
INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the third leading cause of cancer-related death worldwide. It has a yearly fatality ratio of approximately 1, indicating that most patients do not survive one year [1]. HCC develops in cirrhotic patients in up to 90% of cases, mainly related to chronic viral hepatitis and alcohol abuse [2]. Invasion and metastasis are the underlying causes of poor long-term survival after clinical treatment in HCC [3]. HCC metastasis is a complex process that involves multiple factors, and its mechanism is not fully understood [4]. Therefore, in-depth research on the molecular mechanism of HCC metastasis might discover a potential intervention therapy.
Alternative polyadenylation (APA) is a fundamental molecular mechanism that influences the kinetics of gene regulation in diverse physiological and pathological states through the activity of messenger RNA (mRNA) 3′ untranslated regions (3′UTRs) [5, 6].Virtually 90% of human genes have distinct transcripts with variable 3′UTR lengths arising from multiple polyA sites, which is also the cause of transcriptome polymorphism [7–9].
Human cleavage factor Im (CFIm) is an essential component of the pre-mRNA 3′ processing complex that controls polyA site selection through the recognition of UGUA sequences upstream of the polyA site [10]. It contains three polypeptides of 25 kDa, 59 kDa, and 68 kDa that are designated CFIm25 (or CPSF5/NUDT21), CFIm59 (or CPSF7, cleavage and polyadenylation specificity factor 7), and CFIm68 (or CPSF6) [11]. Others have determined that the reduction of CFIm25 not only induces a global switch to the use of the polyA signal most proximal to the stop codon (pPAS) sites but also enhances cell proliferation [12, 13]. Studies have found that the expression of CFIm25 in tumor tissue is lower than in normal tissue in glioblastoma, and the level of CFIm25 is related to the proliferation of tumor cells [13, 14]. However, the function of CFIm25 in the occurrence and development of HCC remains unknown.
We show that CFIm25 inhibits cell invasion and metastasis of HCC though disruption of the epithelial-mesenchymal transition (EMT) process. These results support the hypothesis that CFIm25 prevents tumor invasion and metastasis in HCC. Moreover, we found that CFIm25 blocks the JNK and P38 signal pathways. Our findings indicate that CFIm25is a factor in HCC and is a candidate for directed therapy.
RESULTS
Downregulation of CFIm25 expression in HCC
To determine the significance of CFIm25 expression in benign as well as malignant hepatic lesions, we analyzed CFIm25 levels in 122 HCC patients. We used RT-PCR and Western blotting to analyze the expression of CFIm25 in 62 pairs of tissue samples. Compared with the corresponding HCC tissue sample, CFIm25 was upregulated in pericarcinoma tissue samples (Figures 1A–1C). We applied immunohistochemistry to analyze CFIm25 protein levels in the other 60 HCC informative patients with various hepatic lesions, which mainly included cirrhosis, steatosis, chronic hepatitis, and cancer. CFIm25 protein was detected in the nuclei of hepatocytes, and representative images of immunohistochemical staining for noncancerous and cancerous tissue are shown in Figure 2. We found that CFIm25 in normal liver tissue, steatosis, and cirrhosis, expression was higher than in HCC.

Figure 1: Downregulation of CFIm25 expression in HCC and metastatic HCC cells. (A) PCR shows the expression of CFIm25 in HCC and the corresponding pericarcinoma tissue. N, liver pericarcinoma tissues; C, liver cancer tissues. (B) The expressions of CFIm25 in 62 pairs of HCC tissues and corresponding pericarcinoma tissues were measured by real-time RT-PCR. Data are represented as the mean ± SD. (C) Representative image shows CFIm25 expression in additional four pairs of HCC tissues and corresponding pericarcinoma tissues. N, liver pericarcinoma tissues; C, liver cancer tissues. (D) Real-time RT-PCR and (E) Western blot show the expression of CFIm25 in different HCC cell lines.
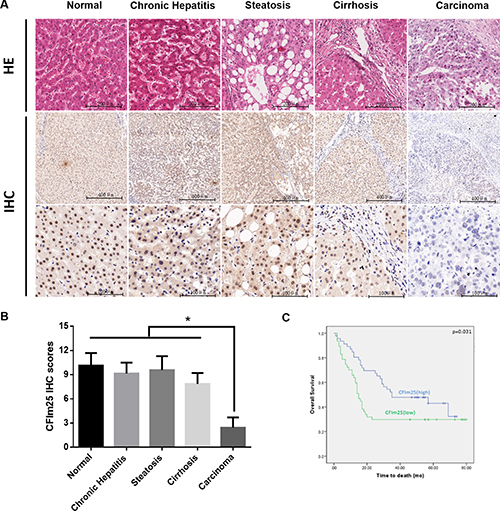
Figure 2: Combined analyses of CFIm25 in benign and malignant hepatic lesions. (A) Representative images of CFIm25 expression in different hepatic lesions are shown (B) with semi-quantitative result displayed as mean ± SE. CFIm25 is present in the nucleus but not in the cytoplasm or on the membrane. (C) Kaplan-Meier survival curve for CFIm25. Median overall survival of the CFIm25-high and CFIm25-low cases was 44 and 31 months, respectively.
