
INTRODUCTION
Hsa-miR-155-5p (miR-155) is one of the first oncogenic miRNAs described in leukemias, breast, lung and colon cancers [1–3]. Several oncogenic roles have been ascribed to this miR, including a contribution to genomic instability through the down-regulation of transcripts implicated in DNA mismatch repair, DNA damage repair, and maintenance of telomere integrity [4–6]. However, genomic instability can also be due to chromosome instability (CIN), which has important implications for tumor initiation [7, 8] and evolution [9, 10].
CIN arises mostly through defects in the spindle assembly checkpoint (SAC), a quality control mechanism that inhibits anaphase onset until all chromosomes are correctly attached to the spindle microtubules via the kinetochores [11–13] and is rarely caused by telomere erosion [14–16]. The SAC depends on proteins of the outer plate of the kinetochore, such as the centromere protein F (CENP-F), the zw10 kinetochore protein (ZW10) and the mitotic checkpoint serine/threonine kinase budding uninhibited by benzimidazoles 1 (BUB1), one of the most important SAC kinases [17]. Loss of these proteins severely affects chromosomal segregation at anaphase, leading to aberrant aneuploid cells that are thought to be selected for proliferative and adaptive advantages linked with transformation [17–20].
Given that CIN is a cancer hallmark and that miR-155, involved in several tumors, affects molecular pathways involved in genomic stability, we hypothesized that this miRNA could interfere also with the molecular complexes of the SAC, leading to aneuploidy in cancer cells.
To demonstrate this, we investigated whether miR-155 could: i) directly influence the expression of the SAC complex proteins BUB1, ZW10 and CENP-F; ii) trigger chromosome alignment defects during cellular transformation; iii) prolong mitosis and increase the rate of aneuploidy at early stages of HDF immortalization. We found that miR-155, acting through the SAC, contributed to generate aneuploidy during cellular transformation.
RESULTS
MiR-155 negatively regulates BUB1 protein expression
To investigate a possible link between miR-155 and CIN we looked for new putative targets of miR-155. In silico analyses provided by the MicroCosm Targets Version 5 database revealed that BUB1 is a predicted target of miR-155. However, this database refers to a discontinued BUB1 sequence (ENST00000389944, Ensembl) with a 3’UTR more extended than the current reference sequences (transcript variant 1: NM_004336/ENST00000302759; transcript variant 2: NM_001278616/ENST00000535254; transcript variant 3: NM_001278617/ENST00000409311; NCBI/Ensembl), that exclude the predicted miR-155 binding site (Figure 1A). We verified whether the BUB1 transcript with the extended 3’UTR was naturally expressed in normal and cancer cells. For this purpose we amplified by RT-PCR two different fragments (Amp~500; Amp~750) (Figure 1A) of the putative 3’UTR from HDF, and from a panel of cancer cells representative of tumors of breast (MCF7), colon (HCT116), of chronic lymphocytic leukemia (CLL: MEC-1), and osteosarcoma (U2OS), neoplasms for which the relevance of miR-155 has been well demonstrated [1–3, 21, 22]. Gel electrophoresis of the RT-PCR products showed the expected ~500 bp fragments in all the analyzed cell types, and the ~750 bp fragments in all the cancer cell lines, confirming the presence of the long transcript isoform (Figure 1B). In HDF cells we detected only the ~500 bp fragment, the ~750 bp fragments was undetectable, which is probably due to the technical issues related to the low expression of the gene in cells replicating slowly. To characterize the 3′end of the long isoform, we performed 3′ Rapid Amplification cDNA Ends (RACE) using RNA from the HG-3 B-CLL cell line. We identified two close 3’UTR ending points at positions 4294 and 4303 of the BUB1 sequence that includes the miR-155 binding region (Figure 1C). Next, we validated BUB1 as a direct target of miR-155 by luciferase assay in U2OS cells. Compared to the respective controls, miR-155 suppressed the luciferase activity of the construct containing the native BUB1 3’UTR by ~30%, while the luciferase activity of the construct containing the mutated BUB1 3’UTR was unaffected. This demonstrates that miR-155 directly interacts with the 3’UTR of BUB1 (Figure 2A and 2B).
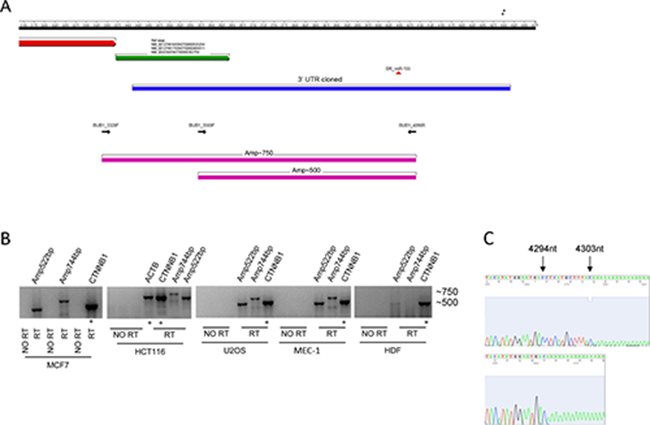
Figure 1: BUB1 3’UTR analysis. (A) Schematic representation of the 3’UTR of the BUB1 gene shows, from top to bottom: nucleotide numbering (RefSeq NM_004336; black bar). Black asterisks indicate the 3′ends of the 3’UTR determined by 3′RACE; 3′end of the coding sequence (red arrow); NCBI/Ensembl reference 3’UTR sequence (NCBI/Ensemble; green arrow); 3’UTR (blue bar) as cloned into psiCHECK-2 reporter vector; predicted miR-155 binding site (red triangle); and PCR primers (black arrows) marking two amplicons of ~500 bp and ~750 bp (magenta bars). (B) Detection of the long mRNA isoform of BUB1 in four human cancer cell lines and normal human dermal fibroblasts (HDF) by RT-PCR followed by gel electrophoresis (1.5% agarose gel in 1X TBE buffer) and ethidium bromide staining. No RT, negative control without reverse transcriptase. CTNNB1 and ACTB, internal positive controls with similar fragment size to the BUB1 amplicons. (C) Sequence chromatograms of two different products obtained by the 3′rapid amplification cDNA ends (RACE) workup of HG-3 cell RNA to determine the 3′end sequence of BUB1 3’UTR. Blacks asterisks in panel A mark the 3′ends of the cloned 3′UTR. The specific primers used are listed in Supplementary Table 2.
