neuro. 2015; 7. https://doi.org/10.1177/1759091415592126.
38. Mullican SE, Gaddis CA, Alenghat T, Nair MG, Giacomin PR, Everett LJ, Feng D, Steger DJ, Schug J, Artis D, Lazar MA. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes & Development. 2011; 25:2480–8. https://doi.org/10.1101/gad.175950.111.
39. Hoeksema MA, Gijbels MJ, Van den Bossche J, van der Velden S, Sijm A, Neele AE, Seijkens T, Stoger JL, Meiler S, Boshuizen MC, Dallinga-Thie GM, Levels JH, Boon L, et al. Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med. 2014; 6:1124–32. https://doi.org/10.15252/emmm.201404170.
40. Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, Mietton F, Matteoli G, Hiebert S, Natoli G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci U S A. 2012; 109:E2865–74. https://doi.org/10.1073/pnas.1121131109.
41. Cao Q, Rong S, Repa JJ, St Clair R, Parks JS, Mishra N. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler Thromb Vasc Biol. 2014; 34:1871–9. https://doi.org/10.1161/ATVBAHA.114.303393.
42. Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, Nguyen D, Wright KL, Atadja PW, Bhalla K, Pinilla-Ibarz J, Seto E, Sotomayor EM. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol. 2009; 10:92–100. https://doi.org/10.1038/ni.1673.
43. Stammler D, Eigenbrod T, Menz S, Frick JS, Sweet MJ, Shakespear MR, Jantsch J, Siegert I, Wolfle S, Langer JD, Oehme I, Schaefer L, Fischer A, et al. Inhibition of Histone Deacetylases Permits Lipopolysaccharide-Mediated Secretion of Bioactive IL-1beta via a Caspase-1-Independent Mechanism. J Immunol. 2015; 195:5421–31. https://doi.org/10.4049/jimmunol.1501195.
44. Yang Q, Wei J, Zhong L, Shi M, Zhou P, Zuo S, Wu K, Zhu M, Huang X, Yu Y, Zhang H, Yin H, Zhou J. Cross talk between histone deacetylase 4 and STAT6 in the transcriptional regulation of arginase 1 during mouse dendritic cell differentiation. Mol Cell Biol. 2015; 35:63–75. https://doi.org/10.1128/MCB.00805-14.
45. Wang B, Liu TY, Lai CH, Rao YH, Choi MC, Chi JT, Dai JW, Rathmell JC, Yao TP. Glycolysis-dependent histone deacetylase 4 degradation regulates inflammatory cytokine production. Mol Biol Cell. 2014; 25:3300–7. https://doi.org/10.1091/mbc.E13-12-0757.
46. Yoshizaki T, Schenk S, Imamura T, Babendure JL, Sonoda N, Bae EJ, Oh DY, Lu M, Milne JC, Westphal C, Bandyopadhyay G, Olefsky JM. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am J Physiol Endocrinol Metab. 2010; 298:E419–28. https://doi.org/10.1152/ajpendo.00417.2009.
47. Ka SO, Song MY, Bae EJ, Park BH. Myeloid SIRT1 regulates macrophage infiltration and insulin sensitivity in mice fed a high-fat diet. J Endocrinol. 2015; 224:109–18. https://doi.org/10.1530/JOE-14–0527.
48. Shen Z, Ajmo JM, Rogers CQ, Liang X, Le L, Murr MM, Peng Y, You M. Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-alpha production in cultured macrophage cell lines. Am J Physiol Gastrointest Liver Physiol. 2009; 296:G1047–53. https://doi.org/10.1152/ajpgi.00016.2009.
49. Belkina AC, Nikolajczyk BS, Denis GV. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol. 2013; 190:3670–8. https://doi.org/10.4049/jimmunol.1202838.
50. Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010; 468:1119–23. https://doi.org/10.1038/nature09589.
51. Neele AE, Van den Bossche J, Hoeksema MA, de Winther MP. Epigenetic pathways in macrophages emerge as novel targets in atherosclerosis. Eur J Pharmacol. 2015; 763:79–89. https://doi.org/10.1016/j.ejphar.2015.03.101.
52. National Center for Biotechnology Information. PubChem Compound Database. Azacytidine, CID=9444. Available at: https://pubchem.ncbi.nlm.nih.gov/compound/5-azacytidine.
