
INTRODUCTION
Head and neck cancers are among the 10 most common cancers worldwide and the great majority of these cancers are squamous cell carcinomas and associated with severe mortality [1–3]. Currently the usual treatment for HNSCC patients remains surgery in combination with radiotherapy and this is further combined with chemotherapy for more advanced disease and for recurrent and metastatic tumours [4–6]. Despite therapeutic advances during the last decade, the 5-year-survival rate for HNSCC remains low with late diagnosis at advanced tumour stages typically associated with local and regional recurrences, and with development of lymph node and distant metastasis [7, 8]. The poor therapeutic responses of HNSCC have encouraged focused efforts towards development of molecular targeted therapies [7, 9, 10].
Overexpression of the epidermal growth factor receptor (EGFR) is a frequent molecular alteration in HNSCC and increased activity of the EGF pathway has been associated with resistance to treatment and poor clinical outcome [5, 11, 12]. The EGFR is a member of the HER tyrosine kinase receptor family and binding of specific ligands, such as epidermal growth factor (EGF) and transforming growth factor alpha (TGF-α), promotes homo- or hetero-dimerization of EGFR family members and activation of intracellular signaling pathways that control growth, differentiation, survival and invasion [13–15]. The EGFR is therapeutically targeted by agents such as Cetuximab and Erlotinib [6] but the modest effects observed on tumour control have been rather disappointing considering the importance of EGFR-initiated signaling pathways. Various mechanisms for primary and acquired resistance to Cetuximab have been suggested and include constitutive activation of EGFR-mediating signaling molecules and activation of alternative ErbB2 and ErbB3 pathways [16, 17].
There is now substantial evidence that HNSCC, like other solid tumours, consist of a heterogeneous population of cells. This results partly from the evolution of clonal heterogeneity [18] but is also due to persistence of a hierarchy of stem and differentiating cells similar to that present in normal tissues [19, 20]. Cancer stem cells (CSCs) are identified as a subpopulation of cells with the ability to self-renew indefinitely whilst also generating daughter cells that differentiate, usually aberrantly, and the self-renewal properties of CSCs are tested by their selective ability to initiate tumours in immune-deficient mice [21]. The CSC subpopulation not only drives tumour growth but is also responsible for the aggressiveness and therapeutic resistance of tumours [22]. CSCs in HNSCC were initially identified and isolated by their high levels of expression of the hyaluronan receptor CD44 [19, 23, 24] and several subsequent reports have described isolation of CSCs from HNSCC using either CD44 or various other markers [25]. CSCs also persist in malignant cell lines and can be identified by high levels of CD44 expression, by morphological characteristics, and by their clonogenicity [23].
Of particular interest, tumour invasion and metastasis have been linked to epithelial-mesenchymal transition (EMT), a process during which cells lose epithelial features of mutual attachment and gain mesenchymal features, including mobility [26–29]. It is proposed that EMT enables CSCs to invade surrounding local and distant tissues where the reverse process of mesenchymal-to-epithelial transition (MET) enables their return to the epithelial phenotype to generate metastatic growth [30]. Biddle and co-workers [31] demonstrated that the self-renewing CD44high CSC fraction of HNSCC contains as at least two distinct cellular phenotypes with marked differences in cell motility, morphology, proliferation, clonogenicity, and sphere forming abilities. Both cell fractions are tumour-initiating and both express high levels of CD44 but the CSCs with an epithelial phenotype express high levels of ESA whereas those that have undergone EMT have low or absent levels of ESA [31]. CSCs have the ability to switch between these phenotypes and, as a result, aggressive tumours consist of at least 3 cell types, epithelial CSCs, mesenchymal CSCs, and a differentiating non-stem cell fraction [31]. A comprehensive understanding of drug actions will therefore depend on information about the individual responses of each of these cell types.
Apart from their EMT-mediated roles in invasion and metastasis, the therapeutic importance of CSCs lies in their greater ability to resist therapeutic killing in response to radio- and chemo-therapies [22, 32, 33]. Differential effects of CSC sub-fractions to the molecularly targeted therapies of agents such as Cetuximab and Erlotinib are therefore of interest [34]. We have now investigated the effects of blocking EGFR function on each of the phenotypically-different cell sub-populations in HNSCC cell lines and find that EGFR inhibition leads to morphological and molecular changes, marked reductions in cell proliferation, but no marked induction of cell death. It leads to reduced CD44 expression and a shift of CSCs into differentiation, a state in which they have enhanced therapeutic sensitivities. This action provides a form of “differentiation therapy” [35–37] and appears related to the greater effectiveness of EGFR inhibition when used as an adjunctive component of therapies. Both Cetuximab and Erlotininib also shift EMT-CSCs back into the epithelial phenotype, a change that may tend to restrict local invasion and metastasis.
RESULTS
Effects of Cetuximab and Erlotinib on cell proliferation and apoptosis in HNSCC cell lines.
Treatment levels were initially determined by treating CA1 and Luc4 cell lines for 3 days with drug concentrations of Cetuximab and Erlotinib ranging from 0–500 μg/ml or 0–500 ng/ml respectively (Figure 1A, 1B). Based on these pilot data, Cetuximab at a dose of 100 μg/ml, and Erlotinib at a dose of 500 ng/ml, were used for subsequent experiments. By day 3, both drugs induced marked morphological changes, including the formation of more compact cell colonies and loss of individual elongated EMT-like cells (Figure 1C). Cell cultures were examined to assess the extent to which the reduced cell numbers present after 3 days of treatment (Figure 1D) were related to reduced proliferation or to increased apoptosis. Cytometry after staining for Annexin V and DAPI showed low (2–4%) levels of apoptosis in untreated cells. The lack of significant increases in counts of Annexin V positive cells after treatment indicated no marked promotion of apoptotic cell death (Figure 1E). Effects of treatment in markedly reducing rates of cell cycling were indicated by significant decreases in IdU labeled cells (Figure 1F, 1G).
