
INTRODUCTION
Amiodarone is an anti-arrhythmic drug that is widely used to treat the most prevalent type of arrhythmia and atrial fibrillation [1–4]. It exerts its phamacologcal function through inhibition of diverse ion channels, including sodium, potassium, and calcium channels. In addition to antiarrhythmic activity, it has also been reported to possess anti-inflammatory and anti-oxidative properties [5, 6]. Amiodarone alters the function of diverse membrane proteins at therapeutic concentration, resulting in complex therapeutic and toxicity profiles [7, 8]. A treatment cocktail containing amiodarone reveals the promise for the treatment of prostate carcinomas and un-resectable hepatocellular carcinomas [9, 10]. However, the detailed mechanism by which amiodarone-induced cancer cell death remains unclear.
Amiodarone has been identified to be involved in the promotion of long-lived protein degradation in three autophagy-induced screening assays [11–13]. In addition, the degradation of autophagy-mediated micro RNA (miR)-224 is confirmed by the autophagy inducer amiodarone [14]. The overexpression of miR-224 promotes cell proliferation, anti-apoptosis, migration, and invasion via directly targeting the TNFα-induced protein 1 (TNFAIP1), SMAD4, caspase 3, and p21 in non-small cell lung cancer (NSCLC), colorectal cancer (CRC), and hepatocellular and prostate cancers [14–19]. Hence, amiodarone has the potential to be developed as an anti-tumor drug via the autophagy-mediated miR-224 degradation or other pathways.
The importance of mRNA splicing is highlighted for tissue homeostasis and disease progression. Alternative splicing might affect all areas of tumor biology, including metabolism, cell cycle control, apoptosis, senescence, and epithelial-mesenchymal transition [20–23]. Upgraded studies have suggested that the human serine and arginine rich splicing factor 3 (SRSF3) gene generates two alternative spliced transcripts: 1. a major mRNA isoform (SRSF3-FL) encoding functional full-length protein; and 2. a premature termination codon (PTC)-containing isoform (SRSF3-PTC), which could be degraded through nonsense-mediated mRNA decay (NMD), i.e. a surveillance mechanism that decomposes PTC-containing mRNAs [24]. Some chemicals, such as caffeine, digoxin, and theophylline, facilitate the translation of a truncated SRSF3 (SRSF3-TC) protein transcribed from SRSF3-PTC mRNA [25–27]. This SRSF3-TC protein is different from full-length SRSF3, which is involved in stressful conditions, such as senescence, hepatocyte differentiation, and metabolic functions [22, 23, 28]. Knockout studies have indicated that SRSF3 is essential for mouse development, hepatocyte differentiation, and metabolic function, as well as tumor cell proliferation and maintenance [23, 28–30]. Cardiac glycosides may inhibit the NMD activity by the elevation of intracellular calcium levels mediated through the binding and inhibiting the sodium-potassium ATPase on the plasma membrane [31].
MiRNAs, a group of small non-coding RNAs, bind to their respective mRNA targets and mediate gene silencing to regulate a range of developmental and physiological processes via the RNA interference mechanism [32, 33]. The dysregulation of miRNAs has been associated with cancers via the role of oncogene or tumor suppressor, depending on the cellular context and the genes targeted. Recently, miRNAs have emerged as promising therapeutic targets mediated through their extraordinary regulatory potential to regulate entire signaling networks within the cells.
Alternative splicing and the RNA interference mechanism have the plasticity to remodel the proteome and, subsequently, to subvert the process to produce proteins for cancer cells to suit the needs of growing and spreading tumors. Recent studies, mechanistically, demonstrated that SLU7 modulates the splicing and expression of SRSF3 and HNF4α genes, which are essential for the preservation of the hepatocyte identity [34]; amiodarone induces the autophagy-preferential degradation of miR-224 in hepatocellular carcinoma tumorigenesis [16]; increased miR-224, directly targeting the TNFAIP1 and SMAD4 expression, functions as a potent oncogenic miRNA to promote cell migration, invasion, and proliferation in NSCLC [18, 19]. We further examined the potential working mechanism of amiodarone via splicing factors and miRs for its anti-tumorigenicity. Our current work and the literature may provide a repurposing function for amiodarone in clinical applications.
RESULTS
Amiodarone downregulated SLU7 and SRSF3 splicing factors in HeLa cells
Recent studies have suggested that amiodarone might have similar effects as caffeine or digoxin on the SRSF3-p53 pathway for senescence functions [7, 8, 22, 23]. Hence, we examined the effects of amiodarone on the alternative splicing of SRSF3 and p53. Our data demonstrated that SRSF3-PTC, the alternative-spliced SFSF3, was observed at the high dose (30 μM) of amiodarone treatment, whereas no alternative splicing p53β form was detected in HeLa cells (Figure 1A). A precious study suggests that Slu7 is responsible for SRSF3 normal transcription [34]. We consistently observed the downregulation of Slu7 mRNA expression by amiodarone in a dose-dependent manner. For SLU7 is also known as a negative regulator of ATF3 mRNA expression, we observed amiodarone dose-dependently enhanced ATF3 mRNA expression in HeLa cells. In addition, cyclin D1 mRNA, as well as Slu7 mRNA, was reduced and p21, COX-2 mRNAs, as well as ATF3 mRNA were induced in an amiodarone dose-dependent manner.
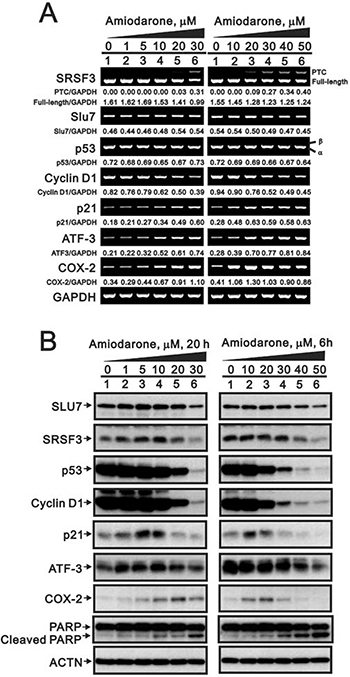
Figure 1: The effects of amiodarone on target gene and protein expression in HeLa cells. HeLa cells were treated with indicated amount of amiodarone for indicated time. The cells were collected and subjected to (A) RT-PCR analysis of SRSF3, Slu7, p53, cyclin D1, p21, ATF3, COX-2, and GAPDH (loading control) mRNA expression; (B) immunoblot analysis for the detection of SLU7, SRSF3, p53, cyclin D1, p21, ATF3, COX-2, PARP, and ACTN (loading control) protein expression. PCR bands (A) were quantified through pixel density scanning and evaluated by ImageJ software, version 1.44a (http://imagej.nih.gov/ij/). The results are representative of two independent experiments.