53. National Center for Biotechnology Information. PubChem Compound Database; Decitabine, CID=451668. Available at: https://pubchem.ncbi.nlm.nih.gov/compound/Decitabine.
54. Jeong HY, Kang WS, Hong MH, Jeong HC, Shin MG, Jeong MH, Kim YS, Ahn Y. 5-Azacytidine modulates interferon regulatory factor 1 in macrophages to exert a cardioprotective effect. Sci Rep. 2015; 5:15768. https://doi.org/10.1038/srep15768.
55. Kim YS, Kang WS, Kwon JS, Hong MH, Jeong HY, Jeong HC, Jeong MH, Ahn Y. Protective role of 5-azacytidine on myocardial infarction is associated with modulation of macrophage phenotype and inhibition of fibrosis. J Cell Mol Med. 2014; 18:1018–27. https://doi.org/10.1111/jcmm.12248.
56. Kim HD, Kang HS, Rimbach G, Park YC. Heat shock and 5-azacytidine inhibit nitric oxide synthesis and tumor necrosis factor-alpha secretion in activated macrophages. Antioxid Redox Signal. 1999; 1:297–304. https://doi.org/10.1089/ars.1999.1.3-297.
57. Cao Q, Wang X, Jia L, Mondal AK, Diallo A, Hawkins GA, Das SK, Parks JS, Yu L, Shi H, Shi H, Xue B. Inhibiting DNA Methylation by 5-Aza-2’-deoxycytidine ameliorates atherosclerosis through suppressing macrophage inflammation. Endocrinology. 2014; 155:4925–38. https://doi.org/10.1210/en.2014-1595.
58. Thangavel J, Samanta S, Rajasingh S, Barani B, Xuan YT, Dawn B, Rajasingh J. Epigenetic modifiers reduce inflammation and modulate macrophage phenotype during endotoxemia-induced acute lung injury. J Cell Sci. 2015; 128:3094–105. https://doi.org/10.1242/jcs.170258.
59. Hams E, Saunders SP, Cummins EP, O’Connor A, Tambuwala MT, Gallagher WM, Byrne A, Campos-Torres A, Moynagh PM, Jobin C, Taylor CT, Fallon PG. The hydroxylase inhibitor DMOG attenuates endotoxic shock via alternative activation of macrophages and IL-10 production by B-1 cells. Shock. 2011; 36:295–302. https://doi.org/10.1097/SHK.0b013e318225ad7e.
60. Jeong SY, Lee JH, Kim HS, Hong SH, Cheong CH, Kim IK. 3-Deazaadenosine analogues inhibit the production of tumour necrosis factor-alpha in RAW264.7 cells stimulated with lipopolysaccharide. Immunology. 1996; 89:558–62.
61. National Center for Biotechnology Information. PubChem Compound Database; Methylthioadenosine, CID=439176. Available at: https://pubchem.ncbi.nlm.nih.gov/compound/Methylthioadenosine.
62. Van den Bossche J, Neele AE, Hoeksema MA, de Heij F, Boshuizen MC, van der Velden S, de Boer VC, Reedquist KA, de Winther MP. Inhibiting epigenetic enzymes to improve atherogenic macrophage functions. Biochem Biophys Res Commun. 2014; 455:396–402. https://doi.org/10.1016/j.bbrc.2014.11.029.
63. Keyel PA, Romero M, Wu W, Kwak DH, Zhu Q, Liu X, Salter RD. Methylthioadenosine reprograms macrophage activation through adenosine receptor stimulation. PLoS One. 2014; 9:e104210. https://doi.org/10.1371/journal.pone.0104210.
64. Ara AI, Xia M, Ramani K, Mato JM, Lu SC. S-adenosylmethionine inhibits lipopolysaccharide-induced gene expression via modulation of histone methylation. Hepatology. 2008; 47:1655–66. https://doi.org/10.1002/hep.22231.
65. Yildirim-Buharalioglu G, Bond M, Sala-Newby GB, Hindmarch CC, Newby AC. Regulation of Epigenetic Modifiers, Including KDM6B, by Interferon-gamma and Interleukin-4 in Human Macrophages. Front Immunol. 2017; 8:92. https://doi.org/10.3389/fimmu.2017.00092.
