
INTRODUCTION
Gastric cancer (GC) is the fourth most common cancer in the world, with an annual diagnosis of 1 million patients and a 700,000 annual mortality rate [1, 2]. The high mortality rate that is seen globally is mainly due to the advanced stage at diagnosis with few biomarkers for early detection and no systematic screening in the majority of the world outside of Japan and Korea.
Whilst the mechanism of gastric carcinogenesis is unknown, a model describing the histological progression of GC was proposed by Correa (Figure 1) [3]. This model predicts that infection with Helicobacter pylori triggers [4, 5] a multi-step cascade, with intestinal metaplasia (IM) as a key precursor lesion of GC usually resulting in intestinal type GC (IGC). IM is easily identified histologically by the presence of goblet cells and can be further subdivided, into Types I (complete), II and III (both termed incomplete) depending on the morphology of the cells as well as the types of mucin(s) they secrete. [6]. It has been suggested that incomplete IM (particularly Type III) is more associated with the development of GC [7], however this hypothesis has been disputed by others [8]. Whilst diffuse GC (DGC), the less differentiated subtype of GC, is not often associated with the distinct histological changes described by Correa there does appear to be an association with H. pylori and IM (Figure 1) [4].
Whilst the Correa model describes the distinct histological transition from IM to GC relatively little is known about the key genetic events which drive the IM to GC transition. A number of molecular events have been implicated in gastric carcinogenesis (reviewed in [9]). Of particular interest is the involvement of the tumour suppressor TP53, the most commonly mutated gene in human cancer [10]. It has been suggested that the frequency of TP53 mutations increases with the progression of GC from normal gastric mucosa, however, the reported frequency of mutations varies widely between studies [11-13]. This in part reflects the methodology used to define TP53 mutations with many studies incorrectly using p53 expression as a surrogate for mutations [14]. TP53 mutations have been shown to be a late event in the progression of some cancer types including colorectal [15]; however this remains to be systematically explored in GC.
In addition to direct mutations of the TP53 gene, a loss of p53 function can occur through elevation in the levels of one or more of its negative regulators. Among these, Mdmx (also known as Mdm4) and Mdm2 (known collectively as Mdm proteins) are the most powerful regulators of TP53. They are frequently altered in human cancer, and their expression is often positively correlated with that of wild-type p53 (reviewed in [16]). Mutations in TP53 usually result in the accumulation of mutant p53 protein, which disrupts the auto-regulatory loops with the Mdm proteins.
The protection of p53 from these two inhibitors has been shown to be an efficient approach for restoration of tumour suppression by p53 in relevant cancers with elevated Mdm proteins (reviewed in [16]). Recent studies have suggested that elevated levels of Mdm2 are detected in GC compared to adjacent normal tissue and correlate with poor prognosis [17], whilst the involvement of Mdmx has not been investigated to date. To our knowledge, the relationships of Mdm proteins have not been studied in premalignant stages of GC.
The aim of this study was to determine whether p53 protein expression and/or TP53 mutation status could be used as a biomarker to allow targeted screening of high risk groups with intestinal metaplasia of the stomach. We compared p53 levels and TP53 mutation status between a cohort of patients with early stage IM (defined as presence of gastric intestinal metaplasia for at least 5 years with no evidence of concurrent GC (IM-GC), and a cohort of IM patients which harboured concurrent GC which we term late stage IM (IM+GC) (Figure 1). This is also the first study to comprehensively analyse the relationship between TP53 mutation status and its negative regulators Mdm2/x in GC and related precursor lesions.
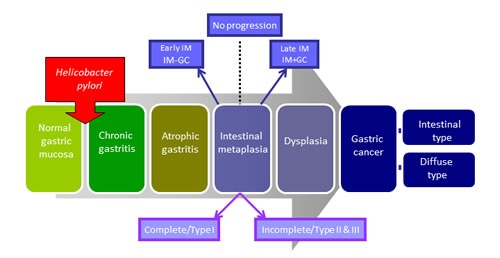
Figure 1: Progressive pathway to Gastric Cancer. First proposed by Correa, this schematic diagram outlines the progressive pathway to gastric cancer characterised by distinct histological changes. Normal gastric mucosa is infected with Helicobacter pylori which initiates an inflammatory response resulting in chronic, and then atrophic, gastritis. This is followed by intestinal metaplasia (IM) which can be further classified into complete and incomplete subtypes. At this point some patients will then proceed to GC via the intermediate stage of dysplasia. Whilst this model typically describes the pathway to intestinal type GC, these histological changes are often also seen in conjunction with the diffuse subtype of GC. For the purpose of this study IM samples collected adjacent to regions of GC during surgical resection are termed IM+GC- or late stage IM. Similarly IM samples collected from patients with no evidence of GC are referred to as IM-GC or early stage IM.