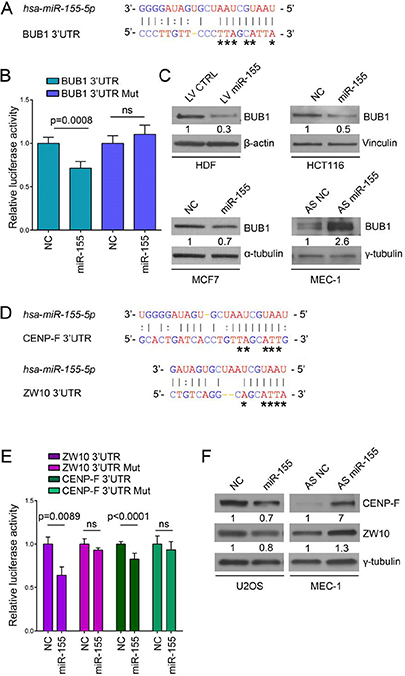
Figure 2: BUB1, CENP-F and ZW10 are targeted by miR-155. (A) Base pairing between miR-155 (top) and predicted binding site in BUB1 3’UTR (bottom) according to MicroCosm Targets version 5). Asterisks indicate nucleotides subject to site-directed mutagenesis. Watson-Crick base pairing (G-C and A-T) is marked by vertical lines while G-T pairs are marked by two dots. (B) Relative luciferase activity in U2OS cells transfected with psiCHECK-2_BUB1 3’UTR with either the predicted miR-155 binding site in reference sequence or mutated (Mut) form and either the miR-155 precursor (miR-155) or the negative control oligonucleotide (NC). Values are means ± SD of five technical replicates, ns: no significant p-value. (C) Western blots of BUB1 protein (120 kDa) after miR-155 lentiviral infection in HDF cells and after transfection with miR-155 precursor, antisense inhibitor oligonucleotide (AS miR-155), or respective negative controls (NC and AS NC) in HCT116, MCF7 and MEC-1 cell lines. (D) Base pairing between miR-155 and predicted binding sites in CENP-F 3’UTR and ZW10 3’UTR, according to MicroCosm Targets version 5. Asterisks indicate nucleotides subject to site-directed mutagenesis. (E) Relative luciferase activity in U2OS cells transfected with psiCHECK-2_CENP-F 3’UTR or psiCHECK-2_ZW10 3’UTR with the predicted miR-155 binding site in reference or mutated form, and either miR-155 or NC. Values are means ± SD of four technical replicates for ZW10 and eight measures from two independent experiments for CENP-F (4 technical replicates from each experiment). (F) Western blots of CENP-F and ZW10 proteins after transfection with miR-155 precursor, antisense of miR-155 or respective negative controls (NC and AS NC) in U2OS and MEC-1 cells as indicated.
To further assess the influence of miR-155 on BUB1 expression, we evaluated changes in BUB1 protein levels by western blotting of HDF infected with a lentiviral expression vector carrying miR-155 (LV miR-155) or a control vector (LV CTRL). Overexpression of miR-155 strongly decreased the BUB1 protein levels (–70%). Concordantly, the BUB1 levels decreased after ectopic miR-155 expression in MCF7 (–30%) and HCT116 (–50%) cells. Furthermore, BUB1 protein expression strongly increased when MEC-1 cells, which present high levels of endogenous miR-155, were transfected with an antisense miR-155 (Figure 2C, Supplementary Figure 1). Overall, these results indicate that miR-155 binds to the 3’UTR of BUB1 and specifically represses BUB1 protein expression.
ZW10 and CENP-F are direct targets of miR-155
To strengthen the relevance of the miR-155 in the mitotic spindle assembly we looked for additional targets among kinetochore proteins and found two additional SAC proteins: ZW10 and CENP-F. (MicroCosm Targets Version 5 database) (Figure 2D). Direct targeting by miR-155 was demonstrated using a luciferase assay, as before. The luciferase activity of constructs containing ZW10 and CENP-F 3′UTRs decreased by ~40% and ~20%, respectively after ectopic expression of miR-155, while these effects were abolished when the 3’UTRs were altered by mutagenesis (Figure 2E). Western blotting showed that induced expression of miR-155 in U2OS cells decreased ZW10 (-20%) and CENP-F (-30%) protein levels, while in MEC-1 cells ZW10 and CENP-F protein expressions increased after modulation of miR-155 with an antisense miR-155 (Figure 2F). Collectively, these results demonstrate that miR-155 directly regulates ZW10 and CENP-F protein levels, which supports a role of this miRNA in the regulation of mitotic progression trough the SAC.
MiR-155 affects chromosome congression and outer plate kinetochore assembly
Chromatids of cells depleted of BUB1 do not align correctly on the spindle equator before anaphase [17], ZW10-deficient cells show aberrant mitoses and abnormal chromosome distribution as well, with formation of chromatin bridges and micronuclei [18] and CENP-F repression induces defects in chromosome alignment [19, 20]. Therefore, we hypothesized that miR-155 could affect the accuracy of mitotic chromosome congression by targeting these kinetochore proteins. To test this hypothesis, we analyzed the morphology of the metaphase plates after induction of miR-155 in U2OS cells infected with LV miR-155 (Supplementary Figure 2A), and found that the percentage of irregular metaphases was higher (~40%) than in control cells infected with LV CTRL (~24%; p = 0.0038) (Figure 3A, right). To further evaluate this mechanism, we transformed normal HDF by sequential infection with retroviruses that transduce first the simian virus 40 (SV40) large T antigen (LT) and then the catalytic subunit of human telomerase (hTERT) (HDFLT/hTERT) [23, 24]. We decided to use this in vitro model because of 1) the possibility to analyze metaphases during the cellular transition from normal to abnormal karyotypes and 2) the possibility to modulate miR-155 expression, since its expression increased starting from the very early passage of infection in HDFLT/hTERT cells (Supplementary Figure 2B). We analyzed the morphology of the metaphase plates in HDFLT/hTERT cells infected with either LV AS miR-155 or LV AS CTRL at low passages (P14 and P17). Metaphases with mal-oriented chromosomes represented ~86% (P14) and ~83% (P17) of the total in HDFLT/hTERT controls, and ~65% (P14) and ~72% (P17) of the total in LV AS miR-155 HDFLT/hTERT cells (P14, p < 0.0001; P17, p = 0.02) (Figure 3A, right). Regulation of BUB1, CENP-F and ZW10 by LV AS miR-155 in HDFLT/hTERT and LV miR-155 in U2OS cells is shown in Supplementary Figure 3.