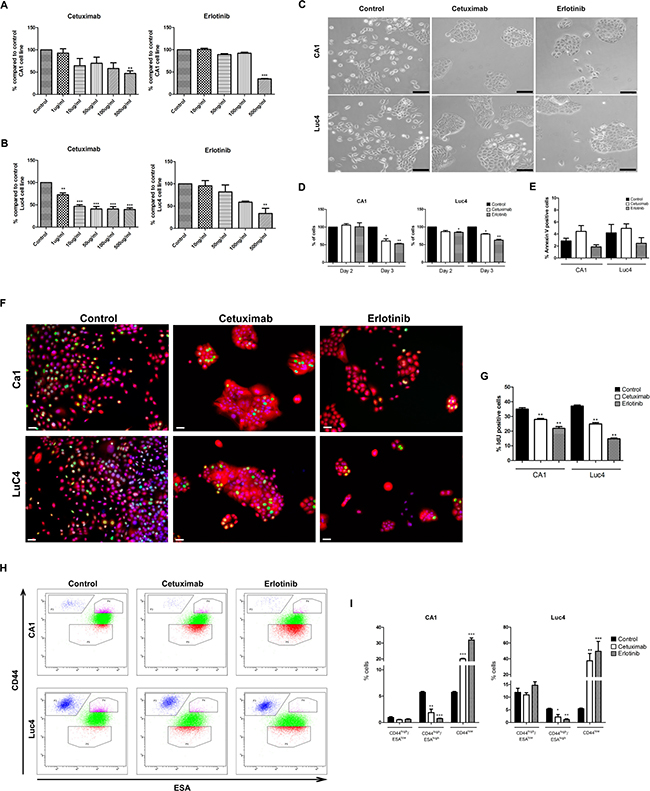
Figure 1: Effects of Cetuximab and Erlotinib on cell proliferation and apoptosis in HNSCC cell lines. Percentage of total number of CA1 (A) and Luc4 (B) cells compared to controls after 3 days of treatment with Cetuximab and Erlotinib at various concentrations. (C) Cell morphology of CA1 and Luc4 after treatment with Cetuximab (100 μg/ml) and Erlotinib (500 ng/ml) for 3 days (Scale = 50μm). (D) Total number of CA1 and Luc4 after each day of treatment. (E) Analysis of Annexin V positive cells after 3 days treatment with either Cetuximab or Erlotinib. (F) Representative images of IdU positive cells (green) in CA1 and Luc4 after treatment with Cetuximab and Erlotinib (Scale = 25μm). (G) Quantification of the number of IdU positive cells. (H) FACS analysis of expression of CD44 and ESA on CA1 and Luc4 cells after 3 days of treatment with Cetuximab or Erlotinib showing decrease of cells with CD44high/ESAhigh expression and increase the CD44high/ESAlow fraction. (I) Quantification of data depicted in H, showing the percentage of CD44high/ESAlow, CD44high/ESAhigh and CD44low fractions. For this and all subsequent figures; *P < 0.05, **P < 0.01, ***P < 0.001.
Cetuximab and Erlotinib decrease the levels of CD44 expression of CA1 and Luc4 cells.
To further assess the effects of EGFR inhibitors, we examined whether there were differential changes in the cell line sub-fractions we have previously identified . Cells were stained for CD44 and ESA and fractionated by flow cytometry into CD44high/ESAlow (EMT-CSC), CD44high/ESAhigh (EPI-CSC) and CD44low (NON-CSC) populations. For both cell lines, treatment significantly decreased the percentage of CD44high/ESAhigh cells and consistently increased the percentage of CD44low cells (Figure 1H, 1I). Following treatment CD44high/ESAlow fractions showed no statistically significant differences in mean CD44 or ESA expression levels, and the proportion of this fraction, expressed as a percentage of the total cells, also remained unchanged.
To determine whether the decreases in CD44 expression indicated by flow cytometry were associated with functional decreases in stemness, treated cell lines were re-plated at low density to assess their colony-forming abilities. Treated cells of both cell lines showed significant reductions in colony forming ability (Figure 2A and 2B) but no significant differences in their ability to form tumour spheres (Figure 2C).
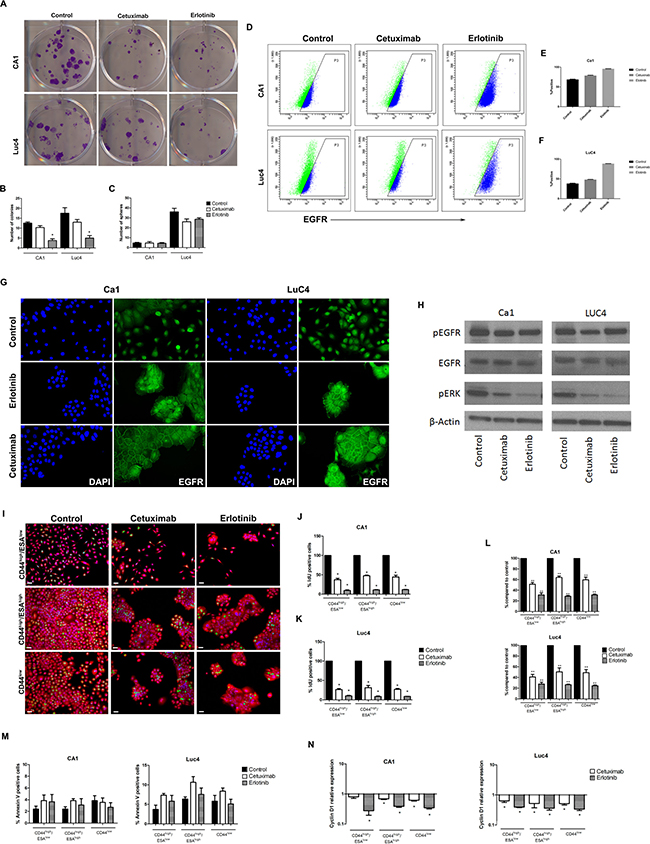
Figure 2: Cetuximab and Erlotinib decrease clonogenicity, proliferation rates and EGFR expression. (A) Effects of treatment on colony forming ability. (B) Quantification of the number of colonies formed in CA1 and Luc4 cell lines. (C) Number of spheres formed after 3 days of treatment. (D) FACS plots of changes in the number of cells expressing cell-surface EGFR. (E, F) Comparisons of percentages of EGFR expressing cells in the two cell lines after treatment. (G) Altered patterns of EGFR staining after treatment. (H) Western blots showing protein levels for EGFR, pEGFR, and pERK. Altered patterns of cell proliferation (I–K), accumulation (L) and apoptosis (M) of cell sub-fractions following treatment. (N) Levels of Cyclin D1 were reduced in all sub-fractions.
Cetuximab and Erlotinib alter EGFR expression patterns
Differences in EGFR expression induced by Cetuximab and Erlotinib were evaluated by flow cytometry and western blotting. Plots of EGFR versus side-scatter indicated that the control populations of both cell lines had similar and substantial levels of total cell surface EGFR and that these levels increased following treatment (Figure 2D–2F). Control populations of both CA1 and Luc4 showed higher expression of EGFR on the CD44high/ESAhigh Epi-CSC subfraction than on either CD44high/ESAlow or CD44low cells and there was a trend for expression in all fractions to increase after treatment. Immunofluorescence showed cytoplasmic staining with higher than control levels of EGFR at the cell peripheries of the cohesive cell colonies formed following treatment (Figure 2G). Following treatment, Western blots indicated little difference in the overall levels of pEGFR or EGFR but showed reduced levels of the downstream target pERK indicated that both inhibitors functioned to interrupt the primary EGFR signaling pathway (Figure 2H).