RESULTS
Subtype classification of IM samples
In order to determine whether incomplete IM is more associated with the development of GC, we used our study cohort to perform a histomorphological analysis. Complete IM, which most closely resembles the small intestine, is characterised by the presence of a brush border and well defined goblet cells which secrete sialomucins and, in some cases, sulfomucins (Figure 2A). Conversely, incomplete IM more closely resembles the large intestine, lacks a brush border, and comprises sialomucin secreting goblet cells and columnar cells which secrete a mixture of neutral and acid mucins (Figure 2B). All IM samples in our study cohort were stained with AB/PAS and H&E and their respective subtypes were identified based on morphology [18].
We observed that many IM samples in our cohort contain regions of both complete and incomplete subtypes. For each case, the proportion of complete and incomplete IM glands present in each sample was determined and is represented in Figure 2C (see also Table S1). This data suggests that early stage IM samples (see Figure 1), derived from cancer free patients (IM-GC), were comprised predominantly of the complete type (p<0.0001; Student’s t-test) whilst late stage IM, from patients with concurrent GC (IM+GC), (see Figure 1) was more closely associated with the incomplete type of IM (p<0.0001; Students t-test).
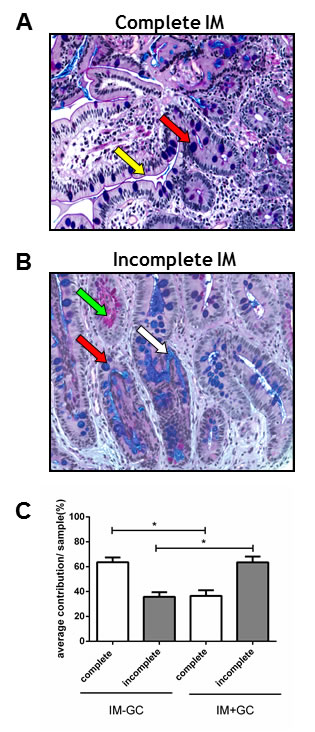
Figure 2: Classification of IM samples. All IM samples were stained with Alcian Blue periodic Acid Schiff (ABPAS) to ascertain the predominant IM subtype. The complete type of IM (A) is characterised by goblet cells (red arrow) and a brush border (yellow arrow) whilst the incomplete type (B) is characterised by the presence of neutral (green arrow) and acidic (white arrow) mucins as well as goblet cells (red arrow). (Magnification x200) (C) The percentage contribution of each IM subtype was determined for each IM sample in the cohort. These results were averaged giving the percent contribution of each IM subtype for both the IM-GC and IM+GC groups. Early IM (IM-GC) is more strongly associated with complete IM (p<0.0001; Student’s t-test) whilst late stage IM (IM+GC) is more correlated with the incomplete type (p<0.0001; Students t-test). Error bars represent SEM.
p53 immunostaining in normal, premalignant and GC tissue
To assess the expression of p53 within the cohort, sections were stained with an anti-p53 monoclonal antibody (D0-7).
Firstly, a direct comparison of p53 staining was made for the IM components of the two cohorts (Table 1) which were collected from patients without evidence of cancer (IM-GC) or patients with concurrent GC (IM+GC). Of the 62 samples in the IM-GC cohort, 32 (52%) showed positive staining as opposed to the IM+GC cohort where 24/32 samples (75%) showed positive staining (p=0.0451; Fisher’s exact test). In both groups, most of the samples only expressed p53 at low levels (82% IM-GC; 78% IM+GC).
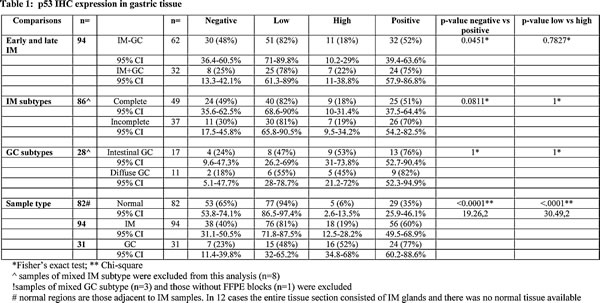
In order to more thoroughly characterise p53 expression levels in the two histological subtypes of IM, all relevant samples were pooled, irrespective of their cohort of origin, and reclassified based on their predominant histological subtype (Table 1; Table S1). Whilst 51% of the predominantly complete IM samples exhibited positive p53 staining this was increased to 70% in the predominantly incomplete IM samples although this did not reach statistical significance (p=0.0811; Fisher’s exact test). Mixed samples, which contained equivalent amounts of complete and incomplete glands, were excluded from this analysis (n=8).
Analysis of this association with respect to intestinal and diffuse subtypes of GC (Table 1) showed that both subtypes of GC exhibit high levels of p53 protein expression with no subtype predilection (p=NS, Fisher’s exact test). For one sample the GC FFPE block had been exhausted and this was not analysed. In addition three GC samples exhibited a combination of IGC and DGC subtypes (mixed) and were not included in this analysis.The overall expression of p53 in benign (normal regions of gastric mucosa, if present in the sample and adjacent to IM; n=82), premalignant (n=94) and GC (n=31) samples within the cohort was assessed (Table 1). This data shows that p53 expression levels incrementally increase with advancing stages of GC progression (p<0.0001; Chi square) and suggests that increased p53 protein expression in IM could be a predictive marker of developing GC.