To further investigate CFIm25 in HCC development, we studied a larger cohort of HCC patients. We recruited another 93 HCC patients with various tumor grades and stages. CFIm25 expression in tumor tissue was classified as high in 47 patients and low in 46 patients, basis for the immunohistochemistry score whose greater than or equal to 5 was defined as high expression, and less than 5 was defined as low expression. Expression of CFIm25 strongly correlated with the tumor progression. In comparing the clinicopathological parameters between groups, including age, sex, lymph node metastasis, tumor grading and TNM staging, we found the level of CFIm25 was significantly associated with lymph node metastasis (Person’s chi-square test, P = 0.011) and TNM staging (Person’s chi-square test, P = 0.029), whereas age, sex, and tumor grade showed no statistical significance with CFIm25 (Table 1). Median overall survival for the CFIm25-high and CFIm25-low cases were 44 and 31 months, respectively, indicating a significant difference in survival (Kaplan Meier log-rank test, P = 0.031; Figure 2C). Low levels of CFIm25 correlate with progression of human HCC and TNM stage correlates with tumor metastasis. Therefore, we investigated the CFIm25 expression in HCC cell lines with different metastatic potentials and found that the expression in Sk-hep-1, MHCC-LM3, and MHCC-97H was lower than in MHCC-97L, Hep-G2, and SMMC-7721 cells, which is contrary to the invasiveness of these cells [15, 16]. CFIm25 was frequently downregulated in HCC samples and metastatic HCC cells (Figure 1D, 1E).
Table 1: Correlations between CFIm25 and clinicopathological features of 93 HCC patients
Variables |
CFIm25 staining |
P value |
|
|---|---|---|---|
high expression( ≥ 5) |
low expression( < 5) |
||
Age |
0.105a |
||
≤ 50 |
14 |
22 |
|
> 50 |
32 |
25 |
|
Gender |
1.000b |
||
Male |
41 |
42 |
|
Female |
5 |
5 |
|
Liver cirrhosis |
0.350a |
||
Absent |
26 |
31 |
|
Present |
20 |
16 |
|
Maximal tumor size |
0.545a |
||
≤ 5 cm |
25 |
26 |
|
> 5 cm |
21 |
21 |
|
Tumor location |
0.139a |
||
Left |
4 |
7 |
|
Right |
16 |
23 |
|
Others |
26 |
17 |
|
Pathological type |
0.119a |
||
Nodular type |
24 |
20 |
|
Giant mass type |
14 |
10 |
|
Others |
8 |
17 |
|
Lymph node metastasis |
0.011a |
||
Yes |
10 |
22 |
|
No |
36 |
25 |
|
Grading |
0.238a |
||
1~2 |
30 |
25 |
|
3~4 |
16 |
22 |
|
TNM staging |
0.029a |
||
I~II |
29 |
19 |
|
III~IV |
17 |
28 |
|
aPerson Chi-square test;
bContinuity correction Chi-square test
CFIm25 suppressed cell migration and invasion in vitro
Cell invasion and migration are critical during the multistep process of tumor metastasis. To assess whether CFIm25 is an important factor in cell invasion and migration, we used RNA interference (RNAi) to suppress CFIm25 expression in SMMC-7721 and Hep-G2 cells, and we used full-length plasmid (pcDNA3.1- CFIm25) to stably express CFIm25 in Sk-hep-1 and MHCC-LM3 cells. Sk-hep-1 and MHCC-LM3 cells stably expressing CFIm25 inhibited the invasion and migration ability compared with control cells (Figure 3E, 3F), and loss of CFIm25function in SMMC-7721 and Hep-G2 cells promoted invasion and migration (Figure 3A, 3B). To confirm this result, we utilized a wound-healing assay to evaluate the effect of CFIm25 on cell migration. Consistent with previous observations, the overexpression of CFIm25 inhibited (Figure 3G, 3H) whereas knockdown CFIm25 enhanced the mobility of HCC cells (Figure 3C, 3D). These results prove that expression of CFIm25 correlates with the invasion and migration ability in HCC cells.
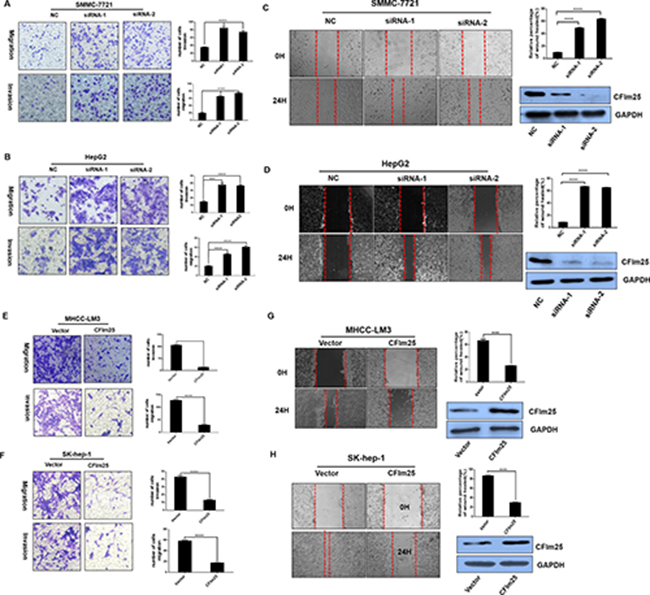
Figure 3: CFIm25 inhibits HCC cell migration and invasion in vitro. CFIm25 knockdown promoted cell migration and invasion by transwell assay in (A) SMMC-7721 and (B) Hep-G2 cells. Overexpression of CFIm25 in (E) MHCC-LM3 and (F) Sk-hep-1 cells inhibits cell migration and invasion by transwell assay. The numbers of migration and invasion cells were quantified. Wound-healing assay was performed with (C) SMMC-7721, (D) Hep-G2, (G) MHCC-LM3, and (H) Sk-hep-1 cells. Cell migration was quantified as percentage of wound-healed area. Data are means ± s.d. *P < 0.05. NC, negative control siRNA; siRNA1& siRNA 2, siRNAs directed at CFIm25.