Cetuximab and Erlotinib decrease the proliferation rates of all cell fractions and prevent G1/S progression
To evaluate proliferative effects of Cetuximab or Erlotinib treatment on the CD44high/ESAlow, CD44high/ESAhigh and CD44low fractions, cells were sorted and re-plated. Treatment resulted in a reduced accumulation of cells for each of the sorted fractions of both cell lines with levels of IdU incorporation significantly and similarly reduced for all sub-fractions (Figure 2I–2L). Counts of Annexin V positive cells indicated low levels of apoptosis in control specimens and treatment-induced changes in apoptosis were small and not significant for any of the 3 cell sub-fractions (Figure 2M). Cyclin D1, which is required for progression of cells through the G1/S cell cycle phase, also showed significant decreases in all cell fractions with both treatments (Figure 2N).
Cetuximab and Erlotinib increase cell differentiation
The observed treatment effects of loss of cells from the CD44high/ESAhigh cell fractions, reduction in colony forming assays, and an increased proportion of CD44low cells indicated a shift of cells from the Epi-CSCs stem cell compartment into differentiation. To assess other indicators of differentiation, we examined changes in cell size and in cytoplasmic-to-nuclear ratio as these are known to increase with differentiation. Following treatment, sorted cell fractions showed trends towards an increased cell size, mean cell area and cytoplasmic to nuclear ratio (Figure 3A–3C). Expression of the epithelial differentiation markers Calgranulin B and Involucrin was found to be lowest in the CD44high/ESAlow EMT cell fractions, and the highest in the CD44low fractions and significant increases in these markers for all cell fractions indicated that treatment resulted in shifts towards differentiation (Figure 3D, 3E).
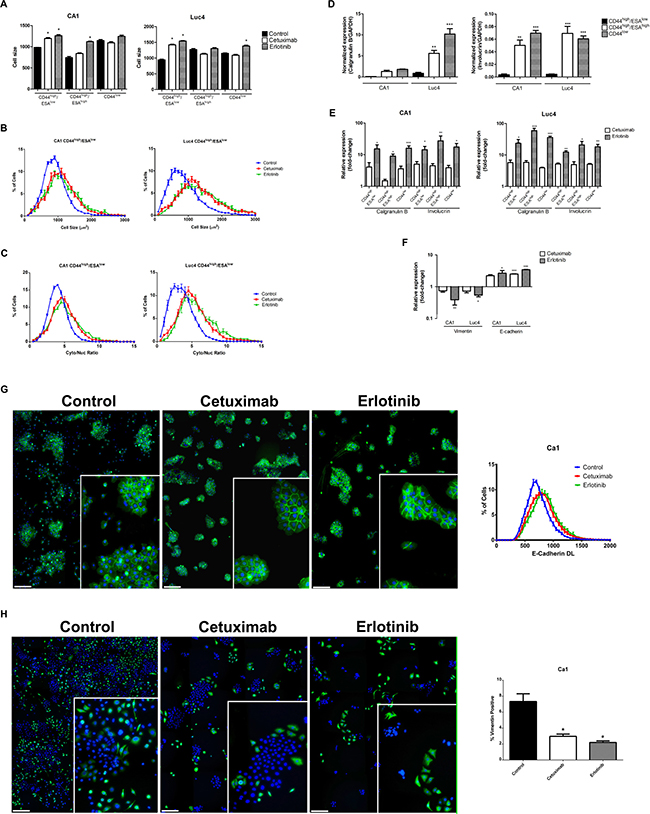
Figure 3: Cetuximab and Erlotinib induce cellular differentiation. (A) Mean cell size of CD44high/ESAlow, CD44high/ESAhigh and CD44low fractions in CA1 and Luc4 cell lines after treatment. Representative graphs showing (B) increased size and (C) increased cytoplasmic/nuclear ratio of CD44high/ESAlow cells. In control cultures, expression of the differentiation markers Calgranulin B and Involucrin is normally lowest in CD44high/ESAlow sub-population (D) but is increased in all fractions (E) after treatment. (F) qPCR indicated reduced Vimentin and increased E-cadherin expression of EMT fractions after treatment. (G) Control cultures show some cohesive colonies staining for E-cadherin (green) surrounded by scattered DAPI-positive E-cadherin negative cells. After treatment with Cetuximab or Erlotinib scattered cells were lost and nearly all cells were E-cadherin positive. (H) Control cultures show numerous scattered vimentin-positive (green) cells which, after treatment are reduced in number and tend to cluster into and around epithelial-like colonies (Scale for G and H = 25 μm) Inserts show colonies at higher magnification.
Cetuximab and Erlotinib effects on the size of EMT cell fractions, rates of cell migration, and cisplatin responses
Control cultures showed populations of scattered cells with an EMT-like appearance and their loss after treatment suggesting a transition of EMT cells back to an epithelial-like phenotype. As little increase in ESA expression was detected by FACS, we examined FACS-isolated CD44high/ESAlow EMT fractions for other evidence of mesenchymal-epithelial transition (MET). QPCR (Figure 3F) and immunofluorescence staining cell indicated significant decreases in vimentin expression and increases in E-cadherin expression in treated populations (Figure 3F–3H). The loss of scattered e-cadherin negative/vimentin positive cells seen in control cultures was again associated with cell clustering to form compact colonies.
A shift of EMT cells towards an epithelial phenotype was assessed in two other ways. When FACS-isolated EMT-CSCs were plated and grown for 3 days, cells from control cultures maintained an EMT-like appearance whereas treated cultures began to transit into cell clusters with an epithelial appearance (Figure 4A, top). The contribution of EMT-CSCs to the compact colonies was apparent when EMT-CSCs were isolated from EGFP-labeled cultures and mixed and plated with unlabeled CD44high/ESAhigh EPI-CSCs. These cultures initially showed individual elongated EGFP-labeled cells that were distinct from the unlabeled epithelial colonies but by 72 hours after treatment clusters of EGFP-labeled cells were present within and around the epithelial colonies (Figure 4A, lower) indicating a morphological transition from EMT-CSCs into an epithelial state. Scratch assays showed marked changes in cell motility as a result of EGFR blocking. Scratches in control wells were essentially closed within 16 hours whereas after EGFR inhibition scratches remained open even at 24 hours (Figure 4B–4D). Cultures treated with Cisplatin were treated with Cetuximab or Erlotinib 3 days later to see whether EGFR blocking enhanced the effects of Cisplatin treatment. Both cell lines showed the enhanced effects on the reduction in cell number when the drugs were used in combination (Figure 4E).
