
INTRODUCTION
Myelodysplastic syndromes (MDS) are oligoclonal disorders of the hematopoietic stem cells (HSC) and are propagated by rare human MDS propagating cells [1, 2]. MDS comprise diverse phenotypes characterized by ineffective hematopoiesis leading to cytopenia, bone marrow dysplasia and transformation to acute myeloid leukemia in 30 to 40% of the cases [3].
Somatic mutations are common in MDS physiopathology and different functional groups are identified: splicing factors, epigenetic regulators, transcription factors and kinase signaling [4-7]. Moreover, the medullar microenvironment especially represented by the mesenchymal stromal cells (MSC) is also implicated in the development and propagation of dysplastic hematopoiesis [8-10]. Extensive data suggest that MDS-derived MSC are functionally altered and harbored epigenetic alterations [11]. To better study the heterogeneity of MDS and to develop new therapeutic drugs, it becomes necessary to create patient derived xenograft (PDX) mice models by engrafting CD34+ cells from MDS patients. Recent studies demonstrated that intramedullary co-transplantation method with HSC and MSC could improve engraftment on MDS in xenograft models [12]. Here, we describe an efficient PDX model by using primary CD34+ and autologous MSC from MDS patients. This model is stable over generations of mice and allows us to decipher the mutational hierarchy amongst different populations of early progenitors generated in mice by longitudinal next generation sequencing. We will use this tool for new therapeutic approaches.
RESULTS
Engraftment of patient-derived myelodysplastic syndrome in NSG mice
We transplanted intramedullary CD34+ cells with autologous mesenchymal stromal cells from bone marrow obtained from patients with MDS in NOD/SCID/IL2rγ−/− (NSG) mice with a ratio of 1/3 respectively for CD34+ and MSC [12]. Twenty-four hours prior to transplantation, mice received one busulfan injection intraperitoneally (25mg/kg). There were one refractory anemia with blast excess (RAEB) 2, two RAEB 1 and one refractory anemia with ring sideroblast (RARS) with karyotype abnormalities and recurrent gene mutations involved in MDS (characteristics are described in Table 1). Each patient sample gave rise to three mice. Six months after transplant, mice were euthanized as already described in Medyouf et al’s work [12]. No mice showed earlier signs of the disease like loss of weight or nose and eye bleeding. The bone marrow was investigated for human chimerism by flow cytometry based on human CD45, human CD33, human CD19 and murine CD45. The average human chimerisms (based on percentage of human CD45 positive cells) were respectively 59.7% (50.92 - 68.48); 0.23% (0.142 - 0.318); 0.13% (0.097 - 0.318) and 0.0175% (0.015 – 0.019) for the 4 patients (Figure 1A, 1B). As already described in previous studies, we have chosen a threshold of 0.01% of human cells to define a human engraftment in mice [13] and according to this threshold all the mice were engrafted. There was a clear myeloid bias in this human engraftment as we did not find any B positive cells.
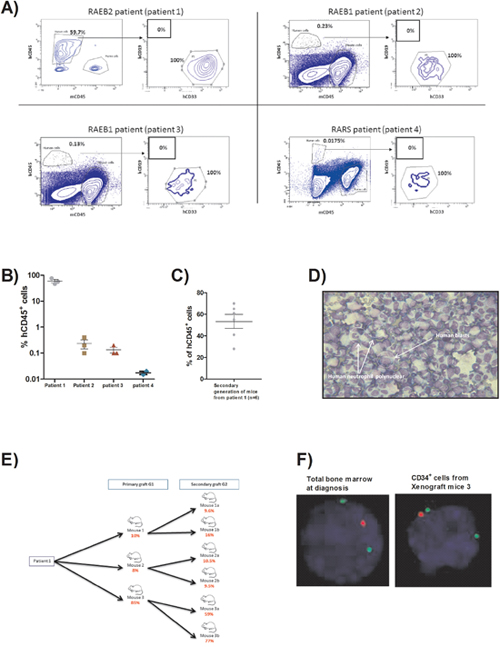
Figure 1: (A) FACS plots showing the percentage of human cells at sacrifice (6 months after transplant) for the four patients engrafted. (B) Scatter plot representing the percentage of human CD45+cells assessed by flow cytometry within the bone marrow retrieved from 1st generation mice for each patient (6 months after transplant). (C) Scatter plot representing the percentage of human CD45+ cells assessed by flow cytometry within the bone marrow retrieved from 2nd generation mice deriving from patient 1 (6 months after transplant). (D) Picture of bone marrow cytospin stained by MGG (x40) from 1st generation mouse of patient 1 showing the human blast infiltration and low granularity of granulocytic lineage. (E) Flow chart representing the number of human blasts counted on bone marrow cytospin from each mouse of 1st and 2nd generations derived from patient 1 (6 months after transplant). (F) FISH detection of del 5q. Nucleus is stained in blue (DAPI), del5q probe in red and control probe in green (x63). Experiment done on total bone marrow from patient 1 at diagnosis and on CD34+ sorted cells from one mouse of 1st generation (6 months after transplant).