66. Du J, Wei N, Guan T, Xu H, An J, Pritchard KA, Shi Y. Inhibition of CDKS by roscovitine suppressed LPS-induced *NO production through inhibiting NFkappaB activation and BH4 biosynthesis in macrophages. Am J Physiol Cell Physiol. 2009; 297:C742–C9. https://doi.org/10.1152/ajpcell.00138.2009.
67. Deng WG, Zhu Y, Wu KK. Role of p300 and PCAF in regulating cyclooxygenase-2 promoter activation by inflammatory mediators. Blood. 2004; 103:2135–42. https://doi.org/10.1182/blood-2003-09-3131.
68. Wang B, Lin L, Ai Q, Zeng T, Ge P, Zhang L. HAT inhibitor, garcinol, exacerbates lipopolysaccharideinduced inflammation in vitro and in vivo. Mol Med Rep. 2016; 13:5290–6. https://doi.org/10.3892/mmr.2016.5189.
69. Hong J, Sang S, Park HJ, Kwon SJ, Suh N, Huang MT, Ho CT, Yang CS. Modulation of arachidonic acid metabolism and nitric oxide synthesis by garcinol and its derivatives. Carcinogenesis. 2006; 27:278–86. https://doi.org/10.1093/carcin/bgi208.
70. Liao CH, Sang S, Liang YC, Ho CT, Lin JK. Suppression of inducible nitric oxide synthase and cyclooxygenase-2 in downregulating nuclear factor-kappa B pathway by Garcinol. Mol Carcinog. 2004; 41:140–9. https://doi.org/10.1002/mc.20050.
71. Gnanaprakasam JN, Estrada-Muniz E, Vega L. The anacardic 6-pentadecyl salicylic acid induces macrophage activation via the phosphorylation of ERK1/2, JNK, P38 kinases and NF-kappaB. Int Immunopharmacol. 2015; 29:808–17. https://doi.org/10.1016/j.intimp.2015.08.038.
72. National Center for Biotechnology Information. PubChem Compound Database; Epigallocatechin gallate, CID=65064. Available at: https://pubchem.ncbi.nlm.nih.gov/compound/_-_-Epigallocatechin_gallate.
73. Ifrim DC, Quintin J, Joosten LA, Jacobs C, Jansen T, Jacobs L, Gow NA, Williams DL, van der Meer JW, Netea MG. Trained Immunity or Tolerance: Opposing Functional Programs Induced in Human Monocytes after Engagement of Various Pattern Recognition Receptors. Clinical and Vaccine Immunology. 2014; 21:534–45. https://doi.org/10.1128/CVI.00688-13.
74. Ji J, Shu D, Zheng M, Wang J, Luo C, Wang Y, Guo F, Zou X, Lv X, Li Y, Liu T, Qu H. Microbial metabolite butyrate facilitates M2 macrophage polarization and function. Sci Rep. 2016; 6:24838. https://doi.org/10.1038/srep24838.
75. Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, Wang J, Ben-Sahra I, Byles V, Polynne-Stapornkul T, Espinosa EC, Lamming D, Manning BD, et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife. 2016; 5. https://doi.org/10.7554/eLife.11612.
76. van den Bosch T, Boichenko A, Leus NG, Ourailidou ME, Wapenaar H, Rotili D, Mai A, Imhof A, Bischoff R, Haisma HJ, Dekker FJ. The histone acetyltransferase p300 inhibitor C646 reduces pro-inflammatory gene expression and inhibits histone deacetylases. Biochem Pharmacol. 2016; 102:130–40. https://doi.org/10.1016/j.bcp.2015.12.010.
77. National Center for Biotechnology Information. PubChem Compound Database; Curcumin, CID=969516. Available at: https://pubchem.ncbi.nlm.nih.gov/compound/curcumin.
78. Zhang X, Tian W, Cai X, Wang X, Dang W, Tang H, Cao H, Wang L, Chen T. Hydrazinocurcumin Encapsuled nanoparticles “re-educate” tumor-associated macrophages and exhibit anti-tumor effects on breast cancer following STAT3 suppression. PLoS One. 2013; 8:e65896. https://doi.org/10.1371/journal.pone.0065896.
