
INTRODUCTION
Signaling through the Epidermal Growth Factor Receptor (EGFR) pathway is a common event in cancer development [1], with activating mutations in KRAS, NRAS and BRAF occurring in approximately 50% of colorectal cancer (CRC) patients [2]. Results from a phase III trial (MRC COIN trial, n = 1630) in metastatic CRC revealed that patients with BRAF mutant (BRAFMT) tumors have a significantly worse prognosis compared to patients with KRAS mutant (KRASMT) tumors or tumors with no detectable mutations in KRAS or BRAF (WT/WT) [3]. The use of a BRAFMT specific inhibitor, vemurafenib, in advanced melanoma, has improved survival rates for patients with this activating mutation [4], and underpinned the rationale for a phase Ib study employing vemurafenib in BRAFMT CRC [5]. Unfortunately, unlike the encouraging results observed in BRAFMT melanoma, the inhibitor did not benefit BRAFMT CRC patients in the advanced disease setting. Mechanistic studies have indicated that resistance to vemurafenib in CRC is due to feedback activation of the EGFR pathway [6], further highlighting the key role played by EGFR signaling in CRC.
To examine the role of BRAF in the adjuvant stage II/III disease setting, Popovici and colleagues performed differential gene expression analysis to identify transcriptional differences between BRAFMT and BRAFWT/KRASWT tumors in a cohort of 688 stage II and III colon cancer (CC) clinical trial samples (PETACC-3) [7]. Their analysis identified the distinct underlying biology of the BRAFMT subgroup. Furthermore, the authors generated a 64-gene classifier, which stratified the cohort into two subgroups. The first subgroup, which accounted for 27% of the cohort, displayed a transcriptional signature similar to BRAFMT tumors (termed “pred-BRAFm”) and had a worse prognosis in terms of overall survival (OS) and survival-after-relapse compared to the second subgroup, which had a signature similar to that of BRAFWT disease (termed “pred-BRAFwt”). Critically however, while both BRAF mutation and the pred-BRAFm signatures could identify subgroups of patients with poorer OS after relapse (i.e. when the patient had progressed to stage IV metastatic disease), the rates of disease relapse in these subgroups were not significantly different to BRAFWT and pred-BRAFwt disease.
There is currently a lack of understanding of the biology that drives disease relapse specifically within stage II/III BRAFMT disease, resolution of which could ultimately inform treatment of a clinically-definable subgroup of BRAFMT patients, who have the worst prognosis when they progress to stage IV, but who still may be potentially curable in stage II/III. Therefore, we aimed to identify novel predictors of relapse for stage II/III BRAFMT CC, employing transcriptomic datasets for in silico discovery/initial corroboration, followed by subsequent validation of promising lead candidate(s) from bioinformatics analyses by immunohistochemistry analysis within a large population-based stage II/III BRAFMT CC study.
RESULTS
Study outline and rationale for risk stratification in BRAFMT CC
We analyzed available transcriptional data from the well-characterized dataset, GSE39582, as outlined in Supplementary Figure 1. Compared to KRASMT and WT/WT patients, BRAFMT patients were significantly more likely to be older (p < 0.001), have proximal tumors (p < 0.001) that exhibited microsatellite instability (MSI, p < 0.001) and to be assigned as Consensus Molecular Subtype 1 (CMS1, p < 0.001) (Table 1). Additionally, patients with BRAFMT tumors were significantly more likely to be female (p = 0.04 and p = 0.001) and to receive no adjuvant chemotherapy (p = 0.001 and p = 0.006) compared to KRASMT and WT/WT respectively (Table 1). Finally, BRAFMT patients were significantly more likely to have later stage disease (stage II v III) compared to WT/WT patients (p = 0.04) (Table 1). Using the 64 gene BRAF classifier identified by Popovici et al. [7] we performed semi-supervised hierarchical clustering of the gene expression profiles of the entire stage II/III patient cohort. We identified a subgroup accounting for 28% (n = 127) of the tumor profiles using this method of clustering, which displayed an expression pattern similar to the pred-BRAFm profile (Supplementary Figure 2A). We found no difference in relapse rates between the pred-BRAFm and the pred-BRAFwt populations in this cohort (Supplementary Figure 2A; HR = 0.95 (95% CI 0.65–1.39)).
Table 1: Characteristics of colon cancer patients and tumors according to BRAF and KRAS status.
Characteristic |
BRAF MT n = 41 |
KRAS MT n = 166 |
p-value |
WT/WT n = 210 |
p-value |
|---|---|---|---|---|---|
Age, years, mean (SD) |
76.0 (7.3) |
67.7 (13.5) |
<0.001 |
65.6 (12.6) |
<0.001 |
Sex, n (%) |
|||||
Male |
14 (34.1) |
86 (51.8) |
130 (61.9) |
||
Female |
27 (65.9) |
80 (48.2) |
0.04 |
80 (38.1) |
0.001 |
Tumour stage, n (%) |
|||||
II |
20 (48.8) |
86 (51.8) |
138 (65.7) |
||
III |
21 (51.2) |
80 (48.2) |
0.73 |
72 (34.3) |
0.04 |
Tumour location, n (%) |
|||||
Proximal |
37 (90.2) |
86 (51.8) |
49 (23.3) |
||
Distal |
4 (9.8) |
80 (48.2) |
<0.001 |
161 (76.1) |
<0.001 |
Adjuvant treatment* receipt, n (%) |
|||||
No |
33 (80.5) |
86 (51.8) |
121 (57.6) |
||
Yes |
8 (19.5) |
80 (48.2) |
0.001 |
89 (42.4) |
0.006 |
MSI status |
|||||
MSI |
27 (65.9) |
15 (9.0) |
15 (7.1) |
||
MSS |
8 (19.5) |
138 (83.1) |
170 (81.0) |
||
Unknown |
6 (14.6) |
13 (7.8) |
<0.001 |
25 (11.9) |
<0.001 |
Consensus Molecular Subtype, n (%) |
32 (78.1) |
17 (10.2) |
26 (12.4) |
||
CMS 1 |
0 (0.0) |
53 (31.9) |
120 (57.1) |
||
CMS 2 |
3 (7.3) |
35 (21.1) |
9 (4.3) |
||
CMS 3 |
3 (7.3) |
45 (27.1) |
40 (19.1) |
<0.001 |
|
CMS 4 |
3 (7.3) |
16 (9.6) |
<0.001 |
15 (7.1) |
|
Unknown |
Characteristics of colon cancer patients and tumours according to BRAF and KRAS status.
MSI: Microsatellite instability; MSS: Microsatellite stable; MT: Mutant; WT/WT: BRAF and KRAS wild-type. *Adjuvant chemotherapy treatment receipt.
Of the 460 tumor profiles within our cohort, 417 have KRAS and BRAF mutational analysis data. Comparative analysis was performed for age, sex, stage, location, treatment, MSI and Consensus Molecular Subtype (CMS). MSI: Microsatellite instability; MSS: Microsatellite stable; MT: Mutant; WT/WT: BRAF and KRAS wild-type. *Adjuvant chemotherapy treatment received.
