
INTRODUCTION
The pathogenesis of colitis-associated cancer (CAC) remains enigmatic. To date, both initiation of colitis, and its progression to dysplasia with a 3-5-fold increased rate of colorectal cancer [1], is likewise unclear. In addition, influences from genetics, the microbiome, and the immune system contribute both to the initiation of colitis and its progression towards CAC. Yet, the disease presentation and course are heterogeneous.
The inflammatory and cancer microenvironments both promote and prevent progression of disease. Within these microenvironments, fibroblasts had previously been believed to be passive members. However, in the last few decades, stromal fibroblasts have been discovered to be active members of these microenvironments [2], either preventing or promoting colitis and colorectal cancer. We have previously reported the contributions of inflammatory and cancer associated fibroblasts to colon cancer stem cell-initiated tumorigenicity [3]. These studies demonstrated that the secretome of stromal fibroblasts includes the inflammatory chemokine CXCL8. In this context, CXCL8 promotes angiogenesis and migration and may contribute to proliferation and tumorigenicity [4–6].
MicroRNAs (miRNAs) are short, non-coding RNAs regulating a myriad of cellular mechanisms including apoptosis, differentiation, and metabolism [7, 8]. In particular, they have been shown to contribute to many aspects of tumorigenesis. The miRNA expression profile is highly predictive for tumor progression [9] and targeting miRNAs has therapeutic benefits in chemoprevention or chemomodulation [10, 11]. The most canonical function of miRNAs is to regulate protein expression and mRNA stability by binding to complementary nucleotide sequences in the 3′UTRs. Most miRNAs are transcribed by RNA polymerase II as part of a much longer primary transcript (pri-miRNA), which becomes processed into the mature ~22 nt duplex miRNA by two consecutive RNA cleavages [12, 13]. The mature miRNAs are then loaded into the RNA-induced silencing complex (RISC), which binds to the 3′UTR of target mRNAs and induces translational inhibition or degradation. Target specificity is determined by the seed sequence (nucleotides 2 to 8 from the 5’ end of a miRNA) and is further strengthened by base pairing of flanking nucleotides [14, 15].
In this study we investigated whether miRNAs play a critical role in the tumor microenvironment by contributing to the transition of stromal fibroblast to colitis- and subsequent cancer-associated fibroblasts. In particular, we identified one miRNA, miR-20a that negatively regulates the expression of stromal fibroblast-derived CXCL8. We show that miR-20a, a member of the miR-17 miRNA family, is part of a regulatory machinery that defines the pro-tumorigenic differentiation of stromal fibroblasts.
RESULTS
Differential secretion of CXCL8 from normal, colitic, and colon cancer-derived stromal fibroblasts is modulated by miRNAs
Previous reports indicate that enhanced secretion of CXCL8 occurs in the inflammatory and oncogenic colon microenvironments compared to grossly and histologically normal colon [3]. To further evaluate this we established primary cultures of normal colon, colitic colon, and cancerous colon associated fibroblasts and measured their CXCL8 concentrations in conditioned media (Figure 1A). As expected, the conditioned media demonstrate that colitis-associated (N = 11) and cancer-associated fibroblasts (N = 5) exhibit increased levels of CXCL8 compared to that of normal colon fibroblasts (N = 6).
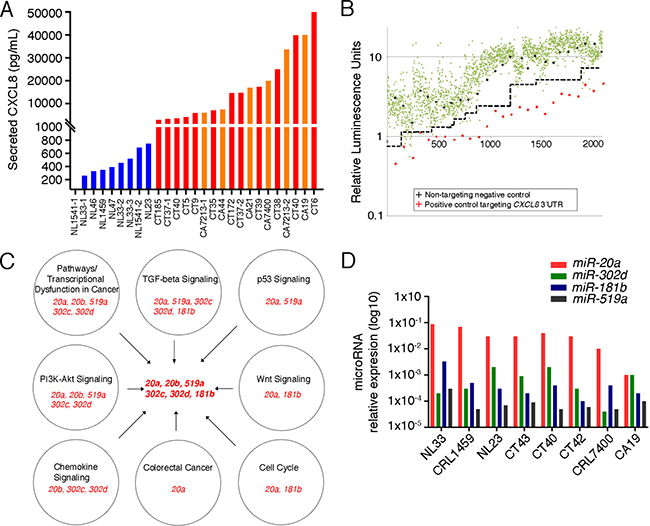
Figure 1: Identification of miRNAs Targeting CXCL8. (A) ELISA analysis of CXCL8 levels present in the conditioned media of stromal fibroblasts isolated from normal (NL, blue, N = 8), colitic (CT, red; N = 9) and cancerous colons (CA, orange; N = 5). See Supplementary Table 2 for patient demographics. (B) Screening of a Miridian human mimic miRNA library using a pMirGLO-CXCL8 3′UTR luciferase construct in primary colitic fibroblasts. Green dots represent luciferase levels from the individual miRNA mimics, red pluses those of a siRNA targeting the 3′UTR of CXCL8 and the black pluses those of a non-targeting negative control. Dashed black line delineates separation between mimics and the negative controls. (C) Pathway analysis of the identified miRNAs. Individual pathways and their associated miRNAs are depicted in the individual circles. miRNAs identified in these processes and selected for subsequent analysis are indicated in the center. (D) q-RT-PCR analysis of the miRNAs identified in (C) for their expression in multiple stromal fibroblasts. (NL: N = 3; CT: N = 3, CA: N = 2).
