
INTRODUCTION
Increasing evidence is implicating synaptic dysfunction in temporal lobe structures, especially the hippocampus, as being causally important to many neuropsychiatric disorders, such as epilepsy, Post Traumatic Stress Disorder (PTSD), schizophrenia, and Alzheimer’s Disease (AD) [1–7]. Though many molecular modifications are likely to account for these disease states, post-transcriptional regulation of local mRNA at synapses has emerged as a key mechanism controlling synaptic plasticity and dendrite morphogenesis in both health and disease [8, 9]. Often, these local post-transcriptional mechanisms rely on RNA-binding proteins (RBP), but detailed accounts of how these proteins affect synaptogenesis are in their infancy.
One important RBP found at excitatory synapses is PUMILIO 2 (PUM2), a member of the Pumilio and FBF (PUF) family, which is evolutionarily conserved from Drosophila to mice and humans [10–14]. PUM proteins bind to the 3′ Untranslated region (3′ UTR) of their target mRNAs, likely halting translation until certain synaptic signals are presented [11, 15, 16]. PUM2 binds to specific RNA sequences known as Nanos response elements (NREs), also called Pumilio Binding Element (PBE) [12, 15, 17, 18], and PUM2 may also be highly regulated by the Noncoding RNA activated by DNA damage (NORAD) [19, 20].
While many studies have shown that mammalian PUM proteins are important for germ cell formation and differentiation during reproductive system development [11, 21], a growing number of studies indicate that PUM proteins may influence motor and neurological disorders, such as epilepsy [7, 22–25]. In conjunction, PUM homologs have been found in neuronal cells and regulate neuronal homeostasis through affecting dendritic structure, synaptic plasticity, and neuronal excitability in Drosophila [26–30] Some of the gene targets of PUM proteins have been identified in the Drosophila nervous system, including certain glutamate receptors [30–33].
Importantly, recent studies indicate that Pum2 is highly expressed in the mammalian brain [23, 34], and the role of PUM2 in mammalian neuronal regulation have been suggested by in vitro studies. In particular, reduction of the Pum2 expression via shRNA accelerates dendrite outgrowth and arborization in rodent primary hippocampal neurons [13]. In addition, PUM2 is a component of dendritically localized ribonucleoparticles (RNPs), suggesting a role of PUM2 in the neuronal response to cellular stress [13, 35]. Mice with Pum2 knockout exhibit hyperactivity, spontaneous spike-wave discharges, and a reduced seizure threshold to chemoconvulsants [22]. Recently PUM2 was shown to regulate neurogenesis together with PUM1, further establishing important roles of PUM proteins in mammalian nervous system [36]. Interestingly, reduced PUM2 was found in the neocortex of patients with drug-refractory Temporal Lobe Epilepsy (TLE) as well rats given seizures through pilocarpine injections [37]. Additionally, pilocarpine-treated rats that developed status epilepticus still had reduced hippocampal and cortical PUM2 up to 60d later, a time point where spontaneous epileptiform activity develops and closely models TLE pathogenesis [37]. These results suggest that decreased PUM2 may promote increased excitability that leads to epileptogenesis [7].
In this study, we aimed to investigate how PUM2 influences dendritic morphology, synaptic density and synaptic proteins in the cortex and hippocampus of mice. In particular, we focused on how PUM2 may influence excitatory transmission through glutamatergic signaling pathways by investigating PUM2’s regulation of Glur2 mRNA translation. Our study provides the first direct evidence for a post-transcriptional role of PUM2 in regulating Glur2 translation and excitatory synapse morphogenesis in mammals.
RESULTS
Pum2 is highly expressed in the temporal lobe
Pum2XE772 mutant mice carry the LacZ gene under the endogenous Pum2 promoter. Hence, X-gal staining can reveal the expression pattern of Pum2 at the cellular level. X-gal staining demonstrated that Pum2 was expressed in many brain areas, including the cortex, amygdala, thalamus, hypothalamus, and cerebellum, with the highest expression in the temporal lobe, especially the hippocampus [22] (Supplementary Figure 1). Although the Pum2-driven LacZ reporter method nicely reflects the expression pattern of Pum2 transcription in the brain, protein localization may not be the same as its mRNA expression, and this method is unable to resolve the subcellular localization of PUM2. Therefore, we conducted immunohistochemical staining to determine PUM2 expression in wild type mice, and demonstrated that PUM2 protein closely follow X-gal staining with high expression of PUM2 in the hippocampus, mainly localized to the cytoplasm and dendritic projections of pyramidal cells. (Figure 1).
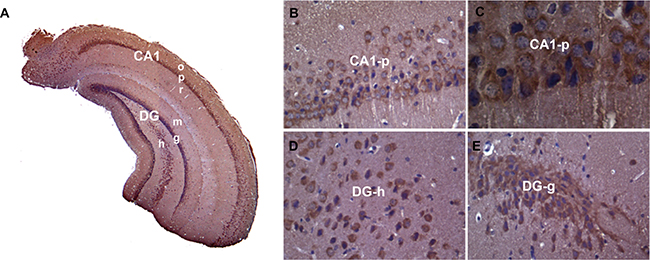
Figure 1: PUM2 protein is highly expressed in the mouse hippocampus. Immunohistochemistry using PUM2 antibody showed that PUM2 proteins are enriched in CA1-3 and dentate gyrus (A). At higher magnification of subregions, PUM2 signals (brown color) were located mainly in the cytoplasm and projections of neuronal cells (B–E), The structures with blue color staining are nuclei. CA1: hippocampal CA1 area; DG: Dentate gyrus; o: stratum oriens; p: striatum pyramidale; r: striatum radiatum; m: dentate molecular layer; g: granular cell layer; h: hilus proper.
Pum2 knockout affects hippocampal spine and synapse densities in vivo
We next investigated how loss of Pum2 affected dendritic spine and synaptic density of pyramidal cells in the cortex and hippocampus of wild-type (WT) and Pum2–/– mice using Golgi Staining. Pyramidal cells in the CA1 layer of the Pum2–/– hippocampus displayed significantly increased primary dendritic branching in apical dendrites as compared to age-matched WT mice (Pum2–/– 3.83 ± 0.20 vs. WT 2.83 ± 0.30; P < 0.01) (Figure 2A, 2B, 2E); however, these increases were restricted to the hippocampus and were not seen in the cortex (Pum2–/– 4.77 ± 0.18 vs. WT 4.72 ± 0.15; P > 0.05) (Figure 2C, 2D, 2F). Primary dendritic branch intersections were also quantified using Sholl analysis in consecutive 30, 60, 90, and 120 μm concentric bands radiating from the center of the soma. Similar to primary branch points, dendritic branch intersections within the 30μm band were significantly increased in the Pum2–/– hippocampus (Pum2–/– 3.83 ± 0.20 vs. WT 2.8 ± 0.30; P < 0.01) (Figure 2G) but not in the frontal cortex (Pum2–/– 4.78 ± 0.18 vs. WT 4.7 ± 0.15, P > 0.05) (Figure 2H) as compared to the same areas of WT mice. Interestingly, in consecutive 60 mm and 90 mm bands, dendritic branch intersections were significantly increased in the frontal cortex of Pum2–/– mice (60 mm: Pum2–/– 1.37 ± 0.18 vs. WT 0.82 ± 0.12, P < 0.01; 90 mm: Pum2–/– 0.34 ± 0.09 vs. WT 0.10 ± 0.05, P < 0.05,) but not the hippocampus at 60 and 90 mm (60 mm: Pum2–/– 1.4 ± 0.19 vs. WT 1.17 ± 0.24, P = 0.44; 90 mm: Pum2–/– 0.13 ± 0.09 vs. WT 0.083 ± 0.08, P > 0.05) compared to WT mice. Golgi staining also revealed significant increases in average spine density in CA1 pyramidal cells in Pum2–/– mice (Pum2–/– 24.86 ± 0.40 vs. WT 20.97 ± 0.26; P < 0.01) (Figure 3A, 3B, 3G) but not in layer IV of the frontal cortex (Pum2–/– 16.44 ± 0.36 vs. WT 16.10 ± 0.35; P > 0.05) (Figure 3C, 3D, 3H) or in CA1 interneurons (Figure 3E, 3F, 3I).