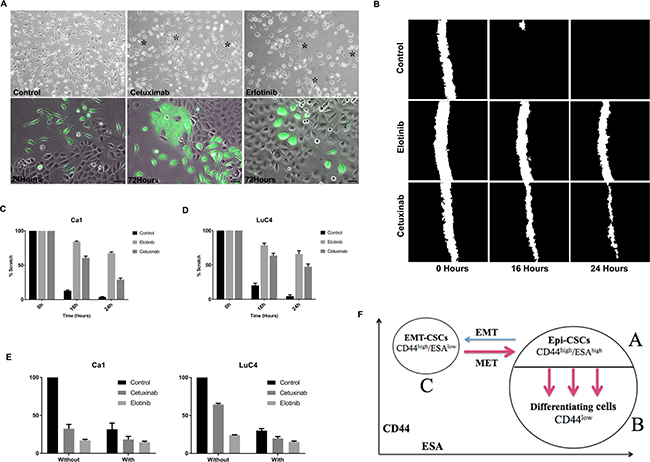
Figure 4: Changes in cell transitions, motility, and response to cisplatin induced by treatment. (A) Isolated populations of CD44high/ESAlow cells retain EMT characteristics 3 days after plating but treated with Cetuximab or Erlotinib begin to form clustered epithelial colonies (*). Three days after plating a mixture of EGFP+ EMT cells with unlabeled parental cells, EGFP+ cells retain their EMT-like morphology but treatment with Cetuximab or Erlotinib induces transition into an epithelial phenotype (Scale = 25μm). (B–D). Images of scratches made in control and treated cultures showed reduced closure of scratches after treatment. (E) Cell counts after treatment with Cisplatin alone or combined with EGFR inhibition indicate more effective action of the combined drugs. (F) Cytometry assessing CD44 and ESA expression identifies 3 cell sub-populations in HNSCC cell lines. The majority cell population is ESAhigh and contains a CD44high/ ESAhigh epithelial stem cell population (A), and a CD44low differentiating non-stem cell population (B) (see Figure 2A). A third population (C) is CD44high/ESAlow and corresponds to cells that have undergone EMT. Three transitions occur – EMT, MET and transition into differentiation. Blocking the EGFR markedly enhanced the transitions indicated by red arrows.
DISCUSSION
Activation of the EGFR has widespread effects on cell proliferation, survival, invasion, DNA repair and drug resistance. This receptor is frequently overexpressed in HNSCC and has been extensively studied as a molecular target [38]. However, blocking EGFR signaling has provided less therapeutic benefit than initially anticipated [39] and this may be largely related to the cellular heterogeneity of tumours and, particularly, to the presence of sub-populations of CSCs and differentiating cells [40]. There is now considerable evidence that elimination of CSCs is required for successful therapy [26, 41] but it has also been found that CSCs generally have greater resistance to therapeutic killing by either radiation or chemotherapy [32, 42]. Therapeutic manipulation of CSCs is further complicated by their presence as heterogeneous populations of epithelial and mesenchymal phenotypes that differ in their therapeutic responses to individual therapies and are able to switch from one phenotype to the other [31, 40]. Information about the particular specificities of individual drugs for each type of cell may therefore be helpful in developing combinations of drugs that provide effective actions upon the total tumour population, potentially also at lower therapeutic concentrations.
CD44 has been widely used as a general marker for CSC identification [23, 25] and has functions related to stem cell maintenance. For example, CD44 knockout mice show reduced epidermal differentiation [43] and CD44 down-regulation in tumour cells is associated with stem cell loss whereas over-expression of CD44 drives tumour progression and promotes CSC properties [44–46]. EGFR activation increases CD44 expression, blocking EGFR tyrosine kinase domains reduces both stem cell maintenance and EMT, and loss of CD44 down-regulates both total and phosphorylated EGFR [46, 47]. Taken together, these findings support the concept that drugs blocking EGFR signalling are likely to affect CSC maintenance, either directly or indirectly, and that information about differential effects of drugs on stem cell fractions is of therapeutic interest.
Cetuximab and Erlotinib interrupt EGFR signalling by different mechanisms but both strongly inhibited cell proliferation and produced distinct and similar morphological changes. Flow cytometry analysis indicated interesting drug effects on the CSC and non-CSC subpopulations in terms of shifts of cells between these sub-fractions. The proportion of cells in CD44high/ESAhigh Epi-CSC fractions was markedly reduced and the proportion of cells in CD44low fractions was correspondingly raised. The rates of cell proliferation and apoptosis for these subpopulations were not differentially affected by treatment with either drug. Therefore, the reduction in CD44high/ESAhigh Epi-CSCs was attributed to their enhanced transit into the differentiating CD44low population. We have previously shown that clonogenic ability, assessed as the formation of adherent colonies, is primarily a property of Epi-CSCs [31] and the markedly reduced colony-forming efficiency of treated cells supports the concept of loss of Epi-CSCs by differentiation. Further evidence for this shift was found in the increased size of cells in this fraction and their increased expression of the Involucrin and Calgranulin B differentiation markers.
Treated cultures showed a striking disappearance of the numerous scattered cells in control cultures that have an EMT-like appearance. Given this apparent loss of EMT cells by morphological criteria, it was surprising that EGFR inhibition induced no statistically significant differences in the number of cells expressing the CD44high/ESAlow cell surface phenotype, typically used as an identifier of EMT-CSCs in FACS assay [48]. Nor was there a change in sphere forming ability. Sphere formation has been used to assess the size of “functional” EMT-CSC fractions [31] and the Luc4 line, which has a larger fraction of CD44high/ESAlow EMT-CSCs than the CA1 cell line, accordingly showed more sphere formation. However, although drug treatment did not induce consistent changes in sphere formation, the decreased Vimentin and increased E-cadherin levels found for sorted CD44high/ESAlow fractions indicated that some degree of transition of EMT-CSCs towards MET had occurred. This was also demonstrated by the treatment-induced shifts of EGFP-labelled EMT cells into epithelial colonies. The treatment-induced EMT changes found therefore differed depending on the assays employed. However, ascribing a simple binary transition to epithelial and mesenchymal cells undergoing EMT/MET processes is perhaps over simplistic [11]. Stable hybrid epithelial/mesenchymal phenotypes that co-express both epithelial and mesenchymal markers are described [31, 49–51] and the EMT-CSC subpopulation itself appears to be heterogeneous with expression of markers such as Aldehyde dehydrogenase 1 marking cells that retain lineage plasticity [31]. The lack of re-expression of ESA and shifts in sphere formation after EGFR blocking may represent partial transition within a hybrid epithelial/EMT phenotype. Alternatively, a 3-day period of treatment may be insufficient for the full transition of all properties.