Dysregulation of miRNAs have been implicated in the function of tumor-associated fibroblasts [16]. Thus, we queried whether posttranscriptional regulation by miRNAs is responsible for the modulation of CXCL8 levels. To test this hypothesis, we used the miRidian miRNA mimic library and screened over 2,500 miRNA mimics for repression of a luciferase reporter containing the 3′UTR of CXCL8 mRNA. This approach identified 66 candidate miRNAs, of which 15 were verified in a secondary screen (Figure 1B and Supplementary Table 3). Next, we interrogated whether any of these miRNAs would have functions that align with their proposed tumor-promoting activity in colon cancer. Evaluating the candidate miRNAs using DIANA mirPath 2.0 [17] followed by KEGG pathway analysis [18] further reduced the number to 6 miRNAs belonging to four miRNA families (Figure 1C). Finally, we performed qRT-PCR using multiple colitis- and colon cancer-associated fibroblasts to determine whether their expression levels are sufficiently high to be relevant in vivo (Figure 1D). Based on these data we decided to focus on miR-20a and miR-20b as prime candidates to modulate CXCL8 expression during tumor progression. As both miR-20a and miR-20b bind to the same seed sequence and are thought to be identical in function, we focused on miR-20a.
Regulation of miR-20a expression in stromal fibroblasts
To confirm that the regulation of miR-20a is relevant in vivo, we queried human colon tissue sections by in situ hybridization for the expression of miR-20a. As shown in Figure 2A–2C, stromal miR-20a staining was stronger in normal colon, when compared to the stroma of colitic and cancerous colon. As previously reported [19], the epithelia in the colon cancer sections showed highly elevated miR-20a levels. Interestingly, this was not yet observed in the colitic epithelia, suggesting that the stromal changes in miR-20a expression precede the changes in the epithelia. Together, these data suggest that miR-20a levels are down regulated in the fibroblasts within the colitic field and is responsible for the up-regulation of CXCL8.
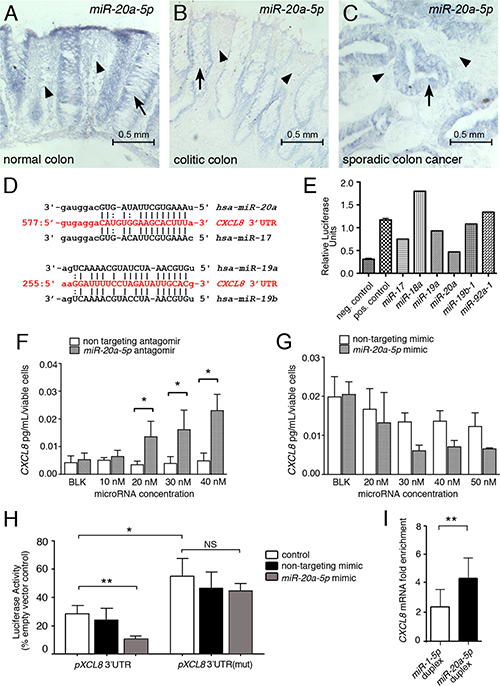
Figure 2: miR-20a directly modulates CXCL8 protein levels. (A–C) miR-20a in situ hybridization on paraplast section of intestinal tissue from healthy individuals (A) and patients with colitis (B) and sporadic cancer (C). miR-20a (blue precipitate) is expressed in the epithelia (indicated by arrows), but also in the stroma between the intestinal crypts (indicated by arrowheads). The latter expression is markedly reduced in colitis and colon cancer. Scale bar 0.5 mm. (D) Sequence alignment of the miR-20a/b and miR-19a/b-1 binding sites in the CXCL8 3′UTR. (E) Luciferase assay for the individual members of the miR17~92 miRNA cluster regulating CXCL8. (F, G) Levels of secreted endogenous CXCL8 are modulated in a concentration dependent manner in miR-20a loss- and gain-of-function studies using antagomirs (F) and mimics (G). Asterisks indicate significance (2-way ANOVA with *p < 0.05) (H) Luciferase assays using the wildtype pMirGLO-CXCL8 3′UTR luciferase construct and one with a mutated miR-20a binding site in the presence and absence of a non-targeting miRNA mimic or miR-20a-5p. Asterisks indicate significance (t-test with *p < 0.05 and **p < 0.01). (I) RNA immuno-precipitation using the miR-Trap system (Takara) in the presence of a miR-20a duplex and as a negative control a miR-1 duplex, which does not have a detectable binding site in the CXCL8 3′UTR. Enrichment scores were calculated. Significance was assessed using t-test (**p < 0.005).
miR-20a directly regulates CXCL8 translation
In silico analysis of the CXCL8 mRNA identified a single miR-20a binding site in its 3′UTR (Figure 2D). miR-20a is encoded in the miR-17~92 cluster, a critical regulator in multiple cancers [20–22]. The cluster contains six miRNAs belonging to three different miRNA families. Using the CXCL8 3′UTR reporter showed two miR-17 family members, miR-20a and miR-17, as well as miR-19a - albeit to a lesser extent - repressed the luciferase activity (Figure 2E). The latter could be traced back to a distinct miR-19 binding site in the 3′UTR of CXCL8, located approximately 300 base pairs downstream of the miR-20 binding site (Figure 2D).
To further address the specificity of the miR-20a/CXCL8 interaction, we added an exogenous miR-20a mimic to in vitro cultures. Increasing the concentrations of the mimic decreased the levels of endogenously secreted CXCL8 accordingly, while a non-targeting mimic had no significant effects (Figure 2G). Conversely, blocking endogenous miR-20a using an antagomir resulted in a dose-dependent increase in CXCL8 secretion (Figure 2F). Mutating the miR-20a binding site in the CXCL8 3′UTR luciferase reporter rendered this construct unresponsive to the addition of exogenous miR-20a mimic (Figure 2H). Moreover, the mutated construct exhibited increased luciferase levels when compared to the un-mutated construct.