In the Western blotting analysis, we observed that the decreasing pattern of SLU7, SRSF3, p53, and cyclin D1 proteins followed by amiodarone treatment in a dose-dependent manner in HeLa cells (Figure 1B). COX-2 and p21 proteins were induced at the narrow dosage window. In contrast to mRNA expression pattern, ATF3 proteins were reduced by amiodarone in a dose-dependent manner in HeLa cells. We also observed amiodarone induced the protein levels of cleaved-PARP in a dose-dependent manner, suggesting that amiodarone might induce apoptosis in HeLa cells.
Amiodarone induced cell death and suppressed cell survival in HeLa cells
To determine the effects of amiodarone on cell cycle profile, we performed cell cycle analysis using flow cytometry. Amiodarone treatment for 20 hours significantly increased populations at sub-G1 and G1 phase (G1 arrest) accompanied by the decreasing populations at S phase and G2/M phase (Figure 2A). However, there was no apparent effect, except for the sub-G1 phase, in the 6 h amiodarone treatment (Figure 2B). We further analyzed the amiodarone-induced cell death, including early- and late-apoptosis and necrosis using the Annexin V apoptosis kit. We observed the maximal percentages of late-apoptosis and necrosis at 20 μM amiodarone. The maximal percentage of early apoptosis was found at 50 μM amiodarone (Figure 3A). We examined the effect of amiodarone on the mitochondrial membrane potential using the JC-1 staining assay (Figure 3B). Our data suggest that amiodarone strongly disrupted the mitochondrial membrane potential via the analysis of FL1-H (monomers) and FL2-H (aggregates), which shows a trend to induce apoptosis in HeLa cells (Figure 3C and Table 1). To determine the effects of amiodarone on the cell survival and anchorage-independent ability, we evaluated the effects of amiodarone on HeLa cells using colony formation analysis and soft agar assay, respectively. We observed that amiodarone inhibited colony formation (Figure 4A) and decreased the anchorage-independent growth of HeLa cells (Figure 4B).
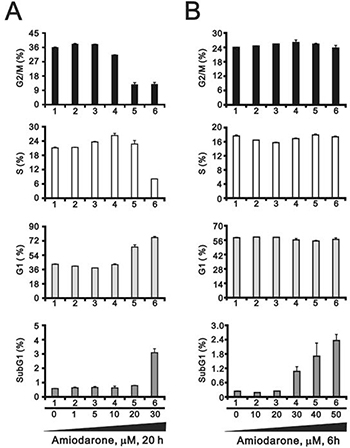
Figure 2: Amiodarone caused cellular apoptosis. HeLa cells were treated with (A) lower concentration of amiodarone for 20 h and (B) higher concentration of amiodarone for 6 h. The cells were stained with PI, and the DNA content was determined by FACS analysis. The results are representative of three independent experiments.
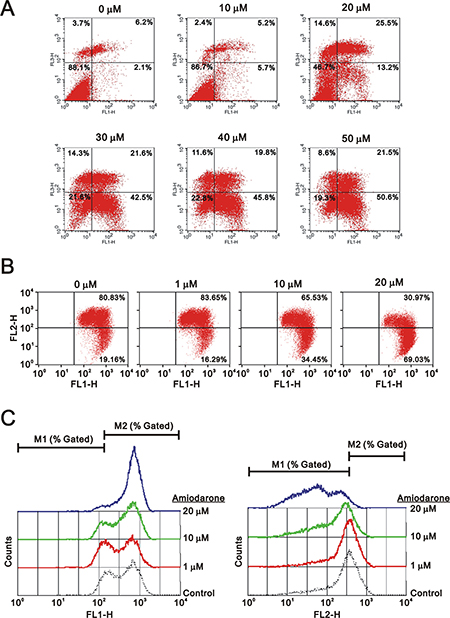
Figure 3: Amiodarone triggered intrinsic apoptosis. HeLa cells were treated indicated amount amiodarone for 20 h. Cells were subject to the flow cytometry analysis using (A) the PE Annexin V Apoptosis Detection Kit (BD Biosciences) (B) the JC-1 staining kit. The results (A and B) are representative of three independent experiments. (C) The data from (B) were further measured the values of FL1-H and FL2-H.
Table 1: The analysis of FL1 and FL2 shift trends from the JC-1 staining assay
amiodarone |
FL1-H |
FL2-H |
||
|---|---|---|---|---|
M1 (% gated) |
M2 (% gated) |
M1 (% gated) |
M2 (% gated) |
|
20 |
5.33 |
94.73 |
96.05 |
4.09 |
10 |
15.68 |
84.52 |
81.02 |
19.39 |
1 |
23.04 |
77.33 |
62.10 |
38.55 |
0 |
14.80 |
85.57 |
61.05 |
39.40 |
right shift |
left shift |
|||
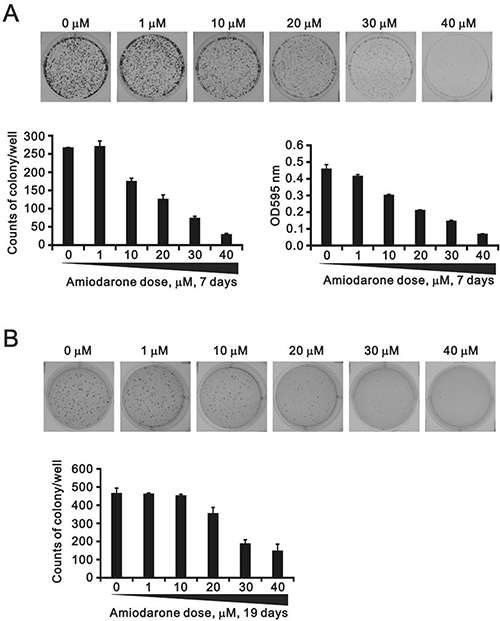
Figure 4: The effects of amiodarone on the cell colony and anchorage-independent growth. (A) HeLa cells were treated indicated amount amiodarone for 7 days and cells were subject to the colony formation. After 7 days incubation, colonies were fixed with methanol and stained with 0.005% crystal violet solution for 1 h and photographed. Colonies were counted and quantified using ImageJ software. (B) HeLa cells were treated indicated amount amiodarone for 19 days and cells were subject to the colony formation in soft agar. After 19 days incubation, colonies were stained with 0.005% Crystal Violet solution for 30~60 min and photographed. Colonies were counted and quantified using ImageJ software. All assays were done in triplicate.