Gene expression associated with risk of relapse in BRAFMT CC
Gene Set enrichment analysis (GSEA) of the discovery subset indicated increased myogenesis, epithelial-to-mesenchymal transition (EMT) and hypoxia pathways in the BRAFMT tumors with the highest-risk of disease relapse (Supplementary Figure 2B). Additionally, using the Microenvironment Cell Populations-counter (MCP), we identified a non-significant trend for increased fibroblasts in high-risk BRAFMT tumors (Supplementary Figure 2C). Using differential gene expression analysis contrasting profiles from high-risk or low-risk BRAFMT tumors in the discovery subset (Supplementary Figure 1), we identified 83 probesets (Supplementary Table 1) corresponding to 67 annotated genes that are prognostic for relapse risk in BRAFMT tumors; high expression of 43 genes were associated with increased risk of relapse, and high expression of 24 genes with decreased risk of relapse (Table 2). Increased expression of endoplasmic reticulum stress-induced transcripts such as PPP1R15A (GADD34), heat shock proteins HSPA6 and DNAJB1, and the stress-related transcription factor DDIT3 were observed in BRAFMT tumours with the highest-risk of disease relapse.
Table 2: Gene list associated with relapse in BRAFMT tumors
Symbol |
Entrez Gene Name |
Symbol |
Entrez Gene Name |
|---|---|---|---|
AEBP1 |
AE binding protein 1 |
AGR2 |
anterior gradient 2, protein disulphide isomerase family member |
ALPP |
alkaline phosphatase, placental |
C2orf72 |
chromosome 2 open reading frame 72 |
ANGPTL1 |
angiopoietin-like 1 |
C3orf70 |
chromosome 3 open reading frame 70 |
BCL2L1 |
BCL2-like 1 |
COBL |
cordon-bleu WH2 repeat protein |
CCDC71L |
coiled-coil domain containing 71-like |
EFNA2 |
ephrin-A2 |
CCL7 |
chemokine (C-C motif) ligand 7 |
GATA6-AS1 |
GATA6 antisense RNA 1 (head to head) |
CDA |
cytidine deaminase |
GMDS |
GDP-mannose 4,6-dehydratase |
CYSRT1 |
cysteine-rich tail protein 1 |
HES6 |
hes family bHLH transcription factor 6 |
DDIT3 |
DNA-damage-inducible transcript 3 |
IMPA2 |
inositol(myo)-1(or 4)-monophosphatase 2 |
DNAJB1 |
DnaJ (Hsp40) homolog, subfamily B, member 1 |
KIAA1324 |
KIAA1324 |
DNTTIP1 |
deoxynucleotidyltransferase, terminal, interacting protein 1 |
KIAA1671 |
KIAA1671 |
DUSP14 |
dual specificity phosphatase 14 |
KREMEN1 |
kringle containing transmembrane protein 1 |
EPYC |
epiphycan |
LARGE |
like-glycosyltransferase |
FST |
follistatin |
NOX1 |
NADPH oxidase 1 |
FXYD5 |
FXYD domain containing ion transport regulator 5 |
NRARP |
NOTCH-regulated ankyrin repeat protein |
GAS1 |
growth arrest-specific 1 |
PER2 |
period circadian clock 2 |
GJB3 |
gap junction protein, beta 3, 31kDa |
PIP5K1B |
phosphatidylinositol-4-phosphate 5-kinase, type I, beta |
GJB5 |
gap junction protein, beta 5, 31.1kDa |
PSMG4 |
proteasome (prosome, macropain) assembly chaperone 4 |
HCFC1R1 |
host cell factor C1 regulator 1 (XPO1 dependent) |
SKP2 |
S-phase kinase-associated protein 2, E3 ubiquitin protein ligase |
HSPA6 |
heat shock 70kDa protein 6 (HSP70B’) |
SLC22A23 |
solute carrier family 22, member 23 |
IER5L |
immediate early response 5-like |
SPRED2 |
sprouty-related, EVH1 domain containing 2 |
IGFBP6 |
insulin-like growth factor binding protein 6 |
TMEM30B |
transmembrane protein 30B |
KLK10 |
kallikrein-related peptidase 10 |
TRIM15 |
tripartite motif containing 15 |
KRT16 |
keratin 16, type I |
TSPAN13 |
tetraspanin 13 |
MFGE8 |
milk fat globule-EGF factor 8 protein |
||
MIR100HG |
mir-100-let-7a-2 cluster host gene |
||
MYH4 |
myosin, heavy chain 4, skeletal muscle |
||
NKIRAS1 |
NFKB inhibitor interacting Ras-like 1 |
||
NPC1L1 |
NPC1-like 1 |
||
NT5E |
5’-nucleotidase, ecto (CD73) |
||
PAEP |
progestagen-associated endometrial protein |
||
PDP1 |
pyruvate dehyrogenase phosphatase catalytic subunit 1 |
||
PHLDA3 |
pleckstrin homology-like domain, family A, member 3 |
||
PPP1R15A |
protein phosphatase 1, regulatory subunit 15A |
||
PRR9 |
proline rich 9 |
||
RBMS2 |
RNA binding motif, single stranded interacting protein 2 |
||
RGS4 |
regulator of G-protein signaling 4 |
||
TAGLN3 |
transgelin 3 |
||
TGFB2 |
transforming growth factor, beta 2 |
||
TNFSF4 |
tumor necrosis factor (ligand) superfamily, member 4 |
||
VEGFB |
vascular endothelial growth factor B |
||
ZFAS1 |
ZNFX1 antisense RNA 1 |
||
ZNF667-AS1 |
ZNF667 antisense RNA 1 (head to head) |
Genes associated with increased (red) and decreased (green) relapse risk from our BRAFMT risk-supervised differential gene expression analysis data.
While the majority of the 67 genes are represented by a single probeset, BCL2L1 (encoding Bcl-xL) and NCRNA00275 (which transcribes ZFAS1) are both represented by 3 different probesets (of the 4 total probesets for each gene), reducing the probability of the single genes themselves being false positives, which could potentially confound the validity of genes identified by a single probeset only (Supplementary Table 1). Gene expression levels of Bcl-xL were increased between 1.76–1.97 fold (Figure 1A) and ZFAS1 by 1.83–1.90 fold (Supplementary Table 1) in the high-risk group compared to the low risk group. Importantly, the 67 BRAFMT prognostic gene list is distinct from the pred-BRAFm classifier reported by Popovici, as only one gene, (Kallikrein-Related Peptidase 10 (KLK10)), overlaps between these 2 gene lists (Supplementary Figure 2D).