Finally, to determine, whether miR-20a targets the endogenous CXCL8 mRNA, we performed RNA immuno-precipitation for GW182, a critical component of the RISC complex. As shown in Figure 2I, adding a synthetic miR-20a duplex significantly enriched immuno-precipitated CXCL8 mRNA when compared to the addition of a miR-1 duplex. Together, these data suggested that miR-20a is an important posttranscriptional regulator of CXCL8 mRNA in colon interstitial fibroblasts.
miR-20a expression levels are inversely correlated with the levels of secreted CXCL8
miRNA expression is regulated by the transcription of its primary transcript and the ensuing processing by the nuclear Drosha/DGCR8 and the cytoplasmic Dicer complex [12, 13]. qPCR analyses from normal, colitis-associated and cancer-associated fibroblasts demonstrated statistically significant differences in the levels of the mature miR-20a upon disease progression (Figure 3A). However, when we interrogated RNAseq data (GSE106119) for the expression of the mRNA encoding miR-20a, the miR17HG, a significant difference was only observed between normal and tumor-associated fibroblasts, while normal and colitis-associated fibroblasts merely exhibited a trend. This was confirmed by qRT-PCR analysis using primers detecting either the unprocessed miR-20a pre-miRNA or its mature counterpart (Figure 3B, 3C). The expression of the miR17HG is regulated by the transcription factor c-MYC [23]. Similar to the miR17HG c-MYC expression levels decreased upon severity of the disease (Figure 3D), suggesting that c-MYC may - at least partially - be responsible for the changes in miR-20a levels. Finally, we also compared CXCL8 mRNA to protein levels (Figure 3E). As expected from a miRNA-mediated process, the CXCL8 protein levels were robustly altered, while the mRNA levels were reduced, but did not reach statistical significance.
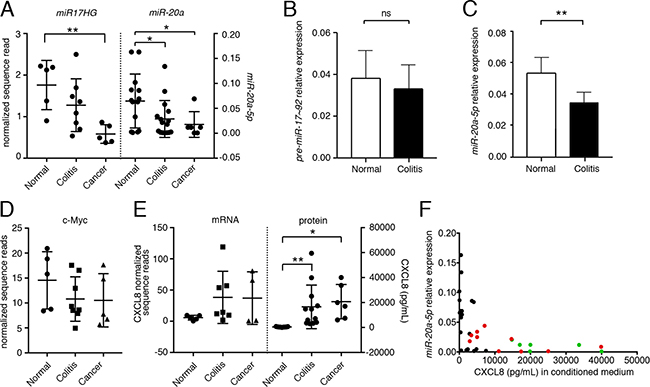
Figure 3: The expression of miR-20a is inversely related to secreted CXCL8. (A) Primary fibroblasts derived from normal, colitic and cancerous stroma were analyzed for the expression levels of the primary transcript encoding miR-20a-5p (miR20HG) and its mature form were quantified by mRNAseq and qRT-PCR, respectively. (B, C) qRT-PCR for the pre-miR-20a-5p (B) and miR-20a-5p (C) comparing normal and colitis-associated fibroblasts. Expression was normalized to U18 RNA. (D) Expression of c-MYC by mRNAseq. (E) Levels of CXCL8 mRNA and protein by mRNAseq and ELISA. Statistical significance in (A–E) was determined using t-test; asterisks indicate significance (*p < 0.05 and **p < 0.01). (F) Spearman’s correlation of miR-20a expression vs. secreted CXCL8 revealing a statistically significant inverse monotonic relationship (p < 0.001; rs = –0.654). Normal colon fibroblasts: black, colitic colon fibroblasts: red, cancer-associated fibroblasts: green.
Based on these results, we hypothesized that the levels of mature miR-20a and CXCL8 protein are reliable indicators for the pro-tumor promoting activity of the interstitial fibroblasts. To this end, we interrogated fibroblasts derived from normal, colitic and cancerous colon measured both for their cellular miR-20a levels and their amount of secreted CXCL8. As expected from our previous work, the levels of CXCL8 varied depending on disease state and statistically significant changes were seen between colitis and cancer compared to normal colon (Figure 3E). miR-20a levels showed the opposite trend being higher in normal cells than in colitis- or cancer-associated fibroblasts (Figure 3A). Most strikingly, measuring the monotonic relationship between both measurements using Spearman’s correlation (Figure 3F) demonstrated a clear inverse relationship (rs = –0.654, p < 0.001).
Differential tumorigenesis of co-inoculations of miR-20ahigh and miR-20alow stromal fibroblasts with colon cancer stem cells
The data so far demonstrated that changes in miR-20a levels are inversely related to the levels of the chemokine, CXCL8, which may influence the tumor promoting activity of colitis-associated fibroblasts. To test the in vivo significance of this observation, we chose to compare the effects of colitis-associated fibroblasts expressing high and low miR-20a levels (miR-20ahigh and miR-20alow) on colon cancer tumorigenicity. Expression of miR-20a as well as the concentration of secreted CXCL8 was confirmed by qRT-PCR and ELISA, respectively (Figure 4A, 4B). These fibroblasts were co-inoculated with colon cancer stem cells as a source of cancerous epithelia into the dorsal subcutaneous flanks of immunocompromised NSG mice. We used colon cancer stem cells since they are enriched in tumor initiating function [3]. The subcutaneous tumors were measured twice-weekly using calipers [3, 24–26]. Serial tumor measurements confirmed that tumor latency was decreased in mice injected with miR-20alow fibroblasts compared to those with the miR-20ahigh fibroblasts or colon cancer stem cells alone (Figure 4C). These differences in tumor volumes were most pronounced at weeks five and six post-inoculation (Figure 4D, 4E; p = 0.012 and p = 0.004, respectively). By week seven, the growth differences were still significant, however, the miR-20ahigh fibroblast/cancer stem cell co-injections revealed increased growth compared to the other groups (Figure 4F; p = 0.02).