Table 1: Table representing the principal biological characteristics of patients used for the xenograft model
MDS |
Percentage of medullary blasts at diagnostis |
Karyotype |
Molecular biology |
|
|---|---|---|---|---|
patient 1 |
RAEB-2 |
14 |
TP53 (G245D), SF3B1(R625H), RUNX1 (R201Q) and KIT (D816V) |
|
patient 2 |
RAEB-1 |
4 |
TP53 (G245D) |
|
patient 3 |
RAEB-1 |
6 |
45,X,-Y,del(11)(q21q25)[1]/45,sl,ins(4;3)(q2?6;q28q24)[2]/90,idemx2 [1]/45,sdl1,dic(11;?)(q13;?)[12]/45,sl,t(6;12)(q25;q12),add(11)(q13)[4]/46,XY [2].ish ins(4;3)(EVI1+) |
TET2 (R1216X) |
patient 4 |
RARS |
1,5 |
46,XX [20] |
SF3B1 (K666R) |
RAEB: Refractory anemia with excess of blast; RARS: Refractory anemia with ring sideroblasts.
We also determined if the human mesenchymal stromal cells injected with CD34+ were still present at the end of the xenograft procedure. By flow cytometry and using a consensual panel of antibodies (CD90, CD73, CD105 and CD45) [14], we did not find any human MSC in mice bone marrow (data not shown) at 6 months of engraftment suggesting that MSC were essentially involved at the beginning of the engraftment process.
Secondary grafts were only performed for mice derived from patient 1 (RAEB2) and each mouse of the first generation gave rise to two mice for the second generation. These secondary grafts were performed by intra bone injection of 1x105 CD34+ cells without autologous MSC to assess the propagating capacity of the cells obtained after the first PDX. For the secondary graft, we hypothesized that the engraftment would be faster so we performed a tibia bone marrow aspiration to monitor the engraftment 3 months after injection and as human engraftment was already evidenced by this intermediate point, the mice were sacrificed at 3 months. The average human chimerism for the six mice of the second generation was 53.33% (36.61 - 70.05), not different from the chimerism obtained at the first engraftment (Figure 1C).
Molecular dissection of the bone marrow compartment of mice engrafted with myelodysplastic syndrome-originated cells
Then, we focused on biological materials from patient 1 graft for all further experiments. First, morphological observations of bone marrow cytospins stained by May-Grünwald Giemsa (MGG) coloration of engrafted mice from first and second generations showed presence of human blasts and human neutrophils and characteristics features of dysplasia already described in the original patient sample (Figure 1D). Moreover, we have realized human blasts count on those cytospins. Interestingly, mice 1 and 2 showed less than 20% of medullar blasts and also for their derived 2nd generation mice (10.6% (9.47 – 11.73)) whereas mouse 3 and the resulting secondary graft mice harbored more than 20% of medullary blasts (73.67% (65.98 – 81.35)) suggesting a more aggressive disease (Figure 1E).
We performed FISH analyses of CD34+ cells from the first generation of mice and found the same cytogenetic abnormality (del5q) in 87.5% of the CD34+ cells harvested from the mice versus 79% in total bone marrow cells at diagnosis of the patient 1 (Figure 1F).