The presence of cells expressing mesenchymal markers has previously been reported in breast, prostate and other carcinoma cell lines [11, 52, 53]. In HNSCC cell lines, EMT generates cells with a CD44high/ESAlow phenotype that are plastic, able to revert to the epithelial phenotype, and relatively resistant to radiation and various chemotherapeutic agents [11, 32, 48]. Mesenchymal cells in HNSCC have been variously identified by a CD44high/EGFRlow cell surface expression pattern [34], by a Vimentinhigh/E-cadherinlow staining pattern [54] and by a CD44high/Aldhhigh staining pattern [55]. Mesenchymal cells are also found in epidermoid and oesophageal carcinoma cell lines [56] and it appears that EMT and MET cell transitions are a common feature of carcinomas [37]. An interesting interpretation of the EMT process is that it “generates cells with the properties of stem cells” [29, 57], a conclusion based mainly on the EMT-induced shift from a CD44high/CD24high to a CD44high/CD24low phenotype in mammary cell lines [57]). Although the latter corresponds to the phenotype of tumour initiating stem cells in breast cancer [58], other evidence shows that the shift of phenotypic properties induced by EMT occurs within the stem cell compartment itself, i.e. between CD44high/CD24high Epi-CSCs and CD44high/CD24low EMT-CSCs [31, 48]. As both Epi-CSCs and EMT-CSCs are self-renewing and capable of initiating and maintaining tumours, both cell types need to be eliminated for successful therapy [41, 59].
In response to environmental stimuli, Epi-CSCs transit into self-renewing CD44high/ESAlow EMT cells and can also revert back to the Epi-CSC phenotype by MET. For a given cell line, balanced rates of the EMT and MET transitions result in a relatively stable size of the EMT-CSC fraction. Typically the rate of these transitions is slow taking, for example, 3 weeks for levels of EMT cells to be greatly increased by hypoxia or by TGFβ, and taking a similar time after withdrawal of stimuli for EMT cells to return to baseline levels [34, 60]. EGFR blocking greatly increased the rate of MET and within 3 days there was loss of cells with an EMT-like appearance and transition of labeled EMT-CSCs back into epithelial colonies. The expected effect of a rapid shift of cells out of the EMT fraction is to reduce the invasive and metastatic potential of the tumour population.
Many reports indicate that the clinical response rate for patients with advanced HNSCC is low when treated with Cetuximab alone, but survival and loco-regional control are improved when patients are treated with Cetuximab in combination with radiotherapy, platinum-based therapy or fluorouracil [5, 12, 51, 61]. Similarly, patients with recurrent or metastatic HNSCC show only a 4.3% response rate to Erlotinib as a monotherapy [62], but have a response rate of 70% for Erlotinib combined with cisplatin or radiotherapy [63, 64]. The present findings of flux between epithelial compartments, each having differing drug responses, may help to explain these observations. EGF has roles in both in initiating EMT and inhibiting epithelial differentiation [65, 66] and the present cytometric analyses of CD44 and ESA expression allow identification of shifts between Epi-CSCs, EMT-CSCs and differentiating cells [31, 32]. Figure 4F is a diagrammatic representation of the altered behaviour of these cell populations observed in response to EGFR blocking. The continuous transit of cells from the self-renewing Epi-CSCs compartment into CD44low/ESAhigh differentiating cells is enhanced by treatment. The increased differentiation of EPI-CSCs, indicated by the increased size of CD44low fractions, decreases the self-renewal of tumour cells and corresponds to a form of “differentiation therapy” [35, 37]. The greater therapeutic sensitivity of the CD44low/ESAhigh cells is likely further to potentiate cell loss in the presence of other therapeutic agents and the combination of EGFR blocking with cisplatin produced such an effect in vitro.
Design of synergistic therapy regimes may enable elimination of CSCs and effective monitoring strategies allowing development of such combinations is imperative [11]. The primary aim of this study was to develop methods capable of assessing differential effects of EGFR blocking agents on recently identified CSC subpopulations. Murine transplantation models for studies of CSCs are typically restricted to examination only of patterns of tumour initiation, growth and histological features. Consequently, such in vivo models have only limited ability to directly image or identify the therapeutic responses of individual cell subtypes. The present results show that in vitro assays allow direct imaging and analysis of individual cell fractions and can therefore accurately detect therapy-relevant shifts in subpopulation compositions. As in vitro studies are adaptable to high throughput assays they can also be used to identify differential changes in CSC subpopulations under a wide range of therapeutic conditions. Such assessment of existing and newly-developed drugs may allow rational development of multiagent combinations able to provide overall control of each of the differing plastic CSC subpopulations that generate tumour growth, spread and therapy resistance.
MATERIALS AND METHODS
Cell lines, growth conditions and exposure to drugs
The CA1 and Luc4 cell lines, previously isolated from oral squamous cell carcinoma were cultivated as previously described [31, 60] in an epithetial growth medium containing EGF (10 ng/ml) and 10% FBS with 5% CO2 at 37°. Cells were plated in triplicate into 96-well plates at 1 × 103 cells per well, cultured overnight and then exposed to Cetuximab (Erbitux, Merck KGaA, Darmstadt, Germany) or Erlotinib (Erlotinib HCL. OSI-744 Cat No. S1023, Selleckchem). Cells were fixed in 4% paraformaldehyde for 15 min, washed in PBS, permeabilized with 0.1% Triton-X for 20 min, and then incubated with Cell Mask Deep Red at a 1:50,000 dilution (Life Technologies) and 1 μg/ml DAPI (Sigma) before image acquisition and analysis. To separately assess drug effects on Epi-EMT and EMT-CSC populations, cells were sorted on the basis of CD44 and ESA staining for investigation by the methods described below.
Colony and sphere formation assays
To test their clonogenic abilities, cells were trypsinized after 3 days of treatment and 1 × 102 cells in 2 ml of medium added to each well of 6 well plates. After 14 days of growth, cells were fixed in 4% paraformaldehyde, stained with crystal violet (0.04% in 1% ethanol) and colonies measuring at least 2mm in diameter were counted visually. To assess growth in suspension as tumour spheres, 24 well plates were coated with 12 mg/ml PolyHEMA (2-hydroxyethyl methacrylate, Sigma) in 95% ethanol to prevent attachment, and then seeded 1 × 103 treated cells/well in 0.5 ml medium containing 1% methylcellulose. After two weeks, the number of tumour spheres larger than 300 μM were counted.
Flow cytometry
For fluorescent activated cell sorting (FACS), cells were detached from cultures using trypsin-EDTA at 37° C (Life Technologies). Cells were stained with anti-CD44 antibody (anti-CD44-FITC, clone G44–26, BD Biosciences) and anti-epithelial specific antigen (anti-ESA-APC, clone HEA-125, Miltenyi Biotec) both at 1:100 in PBS for 15 min in the dark. The DAPI nuclear dye (Sigma) was used at 200 ng/ml to exclude dead cells. For each flow cytometry assay, 100,000 cells were plated and 30,000 live cells counted on a Canto Cytometer (BD Biosciences) and analyzed using FACS Diva software (version 6.1.1, BD Biosciences). Using a FACSAria III (Becton Dickenson), cells were also fractionated (Biddle et al., 2011) as (a) CD44high/ESAlow cells (EMT-CSC phenotype), (b) the top 5% of CD44 expressing cells that also showed ESA expression (Epithelial CSC phenotype - EpiCSC), and (c) the 5% of cells with the lowest CD44 expression also expressing ESA (differentiating cells).