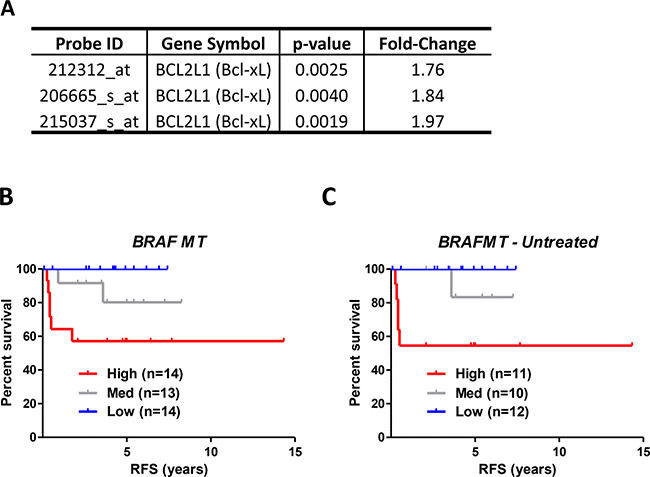
Figure 1: Relapse risk analysis of BRAFMT tumors indicates that Bcl-xL gene expression is associated with prognosis in BRAFMT tumors. (A) BCL2L1 (Bcl-xL) was represented by 3 individual probesets in relapse risk analysis in BRAFMT tumors. (B and C) Relapse-free survival (RFS) curve using Kaplan-Meier estimation in the “Initial Consolidation” dataset comparing tertile stratified Bcl-xL gene expression levels in all BRAFMT (A) and untreated BRAFMT (B) stage II/III CRC patients. Unadjusted and adjusted HR statistics are detailed in Table 3.
Probesets associated with risk in BRAFMT tumors represent distinct novel prognostic biology
As BRAF and KRAS are both key components of the EGFR/MAPK pathway, we performed a similar risk association analysis in KRASMT tumors and identified 139 probesets associated with risk-of-relapse in this genetic subgroup (Supplementary Table 2). We found no overlap between the probesets associated with risk-of-relapse in the KRASMT subgroup and the probesets identified in the BRAFMT analyses (Supplementary Figure 2E), indicating that distinct biologies determine prognosis in these two subgroups, at least in stage II/III disease.
Bcl-xL mRNA expression is associated with poor prognosis in stage II/III BRAFMT CC
To confirm the clinical relevance of elevated Bcl-xL gene expression in our training set, in addition to testing the genotype-specific nature of its prognostic value, we next generated an “Initial Consolidation” dataset (n = 417, Supplementary Figure 1) by removing the filters initially applied to the discovery subset of the GSE39582 cohort, (i.e. we removed the restrictions on chemotherapy treatment and the follow-up criteria as detailed in Methods). This set of 417 patients represents an ideal cohort to assess the prognostic value of Bcl-xL in KRASWT and WT/WT patients that were not used to identify Bcl-xL, in addition to a further 17 BRAFMT patients that were previously excluded from the discovery data. Within BRAFMT tumors (n = 41), the Bcl-xL-high group (Bcl-xLhigh) had a significantly higher risk-of-relapse compared to the Bcl-xL-low (Bcl-xLlow) expression group, using either an unadjusted (HR = 5.83), or adjusted model (HR = 9.63) accounting for potential confounding factors including age, gender, TNM stage and MSI status (confidence intervals could not be calculated due to an absence of events in Bcl-xLlow; Figure 1B and Table 3). When examining untreated patients only, the prognostic value of Bcl-xL mRNA expression in BRAFMT patients was again apparent (Figure 1C); however, the prognostic value of Bcl-xL in the chemotherapy-treated patient subgroup could not be evaluated due to small numbers (n = 8). The Bcl-xL medium expression group (Bcl-xLmed) displayed an intermediate relapse profile compared to the Bcl-xLhigh and Bcl-xLlow, suggesting a dose-response association between relapse risk and Bcl-xL gene expression. Stratification based on the median also demonstrated the prognostic value of Bcl-xL gene expression (HR = 5.24 (95% CI 1.3–21.2)) (Supplementary Figure 3A).
Table 3: Unadjusted and adjusted analyses of relapse-free survival
Bcl-xL |
Non-progressors n |
Progressors n |
Unadjusted Hazard ratios (95% confidence intervals) |
Adjusted** Hazard ratios (95% confidence intervals) |
|---|---|---|---|---|
BRAF MT |
||||
Low |
14 |
0 |
1.00 |
1.00 |
Medium |
11 |
2 |
1.80 (Not calculable) |
3.94 (Not calculable) |
High |
8 |
6 |
5.83 (Not calculable) |
9.63 (Not calculable) |
KRAS MT |
||||
Low |
38 |
18 |
1.00 |
1.00 |
Medium |
33 |
21 |
1.45 (0.75–2.77) |
1.47 (0.76–2.84) |
High |
34 |
22 |
1.25 (0.65–2.40) |
1.32 (0.68–2.56) |
WT/WT |
||||
Low |
57 |
13 |
1.00 |
1.00 |
Medium |
53 |
17 |
1.39 (0.67–2.85) |
1.27 (0.61–2.64) |
High |
50 |
20 |
1.54 (0.77–3.10) |
1.47 (0.72–3.01) |
MT: Mutant; WT/WT: BRAF and KRAS wild-type.
*Cut-offs for low/medium/high Bcl-xl gene expression based on tertile values within each BRAF/KRAS status subgroup.
**Adjustments included age and sex, and were tested for TNM stage, MSI status, adjuvant chemotherapy receipt and tumour location for all models. A backwards elimination model was applied for tested confounders until all were significant at the p < 0.25 level in the model. Final adjustments included age, sex, TNM stage and MSI status (for BRAF MT and WT/WT); age, sex, TNM stage, adjuvant chemotherapy receipt and tumour location (for KRAS MT).
RFS analysis was performed using Cox proportional hazards method in the BRAFMT, KRASMT or WT/WT stratified by Bcl-xL expression levels. Analysis was performed both before and following adjustment. *Cut-offs for low/medium/high Bcl-xL gene expression based on tertile values within each BRAF/KRAS status subgroup. **Adjustments included age and sex, and were tested for TNM stage, MSI status, adjuvant chemotherapy receipt and tumor location for all models. A backwards elimination model was applied for tested confounders, until all were significant at the p < 0.25 level in the model. Final adjustments included age, sex, TNM stage and MSI status (for BRAF MT and WT/WT); age, sex, TNM stage, adjuvant chemotherapy receipt and tumor location (for KRAS MT).
In contrast, although there was a suggestive prognostic trend, no significant associations were observed for Bcl-xL gene expression in either the KRASMT or WT/WT patient groups, using either an adjusted or unadjusted analysis model (Table 3 and Supplementary Figure 3B and 3C).
ZFAS1 mRNA expression is associated with poor prognosis in stage II/III BRAFMT CC
High gene expression of ZFAS1 was associated with a prognostic trend in BRAFMT tumors compared to low gene expression (Supplementary Figure 4A) although given the small number of events in this stratified group, this trend failed to reach significance in either unadjusted HR = 4.69 (95% CI 0.52–42.01), or adjusted HR = 4.71 (95% CI 0.50–44.00) analyses (Supplementary Table 3). There was no prognostic value associated with high ZFAS1 expression in the KRASMT population (adjusted HR = 0.65 (95% CI 0.34–1.24)), although there was a significant association with lower relapse rates in the WT/WT population (adjusted HR = 0.47 (95% CI 0.24–0.92)) (Supplementary Figure 4B, Supplementary Table 3) indicating an opposing prognostic role in these distinct tumor genotypes.