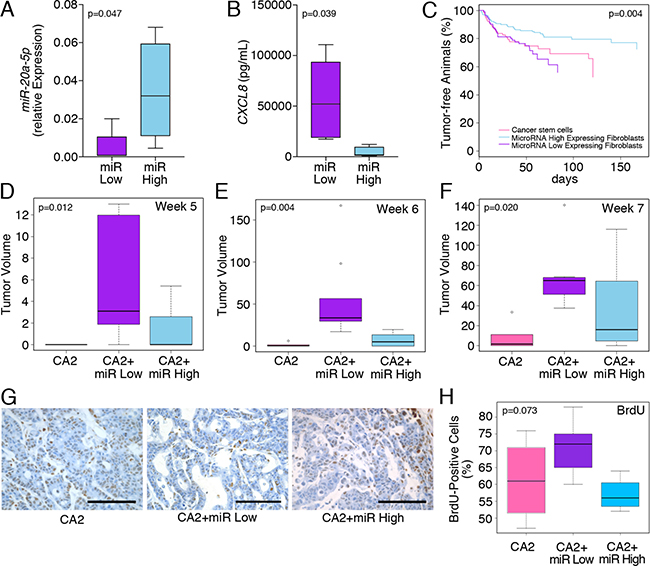
Figure 4: miR-20a/CXCL8 levels of co-inoculated stromal fibroblasts influence tumor latency. (A, B) Confirmation of miR-20a and CXCL8 in the ‘miR High’ and ‘miR Low’ colitis-associated fibroblasts used for the in vivo tumorigenicity experiments. (C–F) Co-injection of colon cancer stem cells (CA2) with and without the miR High and miR Low colitis-associated fibroblasts significantly altered disease progression (C) and tumor volume (D–F) at 5 and 6 weeks, but not 7 weeks after inoculation. (G, H) Images and quantification of the tumors stained for BrdU incorporation upon resection. All xenografts were harvested, when the longest dimension was 5–7 mm. Statistics were preformed using ANOVA and significance levels are indicated in the individual panels. Each group contained n ≥ 9 animals.
When tumors had reached a length of 5–7 mm in at least one dimension, we injected mice with BrdU and harvested the tumors 3 hours later. This harvesting time point was chosen so that tumors were still actively proliferating, but did not yet develop central necrosis. In line with the tumor latency results, the BrdU staining showed a trend towards significance in the levels of BrdU incorporation with the miR-20ahigh fibroblast/cancer stem cell co-injections being the lowest (Figure 4G, 4H). In parallel, we performed immunohistochemistry for CXCL8 on the same tumor samples. However, this comparison was not significant (p = 0.523; Supplementary Figure 1A). Finally, since a downstream effector of the CXCL8 signaling pathway is the pro-angiogenic VEGF [6], we assayed the tumors for angiogenesis by MECA-32 immunostaining. However, like in the case of CXCL8, no differences could be detected between the three groups (p = 0.495; Supplementary Figure 1B). Together, these data support our observation that fibroblasts with different miR-20a levels alter tumor latency, but do not impact the tumor growth once it is established.
DISCUSSION
Inflammation has long been identified as contributing to cancer, as the ‘wound that doesn’t heal’ [27]. Here, we reveal that the tumor promoting activity of the inflammatory stroma is modulated, in part, by miRNAs (Figure 1). Specifically, our studies demonstrate that miR-20a directly regulates the amount of the proinflammatory chemokine CXCL8 secreted from the interstitial fibroblasts (Figure 2). Moreover, miR-20a and CXCL8 show an inverse relationship that models the transitions of the interstitial fibroblasts from normal to colitis-to tumor associated (Figure 3). We propose that low levels of miR-20a result in augmented tumorigenicity due to elevated levels of CXCL8. Conversely, tumor latency is increased, when miR-20a levels are elevated resulting in decreased CXCL8 protein secreted into the tumor microenvironment. Indeed, we can show that in in vivo experiments fibroblasts expressing either high or low levels of miR-20a impact tumorigenicity by influencing the tumor latency of co-inoculated sporadic colon cancer stem cells (Figure 4). The fact that once initiated, the tumors did not exhibit dramatic changes in proliferation or other tumor-associated parameters including angiogenesis suggest that the interstitial fibroblasts provide a supportive microenvironment for the initiation of the epithelial tumorigenicity, but do not alter the ultimate outcome.
miRNAs have been previously connected to colorectal cancer. For example, miR-34b/c and miR-21 are strongly associated with poor prognosis [28]. Similarly, the miR-17~92 cluster, which harbors the miR-20a studied here and is often referred to as ‘oncomir’ [29], has a pronounced role in cancer biology by promoting proliferation, inhibiting differentiation, increasing angiogenesis, and sustaining cell survival. In fact, the six miRNAs encoded in the miR-17~92 cluster target many cancer relevant proteins such as VEGF and c-MYC [23, 30]. In colorectal oncogenesis the miR-17~92 cluster and in particular miR-17, which binds to the same seed sequence as miR-20a, is upregulated early on in tumor formation [31]. This upregulation is confined to the epithelia and has been closely linked to an auto-regulatory loop with c-MYC.