CFIm25 is a factor in the EMT process
EMT is an important mechanism of tumor invasion and metastasis. We further explored whether the CFIm25 blockade of tumor cell invasion and metastasis is through the EMT process. We found that overexpression of CFIm25 increases the expression of E-cadherin, a marker protein of EMT. At the same time, the expression of N-cadherin is decreased. With inactive CFIm25, the result is the opposite (Figure 4A, 4B). To prove that CFIm25 can bind to the cytoskeletons of hepatocarcinoma cells, we used immunofluorescent microscopy to examine the effect of CFIm25 on the F-actin cytoskeletal arrangement. We suppressed CFIm25 expression in SMMC-7721 and Hep-G2 cells. When CFIm25 is inhibited, F-actin fibers in the culture are densely arranged just inside the cell periphery, where they form a circular bundle; slim central fibers are also visible. This organization is characteristic of cells with apical-basolateral polarity, such as epithelial cells. Excessive expression of CFIm25 in Sk-hep-1 and MHCC-LM3 cells yields the opposite result (Figure 4C, 4D).
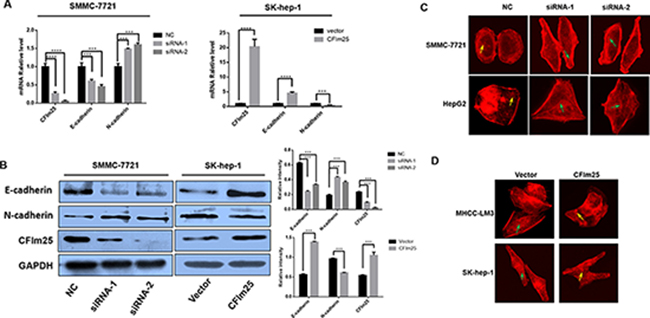
Figure 4: CFIm25 is active in the EMT process. (A) Real-time RT-PCR and (B) Western blot show that CFIm25 suppression decreases the expression of E-cadherin and increases N-cadherin in SMMC-7721, whereas overexpression of CFIm25 in Sk-hep-1 increases the expression of E-cadherin and decreases N-cadherin. (C) and (D) CFIm25 inhibition impairs epithelial phenotype of HCC cells. F-actin cytoskeletal arrangement was examined by fluorescence microscopy in (C) SMMC-7721, Hep-G2, and (D) MHCC-LM3, Sk-hep-1, green arrows: cortical F-actin organized as curvilinear network, yellow arrows: punctate F-actin. Data are means ± s.d. *P < 0.05. NC, negative control siRNA; siRNA1& siRNA 2, siRNAs directed at CFIm25.
CFIm25 inhibits HCC cell migration and invasion by blocking the p38 and JNK/c-Jun signal pathways
To address the molecular mechanism of CFIm25 defense against HCC cell invasion and metastasis, we investigated the MAPKs and Akt signal pathways, which are two main pathways for tumor proliferation and metastasis [17–20]. Inhibition of CFIm25 led to increased levels of phosphorylated JNK, p38, and c-Jun in HCC cells, but no differences were seen in phosphorylated Akt and ERK1/2 (Figure 5A). Activation of c-Jun by CFIm25 knockdown was further confirmed by the activation of its direct downstream effector, c-Jun-driven activating protein-1 (AP-1), as evidenced by luciferase reporter assay (Figure 5B).
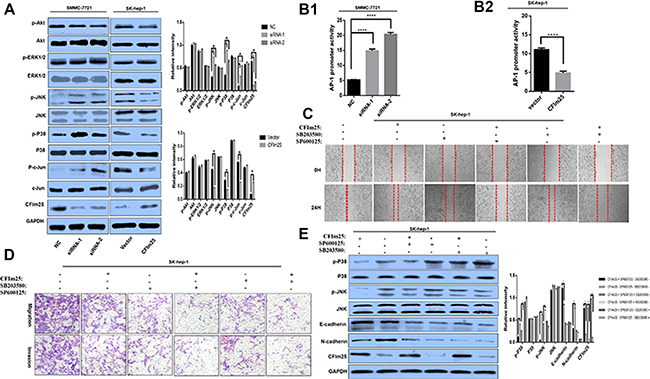
Figure 5: CFIm25 inhibits HCC cell migration and invasion through suppression of the p38 and JNK/c-Jun signal pathways. (A) Protein levels of CFIm25, phosphorylated and total Akt, ERK1/2, p38, JNK, and c-Jun were analyzed by Western blot. (B1) CFIm25 inhibition increases the transcriptional activity of AP-1 in SMMC-7721, as determined by luciferase reporter assay. (B2) Overexpression of CFIm25 decreased the transcriptional activity of AP-1 in Sk-hep-1. Migration and invasion of the HCC cells were disrupted after addition of p38 inhibitor SB203580 (10 μM) and JNK inhibitor SP600125 (10 μM) by (C) wound-healing assay and (D) transwell assay in Sk-hep-1. (E) Protein levels of CFIm25, E-cadherin, N-cadherin, phosphorylated and total p38, and JNK were analyzed with SB203580 (10 μM) and SP600125(10 μM) pretreatment in Sk-hep-1. Data are means ± s.d. *P < 0.05. NC, negative control siRNA; siRNA1& siRNA 2, siRNAs directed at CFIm25.