Bcl-xL gene and protein expression are associated with the epithelial component of the tumor
Given the multiple cell types that constitute the tumor microenvironment (TME) in CC, we analyzed Bcl-xL mRNA expression levels in transcriptional data derived from micro-dissected tumor tissue (detailed in Materials and Methods section). We observed that its expression was bimodal in the epithelial compartment of the TME, with high and low subgroups around the median, whereas stromal expression levels were generally low, with values below the median (Supplementary Figure 5A). Analysis of matched Bcl-xL transcript abundance (by Agilent microarray), and protein level, (by Reverse Phase Protein Array (RPPA)) from 102 CRC tumor samples within The Cancer Genome Atlas (TCGA) indicated a significant correlation between both methodologies (p = 0.001; Supplementary Figure 5B), supporting protein-based assessment as an appropriate methodology to validate our data in an independent cohort. Following optimization of an IHC protocol for Bcl-xL protein expression, the predominantly epithelial-derived nature of Bcl-xL protein expression and neoplastic-specific staining compared to the normal glands in surrounding tissue was confirmed in a series of whole-face CC sections, although there does appear to be some stromal expression, in line with our transcriptional analysis (Figure 2A).
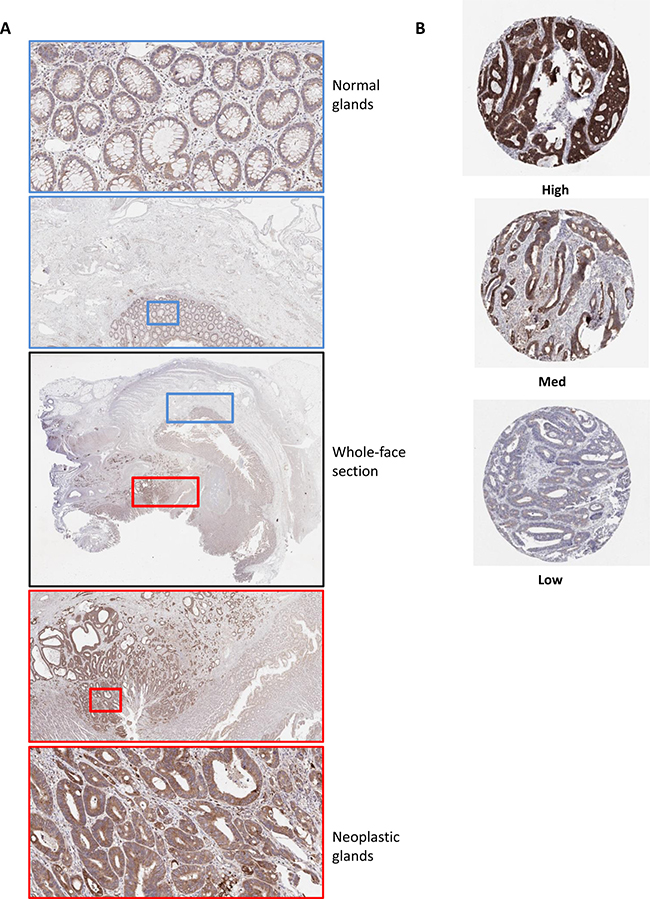
Figure 2: Optimization of immunohistochemistry staining protocol for Bcl-xL protein expression in CC. (A) Whole-face CC tissue sections were used to optimize IHC protocol. A low level of protein expression was observed in the normal glands compared to surrounding stroma (Blue box) Elevated levels of expression were observed in neoplastic glands compared to both the normal glands and surrounding stroma (Red box). Some staining in the stroma is evident in both normal and cancer-associated regions. (B) Representative images of high, medium and low Bcl-xL protein expression by IHC in an independent “Northern Ireland cohort” of stage II/III CRC (Northern Ireland cohort; n = 740).
Independent validation of Bcl-xL as a poor prognostic marker specifically in stage II/III BRAFMT CC
We then independently validated the prognostic value of Bcl-xL protein expression specifically in BRAFMT patient samples from within a Northern Ireland cohort (n = 661) (Supplementary Figure 1 and described in Methods). Employing tertiles defined by protein expression (Figure 2B), we found that Bcl-xLhigh was associated with an increased risk of CRC disease-specific survival (DSS; n = 77) when compared with Bcl-xLlow, in both unadjusted (HR = 3.07 (95% CI 1.24–7.60)) and adjusted models (HR = 5.50 (95% CI 1.71–17.69) (Supplementary Figure 6A and Table 4). Similar findings were evident when using OS (n = 92) as the endpoint (Supplementary Figure 6B).
Table 4: Analyses of disease-specific survival in the independent IHC validation cohort
Bcl-xL |
Alive n |
CRC Death (DSS) N |
Unadjusted Hazard ratios (95% confidence intervals) |
Adjusted** Hazard ratios (95% confidence intervals) |
|---|---|---|---|---|
BRAF MT |
||||
Low (<56.1) |
16 |
7 |
1.00 |
1.00 |
Medium (56.1–<91.1) |
17 |
10 |
1.54 (0.58–4.09) |
1.97 (0.60–6.44) |
High (≥91.1) |
12 |
15 |
3.07 (1.24–7.60) |
5.50 (1.71–17.69) |
KRAS MT |
||||
Low (<53.5) |
44 |
28 |
1.00 |
1.00 |
Medium (53.5–<92.7) |
41 |
24 |
0.98 (0.57–1.69) |
0.93 (0.52–1.66) |
High (≥92.7) |
38 |
33 |
1.37 (0.82–2.27) |
1.00 (0.57–1.77) |
WT/WT |
||||
Low (<66.1) |
57 |
28 |
1.00 |
1.00 |
Medium (66.1–<105.8) |
58 |
29 |
0.99 (0.59–1.68) |
1.05 (0.60–1.84) |
High (≥105.8) |
59 |
31 |
1.07 (0.64–1.78) |
1.18 (0.67–2.09) |
MT: Mutant; WT/WT: BRAF and KRAS wild-type.
*Cut-offs for low/medium/high Bcl-xl gene expression based on tertile values within each BRAF/KRAS status subgroup.
**Adjustments included age, sex, TNM stage, MSI status, adjuvant chemotherapy receipt, ECOG status, family history of colorectal cancer, year of diagnosis and extramural venous invasion for all models.
Bcl-xL |
No Chemotherapy receipt |
Chemotherapy receipt |
||||
|---|---|---|---|---|---|---|
Alive n |
CRC Death (DSS) N |
Adjusted Hazard ratios (95% confidence intervals) |
Alive n |
CRC Death n |
Adjusted Hazard ratios (95% confidence intervals) |
|
BRAF MT |
||||||
Low (<56.1) |
11 |
4 |
1.00 |
5 |
3 |
1.00 |
Medium (56.1–<91.1) |
12 |
6 |
1.99 (0.38–10.29) |
5 |
4 |
2.18 (0.23–20.89) |
High (≥91.1) |
3 |
12 |
12.13 (2.49–59.13) |
9 |
3 |
0.96 (0.08–11.42) |
P for interaction |
0.006 |
|||||
MT: Mutant.