Interestingly, our data demonstrate that in the stromal fibroblasts the miR-17~92 cluster and miR-20a, in particular play the opposite role, when compared to the epithelia. It is expressed highly in normal fibroblasts, but becomes down regulated in the course of their progression to a more tumor-promoting phenotype. This is due to changes in the transcription of the miR-20a host gene and may even involve c-MYC, a transcription factor that is highly associated with tumorigenesis [30]. As such, the fibroblasts and the epithelia appear to follow opposite paths. While the epithelia upregulate c-MYC and the miR-17~92 cluster, the associated fibroblasts do exactly the opposite. How this is attained is currently unknown, but likely involves a yet-to-be-identified regulatory input that is specifically altered in colitis-associated fibroblasts. Its characterization will be very important to better understand how interstitial fibroblasts are undergoing pro-tumorigenic changes, of which miR-20a and CXCL8 are likely just a small aspect.
Based on the results presented here, miRNAs may play an integral part in this transformative process. miRNAs often exhibit rather modulatory functions, by changing e.g. the signal intensity, but not the signaling quality [32, 33]. In our study, CXCL8 expression is present in normal colon-associated fibroblasts, but is kept in check by the repressive activity of miR-20a. Only upon changes in the microenvironment miR-20a levels drop and the repression is relieved. These changes can be rather subtle initially, but worsen the pathogenicity over time. This process probably involves more than just miR-20a/b. It may include several of the other miRNAs that we identified as regulators of CXCL8 translation (Figure 1); it may also include other miRNAs that are up- or down-regulated in colitis-or cancer-associated fibroblasts, but do not impinge on CXCL8 mRNA and were therefore not detected in our screen.
A slow differentiation process for the interstitial fibroblasts is in agreement with the long prodrome that usually exists for colitis-associated cancer. Risk of progressive disease from colitis to dysplasia to cancer occurs over decades and is dominantly associated with both duration and extent of disease. Thus, understanding the processes leading to or supporting carcinogenesis is therapeutically very important. It provides a rather wide window for less drastic treatments, which may delay/supplant the current intervention for medically refractory colitis and colorectal cancer, which is total proctocolectomy. Finally, understanding the pathogenesis of colitis-associated cancer and the contributions of the tumor microenvironment may be a paradigm to understand other inflammatory diseases such as Barrett’s esophagus, for which the pathogenesis is likewise unknown.
In summary, our data support the emerging realization that therapeutic approaches targeting only epithelial cells are likely insufficient to prevent progression from colitis to dysplasia to CAC. Instead, a successful anti-CAC therapy also needs to involve strategies to prevent colitic fibroblasts from secreting tumor-promoting factors such as CXCL8 into the microenvironment.
MATERIALS AND METHODS
Human subjects
Collection and use of human tissues was approved by the Cleveland Clinic Institutional Review Board (IRB 13-1159). Tissues from human subjects were retrieved under pathologic supervision from the Cleveland Clinic. Normal colon tissues were obtained from areas at least 10 cm away from gross tumors. All human tissue samples were obtained from patient tissues that had undergone biopsy or surgery for resection of colon cancer or ulcerative colitis at the Cleveland Clinic. Informed consent in writing was obtained from each patient.
Animals
All animal experiments were approved by the Cleveland Clinic IACUC Committee. Inbred NSG (non-obese diabetic IL-2) 4-6 week-old mice were used for these studies.
Fibroblast isolation
Fibroblasts were isolated from primary tissues using previously described methodologies [3]. Briefly, after mincing, heated collagenase digestion on a shaking water bath was used and the filtrate was cultured on tissue culture plates in DMEM 10% FBS (Atlanta Biologicals) with 1% Pen/Strep (Gibco). Human colon fibroblasts cell lines CRL1541, CRL1459, and CRL7213 were obtained from the American Type Culture Collection (ATCC). Primary colon fibroblasts were isolated from human colon tissue was obtained with informed consent from patients undergoing surgery without preoperative treatment for sporadic colon cancer or colectomy for chronic ulcerative colitis. Primary fibroblasts were cultured from non-dysplastic sections of colon (normal), adenocarcinoma or colitic sections as previously described [3].
Short tandem repeat analysis
Once established, primary isolates of fibroblasts and of the cancer stem cell epithelia were subjected to STR analysis (Duke University Genomics Laboratory) to establish their genetic identity. Prior to final use in assays, a second analysis was completed to confirm the genetic identity of the isolate, and compared it to the original STR for uniqueness (Supplementary Table 1).
CXCL8 ELISA
To quantify the concentration of CXCL8 from the fibroblast conditioned media, 105 stromal fibroblasts were cultured in 1 ml of DMEM base media for 24 h in the absence of serum. The conditioned media was harvested and subjected to quantification per the CXCL8 ELISA manufacturer’s instructions (Raybiotech).
Reporter constructs
The entire 3′-UTR of human CXCL8 mRNA (1,350 bps) was cloned into the pMIRGLO vector (Promega) using SacI and XbaI restriction enzymes. This cloning strategy placed the CXCL8 3′-UTR immediately downstream of the firefly luciferase gene driven by the PGK promoter. Constitutively expressed Renilla luciferase served as a normalizing control. To confirm the in silico predicted miR-20a recognition sequence within the CXCL8 3′-UTR, the nucleotides corresponding to the seed sequence (5′-AAG CAC TTT A-3′; bases 591-600) were mutated by site-directed mutagenesis to 5′-TTC GAC TTT A-3′. The miR-20a seed sequence prediction and its mitigation was performed using the PITA microRNA target prediction algorithm [34].