To clarify stimulation of CFIm25-induced E-cadherin expression by the p38 pathway, the JNK/c-Jun pathway, or both, SB203580 and SP600125 were used inactivate p38 and JNK/c-Jun, respectively. Pretreatment with the JNK inhibitor (SP600125) and the p38 inhibitor (SB203580) reduced CFIm25-induced E-cadherin expression (Figure 5E). Wound-healing and transwell assay results from woverexpression of CFIm25 in Sk-hep-1 cells were confirmed through downregulation of p38 and JNK/c-Jun phosphorylation (Figure 5C, 5D). Our results indicate that CFIm25 upregulate E-cadherin expression in HCC cells, which are dependent on the p38 and JNK/c-Jun signal pathways.
Overexpression of CFIm25 inhibits intrahepatic metastasis and lung metastasis in vivo
Having observed that CFIm25 inhibited HCC cells migration in vitro, we confirmed the anti-metastasis function of CFIm25 in vivo. We injected nude mice with Sk-hep-1 and MHCC-LM3 cells that had high metastatic potential stably expressing CFIm25. The mice injected with Sk-hep-1-CFIm25 and MHCC-LM3- CFIm25 cells showed less intrahepatic and lung metastasis, compared with the control mice (Figure 6A, 6B). These results imply that CFIm25 suppresses HCC metastasis.
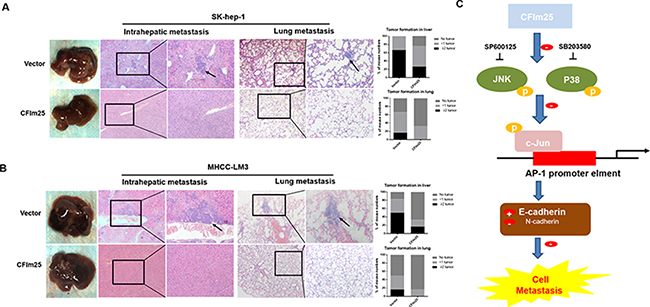
Figure 6: Overexpression of CFIm25 inhibits intrahepatic metastasis and lung metastasis in vivo. Representative H&E staining of livers and lungs after injected (A) Sk-hep-1 and (B) MHCC-LM3 cells that had high metastatic potential stably expressing CFIm25. Black arrow indicates the intrahepatic or lung metastatic tumors. (C) Mechanistic scheme of CFIm25 in HCC metastasis.
DISCUSSION
HCC is prone to early metastasis, which is one of the main causes of cancer-related death. Investigations have demonstrated that normal embryogenesis and neoplasia share many basic processes and molecular pathways. Tumorigenesis may result from the loss of control by normal developmental genes [21]. APA, a post-transcriptional modification implicated in many diseases (hematological disorders [22–24], infection and immunological conditions [25–27], neurological diseases [28–30], and endocrine diseases [31, 32]), including cancer [33, 34]. The closely coupled cleavage and polyadenylation reactions are key steps in the processing of most primary pre-mRNA transcripts into mature mRNAs [35]. Understanding of this process has improved since the initial identification of a common AAUAAA polyadenylation signal (PAS) approximately 20 nt upstream from the cleavage site [36]. The global shortening of messenger RNAs through APA that occurs during enhanced cellular proliferation is a poorly understood mechanism of gene expression [6, 37]. CFIm25 is a key factor in APA [38, 39]. Studies show that CFIm25 promotes the anti-tumor effect in glioblastoma [13]. These findings identify activity of CFIm25 in suppressing APA and reveal a connection between CFIm25 and glioblastoma tumorigenicity [13]. Ospina-Villa et al. [40] observed that Leu135 and Tyr236 residues accelerate RNA binding activity of CFIm25 in Entamoeba histolytica. Fukumitsu et al. [41] showed that knockdown of CFIm25 promotes neurite outgrowth in developing neurons by coordinating activity upstream of NGF-induced RhoA inactivation [41]. However, no studies show that CFIm25 is associated with HCC.
In this study, we found a differential expression of CFIm25 between HCC and non-cancerous tissues We also found that CFIm25 expression is associated with TMN staging and progression of human HCC. This result suggests that CFIm25 might be active in HCC metastasis. Little evidence suggests that APA is associated with tumor metastasis. A study shows that the RhoA guanine exchange factor NET1, which is a factor in the metastasis of gastric cancer, isoforming with short 3′ UTR, promotes cellular migration and invasion in vitro [42], but CFIm25 and metastases in HCC has not been studied.