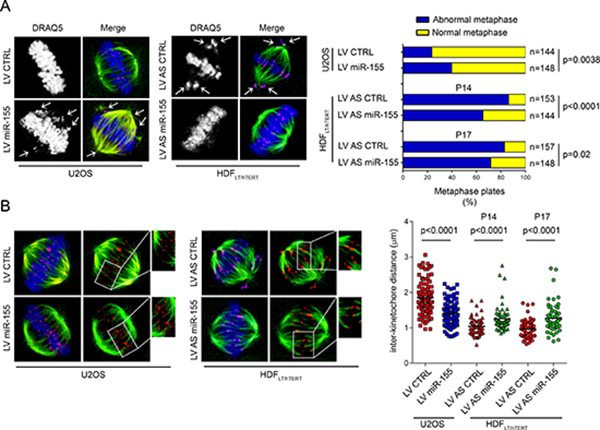
Figure 3: Role of miR-155 in chromosome congression. (A) Left, Immunofluorescence of HDFLT/hTERT and U2OS cells stained for α-tubulin (green), α-centromere (red), and DNA with DRAQ5 (blue). Both cell types, infected as indicated, arrested at the metaphase-anaphase transition upon treatment with the proteasome inhibitor MG132 (U2OS, 10 μM for 2 h) or nocodazole (HDFLT/hTERT, 75 ng/ml for 17 h plus 30 min of release). Chromosomes in metaphase cells were counted as unaligned if they were located outside of the mitotic spindle or if their kinetochores were aligned perpendicularly to the spindle axis (white arrows). Right, Quantification of U2OS and HDFLT/hTERT congression errors in two independent experiments at P14 (Experiment 1) and P17 (Experiment 2). (B) Inter-kinetochore distances measured in prometaphase-metaphase of cells infected as indicated and stained with anti-α-tubulin (green) or anti-centromere (red) antibodies and DRAQ5 (blue). Over 60 kinetochore pairs in more than 10 cells were examined for each condition.
Tension within the kinetochore generated by the microtubules attached to the opposite spindle poles is crucial for proper chromosome configuration [25–27]. To further explain the errors in chromosome alignment associated with the deregulation of miR-155, we measured the inter-kinetochore distances as indirect indicators of tension in the centromere-kinetochore region of the sister chromatids. The inter-kinetochore distances of metaphase chromosomes significantly increased in HDFLT/hTERT infected with LV AS miR-155. On the other hand, in the U2OS cells over-expressing the miR, the distances between kinetochores were significantly reduced (Figure 3B). Next, we examined by immunofluorescence whether overexpression of miR-155 reduced the recruitment of BUB1, CENP-F and ZW10 at the kinetochores. In LV miR-155-infected U2OS cells the BUB1, CENP-F and ZW10 immunofluorescence intensity levels were reduced by – 60% (p < 0.0001), – 38% (p = 0.0211), and – 32% (p = 0.0358), respectively (Figure 4).
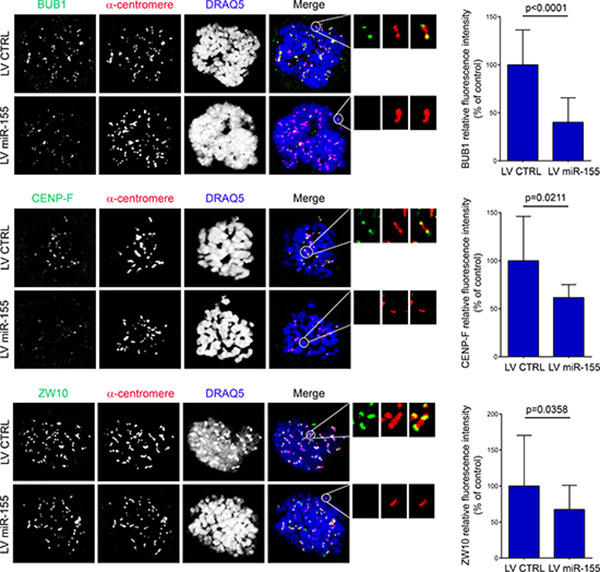
Figure 4: Over-expression of miR-155 affects the recruitment of BUB1, CENP-F and ZW10. U2OS cells infected with LV miR-155 and control were arrested at prometaphase by nocodazole treatment and then fixed and labeled with the indicated antibodies. Enlargements of kinetochores are circled in the merged image and shown in the inserts. Graphs present the fluorescence intensity of the protein normalized to that of anti-centromere staining. The bar graphs were derived from measurements of all the kinetochores in at least 10 cells.
MiR-155 is required to block cell cycle progression at the G2/M phase and to delay mitosis exit
Defects in metaphase plate alignment influence cell cycle progression. Thus, using in U2OS cells, we evaluated the effects of miR-155 on the cell cycle after reversible damage induced by nocodazole. The percentage of miR-155-infected U2OS cells in G2/M was significantly higher than in control U2OS cells (p = 0.0168), and this corresponded to an increased mitotic index (p = 0.0003) (Figure 5A). This finding suggests that cells with high miR-155 expression delay their exit from G2/M after nocodazole-induced damage. We corroborated our results by replicating this experiment in HDFLT/hTERT with or without LV AS miR-155. Compared to controls, the fraction of miR-155-depleted HDFLT/hTERT cells blocked in G2/M was significantly lower (p = 0.0009), which corresponded to a reduction in mitotic index of the metaphases (p = 0.0091) (Figure 5B).