Screening of human mimic microRNA library
A human mimic microRNA library consisting of 2080 unique siRNA corresponding to known human microRNAs (MiRIDIAN, ThermoFisher) was used for screening microRNA that interact with the 3′-UTR of human CXCL8 mRNA. Immortalized colon fibroblasts (CT5 SV40) were seeded at 8000 cells per well in 96-well plates the day before transfection. These cells were immortalized via lentiviral transduction of hTERT SV40 (kind gift from Rosa Hwang, MD, MD Anderson). The next day each well of cells was co-transfected with unique library microRNA mimics (50 nM/well) and pMIRGLO-CXCL8-3′UTR plasmid using Dharmafect Duo (GE Dharmacon) according to the manufacturer protocol. After 48 hours luminescence was measured using DualGlo Luciferase Assay System (Promega) with a Spectra II plate reader (Molecular Devices). A non-targeting negative control and a positive control in form of a siRNA targeting the 3′UTR of human CXCL8 mRNA (5’-AUU CUA GCA AAC CCA UUC AUU-3′, Dharmacon) were included for each individual mimic 96-well plate screen.
microRNA target prediction by in silico analysis
miRNA pathway analysis and the identification of miRNA binding sites in the CXCL8 3′ UTR was performed using four prediction algorithms, DIANA Tools (http://diana.imis.athena-innovation.gr/DianaTools/index.php), microRNA.org, mirWALK (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/) and Target Scan (http://www.targetscan.org/).
Luciferase knockdown assay
Human colon fibroblasts were seeded in a 96-well plate at a concentration of 1 × 104 cells per well. Twenty-four hours after seeding, fibroblasts were transfected with either parental pMIRGLO plasmid, wild type pMIRGLO-CXCL8-3′UTR or mutagenized pMIRGLO-CXCL8-3′UTR(mut) plasmid using Lipofectamine 2000. After 48 hours, luciferase activity was measured with DualGlo (Promega) and normalized to Renilla activity using a Spectra II plate reader (Molecular Devices).
RNA isolation, RNAseq and real-time qPCR
Total RNA was extracted using the miRNeasy kit method (Qiagen). Purity and integrity of the extracted RNA was assessed using the Aligent Bioanalyzer, and only samples with RIN values greater than 8.0 were used for gene expression analysis. Total RNA was processed by Beckman-Coulter Genomics for next generation sequencing using the TruSeq Total RNA kit (Illumina). Total RNA of each sample was depleted of ribosomal RNA using biotinylated oligos, combined with Ribo-Zero rRNA removal beads and subsequent fragmentation of RNA using divalent cations under elevated temperature. After reverse transcription the double stranded cDNA and ligation of sequencing adapter, the cDNA products were purified and enriched with PCR to create the library. Sequencing reads (single 100-bp) generated from the Illumina HiSeq 2500 platform were assessed for quality using FastQC (Tuxedo package retrieved from http://cufflinks.cdcd.umd.edu/igenomes.html). The reads were trimmed for adapter sequences using Trim Galore (https://github.com/FelixKrueger/TrimGalore). Alignment was to the human genome (GRCh19 assembly) using the Tuxedo package [35] followed by analysis for differential expression using a significance cut-off of a false discovery rate (FDR) <0.05 and a log2 fold change of greater than 1.5 or less than –1.5. All files are available at GEO (Accession Number: GSE106119).
The TaqMan microRNA qPCRs were used to perform reverse transcription and quantification of microRNAs. Primer sets specific for the mature and the unprocessed form of hsa-miR-20a-5p as well as small RNA U18 as endogenous control were obtained from Fisher Scientific. miRNA expression was normalized to U18.
In situ hybridization
In situ hybridization on paraplast sections was performed as previously described [36] using a digoxygenin-labeled miR-20a LNA-modified probe from Exiqon.
miRNA mimic and antagomir experiments
Normal adult human colon fibroblasts were seeded at a density of 40,000 cells/well in a 24-well plate in DMEM with 2% fetal bovine serum without antibiotics. The following day cells (approximately 80% confluent) were transfected with either the miR-20a or a control miRNA mimic at concentration ranges between 0-50 nM using Lipofectamine RNAiMAX reagent according to the manufacturer’s instructions (ThermoFisher Scientific). On day 3 after transfection, the medium was changed to plain basal DMEM, which was collected 24 hours later to measure the amount of secreted CXCL8 by ELISA. The adherent cells were washed 2 times with pre-warmed PBS and the number of viable cells in each well was estimated using the CellTiter-Glo cell viability assay (Promega). The antagomir experiments were performed in identical fashion using a miR-20 antagomir or a non-targeting control (Exiqon).
RNA immunoprecipitation
Verification of as CXCL8 mRNA as the target of endogenous miR-20a was performed by RNA immunoprecipitation using the MirTrap System following the manufacturer’s instructions (Takara). Human colon fibroblasts plated in 10 cm dishes (90% confluency) were co-transfected with 500 pmoles of the miR-20a-5p duplex or the miR-1-5p duplex and 30 μg of the pMirTrap plasmid using Lipofectamine 2000. The RNA duplexes were synthesized by IDT (miR-20-5p: passenger strand 5′phos-CUA CCU GCA CUA UAA GCA CUU CAA G-3′ and guide strand 5′phos-UAA AGU GCU UAU AGU GCA GGU AGC G-3′, miR-1-5p: passenger strand 5’phos-AUG GGC AUA UAA AGA AGU AUA UAG-3′ and guide strand 5′phos-ACA UAC UUC UUU AUA UGC CCA UCG-3′). CXCL8 and GAPDH mRNA levels were measured by qRT-PCR using SYBR Green and enrichment scores were calculated as describe in the user manual.
Co-injection tumorigenesis experiments
Xenografts consisting of cancer stem cell line known as CA2 enriched using the Aldefluor® assay [3, 25]. One hundred cancer stem cells (CA2) were inoculated in the absence (baseline) or presence of 1000 stromal fibroblast cells in a 50:50 mixture of Matrigel® (BD) into the subcutaneous right dorsal flank of NSG mice as previously described [3, 24–26] . The cells were re-suspended in media at concentrations of 2 × 103 cells/ml and 2 × 104 cells/ml, epithelia and stromal fibroblasts, respectively. At the time of stromal cell harvest, conditioned media and cells were retained to confirm the miR-20a miRNA levels and CXCL8 concentration. Tumors were measured twice per week using calipers for palpable tumors. Tumor volumes were calculated using the formula: volume = width2 × length (mm, length as the longer measurement). Tumors were harvested when additive total dimensions were approximately 15 mm, both to avoid central necrosis of the tumor and to minimize undue distress to the murine host.