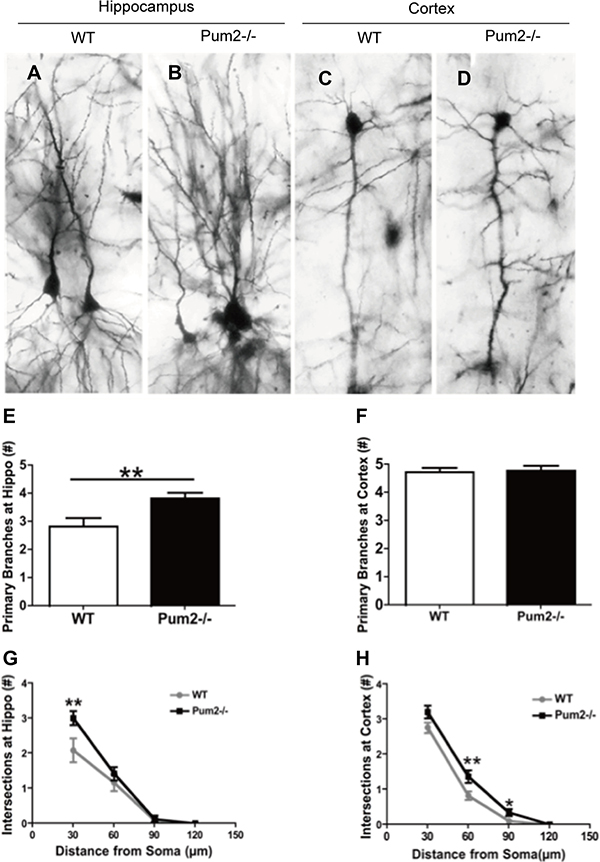
Figure 2: Dendritic branches of Pum2–/– and WT mice via Golgi staining. Representative pyramidal neurons in the CA1 area of the hippocampus (A: WT, l B:Pum2–/–) and the cortex (C:WT and D: Pum2–/–). There is a significant increase in primary dendritic branching in the Pum2–/– mice as compared to WT in the hippocampus (E) but not in the cortex (F). Additional measurements of the dendritic branch intersections using Sholl analysis in consecutive 30, 60, 90, and 120 μm concentric bands radiating from the center of the soma indicated a significant difference between Pum2–/– and WT mice (G and H). *means p < 0.05 and **means p < 0.01. Data represent as mean ± SEM.
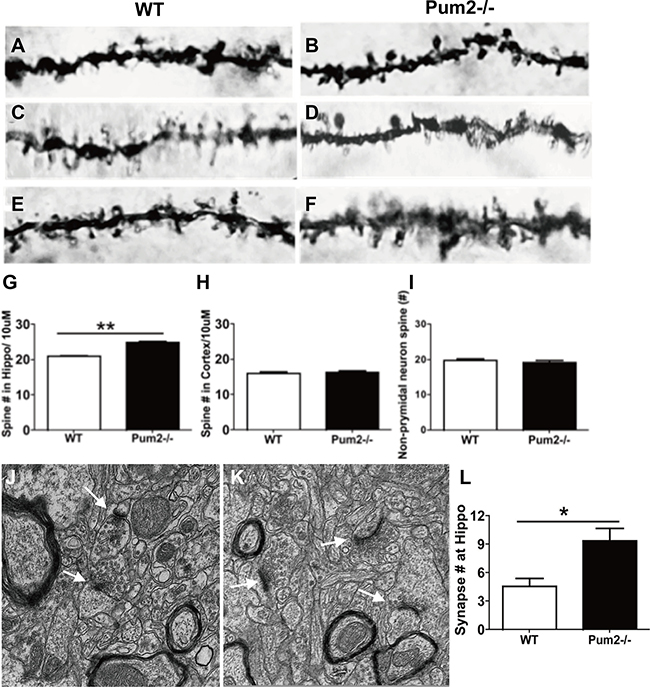
Figure 3: Spine density of Pum2–/– and WT mice by Golgi staining. Spine density significantly increased in the pyramidal cells within the CA1 area of hippocampus (A, B, G) but not in layer V of the frontal cortex (C, D, H) and non-pyramidal cells within the CA1 area of hippocampus (E, F, I). (J) Electron microscopy (EM) quantification of synapses in the stratum radiatum of the CA1. The representative images demonstrated the ultrastructure of synapses in the stratum radiatum in WT mice (J) and Pum2–/– (K) (white arrows indicate synapses). Quantification of synapse number revealed significantly higher number in Pum2–/– than WT mice (L). *means p < 0.05. Data represent as mean ± SEM.
Given the consistent changes in the proximal dendrites of the CA1, we analyzed the synaptic ultrastructure of these cells at the stratum radiatum using electron microscopy (EM). We found that Pum2–/– mice have significantly higher synaptic densities as compared to WT mice (Pum2–/– 9.40 ± 1.3 vs. WT 4.6 ± 0.81, P < 0.05), and interestingly, we found increased length and width of most postsynaptic density projections in the asymmetric but not symmetric synapses in Pum2–/– mice as compared to WT mice (Figure 3J, 3K, 3L). In total, these data further indicate that PUM2 regulates synaptic morphology in pyramidal neurons, and loss of PUM2 led to increased asymmetric but not symmetric synaptogenesis, which may predispose these mice to increased excitability through elevated synaptic transmission at excitatory synapses while inhibitory synapses remain unchanged.
Pum2 affects excitatory synapse proteins in the hippocampus
Pum2 clearly affects excitatory synaptic number and morphology in the hippocampus, suggesting synaptic machinery at excitatory synapses may be affected. Therefore, we measured several proteins located in the post-synaptic density that are linked to synaptic plasticity, particularly the glutamate receptors NR2A, NR2B, and GLUR2 (AMPA), phospho-cAMKII, an important member of the calcium/calmodulin-activated protein kinase family, and PSD95, an important post-synaptic scaffolding protein located at excitatory synapses. In addition, we quantified synaptophysin, a presynaptic protein, to determine if PUM2 has trans-synaptic effects. We found that GLUR2 and phospho-CAMK2 protein levels were significantly increased in the hippocampus of Pum2–/– mice (GLUR2: Pum2–/– 0.79 ± 0.03 vs. WT 0.38 ± 0.03, P < 0.05; phospho-cAMK2: Pum2–/– 0.68 ± 0.002 vs. WT 0.38 ± 0.05, P < 0.05) (Supplementary Figure 2), however, NR2A and NR2B levels were similarly expressed in Pum2–/– and wild-type mice (NR2a: Pum2–/– 0.47 ± 0.09 vs. WT 0.38 ± 0.028, P > 0.05; NR2b: Pum2–/– 0.23 ± 0.00004 vs. WT 0.25 ± 0.01, P > 0.05), suggesting PUM2 affects LTP-associated proteins downstream of NMDAR signaling (Supplementary Figure 2). In addition, we found PSD95 was also significantly increased in the hippocampus of Pum2–/– mice (Pum2–/– 0.84 ± 0.03 vs. WT 0.46 ± 0.061, P < 0.05). Importantly, the absence of PUM2 did not lead to significant changes in synaptophysin levels (Pum2–/– 1.40 ± 0.23 vs. WT 1.49 ± 0.20, P < 0.01), consistent with the postsynaptic function of PUM2. This data suggests that in conjunction with morphological changes in pro-excitatory signaling at the synaptic level, important post-synaptic, LTP-associated proteins were also up-regulated with loss of PUM2.
GLUR2 protein but not mRNA expression is increased in the absence of Pum2
As GLUR2 was up-regulated in the Pum2–/– hippocampus, and previous studies indicate that Drosophila Pumilio protein regulates translation of the Drosophila Glur2 homolog, Glur2a [33], we determined whether Glur2 mRNA in mice is also directly targeted by PUM2. Using immunofluorescence double labeling, we found substantial PUM2 and GLUR2 overlapping expression in the hippocampus. (Figure 4A–4D), with GLUR2 being significantly increased (Figure 4B, 4D, 4E) in Pum2–/– mice compared to WT. However, there was no significant difference in the total amount of Glur2 mRNA (Figure 4F), suggesting a post-transcriptional role for PUM2 in regulating GLUR2.