TP53 mutations are only detected in established GC
In order to determine whether increased p53 expression was associated with somatic mutations in the TP53 gene, HRM analysis for regions covering the entire TP53 gene was performed. Samples were selected based on the availability of fresh frozen tissue for extraction (see Table S2 for clinical characteristics). For all samples needle macrodissection was performed after pathological review to enrich for areas of IM or cancer. As shown in Table 2, 13 of the 23 GC cases (56.5%) tested were positive by HRM. The identified mutations were then validated and characterised by Sanger sequencing. Of the 13 positive samples, four (30.1%) harboured mutations in exon 7, three (23.1%) in exon 6, three (23.1%) in exon 8, two (15.4%) in exon 5 and one (7.7%) in exon 4. The majority of the detected mutations (7/13 (53.8%)) were missense mutations and predicted to cause a non-functional protein. One patient (P#1816) harboured independent TP53 mutations in exons 2 and 5. Of the samples analysed, TP53 mutations were more commonly observed in GC samples of the intestinal subtype (10/15; 66%) compared to the diffuse subtype (2/7; 28.5%).
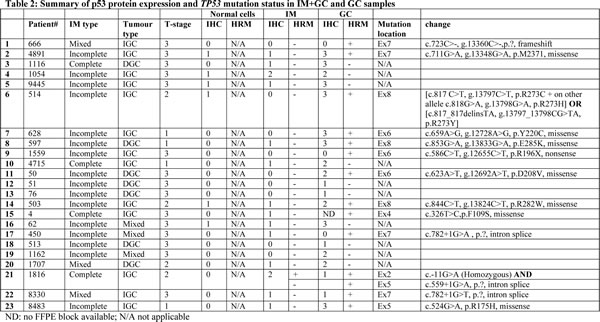
HRM was then performed on the paired IM samples from the GC cases above to determine whether the corresponding mutations were already established in the premalignant stage (Table 3). In most (22/23) IM+GC cases the mutation identified in the GC samples was absent in matched IM. The exception was P#1816 which contained a TP53 mutation at exon 5 in both the tumour and matched IM sample. Further analysis of peripheral blood DNA obtained from this patient showed this to be a novel germline variant. The observation that no somatic mutations were detected in TP53 for the IM component of the IM+GC cases provides strong evidence that TP53 mutation occurs late in the transition event between IM and prior to the development of GC.
Table 3: Summary of p53 protein expression and mutation status in IM-GC samples
IM |
||||
Patient# |
IM type |
IHC |
HRM |
|
1 |
10-038-01 |
Incomplete |
3 |
- |
2 |
2006-009 |
Incomplete |
1 |
- |
3 |
09-032-01 |
Incomplete |
2 |
- |
4 |
10-043-01 |
Incomplete |
2 |
- |
5 |
09-0025-01 |
Incomplete |
1 |
- |
6 |
2007-0015 |
Incomplete |
ND |
- |
7 |
09-0028-01 |
Complete |
0 |
- |
8 |
08-020 |
Complete |
0 |
- |
9 |
08-021 |
Complete |
2 |
- |
10 |
09-021-02 |
Complete |
2 |
- |
11 |
09-024-01 |
Complete |
2 |
- |
12 |
2008-0017 |
Complete |
0 |
- |
13 |
10-037-01 |
Complete |
2 |
- |
14 |
09-033-03 |
Complete |
2 |
- |
ND: no FFPE block available
To further validate this finding, 14 early stage IM samples i.e., from non-cancer patients (IM-GC) were also screened using HRM. There were no mutations in TP53 detected in any of these cases. Thus, although p53 expression may be elevated in IM this is wild type protein and harbours no somatic mutations in early or late IM. Mutations were only found in GC.
Wild-type TP53 is associated with Mdm2/x expression
Sequence analysis of TP53 in the patient samples revealed an absence of mutations in premalignant lesions and a strong selection for mutation in a high proportion of GC cases. Expression of p53 protein however increased incrementally with disease progression even in the premalignant stages. In cancers, where expression of wild- type TP53 is maintained, it is often associated with the expression or amplification of a p53 negative regulator such as Mdm2 or Mdmx. We therefore examined whether Mdm2 and/or MdmX proteins are overexpressed in IM and/or matched GC samples, and how this correlated with p53 expression using serial tissue sections.
Firstly, IM samples were analysed by identifying the region of IM with the highest level of p53 protein expression. The corresponding region was located on the Mdm2 and Mdmx stained serial slides and the respective staining patterns for these proteins were determined (Table S3; representative images in Fig S1). For the purpose of this analysis a sample was considered to have elevated p53 protein expression if greater than 10% of cancer cells were immunopositive (i.e., IHC score of 2+ or 3+; referred to as H in Table S3). A similar 10% immunopositive cutoff was also used to define elevated Mdmx and/or Mdm2 proteins in the same location. Data for Mdm2 and MdmX were then combined and those having elevated levels of one or both proteins classified as positive (Table S3).This data was analysed in combination with the previously generated TP53 mutation data using criteria described previously [19] to further classify the samples. Results are detailed in Table S3 and summarized in Table 4.