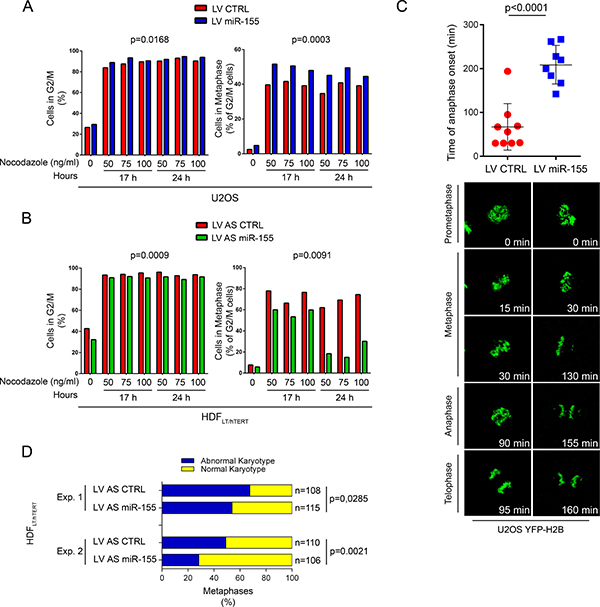
Figure 5: Over-expression of miR-155 blocks cells at G2/M phase and prolongs the duration of metaphase by delaying chromosome congression. Flow citometry of (A) U2OS cells infected with LV miR-155 and LV CTRL and (B) HDFLT/hTERT infected with LV AS miR-155 and LV AS CTRL. As indicated, cells were blocked at different concentrations of nocodazole and for different times. Left, G2/M cells were determined by quantifying nuclear DNA and Right, mitotic cells by anti-phospho-histone-H3 (PH-H3) staining gated on G2/M cells. (C) Time of anaphase onset in U2OS cells stably expressing YFP-H2B and infected with the indicated lentivirus. Time lapse measuring started at prometaphase (T = 0), and images were acquired every 5 min. Individual measurements (dots) with mean and standard deviations are represented in the graph. (D) Karyotype analysis of two independent experiments (Experiment 1, Experiment 2) at P14. HDFLT/hTERT cells were arrested in mitosis by colcemid treatment, harvested for chromosome spread and then stained with Giemsa solution. A minimum of 100 metaphases was considered in each experiment.
The effect of miR-155 during mitosis was also evaluated using live-cell imaging technology. To analyze chromosome dynamics, we generated U2OS cells stably expressing the yellow fluorescent protein-histone H2B fusion protein (YFP-H2B) and either miR-155 (by infection with LV miR-155) or a control (by infection with LV CTRL). In U2OS cells infected with LV CTRL mitosis proceeded from prometaphase to anaphase in a mean of 68 minutes, with chromosomes perfectly aligned on the metaphase plate (Supplementary Movie 1). In U2OS cells overexpressing miR-155 (LV miR-155), progression from prometaphase to anaphase took longer (mean, 209 min) (Supplementary Movie 2), chromosomes failed to align to the metaphase plate and, after a long delay, the cells frequently exited mitosis without clear anaphase and with lagging chromatids (Figure 5C). These results demonstrate a strong activation of the SAC in cells with chromosomal congression defects, even though at the end most of the cells completed cell division (data not shown).
Over-expression of miR-155 promotes aneuploidy at the onset of transformation
BUB1 hypomorphic mice have a high percentage of aneuploid cells and are predisposed to spontaneous tumors [28, 29]. CENP-F expression is associated with poor prognosis and chromosomal instability in primary breast cancer [30], and mutational screening in human colorectal cancers has identified mutations in ZW10, that may contribute to CIN [31]. To determine if miR-155 causes aneuploidy, we analyzed the karyotypes of HDFLT/hTERT cells infected with either LV AS miR-155 or LV AS CTRL at an early passage (P14). Since CIN can affect both entire chromosomes and parts of them, we counted loss and gain of whole chromosomes as well as chromosomal amplifications, unbalanced translocations, and deletions. Transformed HDF cells in which miR-155 was inhibited(HDFLT/hTERT LV AS miR-155) maintained a near-diploid karyotype, while control cells had marked aneuploidy and chromosomal instability (Experiment 1, p = 0.0285; Experiment 2, p = 0.0021) (Figure 5D, Supplementary Table 1).These results strengthen the direct link between miR-155 and CIN/aneuploidy.
DISCUSSION
This study reveals a novel link between miR-155 and aneuploidy. We demonstrated that miR-155 directly targets three key proteins of the mitotic SAC, namely BUB1, CENP-F and ZW10 [32–36]. Enhanced expression of miR-155 in our cellular models caused defects in chromosome alignment associated with altered distances between sister kinetochores and a moderate-to-severe decrease of these proteins levels to the kinetochores after nocodazole-induced damage. Given the abundantly demonstrated role of BUB1, CENP-F and ZW10 in inducing defects of the mitotic spindle after their depletion, we strongly suggest that the mechanism by which miR-155 induces metaphase plate misalignments and in turn aneuploidy is through the direct down-regulation of these proteins. Moreover, this also reflects an important role of miR-155 in the duration of the cell division. Consistently, miR-155 blocked nocodazole-treated cells in metaphase and increased their mitotic index. Indeed, delayed mitosis is the proper reaction of a cell to misaligned chromosomes, as it enables the correction of errors but, in a pre-cancerous context that could involve chronic inflammation and/or viral infection and/or genetic predisposition, cells with up-regulation of miR-155 may acquire an anti-apoptotic support to exit from the G2/M phase, despite chromosome alignment defects at the metaphase plate.