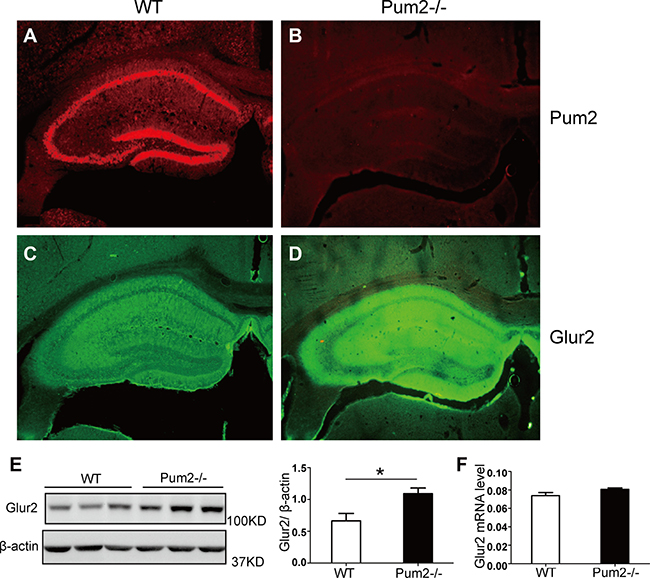
Figure 4: GLUR2 protein level but not mRNA was increased in the absence of Pum2. (A–D) Immunofluorescent staining showed that GLUR2 signaling is much stronger in the mutant hippocampus (n = 3). (E) Western Blot analysis showed that GluR2 protein levels were significantly higher in the hippocampus of Pum2 mutant mice as compared to WT mice, as analyzed by the relative signal intensity of western blots from three different animals. Mean ± SEM, *p < 0.05. (F) mRNA expression of Glur2 is not different between wildtype and Pum2–/– mutant hippocampus, suggesting that the increased GLUR2 protein in Pum2–/– mutant likely resulted from posttranscriptional regulation via PUM2 (n = 3 for each genotype). *means p < 0.05. Data represent as mean ± SEM.
These results were further confirmed in another independently generated Pum2 mutant, Pum2E67 mice, as western blot analysis demonstrated that hippocampal GLUR2 protein is up-regulated in absence of Pum2 (Figure 5A and 6A). Given that the increased number of synapses in Pum2 mutant tissues may also contribute to the increased GLUR2 protein level, we decided to determine if Glur2 translation is specifically increased in the mutants and hence contribute to the increase in GLUR2 protein.
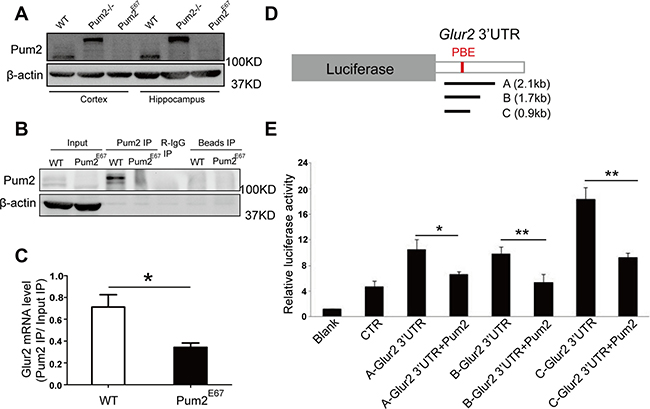
Figure 5: Glur2 mRNA is associated with PUM2, and PUM2 repressed reporter expression carrying Glur2 3′UTR. (A) PUM2 proteins are completely removed in Pum2E67 cortex and hippocampus. Pum2–/– does not have the full-length wildtype PUM2 proteins but still produced a bigger fusion protein. (B) Western blot of RNA immunoprecipitation experiment revealed that PUM2 proteins could be pulled down in PUM2 immunoprecipitates from the hippocampus (6 animals were used for each genotype). (C) Glur2 mRNA is significantly enriched in PUM2 pull-down, supporting Glur2 mRNA as a potential PUM2 target. Glur2 mRNA levels in the pulldown and input were relative to β-actin, and the enrichment of Glur2 mRNA in the pulldown was based on the Glur2 mRNA enrichment of pull-down over input. (D) Three fragments corresponding to areas of the 3′ UTR containing putative PUM binding motifs were cloned into dual luciferase reporter constructs. (E) PUM2 represses translation via the 3′UTR of Glur2 as shown in dual luciferase assays. Luciferase signals were significantly reduced for all three constructed plasmids carrying different lengths of Glur2 3′ UTRs.
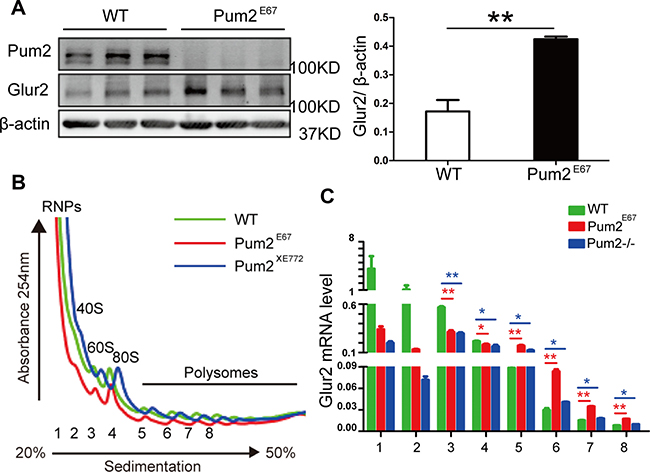
Figure 6: Hippocampal GLUR2 protein translation is significantly up-regulated in two separate loss-of-function Pum2 mutants. (A) Hippocampal GLUR2 protein levels were significantly higher in another Pum2–/– mutant, Pum2E67, while PUM2 protein is completely absent in the mutant hippocampus. On the left is the Western Blot analysis from three pairs of mice, on the right is the quantification of western blot signal intensity (3 animals for each genotype were used). (B) Overlay of polysome profiles from wild type (green solid line) and both Pum2–/– mutants (red and blue solid lines). Tissues from six animals per genotype were used for the experiment. (C) qRT-PCR analysis of Glur2 mRNA from separated fractions showed significantly increased Glur2 mRNAs in multiple polysome fractions (5, 6, 7 and 8) but not in the free RNP fraction (1). Fractions 2, 3 and 4 correspond to 40 S, 60 S and 80 S fractions. n = 6 for each genotype. The Glur2 mRNA expression data was relative to beta-actin expression from each fraction. *means p < 0.05 and ** means p < 0.01. Data represent as mean ± SEM.
Glur2 transcripts were associated with PUM2 protein complex in the hippocampus
As the regulatory role of PUM2 at cellular level is likely dependent on its ability to bind RNA, we determined if Glur2 mRNA is bound by PUM2. Indeed, immunoprecipitation with antibodies specific for PUM2 from hippocampal extracts demonstrated significant binding of PUM2 to Glur2 in WT but not in Pum2 knockout mutant (Pum2E67 mice) (Figure 5B and 5C), whereas non-target controls showed no difference between WT and Pum2 mutant RIP (data not shown). In line with this empirical data, a bioinformatic examination of the Glur2 reference sequence (NM_013540) revealed two PUM binding elements (PBE) in the 3′UTR of Glur2 (Supplementary Figure 3). These PBE sequences were highly conserved from fish to humans, though stricter conservation was seen for the first PBE than the second (Supplementary Figure 3).
PUM2 repressed hippocampal Glur2 translation through its 3′UTR
To determine if PUM2 regulates the expression of Glur2 through the 3′UTR of Glur2, we constructed a dual luciferase reporter construct carrying different lengths of the Glur2 3′UTR and measured the fluorescence of both reporters in the presence and absence of PUM2. As expected, the ratio of Firefly luciferase, which had the Glur2 3′UTR, to the Renilla luciferase, which was the internal control, significantly decreased when PUM2 was expressed (Figure 5D and 5E), demonstrating that PUM2 likely inhibits Glur2 translation via the 3′UTR of Glur2 mRNA. To our surprise, mutations in the two conserved PBEs failed to release PUM2-mediated translational repression (Supplementary Figure 3C), suggesting a mechanism involving sequence outside the two conserved PBEs.