79. Shiri S, Alizadeh AM, Baradaran B, Farhanghi B, Shanehbandi D, Khodayari S, Khodayari H, Tavassoli A. Dendrosomal curcumin suppresses metastatic breast cancer in mice by changing m1/m2 macrophage balance in the tumor microenvironment. Asian Pac J Cancer Prev. 2015; 16:3917–22.
80. Gao S, Zhou J, Liu N, Wang L, Gao Q, Wu Y, Zhao Q, Liu P, Wang S, Liu Y, Guo N, Shen Y, Wu Y, et al. Curcumin induces M2 macrophage polarization by secretion IL-4 and/or IL-13. J Mol Cell Cardiol. 2015; 85:131-9. https://doi.org/10.1016/j.yjmcc.2015.04.025.
81. Zhou Y, Zhang T, Wang X, Wei X, Chen Y, Guo L, Zhang J, Wang C. Curcumin Modulates Macrophage Polarization Through the Inhibition of the Toll-Like Receptor 4 Expression and its Signaling Pathways. Cellular Physiology and Biochemistry. 2015; 36:631–41.
82. Chen FY, Zhou J, Guo N, Ma WG, Huang X, Wang H, Yuan ZY. Curcumin retunes cholesterol transport homeostasis and inflammation response in M1 macrophage to prevent atherosclerosis. Biochemical and Biophysical Research Communications. 2015; 467:872–8. https://doi.org/10.1016/j.bbrc.2015.10.051.
83. Young NA, Bruss MS, Gardner M, Willis WL, Mo X, Valiente GR, Cao Y, Liu Z, Jarjour WN, Wu LC. Oral administration of nano-emulsion curcumin in mice suppresses inflammatory-induced NFkappaB signaling and macrophage migration. PLoS One. 2014; 9:e111559. https://doi.org/10.1371/journal.pone.0111559.
84. Guimaraes MR, Leite FR, Spolidorio LC, Kirkwood KL, Rossa C Jr. Curcumin abrogates LPS-induced pro-inflammatory cytokines in RAW 264.7 macrophages. Evidence for novel mechanisms involving SOCS-1, -3 and p38 MAPK. Arch Oral Biol. 2013; 58:1309–17. https://doi.org/10.1016/j.archoralbio.2013.07.005.
85. Zhong Y, Liu T, Lai W, Tan Y, Tian D, Guo Z. Heme oxygenase-1-mediated reactive oxygen species reduction is involved in the inhibitory effect of curcumin on lipopolysaccharide-induced monocyte chemoattractant protein-1 production in RAW264.7 macrophages. Mol Med Rep. 2013; 7:242–6. https://doi.org/10.3892/mmr.2012.1138.
86. Karuppagounder V, Arumugam S, Thandavarayan RA, Sreedhar R, Giridharan VV, Afrin R, Harima M, Miyashita S, Hara M, Suzuki K, Nakamura M, Ueno K, Watanabe K. Curcumin alleviates renal dysfunction and suppresses inflammation by shifting from M1 to M2 macrophage polarization in daunorubicin induced nephrotoxicity in rats. Cytokine. 2016; 84:1–9. https://doi.org/10.1016/j.cyto.2016.05.001.
87. Sebastián C, Serra M, Yeramian A, Serrat N, Lloberas J, Celada A. Deacetylase Activity Is Required for STAT5-Dependent GM-CSF Functional Activity in Macrophages and Differentiation to Dendritic Cells. The Journal of Immunology. 2008; 180:5898–906. https://doi.org/10.4049/jimmunol.180.9.5898.
88. Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T, Ding XC, Chanson AL, Reymond MK, Miconnet I, Schrenzel J, François P, Calandra T. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood. 2011; 117:1205–17. https://doi.org/10.1182/blood-2010-05-284711.
89. Halili MA, Andrews MR, Labzin LI, Schroder K, Matthias G, Cao C, Lovelace E, Reid RC, Le GT, Hume DA, Irvine KM, Matthias P, Fairlie DP, et al. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J Leukoc Biol. 2010; 87:1103–14. https://doi.org/10.1189/jlb.0509363.