Table 4: Summary of p53 and Mdm alterations at genetic and protein levels in IM+GC amd GC samples
IM+GC (n=21) |
|||
WT TP53 (100%) |
Mutant TP53 (0%) |
||
alterations in p53 protein |
alterations in Mdm2/x gene(s) or protein(s) |
||
+ |
+ |
3 (14.3%) |
0 (0%) |
+ |
- |
0 (0%) |
0 (0%) |
- |
+ |
9 (42.9%) |
0 (0%) |
- |
- |
9 (42.9%) |
0 (0%) |
GC (n=23) |
|||
WT TP53 (100%) |
Mutant TP53 (0%) |
||
alterations in p53 protein |
alterations in Mdm2/x gene(s) or protein(s) |
||
+ |
+ |
6 (26.1%) |
2 (8.7%) |
+ |
- |
2 (8.7%) |
6 (26.1%) |
- |
+ |
2 (8.7%) |
1 (4.3%) |
- |
- |
0 (0%) |
4 (17.4%) |
In the IM component of patients with associated GC (IM+GC), the TP53 gene was exclusively wild-type. Elevation of p53 protein was only observed in conjunction with elevated Mdm protein expression (3/21; 14.3%). In the absence of p53 expression, elevated Mdm2/x protein expression was frequently observed (9/21 samples; 42.9%; Table 4) suggesting that p53 is kept in check as predicted through elevated Mdm2/x.
GC samples were analysed in a similar manner however in addition, quantitative real time PCR was used to determine DNA copy number levels of MDM2 and MDMX in GC cases with wild-type TP53; Table S3). For this analysis a sample was also considered to be altered in MDM2/X if the copy number status for either (or both) of these genes was more than diploid. Again, data for the MDM locus were then combined and those having elevated levels of one or both proteins and/or a copy number increase in one or both genes were classified as positive (Table S3).
During the progression to GC, most samples had elevated p53 protein expression, irrespective of TP53 mutation status. Elevation of p53 protein expression in conjunction with elevated Mdm protein expression and/or MDM copy number alterations were more commonly detected in samples with wild-type TP53 (26.1%; Table 4; Table S3). The majority of the observed Mdm protein changes were due to a preferential elevation of Mdmx rather than Mdm2 expression (Table S3). In cancers harbouring a wild-type TP53 gene, amplification of MDMX was detected in 4/10 (40%; Table S3) samples and was restricted to those of the diffuse or mixed subtypes. Whilst most of these were low level gains of an extra 1-2 copies, one sample harboured high level (>20 copies) copy number gain in both MDM2 and MDMX genes. These results strongly implicate the involvement of MDMX in maintaining the wild-type TP53 status observed in IM.
DISCUSSION
Poor outcome commonly associated with GC can, in part, be attributed to the late stage of diagnosis of the disease. IM is a well-known pre-neoplastic lesion associated with GC, which is able to persist for many years before progressing to neoplasia. This long latency period between IM and GC [9] represents an ideal time for intervention, however not all patients with IM will progress and thus it is important to be able to identify those patients at risk of developing the disease. Here we show that the incomplete type of IM is significantly more likely to be associated with the development of GC and this subtype is more commonly seen in later stages of the GC progression pathway. This histological subtype of IM can be thought to be a more genomically susceptible stage of disease, and consistent with other studies, more at risk of progression to cancer [7, 18, 20].
Due to the long latency period associated with GC it is conceivable that identification of the key molecular changes which drive the progression to GC in a subgroup of patients could aid clinicians in assigning risk to individuals with IM. Mutations in the TP53 gene are associated with many cancer types and often inactivate the tumour suppressor activity of the protein, including loss of transcriptional activation of its negative inhibitor MDM2. Reduced Mdm2 levels, together with altered degradation processes increases p53 half-life, which subsequently results in the nuclear accumulation of p53 [21]. Antibodies utilised for IHC studies cannot easily distinguish between wild-type and missense mutant p53 proteins, due to loss of conformation during fixation. Most studies on GC and associated premalignant conditions have utilised p53 expression as a surrogate marker for TP53 mutation (reviewed in [10]) with only a few sequencing the gene. However, since mutant p53 is not always stable and wild-type p53 is also stabilized under stress conditions; existing studies based on IHC staining alone are insufficient, accounting for much of the discordance between reported studies about p53 in GC.
p53 expression in normal gastric mucosa has been reported in only a few studies and was only detected in a small proportion of cells [22, 23]. This is consistent with our study where only between 0-10% of cells were immunopositive in 94% of samples tested. The disconcordance in published results may, in part, be attributed to the recognition sites of the antibody used but is more likely due to differences in the scoring criteria applied to define those samples with positive staining.