It is commonly accepted that tumor progression is fueled by a gradual accumulation of genetic anomalies, and that higher genetic instability correlates with more aggressive and metastatic forms of disease [9, 37]. However, recent studies showed that aneuploidy occurs early in carcinogenesis followed by a period of genomic stability [38–42]. Accordingly, we confirmed that miR-155 is involved in CIN at an early stage of transformation (14th passage), since in our in vitro model, inhibition of miR-155 decreased the number of cells with abnormal karyotypes after infection with transforming viruses. We took advantage of the ability of the SV40 large T antigen to generate aneuploidy [43], which is the resultant of its direct interaction with the tumor suppressor protein TP53 [44, 45], with members of the retinoblastoma RB proteins family [46, 47], and with BUB1 itself [48] in both HDFLT/hTERT LV AS miR-155 and HDFLT/hTERT LV AS CTRL cells. The inhibition of miR-155 rescued, at least in part, the effect of SV40 LT on BUB1 function by increasing the protein expression of its targets BUB1, CENP-F, and ZW10.
In conclusion, our results provide a novel direct link between miR-155 and CIN, which, together with the already described target genes involved in cell cycle control and DNA repair, strengthens the oncogenic features of miR-155 and highlights its role at the early steps of tumorigenesis.
MATERIALS AND METHODS
Cell culture, transfection
HG-3 and MEC-1 cell lines were acquired from Leibniz Institut DSMZ – German Collection of Microorganisms and Cell Cultures in February 2016 and December 2015 respectively. MCF7 and U2OS cell lines were cultured in Dulbecco’s Modified Eagle’s Medium whereas HG-3 and MEC-1 in RPMI, supplemented with 10% fetal bovine serum, 1% Pen/Strep and 1% L-glutamine (Sigma-Aldrich) at 37 °C in a 5% CO2 incubator. Human adult dermal fibroblasts (HDFa, Thermo-Fisher Scientific) were cultured in Medium 106 (M106) supplemented with Low Growth Serum Supplement (LSGS) (Thermo-Fisher Scientific). Transient transfections of MCF7, U2OS and HCT116 cell lines were performed using Lipofectamine 2000 (Thermo-Fisher Scientific, Release 14 July 2011) in accordance with the manufacturer’s procedures. Transfection of HG-3 or MEC-1 cells was performed using Amaxa™ Nucleofector™ II (Lonza), program U-016. MicroRNA analysis products were from Thermo-Fisher Scientific (Precursor miR-155, ID:PM12601; antisense inhibitor oligonucleotide, AS miR-155: ID:AM12601 or respective negative controls NC and AS NC). U2OS cells, expressing the YFP-H2B histone were generated using pH2B-YFP expression vector (EUROSCARF).
RNA extraction and RT-PCR
Total RNA was isolated from cells using QIAzol Lysis Reagent (Qiagen) and treated with DNase (TURBO DNA-free Kit, Thermo Fischer Scientific) to remove contamination of genomic DNA. RNA quantification was performed by NanoDrop 2000 (Thermo-Fisher Scientific). 700 ng of total RNA was reverse-transcribed with the High Capacity cDNA Reverse Transcription Kit (Thermo-Fisher Scientific). Polymerase Chain Reaction (PCR) was performed using AmpliTaq Gold DNA Polymerase (Thermo-Fischer Scientific).
Plasmids
The 3’UTR region of human BUB1, ZW10 and CENP-F 3′UTRs genes were amplified by PCR from HDF genomic DNA. The amplified fragment was cloned downstream of the Renilla luciferase gene of the psiCHECK-2 luciferase vector (Promega). QuikChange XL Site-Directed Mutagenesis Kit (Agilent technologies) was used for direct mutagenesis.
Luciferase target assays
U20S cells were co-transfected with psiCHECK-2_BUB1 3’UTR, psiCHECK-2_CENP-F 3’UTR and psiCHECK-2_ZW10 3’UTR (in reference or mutated forms; 150 ng/10,000 cells) and precursor miR-155 or NC (100 nM in the culture medium). After 24 h, firefly and Renilla luciferase activities were measured using the Dual-Glo Luciferase Assay System (Promega).
Reverse transcription quantitative PCR (RT-qPCR)
Assay Design Center software (http://lifescience.roche.com/shop/products/universal-probelibrary-system-assay-design) was used to identify primers and UPL probes (Supplementary Table 2). Total RNA (300 ng) was reverse-transcribed with High Capacity cDNA Reverse Transcription Kit (Thermo-Fisher Scientific). MicroRNAs were reverted from 25 ng of total RNA using the specific reverse primer (stem loop RT primers) and the TaqMan Micro-RNA Reverse Transcription Kit (Thermo-Fisher Scientific). Reactions were incubated 30 min at 16°C, followed by pulsed RT [49]. Real-time PCR was performed using the Universal Mastermix (Roche) and the relative expression, normalized to an endogenous reference GAPDH or RNU44, was determined using the 2-Δct method (User Bulletin #2, Applied Biosystems).
Western blotting
Cell lysates having equal protein concentrations were loaded on a Criterion TGX 4–20% precast polyacrylamide gel (Bio-Rad) and transferred onto a PVDF membrane. Primary antibodies: anti-CENP-F (ab5) (Abcam), anti-BUB1 (B3), anti-ZW10 (3363C4a), anti-vinculin (H-300), anti-γ-tubulin (C-20), anti α-tubulin (Santa Cruz Biotechnology) and anti β-actin (Sigma Aldrich). HRP-conjugated secondary antibody (Cell Signaling Technology) was used. ImageJ (Fiji software, https://fiji.sc/) were used to quantify western blot signals.
Flow cytometry
Permeabilized cells (70% ethanol, 30 min) were incubated with antibody anti-phospho-histone H3 (Cell Signaling Technology) and with the secondary antibody (10 μg/ml, Alexa Fluor 633 goat anti-rabbit IgG, Thermo-Fisher Scientific) in accordance with the manufacturer’s procedures. Cells were washed and stained with Vybrant DyeCycle Violet Stain (1 μM, Thermo-Fisher Scientific). Analyses were performed using FlowJo v 8.8.6 software.