Given that GLUR2 is increased in both Pum2 mutant mice brains (Pum2XE772 (Pum2–/–) and Pum2E67) (Figures 4E and 6A), and that in vitro assays suggest a regulatory role for PUM2 at the 3′ UTR of Glur2, we decided to investigate how PUM2 regulates GLUR2 protein expression in vivo. Accordingly, we performed polysome analysis on Glur2 translation in the hippocampus from both our Pum2 mutants (Pum2–/– and Pum2E67) and found that Glur2 transcripts are highly enriched in the polysome fractions but not in RNP fractions of both mutants compared to wildtype controls (Figure 6B and 6C), supporting translational repression of Glur2 by PUM2 through sequestration of Glur2 in RNPs and permissive translation of these transcripts with PUM2 ablation.
DISCUSSION
In this study, we have shown that PUM2 has a dramatic effect on excitatory synapses in pyramidal cells, especially in the hippocampus, at the structural and molecular levels. In particular, loss of PUM2 increases the dendritic branching, spines and synapse density in pyramidal cells, especially in the proximal, apical dendrites of CA1 pyramidal cells. In conjunction, PUM2 regulates the expression of many proteins that are normally up-regulated with post-synaptic mediated LTP but does not affect the upstream NMDARs, perhaps indicating a central role in modifying synaptic strength in response to plasticity-inducing stimuli. Finally, we showed that PUM2 directly regulates Glur2 through binding to the transcript’s 3′ UTR and sequestering it in RNPs, thereby repressing Glur2 expression and likely putting an important check on post-synaptic excitation. In total, our results demonstrate a plausible structural and molecular mechanism whereby PUM2 constrains aberrant excitation and plasticity in pyramidal cells and likely explains its dynamic role in epileptogenesis.
Since the discovery of Drosophila PUM gene in 1987, an increasing number of its homologs have been identified in other species with varying physiological functions [10, 11, 14]. PUM is necessary for embryonic patterning and is involved in multiple stages of germ cell development in Drosophila, and its extraordinary conservation across most, if not all, invertebrate and vertebrate species suggest a critical function in most eukaryotes [30, 38–41]. While it has been hypothesized that the ancestral function of PUM is in regulating germline stem cell proliferation and differentiation, but it’s clear that phylogenetically higher organisms have developed additional regulatory roles for these proteins [11, 14, 42]. In line with this, data suggest that PUM is required for synaptic growth, plasticity, and memory in Drosophila [32, 33, 43]. In mammals, PUM2 also appears to be involved in synaptic plasticity and neuronal activities [7, 13, 22, 35], suggesting that PUF family proteins may also have an integral role in neuronal function and circuit homeostasis. Consistent with this hypothesis, our data presented here depict PUM2 as an important regulator of excitatory transmission through dynamic control of synapse structure and number as well as the molecular machinery involved in synaptic strengthening.
Previously, we reported that the Pum2 is expressed in most brain regions in mice [23], and we’ve further confirmed this expression pattern with more detailed brain subregion distribution, demonstrating particular high expression of PUM2 in important temporal lobe areas, including the cortex, amygdala, and hippocampus, as well as noticeable expression in the thalamus, hypothalamus, and cerebellum (Supplementary Figure 1). Our PUM2 immunohistochemical staining further confirms that PUM2 protein is expressed similarly in these regions.
Intriguingly, Pum2–/– mutant mice exhibited significantly greater outgrowth of primary dendrites, dendritic arborization, spine density, and synaptic numbers in pyramidal neurons in the hippocampus but not in the cortex, suggesting that the impact of the dendritic morphology may be restricted to those areas. In addition, most of these increases in synaptic morphology were most prominent in the proximal dendrites of CA1 pyramidal cells, though the functional significance of this region-dependent regulation will still need closer investigation. These results establish an important role of PUM2 in dendritic morphogenesis and synapse function in a subregion-dependent manner not yet realized by previous in vitro studies [13].
Previous studies in Drosophila show that PUM is involved in long-term memory and alters both pre- and post-synaptic mechanisms of excitability at the neuromuscular junction, partly through regulating Glur2a and eIF-4e [33, 43]. While these experiments in flies are indicative of the role of PUM2 in memory, recent mammalian studies by our group (data not show) and others (Siemen et al. 2011) have not detected similar changes in hippocampus-dependent memory upon PUM2 ablation, making the role of PUM2 in mammals less clear. However, PUM2 ablation in mice leads to spontaneous epileptiform activity in the form of aberrant spike-wave discharges and a decreased seizure threshold to the seizure-inducing drug pentylenetetrazole [7, 22]. In line with this, patients with medically refractory TLE have reduced neocortical PUM2, and rats treated with pilocarpine leading to status epilepticus and subsequent spontaneous seizures days to weeks later also demonstrate a similar reduction in cortical and hippocampal PUM2 [37]. The pyramidal neurons of the hippocampus are the principal glutamatergic neurons [44–49], and the associated neuropil accounts for the most dendritic spines found in the hippocampal CA regions [50–54]. Impaired glutamatergic neurotransmission at the pre- and post-synaptic levels has been suggested to cause epileptic seizures, which are prominent in Pum2–/– mice observed in both our group and others [55–62]. It is well-established that an initial severe seizure may predispose an individual to future seizures – and certainly chemoconvulsant-induced status epilepticus with subsequent spontaneous seizures in rodents is a strong model of this form of epileptogenesis - but the multifaceted mechanism behind this phenomenon is still a hot topic for epilepsy researchers. Therefore, it’s particularly intriguing that seizure-induced loss of PUM2 persists in pilocarpine-treated rats well into the period where spontaneous seizures arise. The increased GLUR2 and phospho-CAMK2 expression in Pum2–/– mice found in our study, along with PUM2’s impact on dendritic morphogenesis and density within the hippocampus, may support a mechanism where translational de-repression of key postsynaptic proteins leads to increased excitation of pyramidal cells within the temporal lobe, leading to favorable conditions for seizure development in Pum2–/– mice. Increased number of synapses in the mutant may also contribute to the increased level of synaptic proteins such as GLUR2, such contribution, however, may be very limited as other synaptic proteins such as synaptophysin (Supplementary Figure 2) did not exhibit similar increase. Therefore, mammalian PUM2 may have a major role in the homeostatic regulation of excitatory signaling that is dysfunctional in TLE. Accordingly, an interesting avenue for future research will be to see if increased activation of the PUM2 regulatory pathway could represent a therapeutic strategy to reverse epileptogenic mechanisms in medically refractory patients.
PUM proteins are known to regulate target gene expression by binding the PBE motif on the 3′UTR of their target transcripts. Hence it is intriguing that mutated PBE in Glur2 3′UTR did not release the repression of PUM2 on luciferase expression, raising the possibility that PUM2 repressed Glur2 translation independent of these two consensus PBE sites. One possibility is that PUM2 regulate Glur2 translation indirectly, via other proteins or miRNAs. Consistent with this possibility, Glur2 was not identified as a direct target by PUM2 iClip [36] and PUM proteins were reported to regulate miRNA-mediated repression of their common targets [63]. Alternatively PUM2 bind to other sites slightly different from concensus PBE sites, further characterization of the smallest region (0.9 kb) subject to repression could distinguish the two possibilities.
Apart from epilepsy, it is reasonable to hypothesize that alterations in PUM2 may be associated with certain neuropsychiatric disorders that are caused by disruption of excitatory synapse homeostasis, especially those specific to hyperactivity or neurodegeneration. In support of this, PUM2 has been found to interact with multiple genes encoding functional proteins that are highly implicated in Alzheimer’s Disease, including amyloid precursor protein (APP), tau protein, and elF-4E [13, 32, 33]. Furthermore, abnormalities in synaptic pruning are thought to be a significant pathological mechanism leading to both schizophrenia and major depressive disorder [64, 65], and PUM2’s contribution to pruning during puberty is considered an important component of the transition from young to mature brain structures during normal development [66]. Our study and others suggest that PUM2 may have a broad effect in the brain function and may directly associate with multiple neuropsychiatric disorders; however, a closer look at the relationship between PUM2 and these specific neuropsychiatric disorders is warranted in future studies.