Elevated p53 protein levels have previously been reported in both IM and GC [22, 24]. We sought to determine whether p53 expression and/or mutation could be utilised as a biomarker to predict progression of patients with IM to GC. Our results show a significant increase in p53 protein expression during the transition from normal gastric mucosa to IM and then to GC. The level of p53 expression in IM varies among studies from 0-42% [14, 22, 25-27] and appears to differ depending on the type of IM and whether the IM was collected from cancer free patients or those with concurrent GC. This is consistent with our data where p53 expression was higher in IM with concurrent GC.
Our analysis also revealed a switch in TP53 mutation status between the IM (0%) to GC (56.5%). The frequency of mutations observed in our GC samples is consistent with existing literature and like other studies we also observed that TP53 mutations were more common in IGC compared to DGC [13, 28]. Our data strongly implicates wild-type p53 as an important checkpoint in the progression of GC suggesting that TP53 mutation is a pivotal event in the neoplastic transition from IM to GC.
Only two previous studies [11, 24] have reported the presence of mutations in IM samples however, in both studies microdisection was not used to isolate IM prior to analysis which may have resulted in contamination by adjacent tumour cells.
The absence of TP53 mutations in IM, despite high protein expression, argued that p53 is present, but ineffective possibly due to suppression of its activity. Key regulators involved in the suppression of p53 activity are Mdm2 and Mdmx. Mdm2 protein is the major E3 ligase of p53, acting as the major regulator of p53 protein stability, maintaining p53 as a labile protein with a short half-life [29]. Mdmx interacts with the transactivation domain of p53 and blocks its transcriptional activity however, Mdmx has also previously been shown to compete with Mdm2 for p53 binding, resulting in the accumulation of wild-type p53 under conditions of stress [30]. In contrast to existing literature [19] we observed negligible levels of Mdm2 staining in all IM and GC samples studied. Intriguingly, when examining IM and its matching tumour we observed substantial increase in Mdmx expression in the majority of IM samples (12/21 samples; 57%; Table S3), which further increased in GC samples with wild-type p53 (8/10; 80%; Table S3). These findings strongly implicate Mdmx in IM and suggest that elevation in Mdmx protein expression may occur in IM, suppressing p53 activity. Elevation in p53 levels due to inflammatory response to H. pylori may select for elevation in Mdmx levels. This is likely to reflect further elevation in p53 levels as the cells progress from IM to GC and further oncogenic stresses that drive this transition. In this particular study the H. pylori status (determined by histology or serology) of the patient did not correlate with the accumulation of TP53 mutations (Table S2).
The novel finding of the alteration in Mdmx expression in the IM to GC transition particularly in those tumour samples harbouring a wild-type TP53 gene opens an opportunity for targeted intervention either in chemoprevention of GC or in GC therapy. Reactivation of p53 has been long considered an attractive therapeutic strategy. The proof of principle of this approach has been successfully demonstrated in mouse models whereby temporal restoration of p53 resulted in tumour regression [31-33]. Specifically, the efficacy of protecting p53 from elevated Mdmx and triggering tumour suppression has been demonstrated in a retinoblastoma model [34] and more recently in melanoma [35]. Several therapeutic approaches for the inhibition of Mdmx are currently under development. These include small molecule and stapled peptides designed to inhibit p53-Mdmx interaction, and compounds intended to suppress Mdmx protein expression (reviewed in[16]). Such treatments are predicted to be suitable for IM patients, which express wild-type p53 with elevated Mdmx, and have a strong likelihood of progressing to GC.
The results described in this study are consistent with the following model involving the Mdmx-p53 axis in disease progression from IM to GC (Figure 3). During IM and in the context of a chronic inflammatory response (possibly due to chronic H. pylori infection), p53 is activated and accumulates. This results in a selection for elevation in Mdmx expression in order to suppress senescence by p53. At late stage of progression, most likely during dysplasia, further stress conditions drive a selection for TP53 mutations, which contributes to the transition to GC. In cases where p53 remains wild-type Mdmx expression remains high and contributes to GC by driving p53-independent pathways.
In conclusion, we describe a model for the frequent overexpression of p53 in premalignant stages of GC and suggest how wild type p53 can be sustained in these non-neoplastic tissues by the expression of the p53 regulator Mdmx. This offers a new target for therapy of GC and a potential avenue for the chemoprevention of GC.
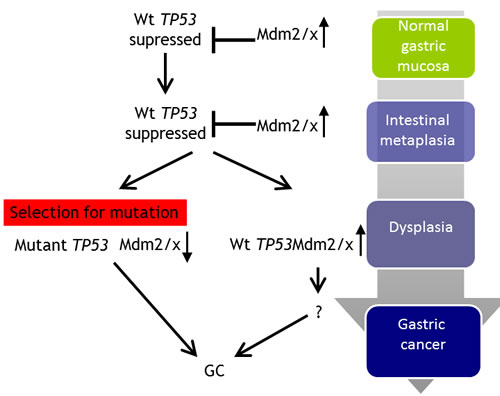
Figure 3: Proposed model for the mechanism of TP53 and MDM2/X gastric cancer. In normal gastric mucosa TP53 is wild-type and its expression is kept in check by Mdm2 and Mdmx. During progression to the intermediate stage of IM the TP53 gene continues to be maintained in the wild-type state but protein expression levels are slightly elevated, possibly due to a decreasing Mdm2/x expression in a small proportion of cases. Mutations in TP53 occur at late stage in the progressive pathway in 56% of GC cases, accompanied by a decrease in Mdm2/x levels. In those GC samples which maintain a wild-type TP53 status Mdm2/x levels continue to remain elevated.