We tried to determine the relation between CFIm25 and HCC metastasis. We identified CFIm25 expressions in HCC cell lines with different metastatic potentials and found that CFIm25 expression is contrary to the invasiveness of HCC cells. This finding implies that CFIm25 might have anti-metastasis properties. We utilized transwell and wound-healing assays to detect the relation between CFIm25 and metastasis in four HCC cells lines (Sk-hep-1, MHCC-LM3, Hep-G2, and SMMC-7721) in vitro. The results were consistent with our expectations.
Based on the previous results, we investigated CFIm25disruption of the metastasis process. EMT is a universal form of tumor metastasis and E-cadherin is an important factor in the EMT process, as is a component in the formation of cell-cell adherens junctions in epithelial tissues [43]. Studies show that suppression of E-cadherin results in mesenchymal morphology and increased cell migration and invasion, as well as metastasis [44]. The experimental and physiological observations regarding the tumor suppressor role of E-cadherin are often interpreted in the context of EMT [45]. The EMT is a reversible and dynamic process that allows the transition between the epithelial and the mesenchymal phenotype during tissue development and repair. It results in the conversion of tightly connected epithelial cells into more loosely adherent, fibroblast-like cells [46]. Research has demonstrated that re-introduction of E-cadherin into cell lines in which it is depleted promotes reversion of poorly differentiated carcinoma phenotypes (i.e., fibroblastic, highly invasive, poorly cell-cell adherent) to well-differentiated, minimally invasive epithelioid phenotypes with well-developed cell-cell junctions [47]. Depletion of E-cadherin activates oncogenic signaling pathways, including mitogen activated kinase (MAPK), rat sarcoma viral oncogene (Ras), and ras-related C3 botulinum toxin substrate (Rac1), and disrupts the Hippo signaling pathway, which leads to cell migration and invasion [44]. Our study found that the expression of E-cadherin was low/high after high/low expression of CFIm25, and the expression of N-cadherin was opposite to that of E-cadherin. This finding suggests that CFIm25 might prevent the metastasis of HCC by suppressing the EMT process.
With further research, we demonstrated that the anti-metastatic effect of CFIm25 in SMMC7721 cells was through blocking the p38 and JNK/c-Jun pathways, but no total ERK or Akt influence. JNK and p38 mitogen-activated protein kinases (MAPKs) have important functions in the signaling mechanisms that orchestrate cellular responses to many types of stresses, but they also control the proliferation, differentiation, survival and migration of specific cell types [48]. Transcription factor AP-1 is associated with JNK, which consists of c-Fos and c-Jun family members [49]. The oncogenic functions of JNK are mostly based on their ability to phosphorylate Jun and to activate AP-1, whereas their tumor-suppressive functions are probably related to their pro-apoptotic activity. Studies show that cross communication between the JNK and p38 MAPK pathways is as a controlling mechanism in many cellular responses [48]. We used siRNA, which diminishes the function of CFIm25, to activate the p38 and JNK/c-Jun signaling cascade, subsequently activating the AP-1 transcription that stimulates the downstream genes involved in promoting HCC cell metastasis.
Previous studies show that CFIm25 inhibits the invasion and metastasis of hepatocellular carcinoma by blocking the JNK/c-Jun signaling pathway and influencing the activity of AP-1 in vitro, and we verified this result in vivo. The results of in vivo experiments compared with our in vitro experiments show the same p38 and JNK/c-Jun signaling cascades.
CFIm25 is crucial for preventing invasion and metastasis of HCC. HCC upregulates AP-1 by stimulating the p38 and JNK/c-Jun signaling pathways. Thus, enhancing CFIm25 and repressing the direct downstream effector of p38 and JNK/c-Jun signaling pathways have a potential clinical application for HCC treatment.
MATERIALS AND METHODS
Cell lines and HCC specimens
All HCC specimens and matched nontumor tissues were collected during surgical resection at Tongji Hospital. Tumor histopathology was confirmed by analysis of hematoxylin and eosin (H&E) stained tissue sections by a qualified neuropathologist. The present study was conducted according to the guidelines of the Ethics Committee of the Tongji Hospital and in accordance with the ethical standards of the World Medical Association Declaration of Helsinki. Human HCC cell lines Huh7, SMMC7721, HepG2, MHCC LM3, and SK-Hep-1were cultured in DMEM medium supplemented with 10% fetal calf serum (Invitrogen Gibco, Carlsbad, CA, USA) and incubated in a 5% CO2 incubator at 37°C.