Generation of viruses and cell infections
MuLV retroviral particles were obtained using HEK 293T as the packaging cell line and pBABE-puro-SV40LT (gift from Thomas Roberts; Addgene plasmid #13970) [50] and pBabe-hygro-hTERT, pUMVC and pCMV-VSV-G (gifts from Bob Weinberg; Addgene plasmids #1773, #8449 and #8454 respectively) [51, 52] in accordance with the protocol of Scott Dessain and Hua-yin Yu, Weinberg Lab (https://www.addgene.org/static/data/84/58/16578800-af64-11e0-90fe-003048dd6500.pdf). LV pmiRZip AS miR-155, LV pmiRZip CTRL, LV pCDH_miR-155 and LV pCDH_CTRL were obtained by using the pPACKH1 HIV Lentivector Packaging Kit (System Biosciences). HDFLT/hTERT LV AS miR-155 and control (LV AS CTRL) cells were developed by sequential infections with polybrene (5 μg/ml). Briefly, normal HDF were infected at passage P4, with RV LTSV40 for 3 hours, then were washed and infected overnight with LV AS miR-155, or LV AS CTRL. After 7 days of puromycin selection (0.5 μg/ml) (Santa Cruz Biotechnology), at P6, cells were infected with RV hTERT for 3 hours and selected with hygromycin (50 μg/ml) (InvivoGen). Since LV AS miR-155 contains the copGFP reporter gene, GFP-positive cells were sorted using the FACSAriaIII, 100 μm nozzle (BD Biosciences).
Immunofluorescence
Cells were incubated with the primary antibody in PBS+ (PBS, 5% FBS, 0.02% sodium azide, and 10 mg/ml BSA) overnight at room temperature. Antibodies: anti-BUB1 (ab9000), anti-ZW10 (ab21582), anti γ-tubulin (ab7291), anti-CENP-F (ab5) (Abcam), and human anti-CREST(Antibodies Inc.). Secondary antibodies were from Bethyl Laboratories. DNA was detected with DRAQ5 (Cell Signaling Technology). Coverslip were mounted with SlowFade® Gold antifade reagent (Thermo-Fisher Scientific). Kinetochore tension was indirectly determined by measuring the inter-kinetochore distance of kinetochore pairs on confocal sections when both sister kinetochores were in the same focal plane. To quantify the amount of protein bound to the kinetochore, the pixel intensities of kinetochores from 10-20 nocodazole-treated cells were measured using ImageJ software. To normalize the fluorescent intensity of the protein of interest, anti-centromere staining was performed in the same region. All the fluorescence intensities were acquired with identical settings and corrected by background subtraction using a Carl Zeiss LSM510 META (Zeiss Axiovert 200 and a 63X Plan Neofluar objective).
Time-lapse experiments
U2OS cells stably expressing YFP-H2B and infected with either LV CTRL or LV miR-155 were seeded into glass-bottomed dishes (Greiner Bio-one) and placed into the incubator of the microscope (system S, Zeiss) set at 37°C in an atmosphere of 5% CO2 and 20% humidity. Images were captured every 5 min with a Plan Neofluar oil-immersion objective (40X). The laser intensity was maintained at a minimum to avoid phototoxicity.
Karyotype analyses
To prepare metaphase spreads, 1–2 x 106 HDFs were seeded (at P14 of two independent experiments) in Amniodishes (Euroclone). Colcemid (Irvine Scientific) treatment (0.5 μg/ml) was carried out after 24 h for 4–5 hours at 37 °C in an atmosphere of 5% CO2 and 20% humidity. Cells were fixed in Carnoy’s solution (75% methanol, 25% acetic acid) and stained with Giemsa solution (5% for 10 min). Digital images were analyzed by Genikon software (Nikon).
3′Rapid Amplification cDNA Ends (RACE)
Total RNA (2 μg) from HG-3 cells was reverse-transcribed using the oligo-dT adapter primer (AP). The first-strand cDNA was then amplified using HotStarTaq DNA Polymerase (Qiagen) using a 3′ abridged universal amplification primer (AUAP), and a gene-specific reverse primer (BUB1_3UTR). A semi-nested PCR was performed using BUB1_3994 forward primer. The PCR products were cloned into pCR4-TOPO TA vector (Thermo-Fisher Scientific) and sequenced using M13 rev (-29) primer by Sanger method.
Statistical analyses
Data regarding luciferase assays, times of anaphase onset, RT-qPCR assays, inter-kinetochore distances and kinetochore recruitments were evaluated using two-tailed Student’s t or Mann-Whitney’s tests, based on the significance obtained by the Shapiro-Wilk Normality Test. The significance of differences between percentages of cells blocked at the G2/M phase was assessed using the one sample t test (two tailed, with theoretical median value = 1.0) after Kolmogorov-Smirnov test with Dallal-Wilkinson-Lillie correction for p value normality. In this experiment, we applied Kolmogorov-Smirnov test because of the small set size. The two-tailed Fisher’s exact test was used for metaphase plate morphology and karyotype analyses. All RT-qPCR assays were performed in triplicate. The number of analyzed samples is shown in the Figures (n), a p-value > 0.05 was considered not significant (ns), significant p-values are indicated in the figures. When indicated, standard deviation (SD) is represented as a scale bar on graphs.
Author contributions
RV and AV designed the study. SP performed experiments with contributions from RV, AV, AR and MDM. RV AV and SP analysed and interpreted the results. CMC contributed to analyze data and participated in the discussion. MVR, AS, PEGF, and GP performed karyotype analysis. PL, PS and MM performed flow cytometry analysis. RMC discussed the experiments and the data analysis. RV, AV, RMC and SP wrote the manuscript. All the authors critically reviewed and edited the paper.
ACKNOWLEDGMENTS AND FUNDING
We thank Dr. Simone Guarnieri for precious guidance using the confocal microscope, Ms. Rosalia De Amicis and Ms. Elvira D’Annunzio for administrative work. This study was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC Start-Up grant 2010, no. 10054 to RV) and partially by the US National Cancer Institute (NCI grant no. R35CA197706 to CMC) and partially by Dalla Zentil, MD and Mr. Guido Milanese private donation to RV.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
REFERENCES
1. Costinean S, Sandhu SK, Pedersen IM, Tili E, Trotta R, Perrotti D, Ciarlariello D, Neviani P, Harb J, Kauffman LR, Shidham A, Croce CM. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-MiR-155 transgenic mice. Blood. 2009; 114:1374–82. https://doi.org/10.1182/blood-2009-05-220814.
2. Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, Croce CM. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A. 2006; 103:7024–9. https://doi.org/10.1073/pnas.0602266103.
3. Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103:2257–61. https://doi.org/10.1073/pnas.0510565103.
4. Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F, Adair B, Vannini I, Fanini F, Bottoni A, Costinean S, Sandhu SK, Nuovo GJ, et al. Modulation of mismatch repair and genomic stability by miR-155. Proc Natl Acad Sci U S A. 2010; 107:6982–7. https://doi.org/10.1073/pnas.1002472107.
5. Dinami R, Ercolani C, Petti E, Piazza S, Ciani Y, Sestito R, Sacconi A, Biagioni F, le Sage C, Agami R, Benetti R, Mottolese M, Schneider C, et al. miR-155 Drives Telomere Fragility in Human Breast Cancer by Targeting TRF1. Cancer Res. 2014; 74:4145–56. https://doi.org/10.1158/0008-5472.CAN-13-2038.
6. Gasparini P, Lovat F, Fassan M, Casadei L, Cascione L, Jacob NK, Carasi S, Palmieri D, Costinean S, Shapiro CL, Huebner K, Croce CM. Protective role of miR-155 in breast cancer through RAD51 targeting impairs homologous recombination after irradiation. Proc Natl Acad Sci U S A. 2014; 111:4536–41. https://doi.org/10.1073/pnas.1402604111.
7. Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004; 432:338–41. https://doi.org/10.1038/Nature03099.
8. Weaver BAA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007; 11:25–36. https://doi.org/10.1016/j.ccr.2006.12.003.
9. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976; 194:23–8.
10. Gao CF, Furge K, Koeman J, Dykema K, Su YL, Cutler ML, Werts A, Haak P, Vande Woude GF. Chromosome instability, chromosome transcriptome, and clonal evolution of tumor cell populations. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:8995–9000. https://doi.org/10.1073/pnas.0700631104.
11. Dick AE, Gerlich DW. Kinetic framework of spindle assembly checkpoint signalling. Nat Cell Biol. 2013; 15:1370–7. https://doi.org/10.1038/ncb2842.
12. Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007; 8:379–93. https://doi.org/10.1038/nrm2163.
13. Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Current Biology. 2012; 22:R966-R80. https://doi.org/10.1016/j.cub.2012.10.006.
14. Guerrero AA, Gamero MC, Trachana V, Futterer A, Pacios-Bras C, Diaz-Concha NP, Cigudosa JC, Martinez AC, van Wely KH. Centromere-localized breaks indicate the generation of DNA damage by the mitotic spindle. Proc Natl Acad Sci U S A. 2010; 107:4159–64. https://doi.org/10.1073/pnas.0912143106.
15. Martinez AC, van Wely KH. Are aneuploidy and chromosome breakage caused by a CINgle mechanism? Cell Cycle. 2010; 9:2275–80. https://doi.org/10.4161/cc.9.12.11865.
16. Martinez AC, van Wely KH. Centromere fission, not telomere erosion, triggers chromosomal instability in human carcinomas. Carcinogenesis. 2011; 32:796–803. https://doi.org/10.1093/carcin/bgr069.
17. Meraldi P, Sorger PK. A dual role for Bub1 in the spindle checkpoint and chromosome congression. EMBO J. 2005; 24:1621–33. https://doi.org/10.1038/sj.emboj.7600641.
18. Kops GJ, Kim Y, Weaver BA, Mao Y, McLeod I, Yates JR 3rd, Tagaya M, Cleveland DW. ZW10 links mitotic checkpoint signaling to the structural kinetochore. J Cell Biol. 2005; 169:49–60. https://doi.org/10.1083/jcb.200411118.
19. Holt SV, Vergnolle MA, Hussein D, Wozniak MJ, Allan VJ, Taylor SS. Silencing Cenp-F weakens centromeric cohesion, prevents chromosome alignment and activates the spindle checkpoint. J Cell Sci. 2005; 118:4889–900. https://doi.org/10.1242/jcs.02614.
20. Feng J, Huang H, Yen TJ. CENP-F is a novel microtubule-binding protein that is essential for kinetochore attachments and affects the duration of the mitotic checkpoint delay. Chromosoma. 2006; 115:320–9. https://doi.org/10.1007/s00412-006-0049-5.
21. Lauvrak SU, Munthe E, Kresse SH, Stratford EW, Namlos HM, Meza-Zepeda LA, Myklebost O. Functional characterisation of osteosarcoma cell lines and identification of mRNAs and miRNAs associated with aggressive cancer phenotypes. Br J Cancer. 2013; 109:2228–36. https://doi.org/10.1038/bjc.2013.549.
22. Chen L, Jiang K, Jiang H, Wei P. miR-155 mediates drug resistance in osteosarcoma cells via inducing autophagy. Exp Ther Med. 2014; 8:527–32. https://doi.org/10.3892/etm.2014.1752.
23. O’Hare MJ, Bond J, Clarke C, Takeuchi Y, Atherton AJ, Berry C, Moody J, Silver AR, Davies DC, Alsop AE, Neville AM, Jat PS. Conditional immortalization of freshly isolated human mammary fibroblasts and endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:646–51. https://doi.org/10.1073/pnas.98.2.646.
24. Fauth C, O’Hare MJ, Lederer G, Jat PS, Speicher MR. Order of genetic events is critical determinant of aberrations in chromosome count and structure. Genes Chromosomes Cancer. 2004; 40:298–306. https://doi.org/10.1002/Gcc.20051.
25. Scholey JM, Brust-Mascher I, Mogilner A. Cell division. Nature. 2003; 422:746–52. https://doi.org/10.1038/nature01599.
26. Nicklas RB. How cells get the right chromosomes. Science. 1997; 275:632–7.
27. Nezi L, Musacchio A. Sister chromatid tension and the spindle assembly checkpoint. Curr Opin Cell Biol. 2009; 21:785–95. https://doi.org/10.1016/j.ceb.2009.09.007.