MATERIALS AND METHODS
Sample cohort
Gastric IM (IM+GC) and matching tumour samples (GC) were collected from patients undergoing curative or palliative resection for GC from Melbourne, Australia. Tumour sections were obtained from viable tumour tissue. Non-malignant mucosa at least 2cm from the macroscopic tumour margin but from the same anatomical region was also resected and, based on pathological review, those were found to contain regions of IM were selected as the IM+GC cohort. Within 30 minutes of resection, samples were divided for storage into either liquid nitrogen (fresh frozen) or fixed in either formalin or 70% ethanol.
Samples that comprise the IM-GC cohort were gastric biopsies collected during routine endoscopies of patients known to have harboured gastric IM for at least 5 years duration without developing gastric cancer. Biopsies were placed immediately into liquid nitrogen or 10% neutral buffered formalin. Written informed consent was obtained from all patients prior to tissue collection. The procedures carried out in this study were reviewed and accepted by the Institutional Review Board at each of the collection centres.
IM and GC subtyping
Ethanol or formalin fixed tissue sections were paraffin embedded for histological use. 5 micron sections of each sample were stained with haematoxylin and eosin (H&E) and Alcian-Blue Periodic Acid Schiff (AB/PAS) and histologically reviewed independently by two pathologists. IM samples were assessed for the proportion of complete and incomplete subtypes and GC samples were classified as IGC, DGC or mixed (combination of IGC and DGC) subtypes. These results were compared to the pathology record prepared by the contributing institution. Any variations were further reviewed until a consensus was reached.
Immunohistochemistry
Immunohistochemistry and scoring of p53 stained individual sections
Immunohistochemistry (IHC) for p53 was performed as described in the Supplementary Methods. Nuclear staining of samples were scored as follows: 0 (completely negative), 1+ (<10% cells positive) 2+ (10-49% cells positive) and 3+ (>50% cells positive). For analysis samples were divided into 4 groups: completely negative (score 0), low (score 0 or 1), high (score 2 or 3) and positive (score 1, 2 or 3). For IM samples p53 protein expression was only assessed in the glands with IM. P53 protein expression was quantitated separately for the adjacent non-IM regions and were classified as normal. For GC samples only regions of tissue containing tumour cells were assessed.
Immunohistochemistry and scoring of p53/mdmx/mdm2 serial sections
IHC for p53, Mdmx and Mdm2 was performed as described in the Supplementary Methods. In this analysis, assessment of all antigens was made by identifying the region with highest p53 expression and determining the percentage of positive tumour cells. Based on methods described previously by Gunther et al.,[19] a sample was considered to have elevated p53 protein expression if greater than 10% of the cells were immunopositive. The matching region was located in the Mdmx (nuclear staining) and Mdm2 (nuclear membrane staining) stained slides and these were scored in the same manner. A cutoff of 10% positive immunostaining was used to identify elevated Mdm protein expression and a copy number change greater than diploid, determined by RT-qPCR in either MDM2 or MDMX was used to identify genomically altered samples based on methods previously described [19] .
HRM and TP53 sequencing
Fresh frozen tissue was sectioned and macro-dissected to enrich for tumour or IM purity after examination by a pathologist (CM). DNA was extracted using DNeasy tissue kit (Qiagen, Germany). High resolution melting analysis (HRM) was used to screen for mutations in exons 2-11 of the TP53 gene using the primers previously reported in [36]. Three different normal controls were included in each run and, when available, a positive control sample was included for each amplicon. DNA samples were tested both directly and spiked with normal control DNA (in a sample/control DNA ratio of 3:1). The practice of testing samples admixed with a normal control DNA enables heteroduplex formation, thus allowing more ready identification of homozygous and hemizygous mutations. PCR and HRM were performed on the LightCycler 480 (Roche, Penzberg, Germany). The reaction mixture included 1x PCR buffer containing 1.5 mM of MgCl2, 1mM of extra MgCl2 (for a final concentration of 2.5 mM MgCl2), 200 nM of each primer, 200 µM of dNTPs, 0.25 U of HotStarTaq polymerase (Qiagen, Hilden, Germany), 5 µM of Syto9 (Invitrogen, Carlsbad, CA), either 5 or 2.5 ng DNA (depending on the amount of sample DNA available) and PCR grade water in a total volume of 10 µL. PCR conditions included an activation step of 15 minutes at 95oC; 65 PCR cycles (95oC for 10 seconds, annealing for 10 seconds comprising 10 cycles of a touchdown from 65 to 55oC at 1oC/cycle, and extension at 72oC for 30 seconds), one cycle of 95oC for 1 minute, one cycle of 45oC for 1 minute, followed by HRM from 72 to 95oC increasing by 0.02oC per second. PCR reactions were performed in duplicate. Samples showing a melting profile different to the controls were directly sequenced from a 1/10 dilution of the HRM product using the BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. Certain samples, which showed heteroduplexes in multiple amplicons, were re-tested by performing the same PCR/HRM assay after treatment with 0.5 U/reaction of uracil-DNA glycosylase (UDG) to eliminate the templates in which cytosine had been deaminated to uracil, as reported in [37]. The IARC TP53 database was used to identify novel variants.