Cut-offs for low/medium/high Bcl-xl gene expression based on tertile values within each BRAF MT subgroup.
Adjustments included age, sex, TNM stage, MSI status, adjuvant chemotherapy receipt, ECOG status, family history of colorectal cancer, year of diagnosis and extramural venous invasion.
(Top) DSS analysis was performed using Cox proportional hazards method in the BRAFMT, KRASMT or WT/WT stratified by Bcl-xL IHC (H-score) protein expression levels. Analysis was performed both before and following adjustment.
*Cut-offs for low/medium/high Bcl-xL gene expression based on tertile values within each BRAF/KRAS status subgroup.
**Adjustments included age, sex, TNM stage, MSI status, adjuvant chemotherapy receipt, ECOG status, family history of colorectal cancer, year of diagnosis and extramural venous invasion for all models. (Bottom) Further adjusted analysis to identify treatment interaction effect of the Bcl-xL-high tertile group of BRAFMT tumors stratified by treatment received.
We next conducted stratified analyses within the Northern Ireland cohort to assess independently the prognostic value of Bcl-xL protein expression in both untreated and chemotherapy-treated BRAFMT patients. In untreated patients, we observed a 12-fold increased DSS risk in patients with the highest Bcl-xL protein expression (adjusted model HR = 12.13 (95% CI 2.49–59.13)) (Figure 3A), which was not observed in treated patients, (adjusted model HR = 0.96 (95% CI 0.08–11.42)) (Supplementary Figure 6C and Table 4). This significant prognostic benefit from adjuvant chemotherapy in BRAFMT patients was only observed in patients with the highest Bcl-xL protein expression (P-value for interaction = 0.006), whereas patients with low Bcl-xL protein expression derived no benefit from the addition of chemotherapy (Figure 3B and 3C, Table 4 and Supplementary Figure 6C). Similar results were evident when using OS as the endpoint (Supplementary Figure 6D–6F). Importantly, in agreement with our initial consolidation cohort, we were again able to confirm that the prognostic value of Bcl-xL protein expression was not observed in KRASMT (HR = 1.00 (95% CI 0.57–1.77) and WT/WT (HR = 1.18 (95% CI 0.67–2.09)) patient samples (Table 4).
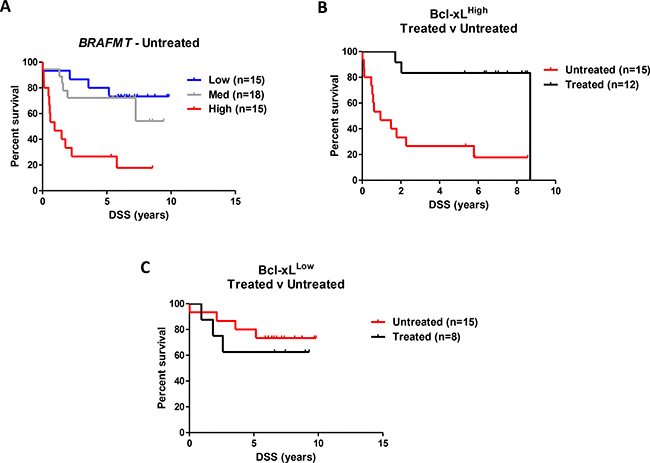
Figure 3: Independent validation of the prognostic value of Bcl-xL protein expression in BRAFMT CC. (A) Colorectal cancer disease-specific survival (DSS) curve using Kaplan-Meier estimation in the “Northern Ireland cohort” comparing tertiles stratification of Bcl-xL protein expression (by IHC H-score) in untreated BRAFMT stage II/III CC patients. (B) DSS of patients in the highest tertile of Bcl-xL protein expression stratified according to adjuvant chemotherapy treatment received. (C) DSS of patients in the lowest tertile of Bcl-xL protein expression stratified according to adjuvant chemotherapy treatment received. Unadjusted and adjusted HR statistics are detailed in Table 4.
DISCUSSION
In this study, we set out to identify factors influencing patient prognosis specifically in tumors harboring an oncogenic BRAF mutation. Stratification of a discovery prognostic cohort based on risk-of-relapse identified the Bcl-2 family member, Bcl-xL, as being upregulated at the transcriptional level in BRAFMT tumors from patients who went on to relapse following surgery, compared to those BRAFMT patients who experienced no disease recurrence. We validated the prognostic value of Bcl-xL specifically in BRAFMT tumors in both a consolidation transcriptional cohort and in an independent population-based stage II/III cohort. Importantly, in each validation series, we also confirmed the BRAFMT-specific nature of this association, as in either KRASMT or WT/WT tumors, the expression of Bcl-xL was not associated with increased risk of disease relapse or death. Interestingly, we observed that although BRAFMT tumors with high Bcl-xL expression have a poor prognosis, this subgroup also appears to benefit the most from standard adjuvant chemotherapy.
The prognostic value of stratifying CC patients based on BRAF mutational status has been well reported, particularly in stage IV metastatic tumors, where patients with BRAFMT tumors have poor survival rates. A previous study identified a transcriptional signature that could stratify stage II and III CRC tumor profiles into subgroups based on their similarity to BRAFMT tumors (pred-BRAFm) [7]. The authors demonstrated the utility of either BRAF mutational status or the pred-BRAFm classifier in identifying patients with shorter survival, although no difference was observed in the initial disease-specific relapse rates between the identified subgroups. This important result suggests that the prognostic power associated with the pred-BRAFm signature, or indeed the presence of the BRAF mutation itself, is due to shorter survival time because of aggressive disease after relapse in stage IV disease; however, initially, BRAFMT stage II and III patients are not at a higher risk of their early-stage disease progressing to metastatic disease. This subtle but crucial point underpins our rationale for performing a stratified analysis to identify factors determining risk-of-relapse specifically within the BRAFTMT genotype. The data presented here identifies for the first time a novel role for Bcl-xL expression in influencing disease relapse, providing a new, important and clinically relevant understanding of the biology underpinning aggressive BRAFMT stage II/III disease. Interestingly, we found almost no overlap between the genes associated with relapse in BRAFMT and KRASMT tumors, suggesting that although there is constitutive activation of the MAP kinase pathway in both these subgroups, there is clearly distinct prognostic tumor biology associated with these different genotypes.
The benefits of combining transcription-array discovery followed by IHC validation in independent patient cohorts, as we have employed in our study, was recently demonstrated in an analysis of stage II/III CC to identify a subgroup of undifferentiated tumors characterized by poor differentiation and low expression of the transcription factor CDX2 [8]. There are a number of parallels between the CDX2 study and our own, as they both use exploratory and retrospective analysis followed by clinically relevant IHC biomarker validation to identify a small subgroup of stage II/III patients with poor prognosis that appear to respond to adjuvant chemotherapy. Poorly differentiated tumors have previously been associated with right-sided MSI, CIMP disease [9]; however, the prognostic value of BRAF mutation was shown to be independent of CDX2 expression [10]. Our analysis did not identify CDX2 gene expression as a driver of disease relapse specifically in BRAFMT tumors and reciprocally Bcl-xL was not identified as prognostic across the entire CC population in the CDX2 study (similar to our data in BRAFWT tumors), thus suggesting that we have identified a unique subgroup of poor prognostic patients. However, as the CDX2 study did not collect or utilize BRAF mutation status, this could not be further assessed using their data.