90. Tran K, Risingsong R, Royce DB, Williams CR, Sporn MB, Pioli PA, Gediya LK, Njar VC, Liby KT. The combination of the histone deacetylase inhibitor vorinostat and synthetic triterpenoids reduces tumorigenesis in mouse models of cancer. Carcinogenesis. 2013; 34:199–210. https://doi.org/10.1093/carcin/bgs319.
91. Edderkaoui M, Xu S, Chheda C, Morvaridi S, Hu RW, Grippo PJ, Mascarinas E, Principe DR, Knudsen B, Xue J, Habtezion A, Uyeminami D, Pinkerton KE, et al. HDAC3 mediates smoking-induced pancreatic cancer. Oncotarget. 2016; 7:7747–60. https://doi.org/10.18632/oncotarget.6820.
92. Leus NG, van den Bosch T, van der Wouden PE, Krist K, Ourailidou ME, Eleftheriadis N, Kistemaker LE, Bos S, Gjaltema RA, Mekonnen SA, Bischoff R, Gosens R, Haisma HJ, et al. HDAC1-3 inhibitor MS-275 enhances IL10 expression in RAW264.7 macrophages and reduces cigarette smoke-induced airway inflammation in mice. Sci Rep. 2017; 7:45047. https://doi.org/10.1038/srep45047.
93. Lohman RJ, Iyer A, Fairlie TJ, Cotterell A, Gupta P, Reid RC, Vesey DA, Sweet MJ, Fairlie DP. Differential Anti-inflammatory Activity of HDAC Inhibitors in Human Macrophages and Rat Arthritis. J Pharmacol Exp Ther. 2016; 356:387–96. https://doi.org/10.1124/jpet.115.229328.
94. Chong W, Li Y, Liu B, Zhao T, Fukudome EY, Liu Z, Smith WM, Velmahos GC, deMoya MA, Alam HB. Histone deacetylase inhibitor suberoylanilide hydroxamic acid attenuates Toll-like receptor 4 signaling in lipopolysaccharide-stimulated mouse macrophages. Journal of Surgical Research. 2012; 178:851–9. https://doi.org/http://dx.doi.org/10.1016/j.jss.2012.07.023.
95. Fang S, Meng X, Zhang Z, Wang Y, Liu Y, You C, Yan H. Vorinostat Modulates the Imbalance of T Cell Subsets, Suppresses Macrophage Activity, and Ameliorates Experimental Autoimmune Uveoretinitis. Neuromolecular Med. 2016; 18:134–45. https://doi.org/10.1007/s12017-016-8383-0.
96. National Center for Biotechnology Information. PubChem Compound Database; Sodium butyrate, CID=5222465. Available at: https://pubchem.ncbi.nlm.nih.gov/compound/sodium_butyrate.
97. Aguilar EC, Leonel AJ, Teixeira LG, Silva AR, Silva JF, Pelaez JM, Capettini LS, Lemos VS, Santos RA, Alvarez-Leite JI. Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFkappaB activation. Nutr Metab Cardiovasc Dis. 2014; 24:606–13. https://doi.org/10.1016/j.numecd.2014.01.002.
98. Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2014; 111:2247–52. https://doi.org/10.1073/pnas.1322269111.
99. Ohira H, Fujioka Y, Katagiri C, Mamoto R, Aoyama-Ishikawa M, Amako K, Izumi Y, Nishiumi S, Yoshida M, Usami M, Ikeda M. Butyrate attenuates inflammation and lipolysis generated by the interaction of adipocytes and macrophages. J Atheroscler Thromb. 2013; 20:425–42.
100. Tai SK, Chang HC, Lan KL, Lee CT, Yang CY, Chen NJ, Chou TY, Tarng DC, Hsieh SL. Decoy receptor 3 enhances tumor progression via induction of tumor-associated macrophages. J Immunol. 2012; 188:2464–71. https://doi.org/10.4049/jimmunol.1101101.
101. Venosa A, Gow JG, Hall L, Malaviya R, Gow AJ, Laskin JD, Laskin DL. Regulation of Nitrogen Mustard-Induced Lung Macrophage Activation by Valproic Acid, a Histone Deacetylase Inhibitor. Toxicol Sci. 2017; 157:222–34. https://doi.org/10.1093/toxsci/kfx032.