Proliferation assays
Total populations of the CA1 and Luc4 cell lines, together with their CD44high/ESAlow, CD44high/ESAhigh, and CD44low fractions were plated in triplicate at 1 × 103 cells per well into 96-well plates. 24 hours later cells were treated with a range of concentrations of Cetuximab or Erlotinib for 1, 2 or 3 days. Cells were then fixed and stained with Cell Mask Deep Red and DAPI and assayed using the GE InCell 1000 imaging system. Based on the results of these assays, for subsequent experiments cells were exposed to Cetuximab at 100 μg/ml and at 500 ng/ml. To assess effects on cell number of EGFR blocking in the presence of Cisplatin, cells were treated with Cetuximab or Erlotinib together with Cisplatin.
After 1, 2 and 3 days of treatment, 2-Deoxy-5-Iodouridine (IdU) was added to the culture medium for 2 hours at a final concentration of 50 μM to label cells in the S phase of the cell cycle. Cells were then washed in PBS before fixing with 4% paraformaldehyde for 15 min, re-washing and permeabilized with blocking solution (0.5% BSA, 0.2% Triton in PBS) for 30 min at room temperature. Cells were rewashed and incubated for 1 hour at RT with DNase (0.5 U/μl), 3 mM MgCl2 and anti-IdU/BrdU antibody (BD Address Cat# 347580) diluted to 100 μl/ml in blocking solution. After washing 3 times in PBS, cells were incubated in PBS containing an anti-mouse-488 secondary antibody (1:500) (Alexafluor, Life Tech), Cell Mask Deep Red and DAPI for 1 hour in the dark at RT.
Apoptosis assays
Cell lines and sub-factions were treated with Cetuximab and Erlotinib before double staining with Annexin V-FITC and DAPI, or staining with anti-ESA-APC (Miltenyi Biotec), anti-CD44-PE (BD Pharmingen), Annexin V-FITC (BD Pharmingen) and DAPI. Samples were examined on a Canto Cytometer (BD Bioscience) and analyzed with the FACS Diva version 6.1.1 (BD Biosciences) software.
RNA extraction, cDNA synthesis and QPCR
RNA was extracted using the RNeasy® micro kit (Qiagen, Netherlands) followed by reverse transcription using the Superscript III first strand synthesis supermix (Invitrogen, USA). RT qPCR was performed in an ABI 7500 real-time PCR system (Applied Biosystems, USA) using Power SYBR® green mix (Applied Biosystems, USA). QPCR cycling conditions were: 95°C for 10 mins, [95° C for 15 seconds, 60° C for 30 seconds, 72° C for 40 seconds] (40 cycles), 95° C for 60 seconds, followed by dissociation curve analysis. Reverse transcribed Human Total Reference RNA (Stratagene) was used to generate a standard curve. Primer sequences were, designed using Primer-BLAST (NCBI).
Protein extraction and western blotting
Cells were grown to 70% confluence before being harvested and treated with 220 μl of pre-heated protein lysis buffer consisting of 1 mM Tris-Hcl, 10% (v/v) ammonium persulphate (APS), 0.5 M sodium orthovanadate (Sigma) at pH 8.4. Cells were removed and heated at 95° C for 2 minutes. Protein was quantified (DC Protein Assay, Bio-Rad) and 50 μl of 5x protein loading dye (Fisher Scientific) was added to the remaining 200 μl of sample. The primary antibodies with a standard western blotting technique were phospho-ERK (9106S, Cell Signaling), phospho-EGFR (04-339, Millipore), PE-EGFR (555997, BD Biosciences) and β-Actin (sc47778, Santa Cruz). Secondary IgG-conjugated horse-radish peroxidase Abs (Dako) were followed by ECL detection (GE Healthcare).
Immunofluorescence
Cells were treated and fixed as above before being permeabilized with 0.1% Triton-X for 20 min, and then incubated with either Vimentin (DAKO 1:500) or E-Cadherin (Abcam 1:100). Wells were then washed 3 times in PBS, before incubated in PBS containing an anti-mouse-488 secondary antibody (Alexafluor, Life Tech 1:500), Cell Mask Deep Red at a 1:50,000 dilution (Life Technologies) and 1 μg/ml DAPI (Sigma).
Labeling of cell transitions and cell motility
To examine directly the transitions occurring between cells of the EMT-CSC and the EPI-CSC compartments, cell lines were transduced with a constitutively expressed GFP plasmid. EMT-GFP and Epithelial Non-GFP cells were then sorted out from their parental populations and combined before replating and exposing to either Cetuximab or Erlotinib. Scratch assays were used to assess enhanced migration associated with a shift to EMT. Cells were plated for 24 hours, scratched, and then washed with PBS before replacing medium with or without the EGFR inhibitors. Cells were then imaged at 0, 16 and 24 hours and the degree of closure of the scratches analysed as outlined below.
Imaging and image analysis
Images were taken on an IN Cell 1000 Analyser (GE Healthcare) automated system. Images were processed and analyzed using GE IN Cell Developer software (Version 1.9.1).
Statistics
All assays were independently repeated a minimum of 3 times and significance of differences calculated using paired t-tests and indicated in the Figures as: P value <0.05 = *, <0.01 = ** or <0.001 = ***. Error bars represent the standard error of the mean (SEM).
Author contributions
Rodrigues MFSS: carried out the literature research, experimental studies, data acquisition and its analysis, manuscript preparation, editing and its review. Gammon L: experimental studies, data acquisition and its analysis, critically reviewed the manuscript. Rahman M: cell culture and transduction. Biddle A: participated in study design and manuscript preparation. Nunes FD: critically reviewed the manuscript. Mackenzie IC: conceived the study, participated in its design and coordination, carried out experimental studies, data acquisition and statistical analysis, manuscript preparation, editing and its review.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
FUNDING
This work was supported by the National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC3Rs, Grant number: NC/L00061X/1), by The São Paulo Research Foundation (FAPESP Grant numbers 2014/12658-3 and 2011/21395-8), and by the Barts and The London Charitable Foundation.
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61:69–90.
2. Haddad RI, Shin DM. Recent advances in head and neck cancer. N Engl J Med. 2008; 359:1143–1154.
3. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67:7–30.
4. Vermorken JB, Specenier P. Optimal treatment for recurrent/metastatic head and neck cancer. Ann Oncol. 2010; 21:vii252–261.
5. Argiris A, Harrington KJ, Tahara M, Schulten J, Chomette P, Ferreira Castro A, Licitra L. Evidence-Based Treatment Options in Recurrent and/or Metastatic Squamous Cell Carcinoma of the Head and Neck. Front Oncol. 2017; 7:72.