RNA extraction and real-time RT-PCR
Total RNA was extracted by use of TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). Reverse-transcribed complementary DNA was synthesized by use of a PrimeScript RT Reagent Kit (TaKaRa, Otsu, Japan). Real-time polymerase chain reaction was performed by use of SYBR Premix ExTaq (TaKaRa, Otsu, Japan) on an ABI StepOne Real-Time PCR System (Applied Biosystem, Carlsbad, CA, USA). The value of 2–ΔΔCt was used to determine fold difference between samples. The primers used for PCR sequences were as follows: GAPDH: 5′-TCATTGACCTCAACTACATGGTTT-3′ (sense) and 5′-GAAGATGGTGATGGGATTTC-3′ (antisense); CFIm25:5′- GGTCACTCAGTTCGGCAACAA-3′ (sense) and 5′- CTCATGCGCTGAAATCTGGC-3′(antisense)
RNA interference and establishment of stable expressing cells
For RNA interference, transfection was performed by use of Lipofectamine 2000 (Life Technologies) following the standard method. The siRNA sequences (Ribo Company, Guangzhou, China) used are the following: CFIm25-siRNA1: forward 5′ GCUCUGUUGCAGCCAGAUU dTdT 3′ and reverse 3′ dTdT CGAGACAACGUCGGUCUAA 5′; CFIm25-siRNA2: forward 5′ GCACCAUUGUUUGAAUUGU dTdT 3′ and reverse 3′ dTdT CGUGGUAACAAACUUAACA 5′. For establishment of stable expressing cells, plasmids (Genechem Company, China) were transfected into cells with Lipofectamine 2000 according to the manufacturer’s instructions. We achieved the stable overexpression transfectant by adding G418 (Sigma Aldrich) for 4 weeks.
In vitro migration and invasion assay
Migration assays were conducted by use of 24-well plate containing inserts (8-μm pores; Costar, USA). Invasion assays were performed similarly by use of matrigel-coated inserts (Costar, USA). The lower chamber was filled with 600-μL medium containing 12% serum, and the top chamber contained 2 × 104 cells suspended in 200-μL medium without serum. After 24 hours of incubation, cells invading into the bottom side of the inserts were fixed, stained, photographed, and quantified by counting them in five random fields. Wound-healing assays were conducted on indicated cells. Cells were plated to confluence in 6-well plates. After the cells had adhered to the plates, streaks were created in the monolayer with a pipette tip. Progression of migration was observed and photographed at 0 and 24 hours after wound.
In vivo metastasis assays
For mouse tail vein injection, 1 × 105 cells in 100 μL of phosphate buffered saline (PBS) were injected into the tail veins of nude mice. The mice were sacrificed on day 30. Liver and lung tissues were resected and fixed with 4% paraformaldehyde, and then strained with H&E.
Western blot analysis
Western blot analysis were performed as previously described [50].
Immunohistochemistry staining
Paraffin-embedded tissues were cut into four μm-thick consecutive sections and were then dewaxed in xylene and rehydrated in graded ethanol solutions. Tissue sample slides were deparaffinized with dimethylbenzene, followed by gradient alcohol dehydration, and incubation with 3% hydrogen peroxide to block endogenous peroxidase, and then to block non-specific binding sites. Primary anti-CFIm25 antibody (1:400, ABcam, USA) was incubated overnight at 4° C. The secondary antibody was added for 1 hour at room temperature, and then developed by peroxidase conjugated streptavidin and diaminobenzidine, and counterstained with hematoxylin.
The intensity of staining was scored on a four-point scale as negative (0), weak (1), moderate (2), and strong (3). The extent of the staining, defined as the percentage of positively stained tissue area in relation to the entire tissue area, was scored on a scale of 0 to 4: 0 (0%), 1 (1%–25%), 2 (26%–50%), 3 (51%–75%), and 4 (76%–100%) [51]. Finally, the immunohistochemistry score was equal to staining intensity multiplied by staining extent [52]. The result greater than or equal to 5 was defined as high expression, and less than 5 was defined as low expression.
Luciferase reporter experiments
AP-1 reporter activity was detected by a Secrete-PairTM Dual Luminescence Assay Kit (GeneCopoeia) according to the manufacturer’s instructions.
Statistical analyses
All experiments were performed in triplicate unless specified. The data are presented as mean ± SD. Statistical analyses were performed by Student’s t-test. The Pearson chi-square was used to analyze the correlation between CFIm25expression and pathogenic conditions of chronic liver disease in human samples. Statistical analyses were performed by Prism 5.0 (GraphPad Software, La Jolla, CA, USA). A value of P < 0.05 was considered statistically significant.
CONFLICTS OF INTEREST
Authors make a joint declaration that they have no conflicts of interests with respect of this work and its publication.
FUNDING
This study was supported by the National Natural Science Foundation of China (No. 81572419, 81401992, 81472311).
REFERENCES
1. Marengo A, Rosso C, Bugianesi E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annual Review of Medicine. 2016; 67:103–117.
2. Pascual S, Herrera I, Irurzun J. New advances in hepatocellular carcinoma. World J Hepatol. 2016; 8:421–438.
3. Tabrizian P, Roayaie S, Schwartz ME. Current management of hepatocellular carcinoma. World J Gastroenterol. 2014; 20:10223–10237.
4. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006; 127:679–695.
5. Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol. 2017; 18:18–30.
6. Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3’ untranslated regions and fewer microRNA target sites. Science. 2008; 320:1643–1647.
7. Xia Z, Donehower LA, Cooper TA, Neilson JR, Wheeler DA, Wagner EJ, Li W. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3’-UTR landscape across seven tumour types. Nat Commun. 2014; 5:5274.
8. Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008; 40:1413–1415.
9. Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008; 456:470–476.
10. Sartini BL, Wang H, Wang W, Millette CF, Kilpatrick DL. Pre-messenger RNA cleavage factor I (CFIm): potential role in alternative polyadenylation during spermatogenesis. Biol Reprod. 2008; 78:472–482.