We then isolated hematopoietic stem cells (HSCs), common myeloid progenitors (CMPs), granulocyte macrophage progenitors (GMPs) and megakaryocyte–erythroid progenitor (MEPs) by FACS cells sorting based on specific antigens (CD34, CD38, CD45RA and CD123) as already described [15] (Figure 2A) from xenografted bone marrow from first and second generations of mice. We performed targeted sequencing for genes with initial mutations found in the patient 1 sample (KIT, SF3B1, TP53 and RUNX1) in all the progenitors by Sanger technique with specific primers. We found these initial mutations in all the different progenitors in 1st and 2nd generation of mice (Figure 2B). We performed also next generation sequencing (NGS) on a panel of 39 genes previously described and involved in MDS physiopathogenesis [16] in the HSC from the first-generation mice to check if the xenograft procedure could induce other mutations than specified in the initial sample of the patient 1, and in HSC derived from mouse 3 to analyze if the excess of blasts number, reflecting probably a more aggressive disease, was associated with new mutations. Concerning HSC, the variant allele frequencies (VAF) of initial mutations were similar between all the mice from the first generation and mice derived from mice 3 (45-50%) (Figure 2C). No new recurrent mutations were found to explain the higher blasts level of mice 3 and its derived mice. NGS was also performed on the initial mesenchymal stromal cells used during the xenograft procedure. Neither the four initial mutations nor other known recurrent mutation in MDS were found in these MSC. We found two polymorphisms, one well-known on TP53 gene and another one on STAG1 gene in both HSC and MSC, strongly suggesting their germinal origin.
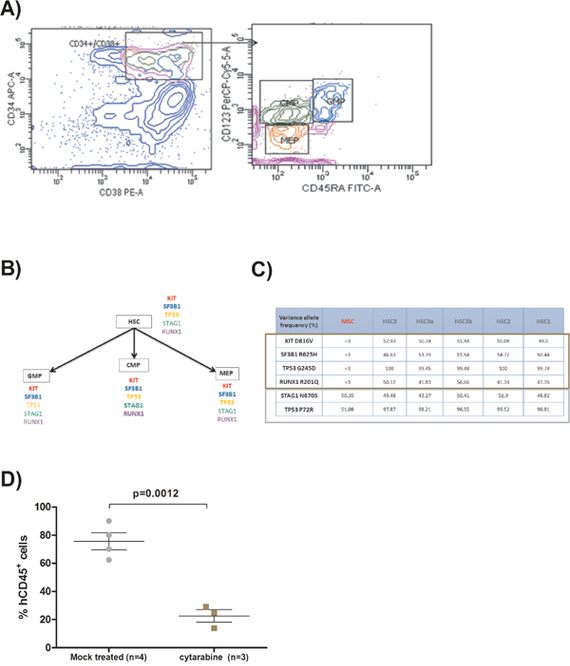
Figure 2: (A) FACS plots showing the method used for hematopoietic progenitors sorting from xenografted bone marrow. Hematopoietic stem cells (HSC) are CD34+/CD38-, common myeloid progenitors (CMP) are CD34+/CD38+/CD123+/CD45RA-, granulocyte macrophage progenitors (GMP) are CD34+/CD38+/CD123+/CD45RA+, and megakaryocyte–erythroid progenitors (MEP) are CD34+/CD38+/CD123-/CD45RA-. Human cells were previously sorted with human CD34+ magnetic beads. (B) Flow chart describing the presence of the different gene mutations in each kind of human progenitors sorted from the bone marrow of the different generations of mice derived from patient 1. (C) Table representing the different variant allele frequency obtained by next generation sequencing on hematopoietic stem cells (HSC) and mesenchymal stromal cells (MSC) from patient 1 xenografted mice. The brown rectangle represents the somatic mutations and the grey one polymorphisms. (D) Scatter plot representing the effect of cytarabine on the percentage of human CD45+cells assessed by flow cytometry within the bone marrow retrieved from xenografted mice deriving from patient 1. Two groups, one with mice treated with cytarabine intraperitoneally (10mg/kg) 5 days/7 during two weeks and another group with mock treated mice.
Chemotherapy validation of the model
Furthermore, we validated our model by using a therapeutic approach with chemotherapy. Indeed, we have treated engrafted mice with intraperitoneally injection of cytarabine (10mg/kg) five days per week during two weeks at the third month post-transplant. The mice were killed one week after the end of cytarabine course. Bone marrow chimerism was analyzed as previously described. Mock treated mice show an average 75.65% (69.4 – 81.66) of human CD45+ cells versus 22.67% (18.69 – 27.15) (p=0.0012) for mice treated with cytarabine (Figure 2D). We have also done cell sorting for the HSCs, CMPs, GMPs and MEPs in order to perform targeted sequencing for initial gene mutations. These mutations were found in all the progenitors, reflecting the fact that although cytarabine reduces human chimerism, all the mutated progenitors remain and will probably be the stem of future relapses.