In summary, our study provides strong evidence that PUM2 plays a significant role in the regulation of synaptic structure, suggesting that neuronal function of PUF proteins may also be highly conserved in addition to their germline function. Future studies of investigating PUM2 during synaptic formation and procession in pathological conditions, such as epilepsy and schizophrenia, with a particular focus on glutamatergic signaling and resultant plasticity, could reveal mechanistic insights into these diseases and potentially lead to novel treatments.
MATERIALS AND METHODS
Animals
Pum2 knockout (Pum2–/–) mice were created from an ESC line carrying a gene trap insertion (XE772) in the Pum2 locus (Pum2XE772 or Pum2–/– mice) [23]. In Pum2–/– mice, no wildtype Pum2 transcripts were detectable [23]. The inserted genetrap vector contains a lacZ reporter under the control of the Pum2 promoter, hence lacZ expression could be used to track Pum2 mRNA expression on a cellular level in vivo. In addition to the Pum2–/– mice, Pum2E67 knockout mice were generated by removing exon 6 and exon 7 of Pum2 [67]. Unlike Pum2XE772 which still produce a truncated and non-functional chimeric protein between PUM2 and LacZ, the brain tissue of Pum2E67 do not have any part of PUM2 protein left (Figure 5A), making this allele an unambiguous null allele of Pum2.
Adult mice (3–8 months older) from both sexes were selected for this study. Each group contained 4–6 animals for morphological and biochemical studies. The animals were maintained in a temperature-controlled facility at 40–50% relative humidity and 20–21° C average room temperature on a 12 h light/dark cycle. Food and water were available ad libitum. Mice were housed and bred in the barrier facility in the Northwestern University Center for Comparative Medicine and at Nanjing Medical University. All procedures involving animals were carried out in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by both the Institutional Animal Care and Use Committees (ACUC) at Northwestern University and at Nanjing Medical University.
X-gal staining
Pum2–/– and wildtype littermates were perfused with 4% paraformaldehyde (PFA) and whole brains were dissected. The brains were in same fixation overnight at room temperature, transferred to 20% sucrose/PBS buffer for 24 hours, then embedded in Tissue-Tek O.C.T. (Electron Microscopy Sciences, Hatfield, PA, USA). Brains were cut in coronal section at 40 μm and mounted on slides. Sections were fixed in 2% PFA/PIPES buffer for 10 min on ice, and rinsed with Rinse Buffer (2 mM MgCl2/PBS) for 10 min on ice, then incubated in X-gal solution at 37° C overnight. Sections were rinsed with Rinse Buffer, 2% PFA/PIPES buffer, PBS + 2 mM MgCl2, (each for 5 min) then counterstained with Neutral Red and dehydrated with ethanol and mounted with Permount. Slides were examined on a Leica MRT compound microscope (Leica Camera AG, Solms, Germany), and images were captured with the Nikon DXM1200 camera and native ACT-1 image software (Nikon Corporation, Tokyo, Japan).
Dendritic number and spine density
Golgi staining was conducted using the Rapid Golgi Stain Kit (FD Neurotechnologies, Ellicott City, MD, USA) according to manufacturer’s instructions. Briefly, the brains were immediately removed and rinsed in 0.1 M phosphate buffer. Brains were immersed in a Golgi-Cox solution, replaced once after 12 hours of initial immersion, and stored at room temperature in darkness for 2–3 weeks. After the immersion period in the Golgi-Cox solution, brains were transferred to a cryoprotectant solution and stored at 4° C for at least 48 hours in the dark before sectioning. Brains were rapidly frozen with dry ice and cut with coronal plain at approximately 150 um thickness on a cryostat. The sections were transferred onto gelatin-coated slides and air dried at room temperature in the dark. After drying, sections were rinsed with distilled water and were subsequently stained in developing solution and dehydrated, cleared, and cover-slipped with Permount. Pyramidal cells from the CA1 layer of the dorsal hippocampus and from Layer IV of cortical sections directly superior to the dorsal hippocampus were compared across age-matched Pum2–/– and WT mice. Dendritic spine density was measured in the on screen live view using the Nikon DXM1200 camera, allowing spines from multiple focal planes to be counted at 1000× magnification by a researcher blind to genotype and experimental conditions. At 5 neurons per section, a total of 15 neurons from 3 sections per animal were selected in corresponding rostral-caudal locations. Spines were counted along the first 15–30 μm of the first primary dendrite branching from the large apical dendrite. Only cells with clearly visible dendrites and easily identifiable secondary structures and soma were included, with the selection of cells otherwise randomized within the chosen location. To allow for comparison of data and determination of relative spine density, the totals were standardized to reflect the number of dendrites per 10 μm span. While it is known that manually counting the number of spines from Golgi-stained slides will lead to gross underestimations, using the same method across all animals allows for a comparison of relative number of spines among the animals.
Overall primary dendritic length and secondary branching were evaluated via Sholl analysis. 30 μm concentric rings were drawn around neurons selected using the same criteria as described above by a researcher blind to genotype and experimental conditions. Measurements were made at 400× magnification. Five neurons per section and a total of three sections per animal were captured with Nikon ACT-1 software and analyzed in Image J (NIH-Image 1.62, Bethesda, MD, USA) by a blinded observer. Total dendritic length and branch intersections represent the sum of the Sholl output up to 150 μm away from the soma. The total number of primary branches was also counted from the same area of the hippocampus and cortex.
Synaptic density
For synaptic density analyzed by electron microscopy, animals were deeply anesthetized and perfused transcardially with 0.01M PBS containing heparin sodium for 2 min, followed by a 30 min perfusion with 2% paraformaldehyde, 2% glutaraldehyde, and 4% sucrose in 0.1M PBS. 250 μm sections were cut in the coronal plane using a vibratome. Fifteen sections encompassing the whole hippocampus were selected from each brain and were rinsed in cold 0.1 M PBS, treated with 2% OsO4 in 0.1M PBS for 90 min at 4° C, and rinsed again in 0.1 M PBS at room temperature. The sections were then dehydrated in a graded series of ethanol solutions, followed by propylene oxide, and left overnight in a 1:1 mixture of propylene oxide-Polybed 812 (Electron Microscopy Sciences, Hatfield, PA, USA). Finally, the sections were flat embedded in Polybed 812 in an oven at 60° C for 48–72 hours. From the 15 embedded sections, three representative sections included the dorsal, medial, and ventral hippocampus, and underlying cortices were selected for semi-thin and ultra-thin sectioning.
Selected embedded sections (250 μm thick) were trimmed and sectioned again using a Reichert Ultracut E Ultramicrotome (Austria). Semi-thin (1 μm) sections that included the hippocampus and underlying cortex were cut and stained with toluidine blue as reference sections for ultra-thin cutting. The sections were trimmed and ultra-cut. The thin (75–90 nm) sections containing the outer molecular layer of the dentate gyrus (dorsal blade) were mounted on 400-mesh grids (62 × 62 μm2; Electron Microscopy Sciences, Hatfield, PA, USA). The sections were stained using 3% uranyl acetate for 20 min followed by lead citrate for 5 min.
At low magnification under the electron microscope, the boundaries of the stratum radiatum of the CA1 subfield were identified according to their characteristic cellular structures. Six-ten photographs per section were taken systematically at 8000× magnification using alternate grid squares and 3 sections from each animal (18–30 photographs per animal), including the dorsal, medial, and ventral hippocampus, were assessed. Synapses were identified in electron micrographs that were enlarged photographically to a final magnification of 29,000×. Synapses were identified by the presence of synaptic vesicles and postsynaptic densities. All asymmetrical and symmetrical synapses were counted. The area of the unbiased counting frame was 247 μm2, the dissector height was 0.085 μm, and the dissector volume was 20.99 μm3. The latter value was used to calculate the density of synapses or degenerating axons (synapses or axons per unit volume) as the quotient of the mean number of synapses or axons counted per dissector, and the mean volume was examined using a JEOL 100 CX electron microscope (JEOL Ltd., Tokyo, Japan).