Tumor growth curves were plotted using Kaplan-Meier survival curves, and differences in growth curves was assessed using the log-rank test. Tumor growth was assessed at week 5, week 6, and week 7, and differences between treatment groups at each time point was conducted using ANOVA. Significance was determined at the 0.05 level. ANOVA was used to assess the mean difference in positive percentage between the BrdU, CXCL8 and MECA-32 measurements in three treatment groups: cancer stem cells (CA2), cancer stem cells co-inoculated with miRNA-20a low expressing fibroblasts (CA2+miR Low), and cancer stem cells co-inoculated with miRNA-20a high expressing fibroblasts (CA2+miR High).
Immunohistochemistry
Mice received IP injections of BrdU (10 μg/kg) 3 hours prior to harvest. Tumor sections were fixed in 4% paraformaldehyde. Tumor sections were deparaffinized in xylene and rehydrated in descending percentages of ethanol. Tissues were fixed in 4% PFA for 24 hours; 4 μm sections were cut and stained for BrdU incorporation using a commercially available kit (BrdU Staining Kit, Invitrogen) following the manufacturer’s instructions. Five random images were documented under bright field using a Leica DMIRB microscope at 40× magnification. Both BrdU positive nuclei and total nuclei for each image were quantified using ImageJ software.
For CXCL8 staining slides were incubated with the anti-CXCL8 antibody (MAB208, R&D Systems) and visualized using the OmniMap anti-Mouse HRP secondary (760-4310, Ventana) and the ChromoMap DAB detection kit (760-159, Ventana). Slides were counterstained with hematoxylin and bluing. Representative images were documented at 40× using a Leica upright microscope. Sections from at least 9 animals per treatment group were evaluated, counting over 3,000 epithelial cells per group. Positivity was quantified as the ratio of DAB-stained colonic epithelia to the total number of nuclei-positive colonic epithelia.
To determine tumor vessel density, deparaffinized, 4 mm paraffin sections from tumor xenografts underwent a 20 minute, 95° C heat retrieval step using Target Retrieval Solution (DakoCytomation). Following water and buffer washes, the sections were blocked for 60 minutes in 2% horse serum followed by a 24 hour incubation at 4° C with a rat anti-MECA 32 (mouse pan-endothelial) monoclonal antibody (1:10; BD Pharmingen) and visualized using an anti-Rat Alexa Fluor 594-coupled secondary antibody (1:500; Invitrogen). Slides were mounted with VectaShield (Vector Laboratories) containing DAPI and five random images per section were documented at 40× magnification. Vessel density was quantified using grid overlay method (ImageJ; grid plugin; 70 μm2 squares) and normalized to mm2.
Author contributions
Conception and design: S. Signs, R. Fisher, O. Wessely, E. Huang; Development of methodology: S. Signs, S. Chakrabarti, R. Fisher, S. Kamali, U. Tran, O. Wessely, E. Huang; Acquisition of data: S. Signs, S. Kamali, S. Xiang, D. Liska, U. Tran, V. Roche, E. Huang; Analysis and interpretation of data: S. Signs, O. Wessely, E. Huang; Writing and revision of the manuscript: S. Signs, O. Wessely, D. Liska, E. Huang.
ACKNOWLEDGMENTS
We would like to thank all members of the Huang and Wessely laboratory for helpful discussions, J. Gerow and E. Schultz from the Flow Cytometry Core for their assistance in FACS sorting and K. Page for help with the murine tumor measurements.
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
FUNDING
This work was supported by National Institute of Health R01 CA142808, R01 CA157663, and U01 CA214300 to E.H. and R01 DK080745 to O.W.
REFERENCES
1. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001; 48:526–35.
2. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004; 432:332–7. https://doi.org/10.1038/nature03096.
3. Carpentino JE, Hynes MJ, Appelman HD, Zheng T, Steindler DA, Scott EW, Huang EH. Aldehyde dehydrogenase-expressing colon stem cells contribute to tumorigenesis in the transition from colitis to cancer. Cancer Res. 2009; 69:8208–15. https://doi.org/10.1158/0008-5472.CAN-09-1132.
4. Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003; 170:3369–76.
5. Li A, Varney ML, Singh RK. Expression of interleukin 8 and its receptors in human colon carcinoma cells with different metastatic potentials. Clin Cancer Res. 2001; 7:3298–304.
6. Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008; 14:6735–41. https://doi.org/10.1158/1078-0432.CCR-07-4843.
7. Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, Yuen ST, Chan TL, Kwong DL, Au GK, Liu CG, Calin GA, Croce CM, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008; 299:425–36. https://doi.org/10.1001/jama.299.4.425.
8. Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006; 103:2257–61. https://doi.org/10.1073/pnas.0510565103.
9. Bullock MD, Pickard K, Mitter R, Sayan AE, Primrose JN, Ivan C, Calin GA, Thomas GJ, Packham GK, Mirnezami AH. Stratifying risk of recurrence in stage II colorectal cancer using deregulated stromal and epithelial microRNAs. Oncotarget. 2015; 6:7262–79. https://doi.org/10.18632/oncotarget.3225.