28. Jeganathan K, Malureanu L, Baker DJ, Abraham SC, van Deursen JM. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J Cell Biol. 2007; 179:255–67. https://doi.org/10.1083/jcb.200706015.
29. Baker DJ, Jin F, Jeganathan KB, van Deursen JM. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell. 2009; 16:475–86. https://doi.org/10.1016/j.ccr.2009.10.023.
30. O’Brien SL, Fagan A, Fox EJ, Millikan RC, Culhane AC, Brennan DJ, McCann AH, Hegarty S, Moyna S, Duffy MJ, Higgins DG, Jirström K, Landberg G, Gallagher WM. CENP-F expression is associated with poor prognosis and chromosomal instability in patients with primary breast cancer. International Journal of Cancer. 2007; 120:1434–43. https://doi.org/10.1002/Ijc.22413.
31. Wang ZH, Cummins JM, Shen D, Cahill DP, Jallepalli PV, Wang TL, Parsons DW, Traverso G, Awad M, Silliman N, Ptak J, Szabo S, Willson JKV, et al. Three classes of genes mutated in colorectal cancers with chromosomal instability. Cancer Res. 2004; 64:2998–3001. https://doi.org/10.1158/0008-5472.Can-04-0587.
32. Bomont P, Maddox P, Shah JV, Desai AB, Cleveland DW. Unstable microtubule capture at kinetochores depleted of the centromere-associated protein CENP-F. EMBO J. 2005; 24:3927–39. https://doi.org/10.1038/sj.emboj.7600848.
33. Karess R. Rod-Zw10-Zwilch: a key player in the spindle checkpoint. Trends Cell Biol. 2005; 15: 386–92. https://doi.org/10.1016/j.tcb.2005.05.003.
34. Meraldi P, Draviam VM, Sorger PK. Timing and checkpoints in the regulation of mitotic progression. Dev Cell. 2004; 7:45–60. https://doi.org/10.1016/j.devcel.2004.06.006.
35. Perera D, Tilston V, Hopwood JA, Barchi M, Boot-Handford RP, Taylor SS. Bub1 maintains centromeric cohesion by activation of the spindle checkpoint. Dev Cell. 2007; 13:566–79. https://doi.org/10.1016/j.devcel.2007.08.008.
36. Yang Z, Guo J, Chen Q, Ding C, Du J, Zhu X. Silencing mitosin induces misaligned chromosomes, premature chromosome decondensation before anaphase onset, and mitotic cell death. Mol Cell Biol. 2005; 25:4062–74. https://doi.org/10.1128/MCB.25.10.4062-4074.2005.
37. Fidler IJ, Hart IR. Biological diversity in metastatic neoplasms: origins and implications. Science. 1982; 217:998–1003.
38. Orsetti B, Selves J, Bascoul-Mollevi C, Lasorsa L, Gordien K, Bibeau F, Massemin B, Paraf F, Soubeyran I, Hostein I, Dapremont V, Guimbaud R, Cazaux C, et al. Impact of chromosomal instability on colorectal cancer progression and outcome. BMC Cancer. 2014; 14:121. https://doi.org/10.1186/1471-2407-14-121.
39. Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, Chen K, Scheet P, Vattathil S, Liang H, Multani A, Zhang H, Zhao R, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014; 512:155–60. https://doi.org/10.1038/nature13600.
40. Gao R, Davis A, McDonald TO, Sei E, Shi X, Wang Y, Tsai PC, Casasent A, Waters J, Zhang H, Meric-Bernstam F, Michor F, Navin NE. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat Genet. 2016; 48:1119–30. https://doi.org/10.1038/ng.3641.
41. Zhao Z, Goldin L, Liu S, Wu L, Zhou W, Lou H, Yu Q, Tsang SX, Jiang M, Li F, McMaster M, Li Y, Lin X, et al. Evolution of multiple cell clones over a 29-year period of a CLL patient. Nat Commun. 2016; 7:13765. https://doi.org/10.1038/ncomms13765.
42. Laughney AM, Elizalde S, Genovese G, Bakhoum SF. Dynamics of Tumor Heterogeneity Derived from Clonal Karyotypic Evolution. Cell Rep. 2015; 12:809–20. https://doi.org/10.1016/j.celrep.2015.06.065.
43. Chang TH, Ray FA, Thompson DA, Schlegel R. Disregulation of mitotic checkpoints and regulatory proteins following acute expression of SV40 large T antigen in diploid human cells. Oncogene. 1997; 14:2383–93. https://doi.org/10.1038/sj.onc.1201196.
44. Lane DP. Cell immortalization and transformation by the p53 gene. Nature. 1984; 312:596–7.
45. Lane DP. Cancer. p53, guardian of the genome. Nature. 1992; 358: 15–6. https://doi.org/10.1038/358015a0.
46. DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH, Marsilio E, Paucha E, Livingston DM. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988; 54:275–83.
47. Zalvide J, DeCaprio JA. Role of pRb-related proteins in simian virus 40 large-T-antigen-mediated transformation. Mol Cell Biol. 1995; 15:5800–10.
48. Cotsiki M, Lock RL, Cheng Y, Williams GL, Zhao J, Perera D, Freire R, Entwistle A, Golemis EA, Roberts TM, Jat PS, Gjoerup OV. Simian virus 40 large T antigen targets the spindle assembly checkpoint protein Bub1. Proc Natl Acad Sci U S A. 2004; 101:947–52. https://doi.org/10.1073/pnas.0308006100.
49. Tang F, Hajkova P, Barton SC, Lao K, Surani MA. MicroRNA expression profiling of single whole embryonic stem cells. Nucleic Acids Res. 2006; 34:e9. https://doi.org/10.1093/nar/gnj009.
50. Zhao JJ, Gjoerup OV, Subramanian RR, Cheng Y, Chen W, Roberts TM, Hahn WC. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell. 2003; 3:483–95. https://doi.org/S1535610803000886.
51. Counter CM, Hahn WC, Wei W, Caddle SD, Beijersbergen RL, Lansdorp PM, Sedivy JM, Weinberg RA. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc Natl Acad Sci U S A. 1998; 95:14723–8.
52. Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, Weinberg RA, Novina CD. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003; 9:493–501.