Quantitative-PCR (qPCR) for MDM2 and MDMX
MDM2 and MDMX gene copy number was determined as described previously [38]. Briefly, 2ng genomic DNA, 1µM of each forward and reverse primer and SYBR green master mix were used to amplify the gene of interest. Two independent primer pairs were used for each gene. Target gene quantity is given as the average log2 ratio value obtained for each primer pair after normalization to the repetitive element Line-1 as a reference gene and a normal DNA reference.
Statistical analysis
Statistical analysis was performed using Graphpad Prism for Windows (Version 5). Statistical tests used are described in the text and figure legends. In all instances the level of significance was set at p<0.05.
CONFLICT OF INTEREST
None
ACKNOWLEDGEMENTS
This work was supported by the NHMRC (AB grant 288714 and YH Fellowship 9628426) and VCA (AD).
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Parkin DM. International variation. Oncogene. 2004; 23(38):6329-6340.
2. Parkin DM, Bray F, Ferlay J and Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005; 55(2):74-108.
3. Correa P. A human model of gastric carcinogenesis. Cancer Res. 1988; 48(13):3554-3560.
4. Boussioutas A, Li H, Liu J, Waring P, Lade S, Holloway AJ, Taupin D, Gorringe K, Haviv I, Desmond PV and Bowtell DD. Distinctive patterns of gene expression in premalignant gastric mucosa and gastric cancer. Cancer Res. 2003; 63(10):2569-2577.
5. Rubio CA, Hirota T, Itabashi M, Mandai K, Yanagisawa A, Kitagawa T, Sugano H and Kato Y. Extended intestinal metaplasia. A survey of 1392 gastrectomies from dwellers of the Pacific basin. Anticancer Res. 2004; 24(5B):3185-3192.
6. Jass JR and Filipe MI. A variant of intestinal metaplasia associated with gastric carcinoma: a histochemical study. Histopathology. 1979; 3(3):191-199.
7. Filipe MI, Potet F, Bogomoletz WV, Dawson PA, Fabiani B, Chauveinc P, Fenzy A, Gazzard B, Goldfain D and Zeegen R. Incomplete sulphomucin-secreting intestinal metaplasia for gastric cancer. Preliminary data from a prospective study from three centres. Gut. 1985; 26(12):1319-1326.
8. Kang KP, Lee HS, Kim N, Kang HM, Park YS, Lee DH, Choe G, Kim JS, Jung HC and Song IS. Role of intestinal metaplasia subtyping in the risk of gastric cancer in Korea. J Gastroenterol Hepatol. 2008.
9. Busuttil RA and Boussioutas A. Intestinal metaplasia: a premalignant lesion involved in gastric carcinogenesis. J Gastroenterol Hepatol. 2009; 24(2):193-201.
10. Fenoglio-Preiser CM, Wang J, Stemmermann GN and Noffsinger A. TP53 and gastric carcinoma: a review. Hum Mutat. 2003; 21(3):258-270.
11. Shiao YH, Palli D, Caporaso NE, Alvord WG, Amorosi A, Nesi G, Saieva C, Masala G, Fraumeni JF, Jr. and Rice JM. Genetic and immunohistochemical analyses of p53 independently predict regional metastasis of gastric cancers. Cancer Epidemiol Biomarkers Prev. 2000; 9(6):631-633.
12. Gleeson CM, Sloan JM, McManus DT, Maxwell P, Arthur K, McGuigan JA, Ritchie AJ and Russell SE. Comparison of p53 and DNA content abnormalities in adenocarcinoma of the oesophagus and gastric cardia. Br J Cancer. 1998; 77(2):277-286.
13. Tolbert D, Fenoglio-Preiser C, Noffsinger A, De Voe G, MacDonald J, Benedetti J and Stemmermann GN. The relation of p53 gene mutations to gastric cancer subsite and phenotype. Cancer Causes Control. 1999; 10(3):227-231.
14. Anagnostopoulos GK, Stefanou D, Arkoumani E, Karagiannis J, Paraskeva K, Chalkley L, Habilomati E, Tsianos E and Agnantis NJ. Immunohistochemical expression of cell-cycle proteins in gastric precancerous lesions. J Gastroenterol Hepatol. 2008; 23(4):626-631.
15. Fearon ER and Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990; 61(5):759-767.
16. Wade M, Li YC and Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013; 13(2):83-96.
17. Ye Y, Li X, Yang J, Miao S, Wang S, Chen Y, Xia X, Wu X, Zhang J, Zhou Y, He S, Tan Y, Qiang F, Li G, Roe OD and Zhou J. MDM2 is a useful prognostic biomarker for resectable gastric cancer. Cancer Sci. 2013; 104(5):590-598.