Amiodarone reduced SRSF3 gene and protein expression in HeLa and glioma cells
As previous study and Figure 1 data suggest that SLU7 is responsible for SRSF3 normal transcription and the downregulation of Slu7 mRNA expression by amiodarone in a dose-dependent manner [34], we further examined the relationship between Slu7 and SRSF3 at the translation and transcription levels by the treatment of respective inhibitor, cycloheximide (CHX) and actinomycin D (Act D). CHX increased the expression of the Slu7 mRNA and subsequently elevated the levels of truncated and full-length SRSF3 mRNAs and p53β mRNA in HeLa cells (Figure 5A, compare lanes 7–12 with lanes 1–6). From the aspect of protein half-life, our data were consistent with previous literature in which indicate p53 is a labile protein, whereas SLU7 and SRSF3 are stable in the presence of CHX (Figure 5B, compare lane 7 with lane 1). The expression trend of SLU7 protein was consistent with its mRNA expression in HeLa cells. The protein levels of full-length SRSF3 were positively correlated with the expression of SLU7 protein in HeLa cells (Figure 5B). Regarding the effects of amiodarone at transcrioptional levels, we observed the instability of Slu7 mRNA in Act D-treated HeLa cells (Figure 5A, compare lane 13 with lane 1), and amiodarone dose-dependantly enhanced the stability of Slu7 mRNA. We also found no truncated SRSF3 mRNA was transcribed by actinomycin D implying that the induction of SRSF3-PTC by amiodarone was derived from the newly synthesized mRNA(Figure 5A, lane 13–18). The effects of amiodarone on SRSF3-PTC induction were enhanced with the addition of calcium chloride (Figure 5A, compare lanes 19–24 with lanes 1–6). The repressive effects of amiodarone on the SLU7 and SRSF3 proteins were potentiated with the addition of calcium chloride treatment (Figure 5B, compare lanes 19–24 with lanes 1–6).
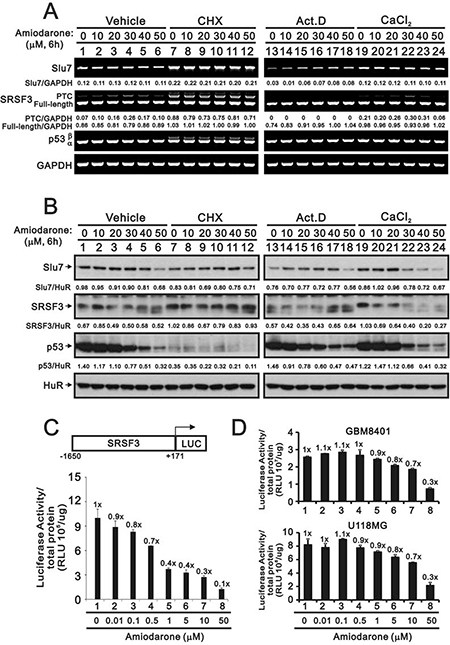
Figure 5: The regulatory mechanism of amiodarone on the SRSF3 expression. HeLa cells were treated indicated amount amiodarone with pre-treated 2 h of vehicle, CHX (50 mg/ml), Act D (1 μM), and calcium chloride (1 mM) for 6 h. Cells were subject to the (A) Western blotting analysis and (B) RT-PCR analysis. The numerical data below each band is to indicate the ratio of specific mRNA (A) or protein (B) with control mRNA (GAPDH) or control protein (HuR) using the ImageJ software. The results (A and B) are representative of three independent experiments. (C and D) HeLa, GBM8401, U118MG cells were transiently transfected with 0.5 mg pGL3.SRSF3 (–1650/+171)-LUC and cells were treated with indicated amount of amiodarone for 20 h. Cells were harvested for luciferase reporter assay with the Promega Luciferase Assay Kit. The numbers (C and D) above the columns indicate the luciferase activity relative to an index of 1 for the reporter alone with vehicle.
We further examined the effect of amiodarone on the SRSF3 promoter activity using the SRSF3(–1650/+171)-LUC reporter system. We observed the suppressive effect on the SRSF3 promoter activity by amiodarone in a dose-dependent manner (Figure 5C). The suppressive effects of amiodarone on the SRSF3 promoter activity were also verified in two glioma cell lines, GBM8401 and U118MG (Figure 5D).
Amiodarone enhanced the efficacy of caffeine and digoxin on cytotoxicity in HeLa cells
Since our previous studies showed that caffeine and digoxin caused cell death via the SRSF3-p53 pathway [26, 27], we further examined the combined effects of amiodarone with caffeine and digoxin in HeLa cells. We found that amiodarone enhanced the alternative splicing effects on SRSF3 (FL and PTC) and p53 (α and β) by caffeine and digoxin, and in turn increased the mRNA levels of p53 target genes, such as cyclin D1, p21, and ATF3 (Figure 6A). In the Western blotting analysis, amiodarone enhanced the degradation of p53 α and cyclin D1 proteins in HeLa cells (Figure 6B). Caffeine and digoxin enhanced the LC3B II levels (autophagy marker) and the cleaved PARP proteins (apoptosis marker) in the context of amiodarone treatment. In addition, amiodarone mitigated cellular DNA damage by monitoring the abundance of γ-H2AX in HeLa cells, whereas caffeine and digoxin reversed the dose-dependent alleviative effect of amiodarone on the DNA damage. Remarkably, Caffeine and digoxin synergistically enhanced the amiodarone-induced sub-G1 populations in cell cycle analysis using flow cytometry. We also found amiodarone alone or cotreated with either caffeine or digoxin, restrained the cell cycle from entering into S phase (Figure 7).
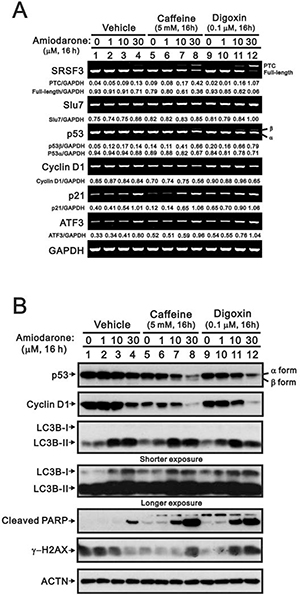
Figure 6: Amiodarone synergistically enhanced caffeine- or digoxin-induced SRSF3 alternative splicing and p53-independent apoptosis in HeLa cells. HeLa cells were treated with 5 mM caffeine or 0.1 μM digoxin and along with indicated amount of amiodarone for 16 h. The cells were collected and subjected to (A) RT-PCR analysis of SRSF3, Slu7, p53, cyclin D1, p21, ATF3, COX-2, and GAPDH (loading control) mRNA expression; (B) immunoblot analysis for the detection of p53, cyclin D1, LC3B, PARP, γ−H2AX, and ACTN (loading control) protein expression. PCR bands (A) were quantified through pixel density scanning and evaluated by ImageJ software, version 1.44a (http://imagej.nih.gov/ij/). The results are representative of two independent experiments.