6. Cohen RB. Current challenges and clinical investigations of epidermal growth factor receptor (EGFR)- and ErbB family-targeted agents in the treatment of head and neck squamous cell carcinoma (HNSCC). Cancer Treat Rev. 2014; 40:567–577.
7. Ausoni S, Boscolo-Rizzo P, Singh B, Da Mosto MC, Spinato G, Tirelli G, Spinato R, Azzarello G. Targeting cellular and molecular drivers of head and neck squamous cell carcinoma: current options and emerging perspectives. Cancer Metastasis Rev. 2016; 35:413–426.
8. Pulte D, Brenner H. Changes in survival in head and neck cancers in the late 20th and early 21st century: a period analysis. Oncologist. 2010; 15:994–1001.
9. Du Y, Peyser ND, Grandis JR. Integration of molecular targeted therapy with radiation in head and neck cancer. Pharmacol Ther. 2014; 142:88–98.
10. Kundu SK, Nestor M. Targeted therapy in head and neck cancer. Tumour Biol. 2012; 33:707–721.
11. Bhatia S, Monkman J, Toh AKL, Nagaraj SH, Thompson EW. Targeting epithelial-mesenchymal plasticity in cancer: clinical and preclinical advances in therapy and monitoring. Biochem J. 2017; 474:3269–3306.
12. Juergens RA, Bratman SV, Tsao MS, Laurie SA, Sara Kuruvilla M, Razak AR, Hansen AR. Biology and patterns of response to EGFR-inhibition in squamous cell cancers of the lung and head & neck. Cancer Treat Rev. 2017; 54:43–57.
13. Wang SJ, Bourguignon LY. Hyaluronan and the interaction between CD44 and epidermal growth factor receptor in oncogenic signaling and chemotherapy resistance in head and neck cancer. Arch Otolaryngol Head Neck Surg. 2006; 132:771–778.
14. Grandis JR, Sok JC. Signaling through the epidermal growth factor receptor during the development of malignancy. Pharmacol Ther. 2004; 102:37–46.
15. Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004; 59:21–26.
16. Chen LF, Cohen EE, Grandis JR. New strategies in head and neck cancer: understanding resistance to epidermal growth factor receptor inhibitors. Clin Cancer Res. 2010; 16:2489–2495.
17. Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013; 19:1389–1400.
18. Zhang XC, Xu C, Mitchell RM, Zhang B, Zhao D, Li Y, Huang X, Fan W, Wang H, Lerma LA, Upton MP, Hay A, Mendez E, et al. Tumor evolution and intratumor heterogeneity of an oropharyngeal squamous cell carcinoma revealed by whole-genome sequencing. Neoplasia. 2013; 15:1371–1378.
19. Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, Weissman IL, Clarke MF, Ailles LE. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007; 104:973–978.
20. Vermeulen L, de Sousa e Melo F, Richel DJ, Medema JP. The developing cancer stem-cell model: clinical challenges and opportunities. Lancet Oncol. 2012; 13:e83–89.
21. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414:105–111.
22. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009; 458:780–783.
23. Harper LJ, Piper K, Common J, Fortune F, Mackenzie IC. Stem cell patterns in cell lines derived from head and neck squamous cell carcinoma. J Oral Pathol Med. 2007; 36:594–603.
24. Locke M, Heywood M, Fawell S, Mackenzie IC. Retention of intrinsic stem cell hierarchies in carcinoma-derived cell lines. Cancer Res. 2005; 65:8944–8950.
25. Major AG, Pitty LP, Farah CS. Cancer stem cell markers in head and neck squamous cell carcinoma. Stem Cells Int. 2013; 2013:319489.
26. Chen C, Zimmermann M, Tinhofer I, Kaufmann AM, Albers AE. Epithelial-to-mesenchymal transition and cancer stem(-like) cells in head and neck squamous cell carcinoma. Cancer Lett. 2013; 338:47–56.
27. Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003; 15:740–746.
28. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014; 15:178–196.
29. Sato R, Semba T, Saya H, Arima Y. Concise Review: Stem Cells and Epithelial-Mesenchymal Transition in Cancer: Biological Implications and Therapeutic Targets. Stem Cells. 2016; 34:1997–2007.
30. Brabletz T. EMT, MET in metastasis: where are the cancer stem cells? Cancer Cell. 2012; 22:699–701.
31. Biddle A, Liang X, Gammon L, Fazil B, Harper LJ, Emich H, Costea DE, Mackenzie IC. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res. 2011; 71:5317–5326.
32. Gemenetzidis E, Gammon L, Biddle A, Emich H, Mackenzie IC. Invasive oral cancer stem cells display resistance to ionising radiation. Oncotarget. 2015; 6:43964–77. https://doi.org/10.18632/oncotarget.6268.
33. Johansson AC, La Fleur L, Melissaridou S, Roberg K. The relationship between EMT, CD44high /EGFRlow phenotype, and treatment response in head and neck cancer cell lines. J Oral Pathol Med. 2016; 45:640–646.
34. La Fleur L, Johansson AC, Roberg K. A CD44high/EGFRlow subpopulation within head and neck cancer cell lines shows an epithelial-mesenchymal transition phenotype and resistance to treatment. PLoS One. 2012; 7:e44071.
35. Sell S. Cancer stem cells and differentiation therapy. Tumour Biol. 2006; 27:59–70.
36. Pham PV, Phan NL, Nguyen NT, Truong NH, Duong TT, Le DV, Truong KD, Phan NK. Differentiation of breast cancer stem cells by knockdown of CD44: promising differentiation therapy. J Transl Med. 2011; 9:209.
37. Pattabiraman DR, Weinberg RA. Targeting the Epithelial-to-Mesenchymal Transition: The Case for Differentiation-Based Therapy. Cold Spring Harb Symp Quant Biol. 2016; 81:11–19.
38. Sacco AG, Worden FP. Molecularly targeted therapy for the treatment of head and neck cancer: a review of the ErbB family inhibitors. Onco Targets Ther. 2016; 9:1927–1943.
39. Echarri MJ, Lopez-Martin A, Hitt R. Targeted Therapy in Locally Advanced and Recurrent/Metastatic Head and Neck Squamous Cell Carcinoma (LA-R/M HNSCC). Cancers (Basel). 2016; 8.
40. Boesch M, Sopper S, Zeimet AG, Reimer D, Gastl G, Ludewig B, Wolf D. Heterogeneity of Cancer Stem Cells: Rationale for Targeting the Stem Cell Niche. Biochim Biophys Acta. 2016; 1866:276–289.
41. Ranji P, Salmani Kesejini T, Saeedikhoo S, Alizadeh AM. Targeting cancer stem cell-specific markers and/or associated signaling pathways for overcoming cancer drug resistance. Tumour Biol. 2016; 37:13059–13075.