A previous study of Bcl-xL protein expression in CRC determined that high expression of this biomarker was associated with poor prognosis across the entire patient cohort [11]. Importantly, this data indicated a potentially confounding variable, as increased Bcl-xL expression was also significantly associated with later stage disease (already a well-established prognostic factor) [12]. In agreement with this earlier study, we also found that Bcl-xL expression was associated with later stage disease; however, using an adjusted analysis to take into account known confounding clinical factors (including stage), we show that Bcl-xL expression can independently predict prognosis, but only in BRAFMT tumors. A recent study using RPPA methodology reported that a mathematical model of Bcl-2 family protein interactions (including Bcl-xL) termed DR_MOMP was prognostic in chemotherapy-treated stage III CRC [13]. Moreover, this study found that Bcl-2 family signaling was particularly important in Consensus Molecular Subtypes (CMS) 1 and 3. As the CMS1 subgroup is enriched for BRAFMT disease, this report appears to be in agreement with our current study. However, the individual contribution of Bcl-xL expression to prognosis in CMS1/BRAFMT CRC was not reported; the study may have been underpowered in that respect.
The reason for the significant benefit from standard chemotherapy of Bcl-xL high BRAFMT CRC is unclear. High Bcl-xL expressing tumors may be “primed” to undergo apoptosis in response to chemotherapy, due to co-expression of pro-apoptotic Bcl-2 family members. [14, 15] A recent high-throughput drug screen aimed at uncovering therapeutic strategies in CRC, revealed the essentiality of MCL1, Bcl-2 and Bcl-xL in BRAFMT-driven disease [16]. Additionally, a further drug screening-based study identified Bcl-xL as a critical regulator of MEK inhibitor resistance, which was synthetically lethal across a broad panel of BRAFMT cell line models [17]. These findings and the findings presented in our study suggest that directly targeting Bcl-xL may be an effective therapeutic strategy for BRAFMT CRC in the adjuvant disease setting.
We also identified high expression of the long noncoding RNA ZFAS1 as a poor prognostic marker in our discovery dataset. ZFAS1 was previously reported to be overexpressed in CRC compared to adjacent non-CRC tissue, with siRNA-mediated targeting revealing its role as a regulator of p53 protein levels, cell proliferation and colony formation in a small panel of CRC cell lines [18]. Validation of this marker, using methodologies such as RNA in situ hybridization, may clarify its role in disease progression and may become increasingly important as our understanding of the biology of long noncoding RNAs increases.
The findings presented both here and by others suggest that BRAFMT driven CRC is more aggressive than BRAFWT disease, but only when the disease has disseminated from the primary site. Interestingly, we observed specific changes in the ER-stress machinery in BRAFMT tumors with the highest-risk of disease relapse, with activation and upregulation of factors including GADD34, heat shock proteins, and stress-related transcription (DDIT3) in our analysis. Additionally, using GSEA, we identify increased hypoxia and EMT signaling in high-risk tumors, again indicating an association with ER stress-activation. Each of these factors have been demonstrated to activate the unfolded protein response (UPR), which in turn has been correlated with a higher risk of metastatic recurrence in breast cancer [19, 20]. In agreement with our findings, upregulation of UPR signaling in disseminated tumor cells from breast cancer, lung cancer and prostate cancer enables both the formation and long term persistence of metastatic lesions [19, 20]. In addition to activation of the UPR machinery, high Bcl-xL expression may promote survival of invasive tumor cells during the metastatic process; for example Bcl-xL has been reported to be a suppressor of anoikis, [15, 21] which would explain its association with increased risk-of-relapse in the BRAFMT subgroup.
This study has several strengths. We have identified Bcl-xL as a novel predictor of response within a poor prognostic group of CC patient samples, using a robust approach that included validation in an independent cohort using a clinically relevant methodology. Importantly, while we do find significant prognostic and predictive value using Bcl-xL gene expression in 2 independent cohorts, final validation of this discovery approach would require transcriptional data, detailed treatment information and clinical follow up from an independent well balanced cohort, preferably in a clinically trial setting, enriched for the specific subtype of interest, in our case BRAFMT stage II/III CRC. The population-based nature of our validation cohort also means that the results should be generalizable to all stage II/III CRC patients, however we do acknowledge that by using a population-based approach for validation of these findings, there may be a selection bias for patients who subsequently received chemotherapy, and this that may have impacted on our results. Additionally, given that IHC and mutational tests for BRAF and KRAS are routinely utilized in the diagnostic work-up of CC patients, the methods we have used here could easily be employed within routine pathology reporting practice. However, we do acknowledge that further work is required to identify an optimal cut-off level of Bcl-xL expression that would allow a more robust classification of low and high expressers for prospective patient stratification.
In conclusion, we have identified and independently validated the prognostic value of Bcl-xL mRNA and protein expression specifically within BRAFMT CC, which should help inform selection of treatment options for high-risk BRAFMT stage II/III patients in the adjuvant disease setting. This approach could prevent the initial relapses, which ultimately contribute to the poor outcomes of patients with this genotype. Data presented here provide compelling evidence that, in addition to BRAF mutational analysis, assessment of Bcl-xL protein expression using routine diagnostic IHC methods can identify both poor prognostic BRAFMT stage II/III CC patients who will benefit from adjuvant therapy and an otherwise good prognostic subgroup of BRAFMT patients who derive no significant advantage from the addition of adjuvant chemotherapy.
MATERIALS AND METHODS
Transcriptional datasets
Gene expression profiles were downloaded from NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/). Accession number GSE39582 contains 566 tumor transcriptional profiles (460 stage II/III) from a CC series and has previously been employed by the CRC subtyping consortium [22, 23]. As detailed in Supplementary Figure 1, the GSE39582 cohort contained 460 stage II/III CC profiles which had relapse data available. For initial biomarker discovery, the “Prognostic Subset” contained untreated stage II/III patients stratified into high-risk (if the patient relapsed within 36 months) or low-risk (if there was no relapse). The “Initial Consolidation” contained all stage II/III patients with relapse information and mutational data (n = 417), which included BRAFMT (n = 41; 24 of which will have been used already in the prognostic subset), KRASMT (n = 166) or WT/WT (n = 210) (Supplementary Figure 1). Accession number GSE35602 contains profiles from 13 CRC cases, which were obtained using laser-microdissected tissue to extract RNA specifically from stroma or epithelium regions separately, followed by gene expression microarray analysis.
Transcriptomics (Agilent; mRNA_Preprocess_Median) and protein (Reverse Phase Protein Array/mda_rppa_core-protein_normalization) data were downloaded from the COAD pipeline in Firehose (https://gdac.broadinstitute.org/). Patient samples which had both mRNA and RPPA data were collated (n = 102) and were analysed with the Pearson’s correlative analysis using GraphPad Prism version 5 for Windows.