DISCUSSION
We demonstrate here the possibility to make a PDX mouse model that perfectly reproduces the MDS founder clone. We chose to add MSC to CD34+ at the first generation of mice as we believe as Medyouf et al [12], that a humanized microenvironment could improve engraftment of MDS CD34+ cells. This method allowed us to get an engraftment success of 100% of our patients although the series is small. There is controversy about the beneficial effect on engraftment of adding mesenchymal stromal cells to CD34+ cells [13]. Muguruma et al have [17] achieved a successful engraftment of myelodysplastic syndrome by adding MSC to CD34+ cells but the efficiency of the engraftment was related to the presence of an abnormality in chromosome 7. Li et al [18] obtained better chimerism by engrafting CD34+ cell with a human marrow stroma cell line (HS27a). Those results were confirmed by Medyouf et al [12] with direct intrabone injection of CD34+ cells and MSC. Like Medyouf et al, we have also chosen some higher risk MDS patients with RAEB1 and RCMD with complex karyotype. Nevertheless patient 1 had a more aggressive disease.
Recently, Rouault-Pierre et al [13] showed that co-injection of CD34+ cells with MSC did not improve the level of engraftment in a cohort of 10 MDS samples. These data suggest that engraftment potential of the MDS CD34+ did not depend on the presence of human MSC. Interestingly, in Medyouf’s study, the secondary grafts were performed without adding MSC and cells successfully engrafted as we have found. We can hypothesize that MSC cells could improve the level of engraftment for primary graft, but for secondary graft, the propagation capacities of the sole cells are sufficient to engraft.
In one patient, all the mutations were present very early in the course of the disease in the HSC, CMP, MEP and GMP. This clone is very stable over time as it does not disappear in the second generation of mice and no new mutations were found neither. Recently, Belderbos et al [19] demonstrated in a xenograft model of lymphoblastic acute leukemia that serial transplantation is leading to a clonal selection with a deterministic pattern. In our model, it seems that serial transplantation increases the potential of reconstitution of the LSC, as we transplanted a lower number of CD34+ in the 2nd generation of mice, but obtained an increased number of blasts in mouse 3, but this was not reflected by a clonal skewing or expansion in the sequential HSC sequencing in our MDS model.
The mutations were still present in the minimal residual disease after cytarabine too, confirming that traditional chemotherapy does not reach the leukemic stem cells and just reduces the bulk blasts. Therefore, we generated a PDX model of MDS where the clonal architecture was not altered. This in vivo model represents an interesting tool to define therapeutic approaches in high-risk MDS models.
MATERIALS AND METHODS
CD34+ and mesenchymal stromal cells (MSC) selection from primary samples
Bone marrows from myelodysplastic patients were collected by bone marrow aspiration after signed consent and approval by our local ethical committee. By density gradient using Ficoll (Eurobio), the mononuclear cells were isolated from total bone marrow. HSPCs (hematopoietic stem progenitor cells) were positively selected by paramagnetic iron-dextran particles directly conjugated to anti-CD34 monoclonal antibodies (CD34 MicroBead Kit, human, Miltenyi Biotec) by using an automatized system (MACS pro, Miltenyi Biotec). Their viability was assessed by trypan blue exclusion (Sigma Aldrich). The purity of all samples was checked by flow cytometry (BD FACS Canto II, BD Biosciences) with a specific CD34 fluorochrome-conjugated antibody (CD34-APC, Miltenyi Biotec) and was superior to 90% for all samples used. The same procedure was used for CD34+ cells sorting from xenografted mice bone marrow.