Immunohistochemistry
Mouse brains were dissected and fixed overnight in Hartman’s fixative (Sigma) and processed for immunohistochemistry according to standard protocols [68]. Immunostaining for PUM2 was performed following citrate buffer antigen retrieval by incubation with anti-PUM2 (Bethyl Lab) primary antibody and detected using Biotin-Streptavidin HRP Detection Systems (ZSGB-BIO). For PUM2 and GLUR2 co-localization in the hippocampus, brains were fixed with 4% paraformaldehyde (PFA) and cryostat cross-sectioned at 40 um thickness. The sections containing the dorsal hippocampus were blocked for 20 min with 1% bovine serum albumin in PBS. Immunoreactivity was detected using a rabbit anti-PUM2 polyclonal antibody (Milipore, 1:200) and mouse- anti-GLUR2 monoclonal antibody. Visualization of the second marker was accomplished using species-specific secondary antibodies conjugated with cyamine dye (Cy3), fluorescein isothiocyanate (1:200, Jackson ImmunoResearch, West Grove, PA, USA), or Alexa 488 (1:100; Molecular Probes, Eugene, OR, USA) for confocal microscopy (Olympus Fluoview).
Western blot
After mice were anesthetized and euthanized, the frontal cortex and hippocampus were quickly dissected on ice, snap-frozen, and stored at –70°C until biochemical analyses were conducted. The frozen brains were allowed to thaw on ice and were then homogenized in 5 volumes of ice-cold homogenization buffer (0.2% NP-40 and protease inhibitor in PBS buffer). Homogenates were centrifuged at 15,000 g for 20 minutes at 4° C, and the supernatant were used to measure protein levels in the brain (BCA method). 25 μg of each sample was further diluted in sample buffer (Bio-Rad, Hercules, CA, USA) and 15% polyacrylamide gels were used for SDS-PAGE. Proteins were transferred to PVDF membranes probed with primary antibodies against mouse PSD95 (1:1000 dilution) (Sigma-Aldrich, St. Louis, MO, USA); synaptophysin (1:500 dilution) (Cell Signaling Technology, Boston, MA, USA); glutamate receptor 2 (1:1000 dilution) (Millipore, Billerica, MA, USA); Rabbit PUM2 (Bethyl Laboratories, A300-202A; 1:500); mouse N-methyl-D-aspartate (NMDA) receptors 2A and 2B (1:500 dilution) (PhosphoSolutions, Aurora, CO, USA); phospho-cAMK2 (1:500) (Millipore); and β-actin (1:1000 dilution) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), followed by horseradish peroxidase (HRP)-conjugated secondary antibody binding (1:20,000) (BD Diagnostic Systems, Sparks, MD, USA). Immunoreactive proteins were visualized using the enhanced chemiluminescence Western blot detection system (Thermo Fisher Scientific, Waltham, MA, USA). Light-emitting bands were detected with X-ray film (Thermo Fisher Scientific). Quantification of individual bands was performed using ImageJ software (NIH-Image 1.62) by plotting density. The relative concentrations of target protein in each sample were measured by comparing target protein band density to β-actin band density in the same well.
Quantitative real-time PCR
Total RNA was isolated using Qiazol (Qiagen) and genomic DNA contamination was eliminated with TURBO DNase (Ambion). Reverse transcription of RNA was carried out with iScriptTM cDNA synthesis kit (Bio-Rad), according to the manufacturer’s protocol. Relative quantification of gene expression through the ΔΔCq method was conducted with the Applied Biosystems 7300 Real Time PCR System (Applied Biosystems) using the iTaq SybrGreen Supermix with ROX (Bio-Rad). PCR results were normalized to the expression of actin.
Immunoprecipitation
Hippocampi of wildtype or Pum2–/– mice were extracted with PLB buffer (100 mM KCL, 5 mM MgCl2, 10 mM Hepes, pH 7.0, 0.5% Nonidet P-40, 1 mM Dithiothrectol (DTT), 100 units/ml RNase OUT (Invitrogen-cat# 10777-019), supplemented with RNAse inhibitors and protease inhibitors. The lysate was pre-cleared with 15 ug of rabbit Ig and 50 μl protein G/A sepharose, then the protein concentration was measured. Hippocampal lysates were immunoprecipitated using protein A Sepharose beads (Amersham Pharmacia Biotech) and pre-incubated with 30 ug of anti-PUM2 (Bethyl Lab). Protein A conjugated Sepharose beads without antibodies were used as negative controls. After immunoprecipitation the beads were extracted with acid phenol-CHCI3 (Ambion) to isolate RNA, followed by qRT-PCR using oligo dT (Promega) and amplification using mouse Glur2 specific primers (Glur2 specific primers forward: 5′GCCGAGGCGAAACGAATGA3′ reverse:5′ CACTCTCGATGCCATATACGTTG3′ and mouse actin primers forward: 5′TGACCCAGATCATGTTTGAG3′ reverse:5′GAGTCCATCACAATGCCTG3′)
Dual fluorescent assay of PUM interaction with 3′UTR of Glur2
Three overlapping fragments from Glur2 3′UTR containing PBEs were subcloned into a Psi-check 2 plasmid (Promega) with XhoI and PmeI restriction enzymes (New England Lab). The 3′UTR fragments were amplified from brain cDNA using Glur2 3′UTR specific primers containing restriction enzyme sites for later cloning. The psi-check 2 plasmids were transfected in NIH3T3 cells in 48-well plates. After 24 hr, Firefly and Renilla luciferase expression were measured respectively using the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions in a Biotek Synergy 2 multi-mode Microplate Reader (Vermont, USA).
Polysome fractionation experiment
Polysome profile analysis was carried out as previously described [69]. Six hippocampus tissue from adult mouse of each genotype were treated with ice-cold PBS containing 100 μg/ml cycloheximide and subsequently lysed in a polysome lysis buffer (100 mM KCl, 0.1%Triton X-100, 50 mM HEPES, 2 mM MgCl2, 10% glycerol, 100 μg/ml cycloheximide, 1 mM DTT, 20 unit/ml RNase Inhibitor(EDTA free) and 1 × cocktail). Lysates were loaded onto 20–50% (w/v) sucrose density gradients (10 mM Tris-HCl [pH 7.5], 5 mM MgCl2, 100 mM NaCl and 1 mM DTT) and centrifuged at 38,000 rpm for 2.5 hr at 4° C in a Beckman SW41 Ti rotor. Gradients were fractionated, and the absorbance at 254 nm was continuously recorded using Gradient Fractionator (BioComp, Canada).
Data analysis
Two-sample comparisons were carried out using Student’s t-test (two-tailed), while multiple comparisons were made using one-way ANOVA followed by Tukey’s post hoc test. All data were presented as mean ± S.E.M., unless noted, and the limit for statistical significance was maintained at P value < 0.05. Values where P < 0.001 are regarded as highly significant.
Author contributions
E.Y.X. and H.D. conceptualized the research, E.Y.X., H.D., M. Z. and L. M. designed the study, M. Z., L. M., Y. D., D. Y., S. Z., W. Q. H.D. and E. Y. X. performed and interpreted experiments. E.Y.X. and H. D. supervised the study, and E.Y.X., H. D. and D. W. F. wrote the paper.
ACKNOWLEDGMENTS AND FUNDING
We would like to thank Yang Yang and Geethi Abraham for technical help. We also acknowledge the Welcome Trust Sanger Institute and Feinberg school of Medicine Transgenic core facility (TTML) for their help with transgenic mouse generation. This work was supported by National Basic Research Program of China (973 program, 2013CB945201 and 2015CB943002); National Science Foundation of China (81270737 and 31771652); Natural Science Foundation of Jiangsu Province (BK2012838); Provincial Innovation and Entrepreneurship Grant as well as NIH grant U01 HD045871. Funding for open access charge: Provincial Shuangchuan Program. W. Qiang was partially supported by NU-PSOC (U54 CA193419) and Robert H. Lurie Comprehensive Cancer Center OncoSET initiatives at the Northwestern University. This work is partially supported by the National Institute of Mental Health (R21 MH100919-01A1, 5R01 MH109466-2) to Hongxin Dong.
CONFLICTS OF INTEREST
None declared.
REFERENCES
1. Forner S, Baglietto-Vargas D, Martini AC, Trujillo-Estrada L, LaFerla FM. Synaptic Impairment in Alzheimer's Disease: A Dysregulated Symphony. Trends Neurosci. 2017; 40:347–57. https://doi.org/10.1016/j.tins.2017.04.002.