Transcriptional analysis
Partek Genomics Suite was employed for dataset analysis. Differentially expressed probesets which had a fold-change +/– 1.75 fold and p-value < 0.005 were defined using analysis of variance (ANOVA) of supervised risk groupings in both the BRAFMT and KRASMT subgroups separately. Genes represented three times by different probesets were selected for further genotype-specific survival analysis. This method inevitably increases false negatives, by ruling out genes represented by fewer probesets, but it increases the confidence in the positive results. In the BRAFMT analysis, these criteria identified BCL2L1 (Bcl-xL) and NCRNA00275 (ZFAS1). Gene Set Enrichment Analysis was accessed (GSEA; http://software.broadinstitute.org/gsea/index.jsp) and the Microenvironment Cell Populations-counter (MCP) was accessed via the https://doi.org/10.5281/zenodo.61372 link.
Bcl-xL Immunohistochemistry (Bcl-xL IHC)
We optimized a protocol for Bcl-xL IHC on sections of CRC tissue using various antibody dilutions and processing parameters. In line with REMARK guidelines, reproducibility and robustness were tested using a TMA block containing 20 cores of CRC tumor from different patient resections. For staining of the control and independent cohort TMAs, sections were cut at 4 μm on a rotary microtome, dried at 37° C overnight, and then used for IHC, which was performed on an automated immunostainer (Leica Bond-Max, Milton Keynes, UK). Antigen-binding sites were detected with a polymer-based detection system (Bond, Newcastle Upon Tyne, UK; cat. no. DS9800). Bcl-xL IHC antibody (Cell Signaling Technology, MA, United States) (Bcl-xL (54H6) Rabbit mAb #2764) was employed at 1:250 dilution with epitope retrieval solution 2 pretreatment for 30 minutes. All sections were visualized with diaminobenzidine, counterstained with hematoxylin, and mounted in DPX.
Independent stage II/III CC Northern Ireland validation cohort
Candidate biomarkers identified from transcriptional datasets were then evaluated within a Northern Ireland population-based cohort of stage II/III CC patient samples (n = 740) using immunohistochemical methods. The Northern Ireland Cancer Registry was used to identify all patients who underwent surgery in Northern Ireland between 2004 and 2008, for a single, primary, stage II or III colon adenocarcinoma (n = 1,539). A detailed clinical case note review was then conducted, to verify diagnosis and stage and to extract clinical information, including the use of adjuvant chemotherapy and outcome data. Following this review, n = 113 cases were excluded (7%), mainly on the basis of inaccurate staging. Of the remaining n = 1,426 patients, n = 740 patients (52%) were diagnosed within the jurisdiction of the Northern Ireland Biobank, of which specimens relating to n = 661 patients (89%) were successfully retrieved. All patients were followed up for occurrence and cause of death via linkage to the Northern Ireland Registrar General’s Office, up to 31st December 2013. Patients were recorded as having a CRC-specific death if any cause of death was listed as ICD-codes C18, C19, C20 and/or C26.
Northern Ireland cohort immunohistochemical and mutational analysis
This cohort was assembled into a tissue microarray, containing 3 cores from epithelial-rich tumor regions per patient. Blocks were retrieved and tumor regions were annotated for subsequent coring (KA, MBL, JJ). One millimeter diameter tissue cores were extracted from donor blocks and inserted into recipient blocks using a manual tissue arrayer (Estigen, Tartu, Estonia). Additionally, mutational analysis was undertaken for KRAS and BRAF on n = 661 (89%) of the TMA cohort using the ColoCarta panel (Agena Bioscience, Hamburg, Germany). This panel includes: BRAF: D594V, V600E, V600K, V600L, V600R. HRAS Q61L. KRAS: A59T, G12A, G12C, G12D, G12F, G12R, G12S, G12V, G13D, G61H, Q61L. Following sequencing, mutational status of BRAF and KRAS was available for a sub-cohort (n = 661; BRAFMT n = 92, KRASMT n = 248, WT/WT n = 321). Using the IHC methodology optimized in line with the REMARK guidelines in whole face CC sections, we assessed Bcl-xL protein expression using digital pathology software, QuPath [24], to give a numerical representation of both the extent and the intensity of staining (H-score), based on the mean expression of all cores (3 cores for each patient). In line with REMARK guidelines, all scoring was performed while blinded to the clinical details of the cohort and the survival endpoints. Using tertile stratification methodology, we assigned patients in each mutational genotype into high, medium or low groups according to their Bcl-xL protein expression H-score.
Ethical approval
Clinical note review was conducted under the auspices of the Northern Ireland Cancer Registry ethical approval from ORECNI (REC: 10/NIR02/53). Ethical (REC:11/NI/0013, project NIB13-0069) and Bcl-xL staining (NIB16-0212) approval was received from the Northern Ireland Biobank.
Statistical analysis
Clinical characteristics were compared using chi-squared tests, according to mutational groupings. Tertile stratification in GSE39582 was performed on the mean biomarker, BCL2L1 (Bcl-xL), expression value from the three probesets used within the BRAFMT (n = 42), KRASMT (n = 166) and the WT/WT (n = 210) subgroups. Similarly, in the Northern Ireland cohort, tertile stratification was performed on the mean Bcl-xL H-score expression value from the multiple tumor cores available (up to 3 per patient) within the BRAFMT (n = 92), KRASMT (n = 248) and the WT/WT (n = 321) subgroups. Cox Proportional hazards analysis was conducted for both the transcriptional dataset and Northern Ireland cohort, prior to and after adjustment for potential confounders, to evaluate the association between Bcl-xL and survival in CC patients, according to BRAF and KRAS mutation status (Stata version 11.2, StataCorp, College Station, TX, USA).
Author contributions
PDD: Design of the study, data acquisition and analysis, interpretation of results, drafting the work and study supervision; HGC: Data acquisition and analysis, interpretation of results and revision of article; PB, MA: Data acquisition and analysis; RTG: Data acquisition, interpretation of results, collection of samples; SMcQ, VB, MBL, JAJ: Collection of samples; AMBMcC, AG, DK: Data analysis; CH, FDiN, ST: Interpretation of results; DMcA, PGJ, DBL: Study supervision, interpretation of results ; ML: Design of the study, interpretation of results, revision of article, study supervision and funding. All authors approved the final version of the submitted work.
ACKNOWLEDGMENTS
The samples used in this research were received from the Northern Ireland Biobank which is funded by HSC Research and Development Division of the Public Health Agency in Northern Ireland and Cancer Research UK through the Belfast CR- UK Centre and the Northern Ireland Experimental Cancer Medicine Centre; additional support was received from the Friends of the Cancer Centre. The Northern Ireland Molecular Pathology Laboratory which is responsible for creating resources for the NIB has received funding from Cancer Research UK, the Friends of the Cancer Centre and the Sean Crummey Foundation. The Northern Ireland Cancer Registry is funded by the Public Health Agency, Northern Ireland. We also thank Ken Arthur (Belfast) for his expertise in TMA construction of the cohort, Enzo Medico (Torino) for his advice and expertise, and all individuals who were involved in the creation and study design of the Northern Ireland cohort.
CONFLICTS OF INTEREST
None.