The CD34 negative fractions or total bone marrows from MDS patients were incubated in culture flask of 75cm2 (BD FalconTM) with DMEM (Dulbecco’s modified eagle medium, Life technologies, Paisley, UK) plus Fetal serum bovine (ThermoFisher Scientific) at 10%, antibiotics (penicillin and streptomycin, Life technologies™) and L.glutamine at 1% (Glutamax, ThermoFisher Scientific) at 37°C. Mesenchymal stromal cells were isolated by adherence to plastic after multiple washes by phosphate buffering saline, and their phenotype was confirmed by flow cytometry (BD FACS Quanto II, BD Biosciences) using CD73 (CD73APC, Miltenyi Biotec), CD90 (CD90PE-CYTM 7, BD Biosciences), CD105 (CD105PE, Miltenyi Biotec) and CD45 (CD45V500, BD horizonTM, BD Biosciences). All MSC were positive > 95% for CD73, CD90 and C105 and negative for CD45. Moreover, we have checked their ability for trilineage adipogenic, osteogenic and chondrogenic differentiation, meeting the conventional criteria for MSC [20]. Osteoblastic, adipogenic and chondrogenic differentiation medium used were commercial solutions (StemMACS™ OsteoDiff Media, StemMACS™ AdipoDiff Media, StemMACS™ ChondroDiff Media, MACS, Miltenyi Biotec). Culture medium were replaced every 4 days for 21 days. Then osteogenic differentiation was evaluated by alkaline phosphatase staining (SIGMA FAST™ 5-Bromo-4-chloro-3-Indonyl phosphate, Sigma-Aldrich), adipogenic differentiation by staining with Oil Red O (Abcam®) and chondrogenic differentiation by sulfated proteoglycans staining with alcian blue (Leica Biosystems) (Supplementary Data, Figure 1). All the MSC used for the xenograft model were inferior to passage 5.
Xenotransplant assays
NOD/SCID/IL2rγ−/− (NSG) mice were maintained in sterile isolator cages in the animal facility of the Grenoble Alpes University. NSG mice used in this study were 8 to 12 weeks old at the beginning of experiments. Twenty-four hours prior to transplantation, mice received one busulfan injection intraperitoneally (25mg/kg). Direct intra bone marrow injection was performed in the tibia with 5x105 CD34+ cells and 1.5x106 MSC from the same patient. Prior to injection, CD34+ cells were thawed. Secondary grafts were performed by intra bone injection of 1x105 CD34+ cells. The mice were monitored everyday according to a specific wellness scoring. The mice were weighted every week and in case of bleeding or asthenia, a retro-orbital blood analysis was done. For the secondary graft, we hypothesized that the engraftment would be faster so we performed a tibia bone marrow aspiration to monitor the engraftment 3 months after injection. All procedures were approved by the Animal care committee of Grenoble Alpes University (agreement number: B385161006).
Human chimerism and morphological analyses
The mice were killed at the end of the xenograft procedure (6 months). Bone marrow contents from tibiae, femora and humeri were collected. The human chimerism was determinated using FACS flow (quanto II, BD) with the following antibodies human CD45 (CD45-Viogreen™, Miltenyi Biotec), murine CD45 (CD45 PeRCP-Vio700, Miltenyi Biotec), human CD33 (CD33-PE, Miltenyi Biotec) and human CD19 (CD19-APC, Miltenyi Biotec). As already described in previous studies, we have chosen a threshold of 0.01% of human cells to define a human engraftment in mice [13]. Aliquot of each bone marrow were stained with May-Grunwald-Giemsa (MGG) by an automatized way (Aerospray®Pro, Elitech). Then, the % of human’s blasts were counted and the morphology of the cells was described by a trained cytologist.
Fluorescence in situ hybridization
For detection of the 5q deletion, FISH targeting the EGR1 gene was performed on CD34+ sorted cells and total bone marrow using a dual-color probe (Del(5q) deletion Cytocell® : EGR1-Tx-Red/control probe 5p15-FITC). Briefly, slides were fixed in methanol/acetic acid 3:1 for 30 minutes, air-dried, and dehydrated through graded alcohols. Both FISH probes and target DNA were denatured simultaneously for 3 minutes at 75°C and incubated for 20 hours at 37°C. The post hybridization washes were performed according to the manufacturer’s instructions (Cytocell®). Two hundred nuclei were scored.
Isolation of MPPs, MLPs, CMPs, GMPs and MEPs
CD34+ human cells obtained at the end of the xenograft procedure were stained with the following antibodies: CD34 (CD34-APC, Miltenyi Biotec), CD123 (CD123-PerCP-CY5.5, BD Bioscineces), CD45RA (CD45RA-FITC, IOTest®) and CD38 (CD38-PE, Miltenyi Biotec). Hematopoietic stem cells (HSCs), common myeloid progenitors (CMPs), granulocyte macrophage progenitors (GMPs) and megakaryocyte–erythroid progenitor (MEPs) were isolated as already described [15]. DAPI (Thermo Scientific) was added to the cell suspension to exclude dead cells. Cells were sorted on a BD FACS Aria (San Jose, CA) operating in 4 way purity sort mode, and collected into 5 ml polypropylene tubes. Then the cells were washed in phosphate-buffered saline solution.