2. Henstridge CM, Pickett E, Spires-Jones TL. Synaptic pathology: A shared mechanism in neurological disease. Ageing Res Rev. 2016; 28:72–84. https://doi.org/10.1016/j.arr.2016.04.005.
3. Huang Z, Walker MC, Shah MM. Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis. J Neurosci. 2009; 29:10979–88. https://doi.org/10.1523/JNEUROSCI.1531-09.2009.
4. O'Sullivan GA, Jedlicka P, Chen HX, Kalbouneh H, Ippolito A, Deller T, Nawrotzki RA, Kuhse J, Kalaidzidis YL, Kirsch J, Schwarzacher SW, Betz H. Forebrain-specific loss of synaptic GABAA receptors results in altered neuronal excitability and synaptic plasticity in mice. Mol Cell Neurosci. 2016; 72:101–13. https://doi.org/10.1016/j.mcn.2016.01.010.
5. Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005; 6:312–24. https://doi.org/10.1038/nrn1648.
6. Liotti M, Mayberg HS, Brannan SK, McGinnis S, Jerabek P, Fox PT. Differential limbic—cortical correlates of sadness and anxiety in healthy subjects: implications for affective disorders. Biol Psychiatry. 2000; 48:30–42. https://doi.org/S0006-3223(00)00874-X.
7. Follwaczny P, Schieweck R, Riedemann T, Demleitner A, Straub T, Klemm AH, Bilban M, Sutor B, Popper B, Kiebler MA. Pumilio2 deficient mice show a predisposition for epilepsy. Dis Model Mech. 2017. https://doi.org/10.1242/dmm.029678.
8. Swanger SA, Bassell GJ. Making and breaking synapses through local mRNA regulation. Curr Opin Genet Dev. 2011. https://doi.org/10.1016/j.gde.2011.04.002.
9. Darnell JC, Richter JD. Cytoplasmic RNA-binding proteins and the control of complex brain function. Cold Spring Harb Perspect Biol. 2012; 4:a012344. https://doi.org/10.1101/cshperspect.a012344.
10. Lehmann R, Nusslein-Volhard C. Involvement of the pumilio gene in the transport of an abdominal signal in the Drosophila embryo. Nature. 1987; 329:167.
11. Wickens M, Bernstein DS, Kimble J, Parker R. A PUF family portrait: 3'UTR regulation as a way of life. Trends Genet. 2002; 18:150–7.
12. Murata Y, Wharton RP. Binding of pumilio to maternal hunchback mRNA is required for posterior patterning in Drosophila embryos. Cell. 1995; 80:747–56.
13. Vessey JP, Schoderboeck L, Gingl E, Luzi E, Riefler J, Di Leva F, Karra D, Thomas S, Kiebler MA, Macchi P. Mammalian Pumilio 2 regulates dendrite morphogenesis and synaptic function. Proc Natl Acad Sci U S A. 2010; 107:3222–7.
14. Quenault T, Lithgow T, Traven A. PUF proteins: repression, activation and mRNA localization. Trends Cell Biol. 2011; 21:104–12.
15. Zamore PD, Bartel DP, Lehmann R, Williamson JR. The PUMILIO-RNA interaction: a single RNA-binding domain monomer recognizes a bipartite target sequence. Biochemistry. 1999; 38:596–604. https://doi.org/10.1021/bi982264s.
16. Zamore PD, Williamson JR, Lehmann R. The Pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins. RNA. 1997; 3:1421–33.
17. Wharton RP, Struhl G. RNA regulatory elements mediate control of Drosophila body pattern by the posterior morphogen nanos. Cell. 1991; 67:955–67.
18. Zhang B, Gallegos M, Puoti A, Durkin E, Fields S, Kimble J, Wickens MP. A conserved RNA-binding protein that regulates sexual fates in the C. elegans hermaphrodite germ line. Nature. 1997; 390:477–84. https://doi.org/10.1038/37297.
19. Tichon A, Gil N, Lubelsky Y, Havkin Solomon T, Lemze D, Itzkovitz S, Stern-Ginossar N, Ulitsky I. A conserved abundant cytoplasmic long noncoding RNA modulates repression by Pumilio proteins in human cells. Nat Commun. 2016; 7:12209. https://doi.org/10.1038/ncomms12209.
20. Lee S, Kopp F, Chang TC, Sataluri A, Chen B, Sivakumar S, Yu H, Xie Y, Mendell JT. Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell. 2016; 164:69–80. https://doi.org/10.1016/j.cell.2015.12.017.
21. Prasad A, Porter DF, Kroll-Conner PL, Mohanty I, Ryan AR, Crittenden SL, Wickens M, Kimble J. The PUF binding landscape in metazoan germ cells. RNA. 2016; 22:1026–43. https://doi.org/10.1261/rna.055871.116.
22. Siemen H, Colas D, Heller HC, Brustle O, Pera RA. Pumilio-2 function in the mouse nervous system. PLoS ONE. 2011; 6:e25932. https://doi.org/10.1371/journal.pone.0025932.
23. Xu EY, Chang R, Salmon NA, Pera RAR. A gene trap mutation of a murine homolog of the Drosophila stem cell factor Pumilio results in smaller testes but does not affect litter size or fertility. Molecular Reproduction and Development. 2007; 74:912–21.
24. Zhang C, Zhu T, Chen Y, Xu EY. Loss of preimplantation embryo resulting from a Pum1 gene trap mutation. Biochem Biophys Res Commun. 2015; 462:8–13. https://doi.org/10.1016/j.bbrc.2015.04.019.
25. Gennarino VA, Singh RK, White JJ, De Maio A, Han K, Kim JY, Jafar-Nejad P, di Ronza A, Kang H, Sayegh LS, Cooper TA, Orr HT, Sillitoe RV, et al. Pumilio1 Haploinsufficiency Leads to SCA1-like Neurodegeneration by Increasing Wild-Type Ataxin1 Levels. Cell. 2015; 160:1087–98. https://doi.org/10.1016/j.cell.2015.02.012.
26. Schweers BA, Walters KJ, Stern M. The Drosophila melanogaster translational repressor pumilio regulates neuronal excitability. Genetics. 2002; 161:1177–85.
27. Baines RA. Neuronal homeostasis through translational control. Mol Neurobiol. 2005; 32:113–21.
28. Mee CJ, Pym EC, Moffat KG, Baines RA. Regulation of neuronal excitability through pumilio-dependent control of a sodium channel gene. J Neurosci. 2004; 24:8695–703.
29. Salazar AM, Silverman EJ, Menon KP, Zinn K. Regulation of synaptic Pumilio function by an aggregation-prone domain. J Neurosci. 2010; 30:515–22.
30. Ye B, Petritsch C, Clark IE, Gavis ER, Jan LY, Jan YN. Nanos and Pumilio are essential for dendrite morphogenesis in Drosophila peripheral neurons. Curr Biol. 2004; 14:314–21.
31. Fiore R, Khudayberdiev S, Christensen M, Siegel G, Flavell SW, Kim TK, Greenberg ME, Schratt G. Mef2-mediated transcription of the miR379-410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. Embo J. 2009; 28:697–710.
32. Menon KP, Andrews S, Murthy M, Gavis ER, Zinn K. The translational repressors Nanos and Pumilio have divergent effects on presynaptic terminal growth and postsynaptic glutamate receptor subunit composition. J Neurosci. 2009; 29:5558–72. https://doi.org/10.1523/JNEUROSCI.0520-09.2009.
33. Menon KP, Sanyal S, Habara Y, Sanchez R, Wharton RP, Ramaswami M, Zinn K. The translational repressor Pumilio regulates presynaptic morphology and controls postsynaptic accumulation of translation factor eIF-4E. Neuron. 2004; 44:663–76. https://doi.org/10.1016/j.neuron.2004.10.028.
34. Spassov DS, Jurecic R. Mouse Pum1 and Pum2 genes, members of the Pumilio family of RNA-binding proteins, show differential expression in fetal and adult hematopoietic stem cells and progenitors. Blood Cells Mol Dis. 2003; 30:55–69.