102. Wu C, Li A, Leng Y, Li Y, Kang J. Histone deacetylase inhibition by sodium valproate regulates polarization of macrophage subsets. DNA Cell Biol. 2012; 31:592–9. https://doi.org/10.1089/dna.2011.1401.
103. Jambalganiin U, Tsolmongyn B, Koide N, Odkhuu E, Naiki Y, Komatsu T, Yoshida T, Yokochi T. A novel mechanism for inhibition of lipopolysaccharide-induced proinflammatory cytokine production by valproic acid. Int Immunopharmacol. 2014; 20:181–7. https://doi.org/10.1016/j.intimp.2014.02.032.
104. Lewis EC, Blaabjerg L, Storling J, Ronn SG, Mascagni P, Dinarello CA, Mandrup-Poulsen T. The oral histone deacetylase inhibitor ITF2357 reduces cytokines and protects islet beta cells in vivo and in vitro. Mol Med. 2011; 17:369–77. https://doi.org/10.2119/molmed.2010.00152.
105. Li Y, Zhao T, Liu B, Halaweish I, Mazitschek R, Duan X, Alam HB. Inhibition of histone deacetylase 6 improves long-term survival in a lethal septic model. J Trauma Acute Care Surg. 2015; 78:378–85. https://doi.org/10.1097/TA.0000000000000510.
106. Wang H, Cheng F, Woan K, Sahakian E, Merino O, Rock-Klotz J, Vicente-Suarez I, Pinilla-Ibarz J, Wright KL, Seto E, Bhalla K, Villagra A, Sotomayor EM. Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J Immunol. 2011; 186:3986–96. https://doi.org/10.4049/jimmunol.1001101.
107. Zhang ZY, Schluesener HJ. HDAC inhibitor MS-275 attenuates the inflammatory reaction in rat experimental autoimmune prostatitis. Prostate. 2012; 72:90–9. https://doi.org/10.1002/pros.21410.
108. Brogdon JL, Xu Y, Szabo SJ, An S, Buxton F, Cohen D, Huang Q. Histone deacetylase activities are required for innate immune cell control of Th1 but not Th2 effector cell function. Blood. 2007; 109:1123–30. https://doi.org/10.1182/blood-2006-04-019711.
109. Usami M, Kishimoto K, Ohata A, Miyoshi M, Aoyama M, Fueda Y, Kotani J. Butyrate and trichostatin A attenuate nuclear factor κB activation and tumor necrosis factor α secretion and increase prostaglandin E2 secretion in human peripheral blood mononuclear cells. Nutrition Research. 2008; 28:321–8. https://doi.org/http://dx.doi.org/10.1016/j.nutres.2008.02.012.
110. Chan CH, Fang C, Yarilina A, Prinjha RK, Qiao Y, Ivashkiv LB. BET Bromodomain Inhibition Suppresses Transcriptional Responses to Cytokine-Jak-STAT Signaling in a Gene-specific Manner in Human Monocytes. European Journal of Immunology. 2015; 45:287–97. https://doi.org/10.1002/eji.201444862.
111. Barrett E, Brothers S, Wahlestedt C, Beurel E. I-BET151 selectively regulates IL-6 production. Biochimica et Biophysica Acta. 2014; 1842:1549–55. https://doi.org/10.1016/j.bbadis.2014.05.013.
112. Fu W, Farache J, Clardy SM, Hattori K, Mander P, Lee K, Rioja I, Weissleder R, Prinjha RK, Benoist C, Mathis D. Epigenetic modulation of type-1 diabetes via a dual effect on pancreatic macrophages and β cells. eLife. 2014; 3:e04631. https://doi.org/10.7554/eLife.04631.
113. Meng S, Zhang L, Tang Y, Tu Q, Zheng L, Yu L, Murray D, Cheng J, Kim SH, Zhou X, Chen J. BET Inhibitor JQ1 Blocks Inflammation and Bone Destruction. Journal of Dental Research. 2014; 93:657–62. https://doi.org/10.1177/0022034514534261.
114. Wienerroither S, Rauch I, Rosebrock F, Jamieson AM, Bradner J, Muhar M, Zuber J, Müller M, Decker T. Regulation of NO Synthesis, Local Inflammation, and Innate Immunity to Pathogens by BET Family Proteins. Molecular and Cellular Biology. 2014; 34:415–27. https://doi.org/10.1128/MCB.01353-13.
115. Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, Yokoyama Y, Kossenkov AV, Bradner JE, Conejo-Garcia JR, Zhang R. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Reports. 2016; 16:2829–37. https://doi.org/10.1016/j.celrep.2016.08.032.
116. Xia M, Liu J, Wu X, Liu S, Li G, Han C, Song L, Li Z, Wang Q, Wang J, Xu T, Cao X. Histone methyltransferase Ash1l suppresses interleukin-6 production and inflammatory autoimmune diseases by inducing the ubiquitin-editing enzyme A20. Immunity. 2013; 39:470–81. https://doi.org/10.1016/j.immuni.2013.08.016.
117. Liu Y, Zhang Q, Ding Y, Li X, Zhao D, Zhao K, Guo Z, Cao X. Histone lysine methyltransferase Ezh1 promotes TLR-triggered inflammatory cytokine production by suppressing Tollip. J Immunol. 2015; 194:2838–46. https://doi.org/10.4049/jimmunol.1402087.
118. Nguyen KD, Fentress SJ, Qiu Y, Yun K, Cox JS, Chawla A. Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science. 2013; 341:1483–8. https://doi.org/10.1126/science.1240636.
119. Chen X, El Gazzar M, Yoza BK, McCall CE. The NF-kappaB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J Biol Chem. 2009; 284:27857–65. https://doi.org/10.1074/jbc.M109.000950.
120. Austenaa L, Barozzi I, Chronowska A, Termanini A, Ostuni R, Prosperini E, Stewart AF, Testa G, Natoli G. The histone methyltransferase Wbp7 controls macrophage function through GPI glycolipid anchor synthesis. Immunity. 2012; 36:572–85. https://doi.org/10.1016/j.immuni.2012.02.016.
121. Li Y, Reddy MA, Miao F, Shanmugam N, Yee JK, Hawkins D, Ren B, Natarajan R. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J Biol Chem. 2008; 283:26771–81. https://doi.org/10.1074/jbc.M802800200.
122. Eames HL, Saliba DG, Krausgruber T, Lanfrancotti A, Ryzhakov G, Udalova IA. KAP1/TRIM28: an inhibitor of IRF5 function in inflammatory macrophages. Immunobiology. 2012; 217:1315–24. https://doi.org/10.1016/j.imbio.2012.07.026.
123. Schliehe C, Flynn EK, Vilagos B, Richson U, Swaminathan S, Bosnjak B, Bauer L, Kandasamy RK, Griesshammer IM, Kosack L, Schmitz F, Litvak V, Sissons J, et al. The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nat Immunol. 2015; 16:67–74. https://doi.org/10.1038/ni.3046.
124. Xu G, Liu G, Xiong S, Liu H, Chen X, Zheng B. The histone methyltransferase Smyd2 is a negative regulator of macrophage activation by suppressing interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-alpha) production. J Biol Chem. 2015; 290:5414–23. https://doi.org/10.1074/jbc.M114.610345.
125. Stender JD, Pascual G, Liu W, Kaikkonen MU, Do K, Spann NJ, Boutros M, Perrimon N, Rosenfeld MG, Glass CK. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol Cell. 2012; 48:28–38. https://doi.org/10.1016/j.molcel.2012.07.020.
126. Arts RJ, Blok BA, van Crevel R, Joosten LA, Aaby P, Benn CS, Netea MG. Vitamin A induces inhibitory histone methylation modifications and down-regulates trained immunity in human monocytes. J Leukoc Biol. 2015; 98:129–36. https://doi.org/10.1189/jlb.6AB0914-416R.
127. van Essen D, Zhu Y, Saccani S. A feed-forward circuit controlling inducible NF-kappaB target gene activation by promoter histone demethylation. Mol Cell. 2010; 39:750–60. https://doi.org/10.1016/j.molcel.2010.08.010.
128. Zhu Y, van Essen D, Saccani S. Cell-type-specific control of enhancer activity by H3K9 trimethylation. Mol Cell. 2012; 46:408–23. https://doi.org/10.1016/j.molcel.2012.05.011.