DNA isolation
The method used for DNA isolation differed with the number of cells. For more than 100 000 cells, and between 100 000 and 10 000 cells, the QIAmp DNA Mini Kit (QIAGEN) and the FlexiGene DNA Kit (QIAGEN) were used respectively following the manufacturer’s instruction. For less than 10 000 cells, cells were lysate with tween 20 and proteinase K.
Sanger and next generation sequencing
A selected panel of 39 genes recurrently mutated in MDS was designed and mutations were identified by next-generation sequencing (NGS) in BM mononuclear cells (MNCs) using a PGM system (ThermoFisher Scientific, Waltham, MA) and the 384-reaction Ion AmpliSeq library kit 2.0 (Life Technologies, Chicago, IL) Base calls were generated by the Torrent Browser software (Life Technologies). The annotated.vcf files were also analyzed using NextGENe software (Softgenetics, State College, PA, USA). Abnormalities identified by both analyses were considered if referenced in UCSC and absent in 1000 genomes databases. For a given mutation, the proportion of mutated cells was twice the VAF of heterozygous mutations, or one-fold the VAF for homozygous mutations or hemizygous mutations of X chromosome genes in males) [21].
Statistics
The significance of the experimental results was determined with an unpaired Student test. p≤0.05 was considered statistically significant. All statistical analyses were done with GraphPad Prism 5 software (http://www.graphpad.com/scientific-software/prism/). Data are mean with SEM.
Author contributions
MM and CD contributed equally. MM, NM, MP, CL, GS and FB contributed to experiments and analysis. CD, OK and ASA contributed to sequencing and data analysis. MM, CD, OK and SP were involved with all aspects of the study’s design and contributed to manuscript preparation.
CONFLICTS OF INTEREST
The authors declare no major conflicts of interest.
FUNDING
MM has received a financial grant from Université Grenoble Alpes (Programme AGIR 2014) and a grant “Espoir Sandrine”.
REFERENCES
1. Woll PS, Kjallquist U, Chowdhury O, Doolittle H, Wedge DC, Thongjuea S, Erlandsson R, Ngara M, Anderson K, Deng Q, Mead AJ, Stenson L, Giustacchini A, et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell. 2014; 25: 794-808. https://doi.org/10.1016/j.ccr.2014.03.036.
2. Mian SA, Rouault-Pierre K, Smith AE, Seidl T, Pizzitola I, Kizilors A, Kulasekararaj AG, Bonnet D, Mufti GJ. SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment. Nat Commun. 2015; 6: 10004. https://doi.org/10.1038/ncomms10004.
3. Gangat N, Patnaik MM, Tefferi A. Myelodysplastic syndromes: Contemporary review and how we treat. Am J Hematol. 2016; 91: 76-89. https://doi.org/10.1002/ajh.24253.
4. Bejar R. Implications of molecular genetic diversity in myelodysplastic syndromes. Curr Opin Hematol. 2017; 24: 73-8. https://doi.org/10.1097/MOH.0000000000000313.
5. Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, Godfrey AL, Rapado I, Cvejic A, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011; 365: 1384-95. https://doi.org/10.1056/NEJMoa1103283.
6. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011; 478: 64-9. https://doi.org/10.1038/nature10496.
7. Malcovati L, Galli A, Travaglino E, Ambaglio I, Rizzo E, Molteni E, Elena C, Ferretti VV, Catricala S, Bono E, Todisco G, Bianchessi A, Rumi E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017; 129: 3371-8. https://doi.org/10.1182/blood-2017-01-763425.
8. Muntion S, Ramos TL, Diez-Campelo M, Roson B, Sanchez-Abarca LI, Misiewicz-Krzeminska I, Preciado S, Sarasquete ME, de Las Rivas J, Gonzalez M, Sanchez-Guijo F, Del Canizo MC. Microvesicles from Mesenchymal Stromal Cells Are Involved in HPC-Microenvironment Crosstalk in Myelodysplastic Patients. PLoS One. 2016; 11: e0146722. https://doi.org/10.1371/journal.pone.0146722.
9. Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, Ebert BL, Al-Shahrour F, Hasserjian RP, Scadden EO, Aung Z, Matza M, Merkenschlager M, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010; 464: 852-7. https://doi.org/10.1038/nature08851.
10. Zambetti NA, Ping Z, Chen S, Kenswil KJ, Mylona MA, Sanders MA, Hoogenboezem RM, Bindels EM, Adisty MN, Van Strien PM, van der Leije CS, Westers TM, Cremers EM, et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell. 2016; 19: 613-27. https://doi.org/10.1016/j.stem.2016.08.021.
11. Mhyre AJ, Marcondes AM, Spaulding EY, Deeg HJ. Stroma-dependent apoptosis in clonal hematopoietic precursors correlates with expression of PYCARD. Blood. 2009; 113: 649-58. https://doi.org/10.1182/blood-2008-04-152686.
12. Medyouf H, Mossner M, Jann JC, Nolte F, Raffel S, Herrmann C, Lier A, Eisen C, Nowak V, Zens B, Mudder K, Klein C, Oblander J, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell. 2014; 14: 824-37. https://doi.org/10.1016/j.stem.2014.02.014.
13. Rouault-Pierre K, Mian SA, Goulard M, Abarrategi A, Tullio AD, Smith AE, Mohamedali A, Best S, Nloga AM, Kulasekararaj AG, Ades L, Chomienne C, Fenaux P, et al. Preclinical modeling of myelodysplastic syndromes. Leukemia. 2017; 31:2702-2708. https://doi.org/10.1038/leu.2017.172.
14. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006; 8: 315-7. https://doi.org/10.1080/14653240600855905.
15. Manz MG, Miyamoto T, Akashi K, Weissman IL. Prospective isolation of human clonogenic common myeloid progenitors. Proc Natl Acad Sci U S A. 2002; 99: 11872-7. https://doi.org/10.1073/pnas.172384399.
16. Kosmider O, Passet M, Santini V, Platzbecker U, Andrieu V, Zini G, Beyne-Rauzy O, Guerci A, Masala E, Balleari E, Bulycheva E, Dreyfus F, Fenaux P, et al. Are somatic mutations predictive of response to erythropoiesis stimulating agents in lower risk myelodysplastic syndromes? Haematologica. 2016; 101: e280-3. https://doi.org/10.3324/haematol.2016.142695.
17. Muguruma Y, Matsushita H, Yahata T, Yumino S, Tanaka Y, Miyachi H, Ogawa Y, Kawada H, Ito M, Ando K. Establishment of a xenograft model of human myelodysplastic syndromes. Haematologica. 2011; 96: 543-51. https://doi.org/10.3324/haematol.2010.027557.
18. Li X, Marcondes AM, Ragoczy T, Telling A, Deeg HJ. Effect of intravenous coadministration of human stroma cell lines on engraftment of long-term repopulating clonal myelodysplastic syndrome cells in immunodeficient mice. Blood Cancer J. 2013; 3: e113. https://doi.org/10.1038/bcj.2013.11.
19. Belderbos ME, Koster T, Ausema B, Jacobs S, Sowdagar S, Zwart E, de Bont E, de Haan G, Bystrykh LV. Clonal selection and asymmetric distribution of human leukemia in murine xenografts revealed by cellular barcoding. Blood. 2017; 129: 3210-20. https://doi.org/10.1182/blood-2016-12-758250.
20. Krampera M, Galipeau J, Shi Y, Tarte K, Sensebe L. Immunological characterization of multipotent mesenchymal stromal cells—The International Society for Cellular Therapy (ISCT) working proposal. Cytotherapy. 2013; 15: 1054-61. https://doi.org/10.1016/j.jcyt.2013.02.010.
21. Chesnais V, Arcangeli ML, Delette C, Rousseau A, Guermouche H, Lefevre C, Bondu S, Diop M, Cheok M, Chapuis N, Legros L, Raynaud S, Willems L, et al. Architectural and functional heterogeneity of hematopoietic stem/progenitor cells in non-del(5q) myelodysplastic syndromes. Blood. 2017; 129: 484-96. https://doi.org/10.1182/blood-2016-03-707745.