10. Cioce M, Valerio M, Casadei L, Pulito C, Sacconi A, Mori F, Biagioni F, Manetti C, Muti P, Strano S, Blandino G. Metformin-induced metabolic reprogramming of chemoresistant ALDHbright breast cancer cells. Oncotarget. 2014; 5:4129–43. https://doi.org/10.18632/oncotarget.1864.
11. Derry MM, Raina K, Balaiya V, Jain AK, Shrotriya S, Huber KM, Serkova NJ, Agarwal R, Agarwal C. Grape seed extract efficacy against azoxymethane-induced colon tumorigenesis in A/J mice: interlinking miRNA with cytokine signaling and inflammation. Cancer Prev Res (Phila). 2013; 6:625–33. https://doi.org/10.1158/1940-6207.CAPR-13-0053.
12. Svobodova E, Kubikova J, Svoboda P. Production of small RNAs by mammalian Dicer. Pflugers Arch. 2016; 468:1089–102. https://doi.org/10.1007/s00424-016-1817-6.
13. Daugaard I, Hansen TB. Biogenesis and Function of Ago-Associated RNAs. Trends Genet. 2017; 33:208–19. https://doi.org/10.1016/j.tig.2017.01.003.
14. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009; 136:215–33. https://doi.org/10.1016/j.cell.2009.01.002.
15. Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biol. 2005; 3:e85. https://doi.org/10.1371/journal.pbio.0030085.
16. Yang F, Ning Z, Ma L, Liu W, Shao C, Shu Y, Shen H. Exosomal miRNAs and miRNA dysregulation in cancer-associated fibroblasts. Mol Cancer. 2017; 16:148. https://doi.org/10.1186/s12943-017-0718-4.
17. Vlachos IS, Kostoulas N, Vergoulis T, Georgakilas G, Reczko M, Maragkakis M, Paraskevopoulou MD, Prionidis K, Dalamagas T, Hatzigeorgiou AG. DIANA miRPath v.2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res. 2012; 40:W498–504. https://doi.org/10.1093/nar/gks494.
18. Kanehisa M. The KEGG database. Novartis Found Symp. 2002; 247:91–101; discussion 101–3, 119–28, 244–52.
19. Zhang GJ, Li Y, Zhou H, Xiao HX, Zhou T. miR20a is an independent prognostic factor in colorectal cancer and is involved in cell metastasis. Mol Med Rep. 2014; 10:283–91. https://doi.org/10.3892/mmr.2014.2144.
20. Fuziwara CS, Kimura ET. Insights into Regulation of the miR-17-92 Cluster of miRNAs in Cancer. Front Med (Lausanne). 2015; 2:64. https://doi.org/10.3389/fmed.2015.00064.
21. Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008; 133:217–22. https://doi.org/10.1016/j.cell.2008.04.001.
22. Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013; 20:1603–14. https://doi.org/10.1038/cdd.2013.125.
23. Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006; 38:1060–5. https://doi.org/10.1038/ng1855.
24. Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, Shelton AA, Parmiani G, Castelli C, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A. 2007; 104:10158–63. https://doi.org/10.1073/pnas.0703478104.
25. Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, Appelman H, Fields JZ, Wicha MS, Boman BM. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009; 69:3382–9. https://doi.org/10.1158/0008-5472.CAN-08-4418.
26. Shenoy AK, Fisher RC, Butterworth EA, Pi L, Chang LJ, Appelman HD, Chang M, Scott EW, Huang EH. Transition from colitis to cancer: high Wnt activity sustains the tumor-initiating potential of colon cancer stem cell precursors. Cancer Res. 2012; 72:5091–100. https://doi.org/10.1158/0008-5472.CAN-12-1806.
27. Dvorak HF. Tumors: wounds that do not heal-redux. Cancer Immunol Res. 2015; 3:1–11. https://doi.org/10.1158/2326-6066.CIR-14-0209.
28. Hiyoshi Y, Schetter AJ, Okayama H, Inamura K, Anami K, Nguyen GH, Horikawa I, Hawkes JE, Bowman ED, Leung SY, Harris CC. Increased microRNA-34b and -34c predominantly expressed in stromal tissues is associated with poor prognosis in human colon cancer. PLoS One. 2015; 10:e0124899. https://doi.org/10.1371/journal.pone.0124899.
29. Olive V, Li Q, He L. mir-17-92: a polycistronic oncomir with pleiotropic functions. Immunol Rev. 2013; 253:158–66. https://doi.org/10.1111/imr.12054.
30. O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005; 435:839–43. https://doi.org/10.1038/nature03677.
31. Knudsen KN, Nielsen BS, Lindebjerg J, Hansen TF, Holst R, Sorensen FB. microRNA-17 Is the Most Up-Regulated Member of the miR-17-92 Cluster during Early Colon Cancer Evolution. PLoS One. 2015; 10:e0140503. https://doi.org/10.1371/journal.pone.0140503.
32. Chiba T, Marusawa H, Ushijima T. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology. 2012; 143:550–63. https://doi.org/10.1053/j.gastro.2012.07.009.
33. Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol. 2010; 11:252–63. https://doi.org/10.1038/nrm2868.
34. Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007; 39:1278–84. https://doi.org/10.1038/ng2135.
35. Jin X, Kim LJY, Wu Q, Wallace LC, Prager BC, Sanvoranart T, Gimple RC, Wang X, Mack SC, Miller TE, Huang P, Valentim CL, Zhou QG, et al. Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat Med. 2017; 23:1352–1361. https://doi.org/10.1038/nm.4415.
36. Tran U, Zakin L, Schweickert A, Agrawal R, Doger R, Blum M, De Robertis EM, Wessely O. The RNA-binding protein bicaudal C regulates polycystin 2 in the kidney by antagonizing miR-17 activity. Development. 2010; 137:1107–16. https://doi.org/10.1242/dev.046045.