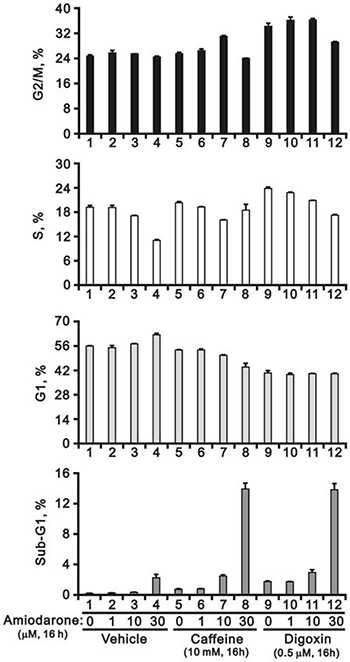
Figure 7: Amiodarone augmented the effects on the increase of sub-G1 population in HeLa cells by caffeine or digoxin. HeLa cells were treated with 5 mM caffeine or 0.1 μM digoxin and along with indicated amount of amiodarone for 16 h. The cells were stained with PI, and the DNA content was determined by FACS analysis. The results are representative of three independent experiments.
Amiodarone induced cellular reactive oxygen species (ROS) and can be synergistically enhanced by caffeine or digoxin
One of the underlying mechanisms by which amiodarone-induced pulmonary toxicity is the generation of ROS [35]. We further examined the status of ROS in the amiodarone treated HeLa cells. Our data demonstrated that the ROS in HeLa cells reached maximal amounts at 30 μM amiodarone treatment (Figure 8). Intriguingly, we found that caffeine and digoxin reduced the amiodarone concentration from 30 μM to 10 μM for the maximal amounts of ROS production (Figure 9). It is noteworthy that digoxin, not caffeine, enhanced the ROS generation in HeLa cells without amiodarone treatment.
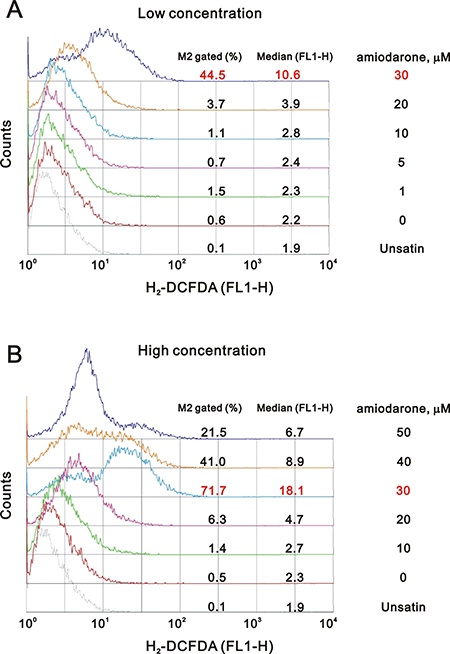
Figure 8: Amiodarone induced the ROS generation in HeLa cell. HeLa cells were treated with (A) lower concentration of amiodarone for 20 h and (B) higher concentration of amiodarone for 16 h. After incubated, we stained live cells with 10 μM DCFH-DA for 30~60 min at 37° C, harvested cells were then subjected to FACS analysis. The results are representative of three independent experiments.
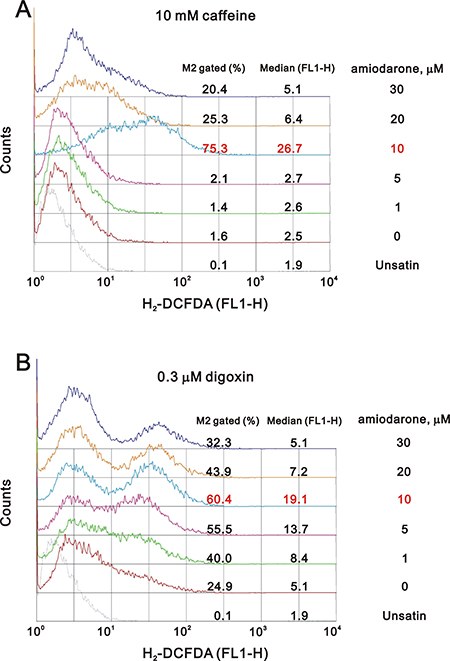
Figure 9: Caffeine or digoxin increased the effects of amiodarone for the elevation of ROS generation in HeLa cell. HeLa cells were treated with (A) 10 mM caffeine or (B) 0.3 μM digoxin and lower concentration of amiodarone for 16 h. After incubation, we stained live cells with 10 μM DCFH-DA for 30~60 min at 37° C, harvested cells were then subjected to FACS analysis. The results are representative of three independent experiments.
Amiodarone reduced miR-224 and other miR expressions in HeLa cells
A recent study suggested that amiodarone-induced autophagy reduced the miR-224 expression at the post-transcriptional level [16], and our data supported that amiodarone did induce autophagy in HeLa cells (Figure 6B). Hence, we analyzed several miRs responsible for p53 proteins, including miR-34a, 145, 125b, 200c, and 504. In addition to miR-224, amiodarone also reduced the expression levels of miR-125b, 200c, and 504 in HeLa cells (Figure 10A–10F). Caffeine exhibited similar effects of abovementioned miRs downregulation with amiodarone (Figure 10C–10F), whereas digoxin showed opposite effects on enhanced miR-34a, 145, and 504 expressions in HeLa cells (Figure 10A, 10B, and 10E).
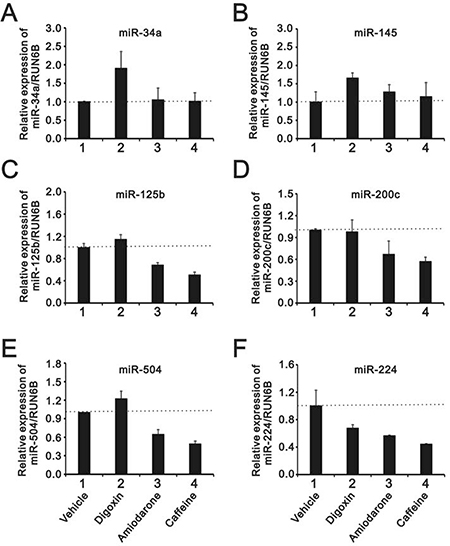
Figure 10: The comparison of the effects of caffeine, amiodarone, and digoxin on the miRs regulation in HeLa cell. HeLa cells were treated with 10 mM caffeine, 0.3 μM digoxin, and 30 mM amiodarone for 16 h. The cells were collected and subjected to the miR analysis for (A) miR-34a; (B) miR-145; (C) miR-125b; (D) miR-200c; (E) miR-504; and (F) miR-224 expression. The results are representative of three independent experiments.
To further examined the effects of miR-224 reduction followed by amiodarone treatment, we detected miR-224 expression in response to various amiodarone concentration. We found that amiodarone repressed the miR-224 expression in a dose-dependent manner for 20 h (Figure 11A). However, we examined three miR-224 target genes: Smad4, caspase 3, and TNFAIP1; and found no apparent increasing effect accompanied by the amiodarone-reduced miR-224 (Figure 11B). We observed the COX-2 gene and protein were increased by amiodarone in a dose-dependent manner. (Figure 11B and 11C). These results suggest that COX-2 gene might be the target gene of miR-224 in response to amiodarone treatment in HeLa cells.