FUNDING SUPPORT
This work was supported by The Entwistle Family Travel Award (PDD), a CRUK Population Research Fellowship (HGC), a CRUK Research Bursary (RG), a CRUK studentship (AMBMcC) a HSC R&D Fellowship (RG), a CRUK Programme Grant (PGJ) and a joint CRUK-MRC Stratified Medicine Programme Grant (S:CORT; PDD, MA, PGJ, ML).
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Mitsudomi T, Yatabe Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. 2010; 277:301–08. https://doi.org/10.1111/j.1742-4658.2009.07448.x.
2. Mao C, Wu XY, Yang ZY, Threapleton DE, Yuan JQ, Yu YY, Tang JL. Concordant analysis of KRAS, BRAF, PIK3CA mutations, and PTEN expression between primary colorectal cancer and matched metastases. Sci Rep. 2015; 5:8065. https://doi.org/10.1038/srep08065.
3. Maughan TS, Adams RA, Smith CG, Meade AM, Seymour MT, Wilson RH, Idziaszczyk S, Harris R, Fisher D, Kenny SL, Kay E, Mitchell JK, Madi A, et al, and MRC COIN Trial Investigators. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011; 377:2103–14. https://doi.org/10.1016/S0140-6736(11)60613-2.
4. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al, and BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011; 364:2507–16. https://doi.org/10.1056/NEJMoa1103782.
5. Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, Morris V, Janku F, Dasari A, Chung W, Issa JP, Gibbs P, James B, et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J Clin Oncol. 2015; 33:4032–38. https://doi.org/10.1200/JCO.2015.63.2497.
6. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012; 483:100–03. https://doi.org/10.1038/nature10868.
7. Popovici V, Budinska E, Tejpar S, Weinrich S, Estrella H, Hodgson G, Van Cutsem E, Xie T, Bosman FT, Roth AD, Delorenzi M. Identification of a poor-prognosis BRAF-mutant-like population of patients with colon cancer. J Clin Oncol. 2012; 30:1288–95. https://doi.org/10.1200/JCO.2011.39.5814.
8. Dalerba P, Sahoo D, Paik S, Guo X, Yothers G, Song N, Wilcox-Fogel N, Forgó E, Rajendran PS, Miranda SP, Hisamori S, Hutchison J, Kalisky T, et al. CDX2 as a Prognostic Biomarker in Stage II and Stage III Colon Cancer. N Engl J Med. 2016; 374:211–22. https://doi.org/10.1056/NEJMoa1506597.
9. Bae JM, Lee TH, Cho NY, Kim TY, Kang GH. Loss of CDX2 expression is associated with poor prognosis in colorectal cancer patients. World J Gastroenterol. 2015; 21:1457–67. https://doi.org/10.3748/wjg.v21.i5.1457.
10. Zlobec I, Bihl MP, Schwarb H, Terracciano L, Lugli A. Clinicopathological and protein characterization of BRAF- and K-RAS-mutated colorectal cancer and implications for prognosis. Int J Cancer. 2010; 127:367–80.
11. Jin-Song Y, Zhao-Xia W, Cheng-Yu L, Xiao-Di L, Ming S, Yuan-Yuan G, Wei D. Prognostic significance of Bcl-xL gene expression in human colorectal cancer. Acta Histochem. 2011; 113:810–14. https://doi.org/10.1016/j.acthis.2011.01.002.
12. Dukes CE, Bussey HJ. The spread of rectal cancer and its effect on prognosis. Br J Cancer. 1958; 12:309–20. https://doi.org/10.1038/bjc.1958.37.
13. Lindner AU, Salvucci M, Morgan C, Monsefi N, Resler AJ, Cremona M, Curry S, Toomey S, O’Byrne R, Bacon O, Stuhler M, Flanagan L, Wilson R, et al. BCL-2 system analysis identifies high-risk colorectal cancer patients. Gut. 2017; 66:2141–2148. https://doi.org/10.1136/gutjnl-2016-312287.
14. Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006; 9:351–65. https://doi.org/10.1016/j.ccr.2006.03.027.
15. Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK, Cantlon A, Fisch S, Golomb-Mello G, Ryan JA, Deng J, Jian B, Corbett C, et al. Developmental Regulation of Mitochondrial Apoptosis by c-Myc Governs Age- and Tissue-Specific Sensitivity to Cancer Therapeutics. Cancer Cell. 2017; 31:142–56. https://doi.org/10.1016/j.ccell.2016.11.011.
16. Faber AC, Coffee EM, Costa C, Dastur A, Ebi H, Hata AN, Yeo AT, Edelman EJ, Song Y, Tam AT, Boisvert JL, Milano RJ, Roper J, et al. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discov. 2014; 4:42–52. https://doi.org/10.1158/2159-8290.CD-13-0315.
17. Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, Asthana S, Neel D, Yan JJ, Lu X, Pham L, Wang MM, Karachaliou N, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet. 2015; 47:250–56. https://doi.org/10.1038/ng.3218.
18. Thorenoor N, Faltejskova-Vychytilova P, Hombach S, Mlcochova J, Kretz M, Svoboda M, Slaby O. Long non-coding RNA ZFAS1 interacts with CDK1 and is involved in p53-dependent cell cycle control and apoptosis in colorectal cancer. Oncotarget. 2016; 7:622–37. https://doi.org/10.18632/oncotarget.5807.
19. Bartkowiak K, Kwiatkowski M, Buck F, Gorges TM, Nilse L, Assmann V, Andreas A, Müller V, Wikman H, Riethdorf S, Schlüter H, Pantel K. Disseminated Tumor Cells Persist in the Bone Marrow of Breast Cancer Patients through Sustained Activation of the Unfolded Protein Response. Cancer Res. 2015; 75:5367–77. https://doi.org/10.1158/0008-5472.CAN-14-3728.
20. Bartkowiak K, Effenberger KE, Harder S, Andreas A, Buck F, Peter-Katalinic J, Pantel K, Brandt BH. Discovery of a novel unfolded protein response phenotype of cancer stem/progenitor cells from the bone marrow of breast cancer patients. J Proteome Res. 2010; 9:3158–68. https://doi.org/10.1021/pr100039d.
21. Tan K, Goldstein D, Crowe P, Yang JL. Uncovering a key to the process of metastasis in human cancers: a review of critical regulators of anoikis. J Cancer Res Clin Oncol. 2013; 139:1795–805. https://doi.org/10.1007/s00432-013-1482-5.
22. Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, Bot BM, Morris JS, Simon IM, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015; 21:1350–56. https://doi.org/10.1038/nm.3967.
23. Marisa L, de Reyniès A, Duval A, Selves J, Gaub MP, Vescovo L, Etienne-Grimaldi MC, Schiappa R, Guenot D, Ayadi M, Kirzin S, Chazal M, Fléjou JF, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013; 10:e1001453. https://doi.org/10.1371/journal.pmed.1001453.
24. Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, James JA, Salto-Tellez M, Hamilton PW. QuPath: Open source software for digital pathology image analysis. Sci Rep. 2017; 7:16878. https://doi.org/10.1038/s41598-017-17204-5.