18. Gonzalez CA, Sanz-Anquela JM, Gisbert JP and Correa P. Utility of subtyping intestinal metaplasia as marker of gastric cancer risk. A review of the evidence. Int J Cancer. 2013; 133(5):1023-1032.
19. Gunther T, Schneider-Stock R, Hackel C, Kasper HU, Pross M, Hackelsberger A, Lippert H and Roessner A. Mdm2 gene amplification in gastric cancer correlation with expression of Mdm2 protein and p53 alterations. Mod Pathol. 2000; 13(6):621-626.
20. Filipe MI, Munoz N, Matko I, Kato I, Pompe-Kirn V, Jutersek A, Teuchmann S, Benz M and Prijon T. Intestinal metaplasia types and the risk of gastric cancer: a cohort study in Slovenia. Int J Cancer. 1994; 57(3):324-329.
21. Lukashchuk N and Vousden KH. Ubiquitination and degradation of mutant p53. Mol Cell Biol. 2007; 27(23):8284-8295.
22. Wu MS, Shun CT, Lee WC, Chen CJ, Wang HP, Lee WJ, Sheu JC and Lin JT. Overexpression of p53 in different subtypes of intestinal metaplasia and gastric cancer. Br J Cancer. 1998; 78(7):971-973.
23. Shiao YH, Rugge M, Correa P, Lehmann HP and Scheer WD. p53 alteration in gastric precancerous lesions. The American journal of pathology. 1994; 144(3):511-517.
24. Gomyo Y, Osaki M, Kaibara N and Ito H. Numerical aberration and point mutation of p53 gene in human gastric intestinal metaplasia and well-differentiated adenocarcinoma: analysis by fluorescence in situ hybridization (FISH) and PCR-SSCP. Int J Cancer. 1996; 66(5):594-599.
25. Brito MJ, Williams GT, Thompson H and Filipe MI. Expression of p53 in early (T1) gastric carcinoma and precancerous adjacent mucosa. Gut. 1994; 35(12):1697-1700.
26. Cesar AC, Borim AA, Caetano A, Cury PM and Silva AE. Aneuploidies, deletion, and overexpression of TP53 gene in intestinal metaplasia of patients without gastric cancer. Cancer genetics and cytogenetics. 2004; 153(2):127-132.
27. Silva TC, Leal MF, Calcagno DQ, de Souza CR, Khayat AS, dos Santos NP, Montenegro RC, Rabenhorst SH, Nascimento MQ, Assumpcao PP, de Arruda Cardoso Smith M and Burbano RR. hTERT, MYC and TP53 deregulation in gastric preneoplastic lesions. BMC gastroenterology. 2012; 12:85.
28. Sud R, Wells D, Talbot IC and Delhanty JD. Genetic alterations in gastric cancers from British patients. Cancer genetics and cytogenetics. 2001; 126(2):111-119.
29. Haupt Y, Maya R, Kazaz A and Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997; 387(6630):296-299.
30. Stad R, Ramos YF, Little N, Grivell S, Attema J, van Der Eb AJ and Jochemsen AG. Hdmx stabilizes Mdm2 and p53. J Biol Chem. 2000; 275(36):28039-28044.
31. Martins CP, Brown-Swigart L and Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006; 127(7):1323-1334.
32. Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R and Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007; 445(7128):661-665.
33. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C and Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007; 445(7128):656-660.
34. Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, Teunisse A, Lam S, Ramos Y, Mohan A, Johnson D, Wilson M, Rodriguez-Galindo C, Quarto M, Francoz S, Mendrysa SM, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006; 444(7115):61-66.
35. Gembarska A, Luciani F, Fedele C, Russell EA, Dewaele M, Villar S, Zwolinska A, Haupt S, de Lange J, Yip D, Goydos J, Haigh JJ, Haupt Y, Larue L, Jochemsen A, Shi H, et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat Med. 2012.
36. Mitchell G, Ballinger ML, Wong S, Hewitt C, James P, Young MA, Cipponi A, Pang T, Goode DL, Dobrovic A and Thomas DM. High frequency of germline TP53 mutations in a prospective adult-onset sarcoma cohort. PLoS One. 2013; 8(7):e69026.
37. Acosta D, Adelman J, Affolder T, Akimoto T, Albrow MG, Ambrose D, Amerio S, Amidei D, Anastassov A, Anikeev K, Annovi A, Antos J, Aoki M, Apollinari G, Arisawa T, Arguin JF, et al. Measurement of the W(+)W(-) production cross section in pp collisions at square root[s]=1.96 TeV using dilepton events. Phys Rev Lett. 2005; 94(21):211801.
38. Ahmed AA, Etemadmoghadam D, Temple J, Lynch AG, Riad M, Sharma R, Stewart C, Fereday S, Caldas C, Defazio A, Bowtell D and Brenton JD. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol. 2010; 221(1):49-56.