42. Naik PP, Das DN, Panda PK, Mukhopadhyay S, Sinha N, Praharaj PP, Agarwal R, Bhutia SK. Implications of cancer stem cells in developing therapeutic resistance in oral cancer. Oral Oncol. 2016; 62:122–135.
43. Bourguignon LY. Matrix hyaluronan-activated CD44 signaling promotes keratinocyte activities and improves abnormal epidermal functions. Am J Pathol. 2014; 184:1912–1919.
44. Kidwai F, Costea DE, Hutchison I, Mackenzie I. The effects of CD44 down-regulation on stem cell properties of head and neck cancer cell lines. J Oral Pathol Med. 2013; 42:682–690.
45. Wang SJ, Bourguignon LY. Role of hyaluronan-mediated CD44 signaling in head and neck squamous cell carcinoma progression and chemoresistance. Am J Pathol. 2011; 178:956–963.
46. Perez A, Neskey DM, Wen J, Pereira L, Reategui EP, Goodwin WJ, Carraway KL, Franzmann EJ. CD44 interacts with EGFR and promotes head and neck squamous cell carcinoma initiation and progression. Oral Oncol. 2013; 49:306–313.
47. Abhold EL, Kiang A, Rahimy E, Kuo SZ, Wang-Rodriguez J, Lopez JP, Blair KJ, Yu MA, Haas M, Brumund KT, Altuna X, Patel A, Weisman RA, et al. EGFR kinase promotes acquisition of stem cell-like properties: a potential therapeutic target in head and neck squamous cell carcinoma stem cells. PLoS One. 2012; 7:e32459.
48. Biddle A, Gammon L, Liang X, Costea DE, Mackenzie IC. Phenotypic Plasticity Determines Cancer Stem Cell Therapeutic Resistance in Oral Squamous Cell Carcinoma. EBioMedicine. 2016; 4:138–145.
49. Jolly MK, Tripathi SC, Jia D, Mooney SM, Celiktas M, Hanash SM, Mani SA, Pienta KJ, Ben-Jacob E, Levine H. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget. 2016; 7:27067–84. https://doi.org/10.18632/oncotarget.8166.
50. Jensen DH, Reibel J, Mackenzie IC, Dabelsteen E. Single cell migration in oral squamous cell carcinoma - possible evidence of epithelial-mesenchymal transition in vivo. J Oral Pathol Med. 2015; 44:674–679.
51. Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol D, Peyrade F, Benasso M, Vynnychenko I, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008; 359:1116–1127.
52. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, Hong S, Adams A, D’Angelo R, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports. 2014; 2:78–91.
53. Lo UG, Lee CF, Lee MS, Hsieh JT. The Role and Mechanism of Epithelial-to-Mesenchymal Transition in Prostate Cancer Progression. Int J Mol Sci. 2017; 18.
54. Basu D, Montone KT, Wang LP, Gimotty PA, Hammond R, Diehl JA, Rustgi AK, Lee JT, Rasanen K, Weinstein GS, Herlyn M. Detecting and targeting mesenchymal-like subpopulations within squamous cell carcinomas. Cell Cycle. 2011; 10:2008–2016.
55. Seino S, Shigeishi H, Hashikata M, Higashikawa K, Tobiume K, Uetsuki R, Ishida Y, Sasaki K, Naruse T, Rahman MZ, Ono S, Simasue H, Ohta K, et al. CD44(high) /ALDH1(high) head and neck squamous cell carcinoma cells exhibit mesenchymal characteristics and GSK3beta-dependent cancer stem cell properties. J Oral Pathol Med. 2016; 45:180–188.
56. Oliveras-Ferraros C, Vazquez-Martin A, Queralt B, Adrados M, Ortiz R, Cufi S, Hernandez-Yague X, Guardeno R, Baez L, Martin-Castillo B, Perez-Martinez MC, Lopez-Bonet E, De Llorens R, et al. Interferon/STAT1 and neuregulin signaling pathways are exploratory biomarkers of cetuximab (Erbitux(R)) efficacy in KRAS wild-type squamous carcinomas: a pathway-based analysis of whole human-genome microarray data from cetuximab-adapted tumor cell-line models. Int J Oncol. 2011; 39:1455–1479.
57. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704–715.
58. Al-Hajj M. Cancer stem cells and oncology therapeutics. Curr Opin Oncol. 2007; 19:61–64.
59. Vig N, Mackenzie IC, Biddle A. Phenotypic plasticity and epithelial-to-mesenchymal transition in the behaviour and therapeutic response of oral squamous cell carcinoma. J Oral Pathol Med. 2015; 44:649–655.
60. Gammon L, Biddle A, Heywood HK, Johannessen AC, Mackenzie IC. Sub-sets of cancer stem cells differ intrinsically in their patterns of oxygen metabolism. PLoS One. 2013; 8:e62493.
61. Ang KK, Zhang Q, Rosenthal DI, Nguyen-Tan PF, Sherman EJ, Weber RS, Galvin JM, Bonner JA, Harris J, El-Naggar AK, Gillison ML, Jordan RC, Konski AA, et al. Randomized phase III trial of concurrent accelerated radiation plus cisplatin with or without cetuximab for stage III to IV head and neck carcinoma: RTOG 0522. J Clin Oncol. 2014; 32:2940–2950.
62. Soulieres D, Senzer NN, Vokes EE, Hidalgo M, Agarwala SS, Siu LL. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J Clin Oncol. 2004; 22:77–85.
63. Siu LL, Soulieres D, Chen EX, Pond GR, Chin SF, Francis P, Harvey L, Klein M, Zhang W, Dancey J, Eisenhauer EA, Winquist E, Princess Margaret Hospital Phase IIC and National Cancer Institute of Canada Clinical Trials Group S. Phase I/II trial of erlotinib and cisplatin in patients with recurrent or metastatic squamous cell carcinoma of the head and neck: a Princess Margaret Hospital phase II consortium and National Cancer Institute of Canada Clinical Trials Group Study. J Clin Oncol. 2007; 25:2178–2183.
64. Herchenhorn D, Dias FL, Viegas CM, Federico MH, Araujo CM, Small I, Bezerra M, Fontao K, Knust RE, Ferreira CG, Martins RG. Phase I/II study of erlotinib combined with cisplatin and radiotherapy in patients with locally advanced squamous cell carcinoma of the head and neck. Int J Radiat Oncol Biol Phys. 2010; 78:696–702.
65. Zuo J, Zhu W, Li M, Li X, Yi H, Zeng G, Wan XX, He QY, Li JH, Qu JQ, Chen Y, Xiao ZQ. Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing EMT-like phenotype change and MMP-9-mediated degradation of E-cadherin. J Cell Biochem. 2011; 11:2508–17.
66. Smith BN, Bhowmick NA. Role of EMT in Metastasis and Therapy Resistance. J Clin Med. 2016; 5.