11. Hardy JG, Norbury CJ. Cleavage factor Im (CFIm) as a regulator of alternative polyadenylation. Biochem Soc Trans. 2016; 44:1051–1057.
12. Gennarino VA, Alcott CE, Chen CA, Chaudhury A, Gillentine MA, Rosenfeld JA, Parikh S, Wheless JW, Roeder ER, Horovitz DD, Roney EK, Smith JL, Cheung SW, et al. NUDT21-spanning CNVs lead to neuropsychiatric disease and altered MeCP2 abundance via alternative polyadenylation. Elife. 2015; 4.
13. Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu AB, Li W, Wagner EJ. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature. 2014; 510:412–416.
14. Han T, Kim JK. Driving glioblastoma growth by alternative polyadenylation. Cell Res. 2014; 24:1023–1024.
15. Tu W, Luo M, Wang Z, Yan W, Xia Y, Deng H, He J, Han P, Tian D. Upregulation of SATB1 promotes tumor growth and metastasis in liver cancer. Liver International. 2012; 32:1064–1078.
16. Liu J, Han P, Li M, Yan W, Liu J, Liu J, He J, Tu W, Xia Y, Zhou Z, Gong J, Liu M, Ding Q, Tian D. The histidine-rich calcium binding protein (HRC) promotes tumor metastasis in hepatocellular carcinoma and is upregulated by SATB1. Oncotarget. 2015; 6:6811–6824. https://doi.org/10.18632/oncotarget.3049.
17. Min L, He B, Hui L. Mitogen-activated protein kinases in hepatocellular carcinoma development. Seminars in Cancer Biology. 2011; 21:10–20.
18. Liu AW, Cai J, Zhao XL, Jiang TH, He TF, Fu HQ, Zhu MH, Zhang SH. ShRNA-targeted MAP4K4 inhibits hepatocellular carcinoma growth. Clinical Cancer Research. 2011; 17:710–720.
19. Fang Y, Xue JL, Shen Q, Chen J, Tian L. MicroRNA-7 inhibits tumor growth and metastasis by targeting the phosphoinositide 3-kinase/Akt pathway in hepatocellular carcinoma. Hepatology. 2012; 55:1852–1862.
20. Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, Destefanis G, Delogu S, Zimmermann A, Ericsson J, Brozzetti S, Staniscia T, Chen X, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. 2011; 140:1071–1083.
21. Chen Y, Hao J, Jiang W, He T, Zhang X, Jiang T, Jiang R. Identifying potential cancer driver genes by genomic data integration. Sci Rep. 2013; 3:3538.
22. Rund D, Dowling C, Najjar K, Rachmilewitz EA, Kazazian HH Jr, Oppenheim A. Two mutations in the beta-globin polyadenylylation signal reveal extended transcripts and new RNA polyadenylylation sites. Proc Natl Acad Sci U S A. 1992; 89:4324–4328.
23. Gehring NH, Frede U, Neu-Yilik G, Hundsdoerfer P, Vetter B, Hentze MW, Kulozik AE. Increased efficiency of mRNA 3’ end formation: a new genetic mechanism contributing to hereditary thrombophilia. Nat Genet. 2001; 28:389–392.
24. Higgs DR, Goodbourn SE, Lamb J, Clegg JB, Weatherall DJ, Proudfoot NJ. Alpha-thalassaemia caused by a polyadenylation signal mutation. Nature. 1983; 306:398–400.
25. Phillips C, Jung S, Gunderson SI. Regulation of nuclear poly(A) addition controls the expression of immunoglobulin M secretory mRNA. EMBO J. 2001; 20:6443–6452.
26. Chuvpilo S, Zimmer M, Kerstan A, Glockner J, Avots A, Escher C, Fischer C, Inashkina I, Jankevics E, Berberich-Siebelt F, Schmitt E, Serfling E. Alternative polyadenylation events contribute to the induction of NF-ATc in effector T cells. Immunity. 1999; 10:261–269.
27. MacMurray JA, Moralejo DH, Kwitek AE, Rutledge EA, Yserloo BV, Gohlke P, Speros SJ, Snyder B, Schaefer J, Bieg S, Jiang J, Ettinger RA, Fuller J, et al. Lymphopenia in the BB Rat Model of Type 1 Diabetes is Due to a Mutation in a Novel Immune-Associated Nucleotide (Ian)-Related Gene. Genome Res. 2002; 12:1029–1039.
28. Rhinn H, Qiang L, Yamashita T, Rhee D, Zolin A, Vanti W, Abeliovich A. Alternative alpha-synuclein transcript usage as a convergent mechanism in Parkinson’s disease pathology. Nat Commun. 2012; 3:1084.
29. Jenal M, Elkon R, Loayza-Puch F, van Haaften G, Kühn U, Menzies FM, Oude Vrielink JA, Bos AJ, Drost J, Rooijers K, Rubinsztein DC, Agami R. The Poly(A)-Binding Protein Nuclear 1 Suppresses Alternative Cleavage and Polyadenylation Sites. Biochem J. 1998; 336:405–11.
30. Tassone F, De Rubeis S, Carosi C, La Fata G, Serpa G, Raske C, Willemsen R, Hagerman PJ, Bagni C. Differential usage of transcriptional start sites and polyadenylation sites in FMR1 premutation alleles. Nucleic Acids Res. 2011; 39:6172–6185.