35. Vessey JP, Vaccani A, Xie Y, Dahm R, Karra D, Kiebler MA, Macchi P. Dendritic localization of the translational repressor Pumilio 2 and its contribution to dendritic stress granules. J Neurosci. 2006; 26:6496–508.
36. Zhang M, Chen D, Xia J, Han W, Cui X, Neuenkirchen N, Hermes G, Sestan N, Lin H. Post-transcriptional regulation of mouse neurogenesis by Pumilio proteins. Genes Dev. 2017. https://doi.org/10.1101/gad.298752.117.
37. Wu XL, Huang H, Huang YY, Yuan JX, Zhou X, Chen YM. Reduced Pumilio-2 expression in patients with temporal lobe epilepsy and in the lithium-pilocarpine induced epilepsy rat model. Epilepsy Behav. 2015; 50:31–9. https://doi.org/10.1016/j.yebeh.2015.05.017.
38. Chen G, Li W, Zhang QS, Regulski M, Sinha N, Barditch J, Tully T, Krainer AR, Zhang MQ, Dubnau J. Identification of synaptic targets of Drosophila pumilio. PLoS Comput Biol. 2008; 4:e1000026.
39. Lin H, Spradling AC. A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development. 1997; 124:2463–76.
40. Macdonald PM. The Drosophila pumilio gene: an unusually long transcription unit and an unusual protein. Development. 1992; 114:221–32.
41. Szakmary A, Cox DN, Wang Z, Lin H. Regulatory relationship among piwi, pumilio, and bag-of-marbles in Drosophila germline stem cell self-renewal and differentiation. Curr Biol. 2005; 15:171–8.
42. Parisi M, Lin H. Translational repression: a duet of Nanos and Pumilio. Curr Biol. 2000; 10:R81-3.
43. Dubnau J, Chiang AS, Grady L, Barditch J, Gossweiler S, McNeil J, Smith P, Buldoc F, Scott R, Certa U, Broger C, Tully T. The staufen/pumilio pathway is involved in Drosophila long-term memory. Curr Biol. 2003; 13:286–96.
44. Carlson ES, Tkac I, Magid R, O'Connor MB, Andrews NC, Schallert T, Gunshin H, Georgieff MK, Petryk A. Iron is essential for neuron development and memory function in mouse hippocampus. J Nutr. 2009; 139:672–9. https://doi.org/10.3945/jn.108.096354.
45. Fried I, MacDonald KA, Wilson CL. Single neuron activity in human hippocampus and amygdala during recognition of faces and objects. Neuron. 1997; 18:753–65. https://doi.org/S0896-6273(00)80315-3.
46. Lee AL, Ogle WO, Sapolsky RM. Stress and depression: possible links to neuron death in the hippocampus. Bipolar Disord. 2002; 4:117–28.
47. Sekiguchi M, Hayashi F, Tsuchida K, Inokuchi K. Neuron type-selective effects of activin on development of the hippocampus. Neurosci Lett. 2009; 452:232–7. https://doi.org/10.1016/j.neulet.2009.01.074.
48. Staba RJ, Wilson CL, Fried I, Engel J Jr. Single neuron burst firing in the human hippocampus during sleep. Hippocampus. 2002; 12:724–34. https://doi.org/10.1002/hipo.10026.
49. Szilagyi T, Orban-Kis K, Horvath E, Metz J, Pap Z, Pavai Z. Morphological identification of neuron types in the rat hippocampus. Rom J Morphol Embryol. 2011; 52:15–20. https://doi.org/520111015020.
50. Bausch SB, He S, Petrova Y, Wang XM, McNamara JO. Plasticity of both excitatory and inhibitory synapses is associated with seizures induced by removal of chronic blockade of activity in cultured hippocampus. J Neurophysiol. 2006; 96:2151–67. https://doi.org/10.1152/jn.00355.2006.
51. Bekkers JM, Stevens CF. NMDA and non-NMDA receptors are co-localized at individual excitatory synapses in cultured rat hippocampus. Nature. 1989; 341:230–3. https://doi.org/10.1038/341230a0.
52. Castro-Alamancos MA, Connors BW. Distinct forms of short-term plasticity at excitatory synapses of hippocampus and neocortex. Proc Natl Acad Sci U S A. 1997; 94:4161–6.
53. Lushnikova I, Skibo G, Muller D, Nikonenko I. Synaptic potentiation induces increased glial coverage of excitatory synapses in CA1 hippocampus. Hippocampus. 2009; 19:753–62. https://doi.org/10.1002/hipo.20551.
54. Salin PA, Scanziani M, Malenka RC, Nicoll RA. Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc Natl Acad Sci U S A. 1996; 93:13304–9.
55. Adesnik H, Li G, During MJ, Pleasure SJ, Nicoll RA. NMDA receptors inhibit synapse unsilencing during brain development. Proc Natl Acad Sci U S A. 2008; 105:5597–602. https://doi.org/10.1073/pnas.0800946105.
56. Farber NB, Newcomer JW, Olney JW. The glutamate synapse in neuropsychiatric disorders. Focus on schizophrenia and Alzheimer's disease. Prog Brain Res. 1998; 116:421–37.
57. Gambrill AC, Barria A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc Natl Acad Sci U S A. 2011; 108:5855–60. https://doi.org/10.1073/pnas.1012676108.
58. Goodwin GM, Martinez-Aran A, Glahn DC, Vieta E. Cognitive impairment in bipolar disorder: neurodevelopment or neurodegeneration? An ECNP expert meeting report. Eur Neuropsychopharmacol. 2008; 18:787–93. https://doi.org/10.1016/j.euroneuro.2008.07.005.
59. Groc L, Choquet D. AMPA and NMDA glutamate receptor trafficking: multiple roads for reaching and leaving the synapse. Cell Tissue Res. 2006; 326:423–38. https://doi.org/10.1007/s00441-006-0254-9.
60. Liu X, Chen C. Different roles for AMPA and NMDA receptors in transmission at the immature retinogeniculate synapse. J Neurophysiol. 2008; 99:629–43. https://doi.org/10.1152/jn.01171.2007.
61. Personius KE, Karnes JL, Parker SD. NMDA receptor blockade maintains correlated motor neuron firing and delays synapse competition at developing neuromuscular junctions. J Neurosci. 2008; 28:8983–92. https://doi.org/10.1523/JNEUROSCI.5226-07.2008.
62. Tackenberg C, Ghori A, Brandt R. Thin, stubby or mushroom: spine pathology in Alzheimer's disease. Curr Alzheimer Res. 2009; 6:261–8.
63. Kedde M, van Kouwenhove M, Zwart W, Oude Vrielink JA, Elkon R, Agami R. A Pumilio-induced RNA structure switch in p27-3' UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol. 2010; 12:1014–20.
64. Faludi G, Mirnics K. Synaptic changes in the brain of subjects with schizophrenia. Int J Dev Neurosci. 2011; 29:305–9. https://doi.org/10.1016/j.ijdevneu.2011.02.013.
65. McEwen BS, Nasca C, Gray JD. Stress Effects on Neuronal Structure: Hippocampus, Amygdala, and Prefrontal Cortex. Neuropsychopharmacology. 2016; 41:3–23. https://doi.org/10.1038/npp.2015.171.
66. Craik FI, Bialystok E. Cognition through the lifespan: mechanisms of change. Trends Cogn Sci. 2006; 10:131–8. https://doi.org/10.1016/j.tics.2006.01.007.
67. Lin K, Zhang S, Chen J, Yang D, Zhu M, Xu EY. Generation and functional characterization of a conditional Pumilio2 null allele. J Biomed Res. 2017 Nov 1st. https://doi.org/10.7555/JBR.32.20170117.
68. Shah C, Vangompel MJ, Naeem V, Chen Y, Lee T, Angeloni N, Wang Y, Xu EY. Widespread presence of human BOULE homologs among animals and conservation of their ancient reproductive function. PLoS Genet. 2010; 6:e1001022.
69. del Prete MJ, Vernal R, Dolznig H, Mullner EW, Garcia-Sanz JA. Isolation of polysome-bound mRNA from solid tissues amenable for RT-PCR and profiling experiments. RNA. 2007; 13:414–21. https://doi.org/10.1261/rna.79407.