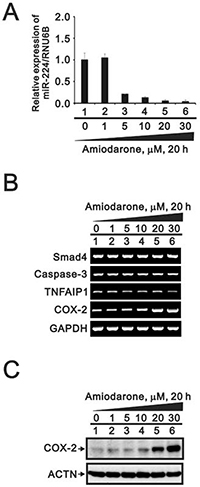
Figure 11: The effects of amiodarone on miR-224 and its target genes in HeLa cell. HeLa cells were treated with lower concentration of amiodarone for 20 h. The cells were collected and subjected to (A) the miR analysis for miR-224 expression; (B) the RT-PCR analysis for caspase3, TNFAIP1, COX-2, and GAPDH (loading control); and (C) the Western blotting analysis using antibodies against COX-2 and ACTN (loading control). The results are representative of three independent experiments.
DISCUSSION
The NMD pathway is well known as a translation-coupled quality control system [36–38]. Human SRSF3 gene generates two alternative spliced transcripts, encoding full-length protein SRSF3-FL and a truncated SRSF3-TC isoform that is degraded through the NMD pathway [28]. Our current findings of the downregulation of SRSF3-FL expression and the upregulation of SRSF3-PTC expression were found by the treatment of caffeine, digoxin, and theophylline in HeLa and other cells [25–27]. Recent reporting system screening for hundreds of clinical drugs from Dr. You’s laboratory have demonstrated that caffeine (positive control in the screening system) and digoxin (the top drug in the listing) may elevate the intracellular cyclic AMP and calcium levels for the inhibitory effects on the NMD pathway [31]. Amiodarone was included in a positive category, but its screening ratio value was around 1 when compared to 30 for digoxin. We observed the SRSF3-PTC expression of at a higher amiodarone concentration, but it failed to alternatively splice p53 from α isoform into β isoform via the downregulation of SRSF3 gene and protein in our current observation window. However, amiodarone acted synergistically with caffeine and digoxin on the alternative splicing of p53 from α isoform into β isoform via the downregulation of SRSF3 gene and protein. Hence, our study revealed that amiodarone, as well as caffeine and digoxin, had common and distinctive working pathways in cells.
Amiodarone has been identified to be an autophagy inducer and autophagy-mediated miR-224 degradation was reconfirmed in this study. Up-regulated miR-224 was identified in several solid tumors including CRC, NSCLC, and hepatocellular carcinoma [14–19]. It subsequently suppressed its targets such as TNFAIP1, p21, caspase 3 and SMAD4. MiR224 might regulate CRC growth by targeting p21 to modulate CRC proliferation and prognosis. In addition, p21 is a well-known p53 target gene [39]. Amiodarone might modulate the expression amount of p21 via the miR-224 and p53 in HeLa cells. The downregulation of miR-224 and p53 by amiodarone has antagonistic effects on the p21 mRNA via defined pathways. However, in some cases, p21 could be regulated and mediated through p53-dependent and/or independent pathway(s). Our previous study demonstrated that the senescence concentration (0.1 μM) of doxorubicin (DXR) induced higher p21 expression than its apoptotic concentration (1 μM), at which the highest p53 abundance was measured in HeLa cells [40]. Correspondingly, we demonstrated that expression of p21 correlates with activation of p53 at low amiodarone concentration but not at high doses. Hence, amiodarone might reduce the miR-224 expression via the autophagy degradation pathway and subsequently to increase the p21 abundance followed by a p53-independent mechanism in our current work. In addition to the expression level in cells, cytoplasm localized p21 blocks the activation of pro-caspase 3 consequently inhibit apoptosis; whereas, the nuclear-localized p21 promotes its degradation and creates favorable conditions for the occurrence of apoptosis in response to DNA damage [41, 42]. Further investigation will be necessary to distinguish the subcellular localization of p21 after various DNA stress conditions.
Our previous study demonstrated that the senescence concentration (0.1 μM) of DXR induced higher percentage of DNA damage than its apoptotic concentration (1 μM) in HeLa cells, since senescence development is mainly controlled by the induction of p21 in cells associated with DNA damage (via the measurement of γ-H2AX) [40]. Amiodarone had the ability to reduce the DNA damage level and induce the autophagy and apoptosis levels, whereas caffeine or digoxin could reverse the amiodarone-reduced DNA damage level and potentiate the amiodarone-induced apoptosis levels. However, the differential levels of DNA damage responsible for cellular apoptosis and/or autophagy by amiodarone remain to be investigated in the future.
Previous studies have demonstrated that increased ROS levels contribute to enhanced tumor cell proliferation and apoptosis suppression, whereas high levels of ROS can also cause apoptosis via the release of pro-apoptotic factors from mitochondria [43, 44]. It is attractive to find targets which are responsible for enhancing mitochondrial ROS production to trigger apoptosis, because this organelle controls the decision for cells to live or die. In this study, we provided supporting data that Amiodarone can also modulate endogenous ROS levels in a dose-dependent manner, and disrupt the mitochondrial membrane potential to induce cellular intrinsic apoptosis. Hence, it may have clinical benefit to use amiodarone with other chemotherapeutic agents from the aspect of the increased ROS produced by triggering DNA damage response pathways or by altering the function of diverse membrane proteins via splicing factors.
An increasing number of studies have indicated that miRs play a critical role in malignant development via the inhibition of the mRNAs stability and/or translation, which can function as tumor suppressors or oncogenes in a tissue specific manner [45, 46]. MiR-224 has been reported to be up-regulated in CRC, hepatocellular, and prostate cancers [14–17]. Nevertheless, the regulation of downstream targets of miR-224 determines its physiological responses, especially affected by different agents A recent study of cervical cancer tissues has shown that up-regulated 195 miRNAs and down-regulated 96 miRs relative to normal cervical tissues [47]. We examined several well-known target genes of miR-224, such as Smad4, caspase 3, and TNFAIP1 and found no change in amiodarone-reduced miR-224 cells in a human papillomavirus (HPV)-infected cervical carcinoma cell line, HeLa cells. COX-2 was the only consistent induction of miR-224 target genes by amiodarone in this study. In addition to further investigate whether COX-2 is a tissue-specific target gene of miR-224, the expression of COX-2 mRNA is also induced by HPV type 16 E5, E6, and E7 oncoproteins and others [48–50]. Despite COX-2 is also a target gene of p53 [51], inversion correlation shown in our results (Figure 1) indicated the induction of COX-2 may be mediated via a p53-independent pathway in our model. The detailed regulatory mechanisms and functional roles of COX-2 in HPV type 18 HeLa cells remain to be investigated in the future.
Summarily, our data revealed that our interested gene expressions are modulated by amiodarone mediated through transcription, post-transcription, and/or translation. Multiple pathways of gene regulation of amiodarone provide a new avenue to define its repurposing functions alone or combined with other clinical drugs.