31. Locke JM, Da Silva Xavier G, Rutter GA, Harries LW. An alternative polyadenylation signal in TCF7L2 generates isoforms that inhibit T cell factor/lymphoid-enhancer factor (TCF/LEF)-dependent target genes. Diabetologia. 2011; 54:3078–3082.
32. Abdel Wahab N, Gibbs J, Mason RM. Regulation of gene expression by alternative polyadenylation and mRNA instability in hyperglycaemic mesangial cells. Biochem J. 1998; 336:405–411.
33. Sakuma K, Sasaki E, Kimura K, Komori K, Shimizu Y, Yatabe Y, Aoki M. HNRNPLL, a newly identified colorectal cancer metastasis suppressor, modulates alternative splicing of CD44 during epithelial-mesenchymal transition. Gut. 2017.
34. Fu Y, Sun Y, Li Y, Li J, Rao X, Chen C, Xu A. Differential genome-wide profiling of tandem 3’ UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res. 2011; 21:741–747.
35. Movassat M, Crabb TL, Busch A, Yao C, Reynolds DJ, Shi Y, Hertel KJ. Coupling between alternative polyadenylation and alternative splicing is limited to terminal introns. RNA Biol. 2016; 13:646–655.
36. Brown KM, Gilmartin GM. A mechanism for the regulation of pre-mRNA 3’ processing by human cleavage factor Im. Mol Cell. 2003; 12:1467–1476.
37. Elkon R, Drost J, van Haaften G, Jenal M, Schrier M, Oude Vrielink JA, Agami R. E2F mediates enhanced alternative polyadenylation in proliferation. Genome Biol. 2012; 13:R59.
38. Kubo T, Wada T, Yamaguchi Y, Shimizu A, Handa H. Knock-down of 25 kDa subunit of cleavage factor Im in Hela cells alters alternative polyadenylation within 3’-UTRs. Nucleic Acids Res. 2006; 34:6264–6271.
39. Gruber AR, Martin G, Keller W, Zavolan M. Cleavage factor Im is a key regulator of 3’ UTR length. RNA Biol. 2012; 9:1405–1412.
40. Ospina-Villa JD, Zamorano-Carrillo A, Lopez-Camarillo C, Castanon-Sanchez CA, Soto-Sanchez J, Ramirez-Moreno E, Marchat LA. Amino acid residues Leu135 and Tyr236 are required for RNA binding activity of CFIm25 in Entamoeba histolytica. Biochimie. 2015; 115:44–51.
41. Fukumitsu H, Soumiya H, Furukawa S. Knockdown of pre-mRNA cleavage factor Im 25 kDa promotes neurite outgrowth. Biochem Biophys Res Commun. 2012; 425:848–853.
42. Lai DP, Tan S, Kang YN, Wu J, Ooi HS, Chen J, Shen TT, Qi Y, Zhang X, Guo Y, Zhu T, Liu B, Shao Z, Zhao X. Genome-wide profiling of polyadenylation sites reveals a link between selective polyadenylation and cancer metastasis. Hum Mol Genet. 2015; 24:3410–3417.
43. Lee BP, Pilling LC, Emond F, Flurkey K, Harrison DE, Yuan R, Peters LL, Kuchel GA, Ferrucci L, Melzer D, Harries LW. Changes in the expression of splicing factor transcripts and variations in alternative splicing are associated with lifespan in mice and humans. Aging Cell. 2016; 15:903–913.
44. Kang Y, Massague J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004; 118:277–279.
45. Tunggal AJ, Helfrich I, Schmitz A, Schwarz H, Günzel D, Fromm M, Kemler R, Krieg T, Niessen CM. E-cadherin is essential for in vivo epidermal barrier function by regulating tight junctions. The EMBO Journal. 2005; 24:1146–1156.
46. Schneider MR, Kolligs FT. E-cadherin’s role in development, tissue homeostasis and disease: Insights from mouse models: Tissue-specific inactivation of the adhesion protein E-cadherin in mice reveals its functions in health and disease. Bioessays. 2015; 37:294–304.
47. Rodriguez FJ, Lewis-Tuffin LJ, Anastasiadis PZ. E-cadherin’s dark side: possible role in tumor progression. Biochim Biophys Acta. 2012; 1826:23–31.
48. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009; 9:537–549.
49. Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003; 3:859–868.
50. Ding Q, Xia Y, Ding S, Lu P, Sun L, Liu M. An alternatively spliced variant of CXCR3 mediates the metastasis of CD133+ liver cancer cells induced by CXCL9. Oncotarget. 2016; 7:14405–14414. https://doi.org/10.18632/oncotarget.7360.
51. Zeng Z, Ren J, O’Neil M, Zhao J, Bridges B, Cox J, Abdulkarim B, Schmitt TM, Kumer SC, Weinman SA. Impact of stem cell marker expression on recurrence of TACE-treated hepatocellular carcinoma post liver transplantation. BMC Cancer. 2012; 12:584.
52. Shan J, Shen J, Liu L, Xia F, Xu C, Duan G, Xu Y, Ma Q, Yang Z, Zhang Q, Ma L, Liu J, Xu S, et al. Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factor pathway in human hepatocellular carcinoma. Hepatology. 2012; 56:1004–1014.