129. Li X, Zhang Q, Shi Q, Liu Y, Zhao K, Shen Q, Shi Y, Liu X, Wang C, Li N, Ma Y, Cao X. Demethylase Kdm6a epigenetically promotes IL-6 and IFN-beta production in macrophages. J Autoimmun. 2017; 80:85–94. https://doi.org/10.1016/j.jaut.2017.02.007.
130. Mounce BC, Mboko WP, Kanack AJ, Tarakanova VL. Primary macrophages rely on histone deacetylase 1 and 2 expression to induce type I interferon in response to gammaherpesvirus infection. J Virol. 2014; 88:2268–78. https://doi.org/10.1128/JVI.03278-13.
131. Yao Z, Zhang Q, Li X, Zhao D, Liu Y, Zhao K, Liu Y, Wang C, Jiang M, Li N, Cao X. Death domain-associated protein 6 (Daxx) selectively represses IL-6 transcription through histone deacetylase 1 (HDAC1)-mediated histone deacetylation in macrophages. J Biol Chem. 2014; 289:9372–9. https://doi.org/10.1074/jbc.M113.533992.
132. Kong X, Fang M, Li P, Fang F, Xu Y. HDAC2 deacetylates class II transactivator and suppresses its activity in macrophages and smooth muscle cells. J Mol Cell Cardiol. 2009; 46:292–9. https://doi.org/10.1016/j.yjmcc.2008.10.023.
133. Poralla L, Stroh T, Erben U, Sittig M, Liebig S, Siegmund B, Glauben R. Histone deacetylase 5 regulates the inflammatory response of macrophages. J Cell Mol Med. 2015; 19:2162–71. https://doi.org/10.1111/jcmm.12595.
134. Yan B, Xie S, Liu Z, Ran J, Li Y, Wang J, Yang Y, Zhou J, Li D, Liu M. HDAC6 deacetylase activity is critical for lipopolysaccharide-induced activation of macrophages. PLoS One. 2014; 9:e110718. https://doi.org/10.1371/journal.pone.0110718.
135. Cheng F, Lienlaf M, Perez-Villarroel P, Wang HW, Lee C, Woan K, Woods D, Knox T, Bergman J, Pinilla-Ibarz J, Kozikowski A, Seto E, Sotomayor EM, et al. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol Immunol. 2014; 60:44–53. https://doi.org/10.1016/j.molimm.2014.02.019.
136. Cheng F, Lienlaf M, Wang HW, Perez-Villarroel P, Lee C, Woan K, Rock-Klotz J, Sahakian E, Woods D, Pinilla-Ibarz J, Kalin J, Tao J, Hancock W, et al. A novel role for histone deacetylase 6 in the regulation of the tolerogenic STAT3/IL-10 pathway in APCs. J Immunol. 2014; 193:2850–62. https://doi.org/10.4049/jimmunol.1302778.
137. Ariffin JK, das Gupta K, Kapetanovic R, Iyer A, Reid RC, Fairlie DP, Sweet MJ. Histone Deacetylase Inhibitors Promote Mitochondrial Reactive Oxygen Species Production and Bacterial Clearance by Human Macrophages. Antimicrobial Agents and Chemotherapy. 2016; 60:1521–9. https://doi.org/10.1128/AAC.01876-15.
138. Shakespear MR, Hohenhaus DM, Kelly GM, Kamal NA, Gupta P, Labzin LI, Schroder K, Garceau V, Barbero S, Iyer A, Hume DA, Reid RC, Irvine KM, et al. Histone deacetylase 7 promotes Toll-like receptor 4-dependent proinflammatory gene expression in macrophages. J Biol Chem. 2013; 288:25362–74. https://doi.org/10.1074/jbc.M113.496281.
139. Vishwakarma S, Iyer LR, Muley M, Singh PK, Shastry A, Saxena A, Kulathingal J, Vijaykanth G, Raghul J, Rajesh N, Rathinasamy S, Kachhadia V, Kilambi N, et al. Tubastatin, a selective histone deacetylase 6 inhibitor shows anti-inflammatory and anti-rheumatic effects. International Immunopharmacology. 2013; 16:72–8. https://doi.org/http://dx.doi.org/10.1016/j.intimp.2013.03.016.
140. U.S. Food & Drug Administration. Drugs@FDA: FDA Approved Drug Products, Pargyline. Available at: https://www.accessdata.fda.gov/scripts/cder/daf/.