MATERIALS AND METHODS
Cell culture and chemicals
HeLa, GBM8401, U118MG cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Invitrogen, USA). Amiodarone, DCFH-DA, CHX, Act D, and calcium chloride were purchased from Sigma-Aldrich (A8423, D6883, C1988, A9415, and C1016, MO, USA).
Western blot analysis
Cell lysates were prepared in lysis buffer (100 mM Tris-HCl of pH 8.0, 150 mM NaCl, 0.1% SDS, and 1% Triton X-100) at 4° C. The extracts were separated by SDS-PAGE, transferred onto a polyvinylidene difluoride membrane (Millipore, USA) and detected using antibodies against PARP, LC3 (Cell Signaling, USA), SRSF3, Slu7, p53, p21, cyclin D1, ATF3, COX-2, α-actinin (ACTN), HuR (Santa Cruz Biotechnology, USA), and γ−H2AX (Epitomics, USA).
Reverse transcription-polymerase chain reaction (RT-PCR)
One microgram of total RNA, isolated using the TRIsure reagent (BIOLINE, UK), was subjected to the reverse transcription reaction using MMLV reverse transcriptase for 60 min at 37° C (Epicentre Biotechnologies, USA). The PCR reactions were run on a Veriti Thermal Cycler (Applied Biosystems, USA). Table 2 shows all PCR primer sequences.
Table 2: Primers were used in this study
Gene name |
Primer sequence (5′→3′) |
|---|---|
p53 |
Forward: 5-CTCTGACTGTACCACCATCCACTA-3′ |
Reverse: 5-GAGTTCCAAGGCCTCATTCAGCTC-3′ |
|
SRSF3 |
Forward: 5-ATGCATCGTGATTCCTGTCCATTG-3′ |
Reverse: 5-CTATTTCCTTTCATTTGACCTAGATC-3′ |
|
cyclin D1 |
Forward: 5-ATGGAACACCAGCTCC-3′ |
Reverse: 5-TCAGATGTCCACGTCCCGC-3′ |
|
p21 |
Forward: 5′-CTGAGCCGCGACTGTGATGCG-3′ |
Reverse: 5′-GGTCTGCCGCCGTTTTCGACC-3′ |
|
ATF3 |
Forward: 5′-GAGGATTTTGCTAACCTGAC-3′ |
Reverse: 5′-TAGCTCTGCAATGTTCCTTC-3′ |
|
COX-2 |
Forward: 5′-CTGAGCCGCGACTGTGATGCG-3′ |
Reverse: 5′-GGTCTGCCGCCGTTTTCGACC-3′ |
|
Slu7 |
Forward: 5-GATGGGAAGAGGGATCGGTGG-3′ |
Reverse: 5-CTGGAGGGGCATCCAAATGTTC-3′ |
|
GAPDH |
Forward: 5-CTTCATTGACCTCAACTAC-3′ |
Reverse: 5-GCCATCCACAGTCTTCTG-3′ |
|
Smad4 |
Forward: 5′-GCATCGACAGAGACATACAGC-3′ |
Reverse: 5′-CAACAGTAACAATAGGGCAGC-3′ |
|
TNFAIP1 |
Forward: 5-GGACAAGAAGGACTCCTACCA-3′ |
Reverse: 5-CCTCCAACAAGGAGTTGTCG-3′ |
|
Caspase 3 |
Forward: 5-CTGGAATGACATCTCGGTCTG-3′ |
Reverse: 5-ACCAGGTGCTGTGGAGTATG-3′ |
Fluorescence-activated cell sorting (FACS) analysis
FACS analysis was based on the measurement of DNA content for nuclei labeled with propidium iodide (PI). For cell cycle evaluation, cells were treated using procedures for the proliferation experiments, washed with ice-cold PBS, and incubated with PI solution (0.05% mg/ml in PBS, 0.1% Triton X-100, and 0.01% RNase) for 15 min at room temperature in the dark. Cells were then subjected to FACS and cell cycle analysis was performed using a FACSCalibur flow cytometer (BD Biosciences, USA). For early and late apoptosis analysis, the cells were treated and measured by PE Annexin V Apoptosis Detection Kit (BD Biosciences) according to the manufacturer’s instructions.
Mitochondrial membrane potential measurements
The mitochondrial potential was performed using BDTM MitoScreen Flow Cytometry Mitochondrial Membrane Potential Detection Kit (BD Biosciences), according to the manufacturer’s instructions. In brief, HeLa cells were seeded on a 6 cm culture plate and incubated with the amiodarone dose. After 18 h treatment, cells were trypsinized and collected cell pellets at 1000 rpm, resuspended the cells in PBS and to count cell number, cell counting should not exceed 1 × 106 cells per ml. All the treated cells were the stained with the JC-1 dye for 10~15 min at 37° C in a CO2 incubator. After washing twice with 1x Assay Buffer, the pellets were resuspended in 1x Assay Buffer (0.5 ml) and the fluorescence of the JC-1 dye was measured by flow cytometric analysis by exciting the dye at 488 nm and detecting the JC-1 monomer through its emission at 530 nm (FL1 channel) with aggregates of JC-1 being detected at 580 nm (FL2 channel).
Immortalization and transformation assays
For colony formation at low density to assess immortalization, a total of 3,000 HeLa cells were seeded per well (six-well plate) and treated with amiodarone dose. Subsequently, the culture plates were kept in culture incubator maintained at 37° C and 5% CO2 for 7 days to allow for colony growth. After 7 days incubation, colonies were fixed with methanol and stained with 0.005% crystal violet solution for 1 h and photographed. Colonies were counted and quantified using ImageJ software. For colony formation in soft agar, a total of 3,000 HeLa cells were resuspended in 10% FBS DMEM medium containing 0.35% low melting temperature agarose (SeaPlaque® Agarose; Lonza) and seeded onto six-well plates previously coated with 0.5% low melting temperature agarose. After 19 days incubation, colonies were stained with 0.005% Crystal Violet solution for 30~60 min. Colonies were counted and quantified using ImageJ software.
SRSF3 promoter reporter construction, transfection, and reporter assay
The reporter vector for SRSF3 promoter from –1650 to +171 was constructed into the pGL3-LUC vector, pGL3.SRSF3(–1650/+171)-LUC, via the BamHI/XhoI site of a PCR fragment. HeLa, GBM8401, U118MG cells were transfected into 24-well plates with jetPEI (Polyplus Transfection Inc., New York, NY) according to the manufacturer’s protocol. The total DNA was adjusted to 1.0 μg by the addition of the empty vector. The cells were treated with vehicle or indicated amount of amiodarone for 20 h and were harvested for luciferase reporter assay, which was performed with the Promega Luciferase Assay Kit; the values are expressed numerically as relative light units normalized by total proteins. The luciferase activities are presented as the mean ± SD of three transfected wells and are representative of at least three independent experiments.
Reactive oxygen species (ROS) measurement
Intracellular ROS levels were determined using the fluorescent marker DCFH-DA according to the manufacturer’s instructions. The cells were treated with indicated drugs (amiodarone, caffeine, and/or digoxin) for 16 h and then live cells were stained with 10 μM DCFH-DA for 30~60 min at 37° C. The harvested cells and washed twice with PBS and then were subjected to FACS. The 2′,7′-dichlorofluorescein (DCF) fluorescence intensity analysis was performed using a FACSCalibur flow cytometer and the Cell Quest Pro software (BD Biosciences, CA, USA).
MiR extraction and quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from growing cells using the TRIsure reagent (Bioline Reagents, London, UK) and the miRNA reverse transcription reactions were using miScript II RT kit (Qiagen) according to the manufacturer’s instructions. Expression of miRNA was quantified by qRT-PCR with miScript SYBR Green PCR Kit (Qiagen). Small endogenous nucleolar U6 snRNA was used as control for normalization of miRNA. Target miR primers were purchased from miScript Primer Assay (Qiagen).
Abbreviations
ACTN: α-actinin; CRC: colorectal cancer; DCF: 2′,7′-dichlorofluorescein; DCFH-DA: 2′,7′- dichlorofluorescin diacetate; DXR: doxorubicin; FACS: fluorescence-activated cell sorting; miR: HPV: human papillomavirus; micro RNA; NMD: nonsense-mediated mRNA decay; NSCLC: non-small cell lung cancer; PI: propidium iodide; PTC: premature termination codon; qPCR: quantitative polymerase chain reaction; ROS: reactive oxygen species; RT-PCR: reverse transcription-polymerase chain reaction; SRSF3: serine and arginine rich splicing factor 3; SRSF3-TC: truncated SRSF3; TNFAIP1: TNFα-induced protein 1.
Author contributions
YLC conceived and carried out experiments, analyzed data and wrote the paper. STL carried out experiments and analyzed data. YWW analyzed data and wrote the paper. WSL and SMH conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interests related to this study.
FUNDING
This work was supported by grants from the National Defense Medical Bureau [MAB–105–009 to S-M HUANG and MAB–105–014 and MAB–106–020 to W-S LIN] and Teh-Tzer Study Group for Human Medical Research Foundation [B1061050 to W-S LIN], Taiwan, ROC.
REFERENCES
1. Ellison KE, Stevenson WG, Sweeney MO, Epstein LM, Maisel WH. Management of arrhythmias in heart failure. Congest Heart Fail. 2003; 9:91–99.
2. Singh BN. Current antiarrhythmic drugs: an overview of mechanisms of action and potential clinical utility. J Cardiovasc Electrophysiol. 1999; 10:283–301.
3. Singh BN. Amiodarone as paradigm for developing new drugs for atrial fibrillation. J Cardiovasc Pharmacol. 2008; 52:300–305.
4. Kathofer S, Thomas D, Karle CA. The novel antiarrhythmic drug dronedarone: comparison with amiodarone. Cardiovasc Drug Rev. 2005; 23:217–230.
5. Karavelioglu Y, Karapinar H, Yuksel M, Memic K, Sarak T, Kurt R, Yilmaz A. Neutrophil to lymphocyte ratio is predictor of atrial fibrillation recurrence after cardioversion with amiodarone. Clin Appl Thromb Hemost. 2015; 21:5–9.
6. Cheng SM, Lin WH, Lin CS, Ho LJ, Tsai TN, Wu CH, Lai JH, Yang SP. Modulation of both activator protein-1 and nuclear factor-kappa B signal transduction of human T cells by amiodarone. Exp Biol Med (Maywood). 2015; 240:99–108.
7. Maresova L, Muend S, Zhang YQ, Sychrova H, Rao R. Membrane hyperpolarization drives cation influx and fungicidal activity of amiodarone. J Biol Chem. 2009; 284:2795–2802.
8. Stahl E, Ellis J. Novel allosteric effects of amiodarone at the muscarinic M5 receptor. J Pharmacol Exp Ther. 2010; 334:214–222.
9. Theodossiou TA, Galanou MC, Paleos CM. Novel amiodarone-doxorubicin cocktail liposomes enhance doxorubicin retention and cytotoxicity in DU145 human prostate carcinoma cells. J Med Chem. 2008; 51:6067–6074.
10. Favoulet P, Cercueil JP, Faure P, Osmak L, Isambert N, Beltramo JL, Cognet F, Krause D, Bedenne L, Chauffert B. Increased cytotoxicity and stability of Lipiodol-pirarubicin emulsion compared to classical doxorubicin-Lipiodol: potential advantage for chemoembolization of unresectable hepatocellular carcinoma. Anticancer Drugs. 2001; 12:801–806.
11. Mahavadi P, Knudsen L, Venkatesan S, Henneke I, Hegermann J, Wrede C, Ochs M, Ahuja S, Chillappagari S, Ruppert C, Seeger W, Korfei M, Guenther A. Regulation of macroautophagy in amiodarone-induced pulmonary fibrosis. J Pathol Clin Res. 2015; 1:252–263.
12. Morissette G, Ammoury A, Rusu D, Marguery MC, Lodge R, Poubelle PE, Marceau F. Intracellular sequestration of amiodarone: role of vacuolar ATPase and macroautophagic transition of the resulting vacuolar cytopathology. Br J Pharmacol. 2009; 157:1531–1540.
13. Balgi AD, Fonseca BD, Donohue E, Tsang TC, Lajoie P, Proud CG, Nabi IR, Roberge M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One. 2009; 4:e7124.
14. Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ, Tsai TF, Lin YJ, Wu CT, Liu HS. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology. 2014; 59:505–517.
15. Wan Y, Zeng ZC, Xi M, Wan S, Hua W, Liu YL, Zhou YL, Luo HW, Jiang FN, Zhong WD. Dysregulated microRNA-224/apelin axis associated with aggressive progression and poor prognosis in patients with prostate cancer. Hum Pathol. 2015; 46:295–303.
16. Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ, Tsai TF, Lin YJ, Wu CT, Liu HS. Autophagy-preferential degradation of MIR224 participates in hepatocellular carcinoma tumorigenesis. Autophagy. 2014; 10:1687–1689.
17. Zhang X, Zhang X, Liu C, Jia N, Li X, Xiao J. MiR 224 promotes colorectal cancer cells proliferation via downregulation of P21WAF1/CIP1. Mol Med Rep. 2014; 9:941–46.
18. Cui R, Kim T, Fassan M, Meng W, Sun HL, Jeon YJ, Vicentini C, Tili E, Peng Y, Scarpa A, Liang G, Zhang YK, Chakravarti A, Croce CM. MicroRNA-224 is implicated in lung cancer pathogenesis through targeting caspase-3 and caspase-7. Oncotarget. 2015; 6:21802–15. https://doi.org/10.18632/oncotarget.5224.
19. Cui R, Meng W, Sun HL, Kim T, Ye Z, Fassan M, Jeon YJ, Li B, Vicentini C, Peng Y, Lee TJ, Luo Z, Liu L, et al. MicroRNA-224 promotes tumor progression in nonsmall cell lung cancer. Proc Natl Acad Sci USA. 2015; 112:E4288–4297.
20. David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010; 24:2343–2364.
21. Walsh CM, Suchanek AL, Cyphert TJ, Kohan AB, Szeszel-Fedorowicz W, Salati LM. Serine Arginine Splicing Factor 3 Is Involved in Enhanced Splicing of Glucose-6-phosphate Dehydrogenase RNA in Response to Nutrients and Hormones in Liver. J Biol Chem. 2013; 288:2816–2828.
22. Tang Y, Horikawa I, Ajiro M, Robles AI, Fujita K, Mondal AM, Stauffer JK, Zheng ZM, Harris CC. Downregulation of splicing factor SRSF3 induces p53beta, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene. 2013; 32:2792–2798.
23. Sen S, Jumaa H, Webster NJ. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat Commun. 2013; 4:1336.
24. McGlincy NJ, Smith CW. Alternative splicing resulting in nonsense-mediated mRNA decay: what is the meaning of nonsense? Trends Biochem Sci. 2008; 33:385–393.
25. Lu GY, Liu ST, Huang SM, Chang YL, Lin WS. Multiple effects of digoxin on subsets of cancer-associated genes through the alternative splicing pathway. Biochimie. 2014; 106:131–139.
26. Lu GY, Huang SM, Liu ST, Liu PY, Chou WY, Lin WS. Caffeine induces tumor cytotoxicity via the regulation of alternative splicing in subsets of cancer-associated genes. Int J Biochem Cell Biol. 2014; 47:83–92.
27. Chang YL, Hsu YJ, Chen Y, Wang YW, Huang SM. Theophylline exhibits anti-cancer activity via suppressing SRSF3 in cervical and breast cancer cell lines. Oncotarget. 2017; 8:101461–74. https://doi.org/10.18632/oncotarget.21464.
28. Kano S, Nishida K, Kurebe H, Nishiyama C, Kita K, Akaike Y, Kajita K, Kurokawa K, Masuda K, Kuwano Y, Tanahashi T, Rokutan K. Oxidative stress-inducible truncated serine/arginine-rich splicing factor 3 regulates interleukin-8 production in human colon cancer cells. Am J Physiol Cell Physiol. 2014; 306:C250–262.
29. Sen S, Langiewicz M, Jumaa H, Webster NJ. Deletion of serine/arginine-rich splicing factor 3 in hepatocytes predisposes to hepatocellular carcinoma in mice. Hepatology. 2015; 61:171–183.
30. Kurokawa K, Akaike Y, Masuda K, Kuwano Y, Nishida K, Yamagishi N, Kajita K, Tanahashi T, Rokutan K. Downregulation of serine/arginine-rich splicing factor 3 induces G1 cell cycle arrest and apoptosis in colon cancer cells. Oncogene. 2014; 33:1407–1417.
31. Nickless A, Jackson E, Marasa J, Nugent P, Mercer RW, Piwnica-Worms D, You Z. Intracellular calcium regulates nonsense-mediated mRNA decay. Nat Med. 2014; 20:961–966.
32. Kong YW, Ferland-McCollough D, Jackson TJ, Bushell M. microRNAs in cancer management. Lancet Oncol. 2012; 13:e249–258.
33. Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011; 12:861–874.
34. Elizalde M, Urtasun R, Azkona M, Latasa MU, Goni S, Garcia-Irigoyen O, Uriarte I, Segura V, Collantes M, Di Scala M, Lujambio A, Prieto J, Avila MA, et al. Splicing regulator SLU7 is essential for maintaining liver homeostasis. J Clin Invest. 2014; 124:2909–2920.
35. Leeder RG, Rafeiro E, Brien JF, Mandin CC, Massey TE. Evaluation of reactive oxygen species involvement in amiodarone pulmonary toxicity in vivo and in vitro. J Biochem Toxicol. 1996; 11:147–160.
36. Karam R, Wengrod J, Gardner LB, Wilkinson MF. Regulation of nonsense-mediated mRNA decay: implications for physiology and disease. Biochim Biophys Acta. 2013; 1829:624–633.
37. Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999; 8:1893–1900.
38. Khajavi M, Inoue K, Lupski JR. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet. 2006; 14:1074–1081.
39. El-Deiry WS, Harper JW, O’Connor PM, Velculescu VE, Canman CE, Jackman J, Pietenpol JA, Burrell M, Hill DE, Wang Y, Wiman KG, Mercer WE, Kastan MB, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994; 54:1169–74.
40. Chang YL, Lee HJ, Liu ST, Lin YS, Chen TC, Hsieh TY, Huang HS, Huang SM. Different roles of p53 in the regulation of DNA damage caused by 1,2-heteroannelated anthraquinones and doxorubicin. Int J Biochem Cell Biol. 2011; 43:1720–1728.
41. Cmielová J, Rezáčová M. p21Cip1/Waf1 protein and its function based on a subcellular localization [corrected]. J Cell Biochem. 2011; 112:3502–06.
42. Liu S, Bishop WR, Liu M. Differential effects of cell cycle regulatory protein p21(WAF1/Cip1) on apoptosis and sensitivity to cancer chemotherapy. Drug Resist Updat. 2003; 6:183–195.
43. Wei YH, Lu CY, Lee HC, Pang CY, Ma YS. Oxidative damage and mutation to mitochondrial DNA and age-dependent decline of mitochondrial respiratory function. Ann N Y Acad Sci. 1998; 854:155–170.
44. Lee HC, Wei YH. Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp Biol Med (Maywood). 2007; 232:592–606.
45. Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007; 302:1–12.
46. Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006; 6:259–269.
47. Li MY, Hu XX. Meta-analysis of microRNA expression profiling studies in human cervical cancer. Med Oncol. 2015; 32:510.
48. Subbaramaiah K, Dannenberg AJ. Cyclooxygenase-2 transcription is regulated by human papillomavirus 16 E6 and E7 oncoproteins: evidence of a corepressor/coactivator exchange. Cancer Res. 2007; 67:3976–3985.
49. Kim HS, Kim T, Kim MK, Suh DH, Chung HH, Song YS. Cyclooxygenase-1 and -2: molecular targets for cervical neoplasia. J Cancer Prev. 2013; 18:123–134.
50. Gadducci A, Guerrieri ME, Greco C. Tissue biomarkers as prognostic variables of cervical cancer. Crit Rev Oncol Hematol. 2013; 86:104–129.
51. Han JA, Kim JI, Ongusaha PP, Hwang DH, Ballou LR, Mahale A, Aaronson SA, Lee SW. P53-mediated induction of Cox-2 counteracts p53- or genotoxic stress-induced apoptosis. EMBO J. 2002; 21:5635–5644.